

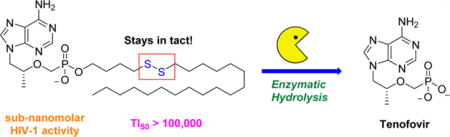
Abstract
The pharmacokinetic properties of tenofovir (TFV) and other charged nucleoside analogues are dramatically improved upon conjugation to a lipid prodrug. We previously prepared reduction-sensitive lipid conjugates of TFV that demonstrate superior antiviral activity compared to other lipid conjugates including the clinically approved formulation, tenofovir disoproxil fumarate (TDF). In continuation of that work, we have synthesized next-generation conjugates with reduced cytotoxicity that retain potent antiviral activity against HIV-1 and HBV with a therapeutic index >100000 for our most potent conjugate. We also show that disulfide reduction is not responsible for prodrug cleavage unless 3-exo-tet intramolecular cyclization can occur, suggesting that enzymatic hydrolysis is predominantly responsible for activity of our prodrugs in vitro.
Graphical abstract
INTRODUCTION
Many drug candidates and natural products are flanked with structural features that may limit their therapeutic potential in vivo.1–3 These tend to include carboxylic acids,4 amines,5 dianionic phosphates,6 and polyhydroxylated aromatic rings,7,8 which often require temporary protection with a prodrug to improve the absorption, distribution, metabolism, and excretion (ADME) properties of a pharmacologically active compound within the body.9,10
Tenofovir (TFV) is an acyclic antiviral nucleoside analogue that has achieved renowned clinical success for the treatment of hepatitis B virus (HBV)11,12 and human immunodeficiency virus (HIV)13,14 when administered as its prodrug, tenofovir disoproxil fumarate (TDF), which contains two isopropyloxymethyl carbonate prodrugs esterified to the phosphonate moiety of TFV. These masking groups are cleaved both extracellularly and intracellularly by nonspecific carboxyesterases, releasing TFV, which is subsequently phosphorylated to its active diphosphate to arrest viral replication. In the absence of prodrug conjugation, the considerable dianionic character of TFV at physiological pH reduces its oral bioavailability to <10% in various animal models.14 Although TDF largely overcomes this limitation, continuous exposure to TDF has been linked to a series of conditions including lactic acidosis, Fanconi syndrome, acute renal failure, and bone loss, due in part to its cleavage in the bloodstream.15,16
Systemic exposure to TFV may be reduced by altering the identity of the prodrug. For instance, tenofovir alafenamide (TAF) features a phosphoramidate prodrug that is cleaved by cathepsin A to selectively release TFV within the cytosol and demonstrates little to no nephrotoxicity.17,18 CMX-157 is a lipid prodrug formulation of TFV that is also cleaved by intracellular enzymes to minimize plasma exposure and reduce nephrotoxicity. CMX-157 is endowed with interesting pharmacokinetic properties and retains significant antiviral activity for up to a week after a single administration.19,20 The potential for reduced dosing frequency is highly attractive and inspired us to develop novel reduction sensitive lipid conjugates of TFV that rival CMX-157.21 We previously reported that our first-generation conjugates shown in Figure 1 release TFV in vitro to potently inhibit HIV-1 and HBV replication in PBMCs and hepatocytes, respectively. These compounds demonstrate subnanomolar HIV-1 activity, have a wider therapeutic index (TI) than TDF and CMX-157 at the EC50, and are stable in human plasma for more than 24 h. Despite these therapeutic gains, we speculated that the β-mercaptoethyl spacer is a precursor for thiirane, which has been implicated in the decomposition of S-acyl-2-thioethyl (SATE) and dithioethanol (DTE) prodrugs. The mutagenic potential of thiirane has largely precluded the clinical use of SATE and DTE-bound nucleosides, which prompted us to prepare next-generation reduction-sensitive TFV conjugates that exhibit potent anti-HIV-1 and anti-HBV activity with reduced cytotoxicity. Herein, we disclose the details of these efforts and also probe the mechanism of prodrug release.
Figure 1.
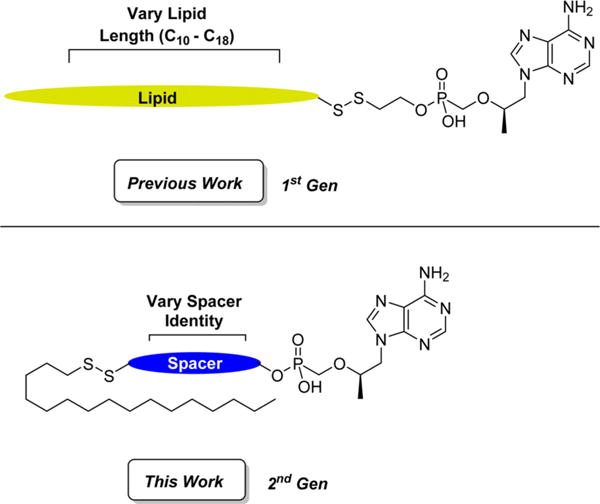
Comparison of structural features previously examined with those investigated in the current study.
RESULTS AND DISCUSSION
Chemistry
Conjugates 1–5b, 6, and 7 were readily prepared using our previous protocol outlined in Scheme 1. Briefly, hexadecanethiol or dodecanethiol was oxidized with select thiols in the presence of iodine to furnish disulfides 1–5a as low-melting-point solids that were purified by normal phase column chromatography. Compounds 1–5a were then coupled to TFV with oxalyl chloride and DMF to afford the corresponding conjugates 1–5b after formimidine deprotection and purification on silica gel using a DCM:MeOH:NH4OH gradient. Both 6 and 7 were purified on a C18 reverse phase column and were isolated as free acids following lyophilization. The structures of 1–5b, 6, and 7 are presented in Table 1.
Scheme 1.
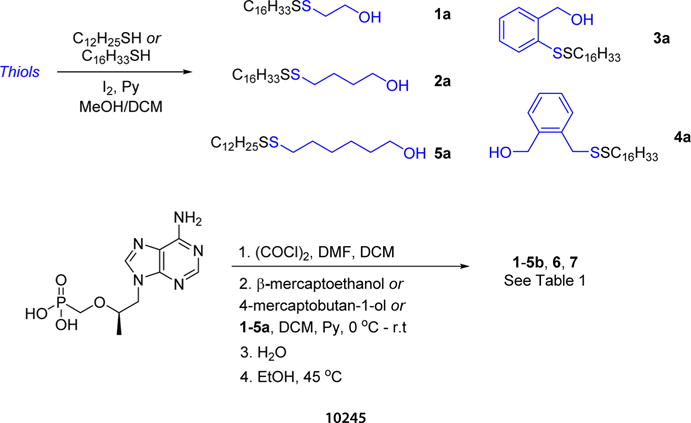
Synthesis of Conjugates 1–5b, 6, and 7
Table 1.
HIV-1 and HBV Activity of Conjugates 1–5b, 6, and 7 Compared to TFV and TDFa
| HIV-l
|
HBV
|
||||||
|---|---|---|---|---|---|---|---|
| Ident. | Structure | EC50b (PBMCs) | CC50b(PBMCs) | TI (CC50/EC50) | EC50b (HepG2) | CC50b (HepG2) | TI (CC50/EC50) |
|
|
|
||||||
| TFV |
|
0.320 | > 100 | >300 | – | – | – |
| TDF |
|
0.0045 | 44.0 | 9500 | 0.34 | 64.5 | 190 |
| 1b |
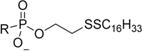
|
0.00065 | 14.3 | 22000 | 0.020 | >25 | > 1200 |
| 2b |
|
< 0.0005 | >50 | >100000 | 0.248 | >50 | >200 |
| 3b |
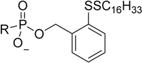
|
0.0229 | 17.2 | 751 | 4.48 | 17.5 | 3.9 |
| 4b |
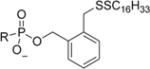
|
< 0.0005 | 15.9 | >31800 | 0.152 | 32.5 | 214 |
| 5b |
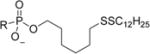
|
0.007 | >50 | >7000 | 1.05 | >50 | >47 |
| 6 |
|
18.6 | > 100 | >5.38 | 41.9 | >100 | >2 |
| 7 |
|
5.13 | > 100 | > 18 | > 100 | > 100 | > 1 |
All data represent an average of triplicate experiments. R = tenofovir.
EC50, effective concentration (in μM) required to inhibit HIV-1 or HBV by 50%.
CC50, effective concentration (in μM) required to reduce the viability of uninfected cells by 50%.
Kinetic Studies and Antiviral Activity
Conjugates 1b, 2b, and 5b feature the disulfide moiety at various positions along the lipid tail with increasing distance from the phosphonic acid whereas 3b and 4b are endowed with a benzylic ring to restrict the conformational flexibility of the spacer. The influence of these modifications on anti-HIV and anti-HBV activity is shown in Table 1 and is discussed herein. The first question we sought to address from our previous work was the mechanism by which first-generation conjugate 1b liberates TFV. Presumably 1b undergoes glutathione (GSH)-mediated reduction within the cytosol to reveal transient intermediate 8 shown in Scheme 2. This intermediate is thought to collapse upon itself to generate thiirane with concomitant release of TFV. To validate this mechanism, we initially treated 1b with GSH in PBS at 37.4 ± 0.2 °C and monitored decomposition by LC-MS. Unfortunately, the poor aqueous solubility of 1b precluded accurate spectral measurements and necessitated an alternative model system. We therefore chose to access thiolate 8 via deprotonation of 6 rather than subjecting 1b to GSH with the underlying assumption that intramolecular cyclization governs TFV release. Thiol 6 was incubated in various aqueous solutions buffered at pH 2.0, 9.19, and 11.56 spiked with 0.1 M DTT at 37.4 °C. 6 demonstrated high stability in glycine/HCl buffer (pH = 2.0) with no observed decomposition. Moderate stability was noted in PBS (pH = 7.43) with the appearance of a new UV signal that faintly presented itself at a significantly polar retention time (tr = 0.66 min). This species was identified as TFV when compared to an authentic sample of the parent nucleoside. When 6 was incubated in aqueous carbonate/bicarbonate buffer (pH = 9.19), significant decomposition occurred and complete consumption of starting material was noted within 8 min at pH 10.16. Figure 2 graphically reveals a pseudo-first-order rate dependence wrt 6 and clearly illustrates the link between pH and rate of reaction. Using the method of initial rates, the differential rate law was found to be first order wrt [OH−], resulting in the overall second-order rate expression for the decomposition of 6.
| (1) |
Scheme 2.
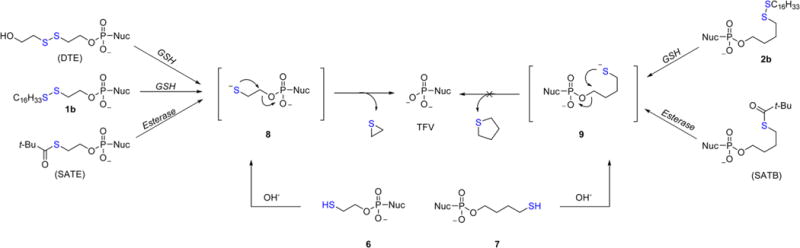
Posited Decomposition Mechanism of Relevant Prodrug Strategies in Vitro
Figure 2.
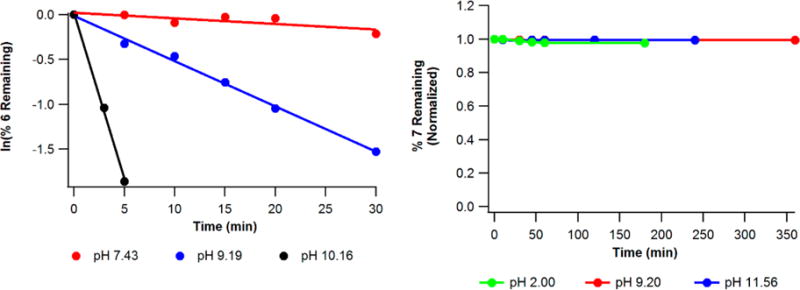
Decomposition profile of 6 and 7 in various buffer solutions containing 0.1 M DTT at 37.4 °C.
The [OH−] dependence in eq 1 suggests that thiolate 8 performs the 3-exo-tet cyclization to release TFV rather than thiol 6, which is consistent with the mechanism presented in Scheme 2. These results are also in agreement with previous work22,23 and confirm that thiolates of SATE, DTE, and 1b have the capacity to cyclize and release the parent nucleoside. When assayed against HIV-infected PBMCs, 6 is 58-fold less active than TFV, which indicates that the mercaptoethyl spacer is not cleaved enzymatically and reinforces the notion that a single proton governs TFV release.
In light of the fact that 1b has the potential to generate thiirane, we prepared conjugate 2b whose predicated mechanism of cleavage involves a 5-exo-tet cyclization to yield nonelectrophilic tetrahydrothiophene and TFV (Scheme 2). To our delight, 2b exhibited subnanomolar anti-HIV activity comparable to 1b with reduced toxicity and a TI50 > 100000. Unfortunately, the poor aqueous solubility of 2b prevented accurate CC50 measurements beyond 50 μM. To determine if 2b passes through intermediate 9 to release TFV, thiol 7 was synthesized and subjected to various aqueous buffers in a similar manner done for 6. Surprisingly, 7 was stable at all tested pH values with no detectable decomposition after several hours at 37 °C (Figure 2). Clearly 9 does not cyclize to liberate TFV as shown in Scheme 2. Furthermore, 7 demonstrates dismal anti-HIV (EC50 = 5.13 μM) and anti-HBV (EC50 > 100 μM) activity, which suggests poor enzymatic hydrolysis of the bare thiobutyl spacer. A search of the literature revealed that Gosselin and collaborators reported similar results for their S-pivaloyl-4-thiobutyl (SATB) prodrug whose structure is shown in Scheme 2.24 In that study, the authors conjugated SATB to azidothymidine (AZT) and dideoxyadenosine (ddA) and noted a significant reduction in HIV-1 activity relative to the parent nucleoside. The compromised activity of SATB conjugates can be traced to either poor enzymatic cleavage of the thiopivaloyl ester or the robust nature of the intermediate thiol/ate (7/9). The authors comment that their bis(SATB) nucleoside conjugates are suitable substrates for esterases and speculate that the source of inactivity is due to the persistence of the resultant 4-thiobutyl linker in vitro. Our results confirm this to be the case. However, in contrast to SATB prodrug conjugates, 2b demonstrates potent antiviral activity that appears to rely on the structural integrity of the entire lipid. This leads us to believe that 2b itself is a substrate for cellular enzymes, specifically phospholipase C and/or sphingomyelinase, which have been implicated in the cleavage of CMX-157 and CMX-001.19,25 The kinetic data and antiviral activity collected for 7 supports this hypothesis and suggest that the P–O bond is severed before disulfide reduction. Additional evidence for enzymatic hydrolysis is provided by the observed anti-HIV activity for 5b (7.0 nM), which features the disulfide moiety eight atoms away from the phosphonate headgroup to abolish the potential for kinetic and thermodynamic cyclization following reduction. Translocation of the disulfide away from the phosphonate reduces the antiviral activity of 5b when compared to 1b (note that 5b and 1b are isomers). This effect may be countered when translocation is accompanied by a concomitant increase in lipid chain length, such as is the case for 2b, which bears a C16 lipid and features the S–S moiety five atoms away from the phosphonate. Even though 5b is less active than congeners 1b and 2b, all three compounds are more potent than CMX-157 against HIV-1 (EC50 = 20 nM),26 thereby demonstrating an advantage of our disulfide prodrugs over hexadecyloxypropyl lipids. Although the mechanistic rationale for this advantage is unclear at this time, it is possible that the “kinked” S–S bond (i.e., its 90° dihedral angle) increases membrane fluidity to accelerate translocation of the conjugate from the outer leaflet of the plasma membrane to the inner leaflet where it is then enzymatically cleaved to release TFV within the cytosol.
In light of these findings, the following question arises: how significant is enzymatic hydrolysis of 1b over the reductive mechanism shown in Scheme 2? This question remains largely unanswered. It is likely that 1–5b produce micelles in solution that hinder GSH and other hydrophilic reductants from approaching the S–S bond. This would imply that enzymatic hydrolysis is exclusive mechanism of release, however, future experiments will characterize the aggregation properties of 1–5b and tease out these mechanistic details.
The final goal of this study was to probe the rigidity of the spacer which resulted in the preparation of compounds 3b and 4b. Conjugate 3b demonstrated unremarkable antiviral activity against HIV and HBV, whereas 4b exhibited potency comparable to 2b at the expense of increased cytotoxicity. 3b was tailored to cleave via an o-thioquinone methide elimination following disulfide reduction, whereas 4b was designed to accelerate the rate of 5-exo-tet cyclization post reduction. However, in the wake of our previous findings, these cleavage mechanisms may be insignificant. Nonetheless, it is clear that other spacers such as (2-(mercaptomethyl)phenyl)methanol can be used in place of undecorated aliphatic thioalcohols to furnish potent disulfide lipid conjugates of TFV.
CONCLUSIONS
We have successfully synthesized a second-generation disulfide lipid conjugate of TFV (2b) that demonstrates potent antiviral activity against HIV-1 and HBV with reduced cytotoxicity. The data suggest that 2b does not cleave by chemical means, and the structural integrity of the disulfide linkage is required to release TFV in vitro. When taken together, we conclude that enzymatic hydrolysis is central to the antiviral activity of 2b and speculate that phospholipase C and/or sphingomyelinase may be responsible for prodrug cleavage, which have also been implicated in the decomposition of other lipid nucleoside conjugates.19,20,27 We further demonstrate that 1b has the potential to undergo intramolecular prodrug cleavage following disulfide reduction, however, the significance of this mechanism remains to be determined in vitro. Future studies will probe the aggregation of 1–5b and elucidate the enzyme(s) responsible for prodrug cleavage. Additional efforts will focus on the design of novel disulfide prodrugs bound to antiviral cargo which may exhibit compounded activity when conjugated to TFV or other phosphonate nucleoside analogues.
EXPERIMENTAL SECTION
Chemical Synthesis
All reagents were obtained from commercial suppliers and used without further purification. Reaction progress was monitored by either thin layer chromatography (TLC) using precoated aluminum-backed silica gel plates (60 F254 Merck, article 5554) or liquid chromatography–mass spectrometry (LC-MS) on an Agilent Technologies 6100 quadrupole instrument equipped with UV detection at 254 and 210 nm and a Varian C8 analytical column. Hanes reagent and UV detection at 254 nm were the preferred visualization agents for TLC. LC-MS analysis was performed using a stepwise H2O/MeOH gradient with the % MeOH increasing from 75% to 95% over the course of 3 min unless otherwise specified. Flash column chromatography was conducted using CombiFlash Rf 200 (Telendyne-Isco) automated flash chromatography system with hand-packed RediSep columns. Evaporation of solvents was carried out on a rotary evaporator under reduced pressure and under ultrahigh vacuum (UHV) where appropriate. 1H NMR and 13C NMR spectra were recorded at ambient temperature on a Varian 400 spectrometer. 31P spectra were recorded at ambient temperature on either a Mercury 300 or Varian 400 spectrometer. Unless otherwise specified, all NMR spectra were obtained in deuterated chloroform (CDCl3) and referenced to the residual solvent peak. Chemical shifts are given in δ values and coupling constants are reported in hertz (Hz). Coupling constants are not reported in the 13C spectrum of diastereomeric mixtures. Melting points were determined on a MelTemp melting apparatus and are uncorrected. High resolution mass-spectra (HRMS) were acquired on a VG 70-S Nier Johnson or JEOL mass spectrometer. Elemental analyses were performed by Atlantic Microlabs (Norcross, GA) for C, H, N analysis and are in agreement with the proposed structures with purity ≥95%.
General Procedure A
To a stirring solution of dry tenofovir (0.05 g, 0.174 mmol) in anhydrous DCM (6 mL) and N,N-dimethylformamide (0.016 mL, 0.209 mmol) was gradually added excess oxalyl chloride (0.075 mL, 0.870 mmol). The mixture stirred exposed to atmosphere for 1 h at room temperature until a colorless, transparent solution was observed and no starting material coated the walls of the vessel. The solvent and excess oxalyl chloride was evaporated under reduced pressure to produce a pale-yellow foam, which was redissolved in anhydrous DCM (5 mL) and placed under argon. The vessel was equipped with a magnetic stir bar and chilled to 0 °C. Then, a solution of 2-(hexadecyldisulfanyl)ethanol (0.070 g, 0.209 mmol) and pyridine (0.084 mL, 1.045 mmol) in anhydrous DCM was slowly added dropwise. After stirring for 10 min at this temperature, the mixture was naturally warmed to room temperature and stirred for an hour.
General Procedure B
To a stirring solution of dry tenofovir (0.1 g, 0.348 mmol) in anhydrous DCM (6 mL) and N,N-dimethylformamide (0.032 mL, 0.418 mmol) was gradually added excess oxalyl chloride (0.149 mL, 1.741 mmol). The mixture stirred at rt exposed to air for 15 min or until complete dissolution of starting material was observed. The solvent and excess oxalyl chloride was evaporated under reduced pressure, and the resulting residue was placed under argon and redissolved in anhydrous DCM (5 mL) to afford a clear colorless solution. The mixture was then chilled to 0 °C.
2-(Hexadecyldisulfanyl)ethanol (1a)
To a stirring solution of hexadecane-1-thiol (8.74 mL, 28.4 mmol) and 2-mercaptoethanol (2 mL, 28.4 mmol) in MeOH/DCM (50:50, 200 mL) was added pyridine (4.94 mL, 56.8 mmol) followed by the gradual added diiodine (7.21 g, 28.4 mmol) until the color of the solution remained brown. The solution was stirred for 2 h at room temperature and then the resulting suspension was filtered and the supernatant collected. The solvents were evaporated under reduced pressure, and the resulting solid was washed with water and extracted into DCM. The organic layer was concentrated and the residue purified on a silica column using hexanes/EtOAc (0–8%) gradient to afford the title compound 2-(hexadecyldisulfanyl)-ethanol (3.98 g, 11.89 mmol, 41.9% yield) as a fluffy white powder. 1H NMR (400 MHz, CDCl3) δ 3.90 (q, J = 5.6 Hz, 2H), 2.95–2.80 (m, 2H), 2.77–2.63 (m, 2H), 2.01 (t, J = 6.0 Hz, 1H), 1.69 (dt, J = 14.9, 7.3 Hz, 2H), 1.46–1.17 (m, 26H), 0.89 (t, J = 6.9 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 60.29, 41.16, 39.05, 31.92, 29.69 (4), 29.66, 29.63, 29.58, 29.49, 29.36, 29.21, 29.13, 28.51, 22.69, 14.13. HRMS (ESI) m/z calculated for C18H38OS2Na [M + Na]+, 357.22563; found, 357.22533. Melting point: 50–51 °C.
2-(Hexadecyldisulfanyl)ethyl Hydrogen ((((R)-1-(6-Amino-9H-purin-9-yl)propan-2-yl)oxy)methyl)phosphonate (1b)
Following general procedure A, a solution of 2-(hexadecyldisulfanyl)ethanol (0.070 g, 0.209 mmol) and pyridine (0.084 mL, 1.045 mmol) in anhydrous DCM was slowly added dropwise to the mixture. After stirring for 10 min at this temperature, the solution was naturally warmed to room temperature and stirred for an hour. The reaction was then quenched with excess water and stirred for 30 min at room temperature followed by HCl and methanol. Complete hydrolysis of formimidine was confirmed by LC-MS (isocratic 95% MeOH, 5% H2O, 7 min) after several hours. The mixture was washed with brine and the crude extracted into DCM. The organic layer was collected, concentrated, and purified on a silica column using a DCM/DCM:MeOH:NH4OH (90:10:0.1) gradient (0–65%) to afford the title compound 2-(hexadecyldisulfanyl)ethyl hydrogen ((((R)-1-(6-amino-9H-purin-9-yl)propan-2-yl)oxy)methyl)phosphonate (29.8 mg, 0.049 mmol, 28.3% yield) as a white solid. 1H NMR (400 MHz, CD3OD) δ 8.32 (s, 1H), 8.20 (s, 1H), 4.43–4.33 (m, 1H), 4.23 (dd, J =J = 14.4, 6.7 Hz, 1H), 4.00 (qd, J = 7.0, 1.7 Hz, 2H), 3.90 (pd, J = 6.3, 3.1 Hz, 1H), 3.73 (dd, J = 12.8, 9.4 Hz, 1H), 3.49 (dd, J = 12.7, 10.1 Hz, 1H), 2.86–2.73 (m, 2H), 2.64 (dd, J = 7.7, 6.8 Hz, 2H), 1.68–1.55 (m, 2H), 1.39–1.25 (m, 26H), 1.17 (d, J = 6.2 Hz, 3H), 0.94–0.85 (m, 3H). 13C NMR (101 MHz, CD3OD) δ 155.36, 151.48, 149.41, 142.91, 139.91, 118.10, 75.52 (d, J = 13.1 Hz), 64.05 (d, J = 160.2 Hz), 62.96 (d, J = 5.7 Hz), 38.87 (d, J = 6.2 Hz), 38.46, 31.65, 29.38, 29.35, 29.34, 29.28, 29.23, 29.06, 28.93, 28.71, 28.06, 22.32, 15.41, 13.04. 31P NMR (162 MHz, CD3OD) δ 16.87. HRMS (ESI) m/z calculated for C27H51O4N5PS2 [M + H]+, 604.31146; found, 604.31149. Anal. Calculated for C27H53O5N6PS2 (as an ammonium salt monohydrate): C, 50.76; H, 8.68; N, 13.15. Found: C, 51.45; H, 8.42; N, 12.79. Melting point: 138–142 °C
4-(Hexadecyldisulfanyl)butan-1-ol (2a)
To a stirring solution of hexadecane-1-thiol (5.94 mL, 19.30 mmol) and 4-mercaptobutan-1-ol (1.990 mL, 19.30 mmol) in MeOH/DCM (1:2, 150 mL) was added pyridine (3.36 mL, 38.6 mmol) followed by the gradual addition of diiodine (4.90 g, 19.30 mmol) at room temperature. The solution stirred for 3 h at room temperature, and reaction progress was monitored by TLC (hexanes/EtOAc 4:1, PMA stain). Then the solvents were evaporated under reduced pressure to afford a white residue, which was redissolved in DCM and partitioned with water. The organic layer was collected, dried over anhydrous sodium sulfate, filtered, and purified on a silica column using a hexanes/EtOAc gradient (0–16% EtOAc) to afford the title compound 4-(hexadecyldisulfanyl)butan-1-ol (2.7 g, 7.44 mmol, 38.6% yield) as a pearlescent white powder. 1H NMR (400 MHz, CDCl3) δ 3.68 (t, J = 6.3 Hz, 2H), 2.78–2.65 (m, 4H), 1.85–1.73 (m, 2H), 1.73–1.62 (m, 4H), 1.43–1.22 (m, 26H), 0.88 (t, J = 6.9 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 62.42, 39.10, 38.65, 31.90, 31.42, 29.67 (2), 29.66 (2), 29.64 (2), 29.58, 29.50, 29.35, 29.23, 29.20, 28.51, 25.42, 22.68, 14.12. HRMS (ESI) m/z calculated for C20H42OS2 [M + Na]+, 385.25693; found, 385.25621. Melting point: 51–52 °C.
(R)-4-(Hexadecyldisulfanyl)butyl (((1-(6-Amino-9H-purin-9-yl)-propan-2-yl)oxy)methyl)phosphonate (2b)
Following general procedure B, a mixture of 4-(hexadecyldisulfanyl)butan-1-ol (0.152 g, 0.418 mmol) and pyridine (0.168 mL, 2.089 mmol) in anhydrous DCM was slowly added dropwise to the solution. The mixture stirred at 0 °C for 15 min and then naturally warmed to room temperature and stirred for 1 h. Water (0.094 mL, 5.22 mmol) was added and the mixture continued stirring for an additional 30 min. The solvent was evaporated under reduced pressure, and the resulting residue was dried under UHV and redissolved in EtOH (5 mL) and stirred at 40 °C overnight. Reaction progress could be monitored by TLC (80:20:1 DCM/MeOH/NH4OH). The solvent was evaporated under reduced pressure, and the crude was dissolved in minimal DCM and purified on a silica column using a DCM/DCM:MeOH:NH4OH (80:20:1) gradient (0–45%) to afford the title compound (R)-4-(hexadecyldisulfanyl)butyl (((1-(6-amino-9H-purin-9-yl)propan-2-yl)oxy)methyl)phosphonate (140 mg, 0.222 mmol, 63.7% yield) as a waxy solid. 1H and 13C spectra referenced to CD3OD (3.31 δ and 49.15 δ, respectively). 1H NMR (400 MHz, CDCl3/CD3OD) δ 8.30 (s, 1H), 8.20 (s, 1H), 4.37 (dd, J = 14.4, 3.1 Hz, 1H), 4.22 (dd, J = 14.4, 6.8 Hz, 1H), 3.94–3.82 (m, 1H), 3.82–3.65 (m, 3H), 3.45 (dd, J = 12.7, 10.2 Hz, 1H), 2.68–2.58 (m, 4H), 1.74–1.54 (m, 6H), 1.42–1.20 (m, 25H), 1.18 (d, J = 6.2 Hz, 3H), 0.88 (t, J = 6.9 Hz, 3H). 13C NMR (101 MHz, CDCl3/CD3OD) δ 156.92, 153.23, 150.78, 144.12, 119.53, 76.96 (d, J = 13.2 Hz), 65.40 (d, J = 160.3 Hz), 65.37 (d, J = 5.9 Hz), 66.20, 65.40, 65.34, 64.61, 39.71, 39.29, 33.08, 30.81, 30.80, 30.79, 30.78, 30.74, 30.69 (d, J = 5.9 Hz), 30.49, 30.37, 30.22, 29.53, 26.57, 23.76, 16.95, 14.64. 31P NMR (162 MHz, CDCl3/CD3OD) δ 16.54. HRMS (ESI) m/z calculated for C29H55N5O4PS2 [M + H]+, 632.34276; found, 632.34515. Anal. Calculated for C29H58N6O5PS2 (NH4+ monohydrate): C, 52.31; H, 8.78; N, 12.62. Found: C, 52.49; H, 8.78; N, 12.64. Melting point: 140–150 °C. Solid is amorphous.
(2-(Hexadecyldisulfanyl)phenyl)methanol (3a)
To a stirring solution of hexadecane-1-thiol (5.43 mL, 17.65 mmol) and (2-mercaptophenyl)methanol (2.474 g, 17.65 mmol) in MeOH/DCM (1:2, 150 mL) was added pyridine (3.07 mL, 35.3 mmol), followed by the gradual addition of diiodine (4.48 g, 17.65 mmol) at room temperature. The solution stirred for 4 h at room temperature, and reaction progress was monitored by TLC (hexanes/EtOAc 4:1, UV). Then, the solvents were evaporated under reduced pressure to afford a white residue which was redissolved in DCM and partitioned with water. This afforded a third layer between the organic and aqueous interface and was identified as the homodimer of (2-mercaptophenyl)methanol by LC-MS. The organic layer was collected and dried over sodium sulfate. The mixture was filtered and the solvents evaporated under reduced pressure. The pale-yellow residue was redissolved in minimal DCM. A substantial quantity of the solid resisted dissolution and was subsequently filtered over a fine glass frit. The supernatant was collected, concentrated, and purified on a silica column via flash chromatography using a hexanes/EtOAc gradient (0–6% EtOAc) to afford the title compound (2-(hexadecyldisulfanyl)phenyl)methanol (3.45 g, 8.70 mmol, 49.3% yield) as an off-white solid. 1H NMR (400 MHz, CDCl3) δ 7.78–7.72 (m, 1H), 7.46–7.40 (m, 1H), 7.33–7.25 (m, 2H), 4.84 (s, 2H), 2.75–2.69 (m, 2H), 1.65 (dt, J = 14.9, 7.3 Hz, 2H), 1.39–1.17 (m, 26H), 0.88 (t, J = 6.9 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 140.23, 135.65, 129.78, 128.51, 128.24, 127.67, 63.26, 38.73, 31.92, 29.69 (3), 29.67 (2), 29.63, 29.57, 29.46, 29.37, 29.16, 28.68, 28.44, 22.69, 14.14. HRMS (ESI) m/z calculated for C23H40S2O [M + Na]+, 419.24128; found, 419.24099. Melting point: 40–42 °C.
(R)-2-(Hexadecyldisulfanyl)benzyl (((1-(6-Amino-9H-purin-9-yl)-propan-2-yl)oxy)methyl)phosphonate (3b)
Following general procedure B, a mixture of (2-(hexadecyldisulfanyl)phenyl)methanol (0.166 g, 0.418 mmol) (KEG-4-164) and pyridine (0.168 mL, 2.089 mmol) in anhydrous DCM was slowly added dropwise to the solution. The mixture stirred at this temperature for 15 min and then naturally warmed to room temperature and stirred for 3 h. Then water (0.094 mL, 5.22 mmol) was added and the mixture continued stirring for an additional 30 min. The solvent was evaporated under reduced pressure, and the resulting residue was dried under UHV. Then, the residue was redissolved in EtOH (5 mL) and stirred at 40 °C overnight. The product precipitated from the reaction mixture and was filtered with additional EtOH and dried under UHV. The solid was dissolved in chloroform and further purified on a silica column using a DCM/DCM:MeOH:NH4OH (80:20:1) gradient (0–66%) to afford the title compound (R)-2-(hexadecyldisulfanyl)benzyl (((1-(6-amino-9H-purin-9-yl)propan-2-yl)oxy)methyl)phosphonate (94.5 mg, 0.142 mmol, 40.8% yield) as an off-white solid. 1H NMR (400 MHz, CD3OD/CDCl3, referenced to CD3OD) δ 8.26 (s, 1H), 8.18 (s, 1H), 7.67 (dd, J = 7.7, 1.3 Hz, 1H), 7.46 (dd, J = 7.5, 1.3 Hz, 1H), 7.22 (dtd, J = 21.2, 7.4, 1.5 Hz, 2H), 5.13–5.03 (m, 2H), 4.32 (dd, J = 14.4, 3.1 Hz, 1H), 4.17 (dd, J = 14.4, 6.5 Hz, 1H), 3.88–3.81 (m, 1H), 3.77 (dd, J = 12.7, 9.4 Hz, 1H), 3.53 (dd, J = 12.7, 10.0 Hz, 1H), 2.65 (t, J = 7.2 Hz, 2H), 1.57 (dt, J = 14.8, 7.2 Hz, 2H), 1.34–1.14 (m, 26H), 1.11 (d, J = 6.3 Hz, 3H), 0.86 (t, J = 6.9 Hz, 3H). 13C NMR (101 MHz, CD3OD/CDCl3, referenced to CD3OD) δ 156.16, 152.29, 150.44, 143.92, 138.79 (d, J = 6.9 Hz), 135.94, 129.88, 128.86, 128.77, 128.13, 119.21, 76.79 (d, J = 13.2 Hz), 65.46 (d, J = 159.8 Hz), 65.23 (d, J = 5.0 Hz), 48.88, 39.57, 32.85, 30.60, 30.59, 30.59, 30.56, 30.54, 30.48, 30.39, 30.28, 30.08, 29.57, 29.23, 23.55, 16.77, 14.53. 31P NMR (121 MHz, CDCl3/CD3OD) δ 16.49. HRMS (ESI) m/z calculated for C32H51O4N5PS2 [M – H]−, 664.31146; found, 664.31306. Anal. Calculated for C32H57N6O5PS2 (NH4+ monohydrate): C,54.83; H, 8.20; N, 11.99. Found: C, 54.23; H, 8.16; N, 12.38. Melting point: 177–180 °C.
Methyl 2-((Acetylthio)methyl)benzoate
To a stirring solution of methyl 2-(bromomethyl)benzoate (7 g, 30.6 mmol) in THF (100 mL) and DMF (5 mL) at 0 °C was added potassium ethanethioate (3.84 g, 33.6 mmol), followed by catalytic tetrabutylammonium iodide (2.257 g, 6.11 mmol). The mixture stirred at this temperature for 15 min and then naturally warmed to room temperature and stirred for 12 h. Progress was monitored by TLC (hexanes/EtOAc, 4:1). Upon completion, the organic solvent was evaporated under reduced pressure and the resulting oil was partitioned between EtOAc and brine (3×). The organic layer was collected, dried over anhydrous sodium sulfate, and purified on a silica column using a hexanes/EtOAc gradient (0–6% EtOAc) to afford the title compound methyl 2-((acetylthio)methyl)benzoate (6.53 g, 29.1 mmol, 95% yield) as a foul-smelling yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.95 (d, J = 7.8 Hz, 1H), 7.51 (d, J = 7.7 Hz, 1H), 7.47–7.39 (m, 1H), 7.35–7.26 (m, 1H), 4.47 (s, 2H), 3.90 (s, 3H), 2.29 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 195.64, 167.30, 140.04, 132.54, 131.64, 131.00, 128.48, 127.43, 52.11, 32.24, 30.15. HRMS (ESI) m/z calculated for C11H13O3S [M + H]+, 225.05799; found, 225.05809.
(2-(Mercaptomethyl)phenyl)methanol
To a stirring solution of lithium aluminum hydride (82 mL, 163 mmol) in THF under inert atmosphere at 0 °C was added methyl 2-((acetylthio)methyl)benzoate (9.15 g, 40.8 mmol), dropwise. The solution stirred at this temperature for 15 min and then naturally warmed to room temperature. Progress was monitored by LC-MS (H2O/MeOH gradient, 75–95% MeOH, 3 min). The reaction reached completion after stirring for 1 h and was subsequently chilled to 0 °C. Then acetone (8.99 mL, 122 mmol) was added slowly dropwise, followed by the slow addition of 15% aqueous NaOH (37 mL) with vigorous stirring under inert atmosphere. The reaction mixture was then diluted with a saturated solution of sodium potassium tartrate and vigorously stirred for 2 h. After stirring, the pH was adjusted to 9 with solid ammonium chloride and the reaction mixture was allowed to settle. The supernatant was collected and concentrated under reduced pressure, and the resulting aqueous mixture was partitioned with DCM. The solids were also partitioned with DCM and brine. The organic layers were collected, dried over anhydrous sodium sulfate, and purified via flash chromatography on a silica column using a hexanes/EtOAc gradient (0–41% EtOAc) to afford the title compound (2-(mercaptomethyl)phenyl)methanol (3.76 g, 24.38 mmol, 59.8% yield) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.41–7.33 (m, 1H), 7.30–7.23 (m, 3H), 4.76 (s, 2H), 3.83 (d, J = 7.2 Hz, 2H), 1.87 (t, J = 7.2 Hz, 1H). 13C NMR (101 MHz, CDCl3) δ 139.37, 138.15, 129.37, 129.33, 128.51, 127.75, 63.12, 26.10. HRMS (ESI) m/z calculated for C8H10SOCl [M + Cl]−, 189.01464; found, 189.01465.
(2-((Hexadecyldisulfanyl)methyl)phenyl)methanol (4a)
To a stirring solution of hexadecane-1-thiol (2.99 mL, 9.73 mmol) and (2-(mercaptomethyl)phenyl)methanol (1.5 g, 9.73 mmol) in MeOH/DCM (1:3, 150 mL) was added pyridine (1.691 mL, 19.45 mmol), followed by the gradual addition of diiodine (2.468 g, 9.73 mmol) at room temperature. The solution stirred for 3 h at room temperature and reaction progress was monitored by TLC (DCM:MeOH:NH4OH, 95:5:0.1, PMA stain). The reaction mixture was diluted with methanol (25 mL), and the resulting precipitate was filtered and discarded. The supernatant was collected and the solvent evaporated under reduced pressure to afford a pale-orange residue that was redissolved in DCM and partitioned with water. The organic layer was collected, dried over anhydrous sodium sulfate, filtered, and purified on a silica column using a hexanes/EtOAc gradient (0–7% EtOAc) to afford the title compound (2-((hexadecyldisulfanyl)methyl)phenyl)methanol (1.79 g, 4.36 mmol, 44.8% yield) as a light-orange solid. 1H NMR (400 MHz, CDCl3) δ 7.44–7.37 (m, 1H), 7.34–7.21 (m, 3H), 4.79 (d, J = 5.6 Hz, 2H), 4.01 (s, 2H), 2.42–2.33 (m, 2H), 2.08 (t, J = 5.8 Hz, 1H), 1.53 (dt, J = 14.8, 7.3 Hz, 2H), 1.43–1.02 (m, 26H), 0.88 (t, J = 6.9 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 139.03, 135.22, 131.02, 129.11, 128.06, 127.97, 63.03, 40.45, 38.69, 31.92, 29.69 (2), 29.68 (2), 29.66, 29.65, 29.59, 29.48, 29.36, 29.16, 28.98, 28.46, 22.70, 14.14. HRMS (ESI) m/z calculated for C24H42OS2 [M + Na]+, 433.25693; found, 433.25619. Melting point: 38–40 °C.
(R)-2-((Hexadecyldisulfanyl)methyl)benzyl (((1-(6-Amino-9H-purin-9-yl)propan-2-yl)oxy)methyl)phosphonate (4b)
Following general procedure B, a mixture of (2-((hexadecyldisulfanyl)methyl)-phenyl)methanol (0.172 g, 0.418 mmol) and pyridine (0.168 mL, 2.089 mmol) in anhydrous DCM (2 mL) was slowly added dropwise to the reacting solution. The mixture stirred at this temperature for 15 min and then naturally warmed to room temperature and stirred for 3 h. Water (0.094 mL, 5.22 mmol) was added, and the mixture continued stirring for an additional 30 min. The solvent was evaporated under reduced pressure, and the resulting residue was dried under UHV. The residue was redissolved in 190 proof EtOH (5 mL) and stirred at 40 °C overnight. The mixture was diluted with water (5 mL), and the precipitate was filtered over a glass frit. The resulting solid was dried under UHV and purified via flash chromatography on a silica column using a DCM/DCM/MeOH/NH4OH (80:20:1) gradient (0–62%) to afford the title compound (R)-2-((hexadecyldisulfanyl)methyl)benzyl (((1-(6-amino-9H-purin-9-yl)propan-2-yl)oxy)methyl)phosphonate (114 mg, 0.168 mmol, 48.2% yield) as a white solid. Proton spectrum referenced to CD3OD (3.31 ppm). 1H NMR (400 MHz, CDCl3/CD3OD) δ 8.23 (s, 1H), 8.19–8.15 (m, 1H), 7.40–7.33 (m, 1H), 7.26–7.15 (m, 3H), 5.07 (d, J = 6.9 Hz, 2H), 4.29 (dd, J = 14.4, 3.0 Hz, 1H), 4.11 (dd, J = 14.4, 6.7 Hz, 1H), 3.92 (s, 2H), 3.74 (ddd, J = 17.0, 10.3, 6.2 Hz, 2H), 3.42 (dd, J = 12.7, 10.3 Hz, 1H), 2.31–2.22 (m, 2H), 1.44 (dd, J = 14.2, 7.1 Hz, 2H), 1.31–1.13 (m, 26H), 1.09 (d, J = 6.3 Hz, 3H), 0.84 (t, J = 6.9 Hz, 3H). Carbon referenced to CD3OD (49.00 ppm). 13C NMR (101 MHz, CD3OD/CDCl3) δ 155.54, 151.93, 149.95, 143.42, 136.93 (d, J = 6.5 Hz), 135.78, 131.22, 129.17, 128.22, 128.07, 118.74, 76.34 (d, J = 13.3 Hz), 64.96 (d, J = 5.3 Hz), 64.90 (d, J = 159.4 Hz), 48.44, 40.54, 39.02, 32.37, 30.13 (3), 30.09 (3), 30.03, 29.94, 29.80, 29.61, 29.36, 28.88, 23.11, 16.62, 14.36. 31P NMR (162 MHz, CDCl3/CD3OD) δ 16.30. HRMS (ESI) m/z calculated for C33H53O4N5PS2 [M – H]−, 678.32821; found, 678.32746. Anal. Calculated for C33H59N6O5PS2 (NH4+ monohydrate): C, 55.44; H, 8.32; N, 11.75. Found: C, 56.09; H, 8.10; N, 11.42. Melting point: decomposes at 150 °C.
6-(Dodecyldisulfanyl)hexan-1-ol (5a)
To a stirring solution of dodecane-1-thiol (3.50 mL, 14.62 mmol) and 6-mercaptohexan-1-ol (2 mL, 14.62 mmol) in MeOH/DCM (25:75, 70 mL) was added pyridine (2.54 mL, 29.2 mmol) followed by the gradual addition of iodine (4.08 g, 16.08 mmol). The solution stirred for 5 h at room temperature under nitrogen. Then the solvents were evaporated under reduced pressure and the residue was partitioned between brine and DCM. The organic layer was concentrated and purified on a silica column using a hexanes/EtOAc (0–10% EtOAc) gradient to afford the title compound 6-(dodecyldisulfanyl)hexan-1-ol (2.27 g, 6.78 mmol, 46.4% yield) as a white solid. 1H NMR (400 MHz, CDCl3) δ 3.65 (t, J = 6.6 Hz, 2H), 2.71–2.66 (m, 4H), 1.75–1.63 (m, 4H), 1.63–1.54 (m, 2H), 1.48–1.35 (m, 6H), 1.34–1.21 (m, 16H), 0.88 (t, J = 6.9 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 62.87, 39.14, 38.95, 32.58, 31.90, 29.64, 29.61, 29.58, 29.50, 29.33, 29.23, 29.20, 29.10, 28.51, 28.24, 25.36, 22.68, 14.12. HRMS (ESI) m/z calculated for C18H38OS2 [M + H]+, 335.24368; found, 335.24391. Melting point: 38–39 °C.
6-(Dodecyldisulfanyl)hexyl Hydrogen ((((R)-1-(6-Amino-9H-purin-9-yl)propan-2-yl)oxy)methyl)phosphonate (5b)
Following general procedure B, a solution of 6-(dodecyldisulfanyl)hexan-1-ol (0.117 g, 0.348 mmol) and pyridine (0.337 mL, 4.18 mmol) in DCM (2 mL) was slowly added dropwise to the solution stirring at 0 °C. The mixture stirred at this temperature for 15 min and then naturally warmed to room temperature and stirred for 3 h. Then water (0.094 mL, 5.22 mmol) was added and the mixture was continued stirring for an additional 30 min and a 30% solution of NH4OH was added. Stirring continued for 1 h and then the solvents were evaporated under reduced pressure and the resulting residue was dried under UHV and purified directly on silica gel using a DCM/DCM:MeOH:NH4OH (80:20:3) gradient (0–50%) to afford the title compound 6-(dodecyldisulfanyl)-hexyl hydrogen ((((R)-1-(6-amino-9H-purin-9-yl)propan-2-yl)oxy)-methyl)phosphonate (40 mg, 0.066 mmol, 19.03% yield) as a waxy solid. 1H NMR (400 MHz, CD3OD/CDCl3 referenced to TMS) δ 8.31 (s, 1H), 8.24 (s, 1H), 4.39 (dd, J = 14.4, 3.0 Hz, 1H), 4.20 (dd, J = 14.4, 6.9 Hz, 1H), 3.85 (ddd, J = 9.9, 6.4, 3.1 Hz, 1H), 3.83–3.71 (m, 3H), 3.45 (dd, J = 12.6, 10.2 Hz, 1H), 2.67 (dt, J = 10.8, 3.8 Hz, 4H), 1.72–1.61 (m, 4H), 1.56 (dt, J = 13.5, 6.8 Hz, 2H), 1.44–1.23 (m, 22H), 1.20 (d, J = 6.2 Hz, 3H), 0.89 (t, J = 6.9 Hz, 3H). 13C NMR (101 MHz, CDCl3/CD3OD referenced to CD3OD) δ 155.82, 152.46, 150.01, 143.31, 118.77, 76.26 (d, J = 13.2 Hz), 65.23 (d, J = 6.0 Hz), 64.63 (d, J = 159.7 Hz), 48.47, 39.34, 39.17, 34.90, 32.33, 31.27 (d, J = 6.3 Hz), 30.05, 30.03, 29.99, 29.92, 29.75, 29.65, 29.58, 29.47, 28.89, 28.58, 25.80, 23.07, 16.61, 14.30. 31P NMR (162 MHz, CDCl3/CD3OD) δ 16.36. HRMS (ESI) m/z calculated for C27H49O4N5PS2 [M – H]−, 602.29691; found, 602.29703. Elemental Analysis Calculated for C27H53N6O4PS2 (as an ammonium salt): C, 52.23; H, 8.60; N, 13.54. Found: C, 52.21; H, 8.65; N, 12.42.
2-Mercaptoethyl Hydrogen ((((R)-1-(6-Amino-9H-purin-9-yl)-propan-2yl)oxy)methyl)phosphonate (6)
To a stirring solution of dry tenofovir (1 g, 3.48 mmol) in anhydrous DCM (100 mL) and DMF (0.537 mL, 6.96 mmol) was gradually added excess oxalyl chloride (1.19 mL, 13.93 mmol) at room temperature. After stirring for 1 h, the mixture was then cooled to 0 °C and quenched with excess 2-mercaptoethanol (2.451 mL, 34.8 mmol). Progress was monitored by LC-MS (H2O/MeOH gradient, 50–95% MeOH, 3 min). The mixture stirred for an additional hour at this temperature. Aqueous HCl was then added (3 mL), followed by 15 mL of methanol (pH = 1). The mixture stirred at room temperature overnight, and then the pH was gradually raised to 5 using saturated aqueous sodium bicarbonate. The organic solvents were evaporated under reduced pressure at 30 °C, and the resulting residue was purified on a C18 reverse phase column using a H2O/MeOH gradient (isocratic 10% MeOH) to afford the title compound 2-mercaptoethyl hydrogen ((((R)-1-(6-amino-9H-purin-9-yl)propan-2-yl)oxy)methyl)phosphonate (0.6043 g, 1.740 mmol, 50.0% yield) as a white solid. 1H NMR (400 MHz, D2O) δ 8.38 (s, 1H), 8.37 (s, 1H), 4.44 (dd, J = 14.7, 3.1 Hz, 1H), 4.26 (dd, J = 14.7, 7.7 Hz, 1H), 4.04–3.92 (m, 1H), 3.77–3.66 (m, 3H), 3.48 (dd, J = 13.4, 9.6 Hz, 1H), 2.53 (td, J = 6.4, 4.2 Hz, 2H), 1.19 (d, J = 6.3 Hz, 3H). 13C NMR (101 MHz, D2O) δ 149.73, 148.53, 145.45, 144.35, 117.52, 75.55 (d, J = 12.5 Hz), 66.10 (d, J = 5.6 Hz), 62.92 (d, J = 159.5 Hz), 48.37, 24.40 (d, J = 6.5 Hz), 15.58. 31P NMR (162 MHz, D2O) δ 20.77. HRMS (ESI) m/z calculated for C11H19O4N5PS [M + H]+, 348.08899; found, 348.08929. Anal. Calculated for C11H20N5O5PS (as a monohydrate): C, 36.16; H, 5.52; N, 19.17. Found: C, 35.73; H, 5.53; N, 19.07. Melting point: decomposes at 100 °C.
4-Mercaptobutyl Hydrogen ((((R)-1-(6-Amino-9H-purin-9-yl)-propan-2-yl)oxy)methyl)phosphonate (7)
To a stirring solution of tenofovir (1 g, 3.48 mmol) in anhydrous DCM (34.8 mL) and N,N-dimethylformamide (0.322 mL, 4.18 mmol) was added excess oxalyl chloride (1.493 mL, 17.41 mmol) at room temperature. The mixture stirred for 15 min with progress monitored with LC-MS (H2O/MeOH gradient, 35–75% MeOH, 3 min) by quenching an aliquot of the reaction mixture with MeOH. Upon completion, the solvents and excess oxalyl chloride were evaporated under reduced pressure and the resulting yellow foam was further dried under UHV. The foam was redissolved in anhydrous DCM (34 mL), and the mixture was chilled to 0 °C. Then a solution of 4-mercaptobutan-1-ol (0.395 mL, 3.83 mmol) and pyridine (1.683 mL, 20.89 mmol) in DCM (2 mL) was added dropwise and the reaction stirred at this temperature for 15 min and then naturally warmed to room temperature with progress monitored by LC-MS. After stirring for 2 h, the mixture was quenched with water (0.25 mL) and stirring continued at room temperature for 30 min. The solution was then acidified with HCl (1.2 M) and homogenized with MeOH until the aqueous/organic interface disappeared and stirred overnight to facilitate the cleavage of formimidine. The solvents were then evaporated under reduced pressure, and the resulting yellow oil was purified on a C18 reverse phase column using a H2O/MeOH gradient with 0.1% formic acid (gradient 0–25% MeOH) to afford the title compound 4-mercaptobutyl hydrogen ((((R)-1-(6-amino-9H-purin-9-yl)propan-2-yl)oxy)methyl)phosphonate (0.7075 g, 1.885 mmol, 54.1% yield) as a white foam. 1H NMR (400 MHz, CD3OD) δ 8.40 (s, 1H), 8.27 (s, 1H), 4.46 (dd, J = 14.4, 3.0 Hz, 1H), 4.27 (dd, J = 14.5, 7.1 Hz, 1H), 4.08–3.98 (m, 1H), 3.83 (ddd, J = 22.4, 12.6, 7.7 Hz, 3H), 3.63 (dd, J = 13.0, 9.5 Hz, 1H), 2.99 (t, J = 6.9 Hz, 1H), 2.49 (t, J = 6.7 Hz, 2H), 1.72–1.58 (m, 4H), 1.19 (d, J = 6.3 Hz, 3H). 13C NMR (101 MHz, CD3OD) δ 152.23, 150.40, 146.30, 146.15, 119.17, 76.58 (d, J = 12.2 Hz), 65.56 (d, J = 6.0 Hz), 64.72 (d, J = 162.9 Hz), 31.47, 30.63 (d, J = 6.1 Hz), 27.40, 24.67, 16.96. 31P NMR (162 MHz, CD3OD) δ 17.70. HRMS (ESI) m/z calculated for C13H23N5O4PS [M + H]+, 376.12029; found, 376.11987. Anal. Calculated for C13H24O5N5PS (as a monohydrate): C, 39.69; H, 6.15; N, 17.80. Found: C, 39.88; H, 5.73; N, 17.76. Melting Point: 151–152 °C.
Supplementary Material
Acknowledgments
The authors would like to acknowledge Manohar Saindane for transporting numerous samples from Emory to the EIDD for analysis. Work presented herein was funded internally.
ABBREVIATIONS
- DCM
dichloromethane
- DMF
dimethylformamide
- TFV
tenofovir
- TDF
tenofovir disoproxil fumarate
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.6b01292.
Assay details, kinetic studies, and associated spectra for synthesized compounds (PDF)
Molecular formula strings (CSV)
Notes
The authors declare no competing financial interest.
References
- 1.Walle T, Hsieh F, DeLegge MH, Oatis JE, Walle UK. High Absorption but Very Low Bioavailability of Oral Resveratrol in Humans. Drug Metab Dispos. 2004;32:1377–1382. doi: 10.1124/dmd.104.000885. [DOI] [PubMed] [Google Scholar]
- 2.Anand P, Kunnumakkara AB, Newman RA, Aggarwal BB. Bioavailability of Curcumin: Problems and Promises. Mol Pharmaceutics. 2007;4:807–818. doi: 10.1021/mp700113r. [DOI] [PubMed] [Google Scholar]
- 3.Veber DF, Johnson SR, Cheng HY, Smith BR, Ward KW, Kopple KD. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J Med Chem. 2002;45:2615–2623. doi: 10.1021/jm020017n. [DOI] [PubMed] [Google Scholar]
- 4.Li WX, Escarpe PA, Eisenberg EJ, Cundy KC, Sweet C, Jakeman KJ, Merson J, Lew W, Williams M, Zhang LJ, Kim CU, Bischofberger N, Chen MS, Mendel DB. Identification of Gs 4104 as an Orally Bioavailable Prodrug of the Influenza Virus Neuraminidase Inhibitor Gs 4071. Antimicrob Agents Chemother. 1998;42:647–653. doi: 10.1128/aac.42.3.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Simplicio AL, Clancy JM, Gilmer JF. Prodrugs for Amines. Molecules. 2008;13:519–547. doi: 10.3390/molecules13030519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pradere U, Garnier-Amblard EC, Coats SJ, Amblard F, Schinazi RF. Synthesis of Nucleoside Phosphate and Phosphonate Prodrugs. Chem Rev. 2014;114:9154–9218. doi: 10.1021/cr5002035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thilakarathna SH, Rupasinghe HP. Flavonoid Bioavailability and Attempts for Bioavailability Enhancement. Nutrients. 2013;5:3367–3387. doi: 10.3390/nu5093367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Manach C, Williamson G, Morand C, Scalbert A, Remesy C. Bioavailability and Bioefficacy of Polyphenols in Humans. I. Review of 97 Bioavailability Studies. Am J Clin Nutr. 2005;81:230S–242S. doi: 10.1093/ajcn/81.1.230S. [DOI] [PubMed] [Google Scholar]
- 9.Rautio J, Kumpulainen H, Heimbach T, Oliyai R, Oh D, Jarvinen T, Savolainen J. Prodrugs: Design and Clinical Applications. Nat Rev Drug Discovery. 2008;7:255–270. doi: 10.1038/nrd2468. [DOI] [PubMed] [Google Scholar]
- 10.Kang MJ, Cho JY, Shim BH, Kim DK, Lee J. Bioavailability Enhancing Activities of Natural Compounds from Medicinal Plants. J Med Plant Res. 2009;3:1204–1211. [Google Scholar]
- 11.Kuo A, Dienstag JL, Chung RT. Tenofovir Disoproxil Fumarate for the Treatment of Lamivudine-Resistant Hepatitis B. Clin Gastroenterol Hepatol. 2004;2:266–272. doi: 10.1016/s1542-3565(04)00017-5. [DOI] [PubMed] [Google Scholar]
- 12.Heathcote EJ, Marcellin P, Buti M, Gane E, De Man RA, Krastev Z, Germanidis G, Lee SS, Flisiak R, Kaita K, Manns M, Kotzev I, Tchernev K, Buggisch P, Weilert F, Kurdas OO, Shiffman ML, Trinh H, Gurel S, Snow-Lampart A, Borroto-Esoda K, Mondou E, Anderson J, Sorbel J, Rousseau F. Three-Year Efficacy and Safety of Tenofovir Disoproxil Fumarate Treatment for Chronic Hepatitis B. Gastroenterology. 2011;140:132–143. doi: 10.1053/j.gastro.2010.10.011. [DOI] [PubMed] [Google Scholar]
- 13.Fung HB, Stone EA, Piacenti FJ. Tenofovir Disoproxil Fumarate: A Nucleotide Reverse Transcriptase Inhibitor for the Treatment of Hiv Infection. Clin Ther. 2002;24:1515–1548. doi: 10.1016/s0149-2918(02)80058-3. [DOI] [PubMed] [Google Scholar]
- 14.Kearney BP, Flaherty JF, Shah J. Tenofovir Disoproxil Fumarate - Clinical Pharmacology and Pharmacokinetics. Clin Pharmacokinet. 2004;43:595–612. doi: 10.2165/00003088-200443090-00003. [DOI] [PubMed] [Google Scholar]
- 15.Lewis W, Day BJ, Copeland WC. Mitochondrial Toxicity of Nrti Antiviral Drugs: An Integrated Cellular Perspective. Nat Rev Drug Discovery. 2003;2:812–822. doi: 10.1038/nrd1201. [DOI] [PubMed] [Google Scholar]
- 16.Herlitz LC, Mohan S, Stokes MB, Radhakrishnan J, D’Agati VD, Markowitz GS. Tenofovir Nephrotoxicity: Acute Tubular Necrosis with Distinctive Clinical, Pathological, and Mitochondrial Abnormalities. Kidney Int. 2010;78:1171–1177. doi: 10.1038/ki.2010.318. [DOI] [PubMed] [Google Scholar]
- 17.Ruane PJ, DeJesus E, Berger D, Markowitz M, Bredeek UF, Callebaut C, Zhong LJ, Ramanathan S, Rhee MS, Fordyce MW, Yale K. Antiviral Activity, Safety, and Pharmacokinetics/Pharmacodynamics of Tenofovir Alafenamide as 10-Day Monotherapy in Hiv-1-Positive Adults. JAIDS, J Acquired Immune Defic Syndr. 2013;63:449–455. doi: 10.1097/QAI.0b013e3182965d45. [DOI] [PubMed] [Google Scholar]
- 18.Markowitz M, Zolopa A, Squires K, Ruane P, Coakley D, Kearney B, Zhong LJ, Wulfsohn M, Miller MD, Lee WA. Phase I/Ii Study of the Pharmacokinetics, Safety and Antiretroviral Activity of Tenofovir Alafenamide, a New Prodrug of the Hiv Reverse Transcriptase Inhibitor Tenofovir, in Hiv-Infected Adults. J Antimicrob Chemother. 2014;69:1362–1369. doi: 10.1093/jac/dkt532. [DOI] [PubMed] [Google Scholar]
- 19.Hostetler KY. Alkoxyalkyl Prodrugs of Acyclic Nucleoside Phosphonates Enhance Oral Antiviral Activity and Reduce Toxicity: Current State of the Art. Antiviral Res. 2009;82:A84–98. doi: 10.1016/j.antiviral.2009.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Painter GR, Almond MR, Trost LC, Lampert BM, Neyts J, De Clercq E, Korba BE, Aldern KA, Beadle JR, Hostetler KY. Evaluation of Hexadecyloxypropyl-9-R-[2-(Phosphonomethoxy)-Propyl]-Adenine, Cmx157, as a Potential Treatment for Human Immunodeficiency Virus Type 1 and Hepatitis B Virus Infections. Antimicrob Agents Chemother. 2007;51:4538–4538. doi: 10.1128/AAC.00460-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Giesler KE, Marengo J, Liotta DC. Reduction Sensitive Lipid Conjugates of Tenofovir: Synthesis, Stability, and Antiviral Activity. J Med Chem. 2016;59:7097–7110. doi: 10.1021/acs.jmedchem.6b00428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Iyer S, Hengge AC. The Effects of Sulfur Substitution for the Nucleophile and Bridging Oxygen Atoms in Reactions of Hydroxyalkyl Phosphate Esters. J Org Chem. 2008;73:4819–4829. doi: 10.1021/jo8002198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xia F, Zhu H. Density Functional Calculations on the Effect of Sulfur Substitution for 2′-Hydroxypropyl-P-Nitrophenyl Phosphate: Co Vs. Po Bond Cleavage. Bioorg Chem. 2012;40:99–107. doi: 10.1016/j.bioorg.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 24.Egron D, Perigaud C, Aubertin AM, Imbach JL, Gosselin C. Effect of the Thioalkyl Chain Variation in the Efficiency of Sate Pronucleotides. Nucleosides Nucleotides. 1998;17:1725–1729. [Google Scholar]
- 25.Pertusat F, Serpi M, McGuigan C. Medicinal Chemistry of Nucleoside Phosphonate Prodrugs for Antiviral Therapy. Antivir Chem Chemother. 2012;22:181–203. doi: 10.3851/IMP2012. [DOI] [PubMed] [Google Scholar]
- 26.Giesler KE, Marengo J, Liotta DC. Reduction Sensitive Lipid Conjugates of Tenofovir: Synthesis, Stability, and Antiviral Activity. J Med Chem. 2016;59:7097–7110. doi: 10.1021/acs.jmedchem.6b00428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ray AS, Hostetler KY. Application of Kinase Bypass Strategies to Nucleoside Antivirals. Antiviral Res. 2011;92:277–291. doi: 10.1016/j.antiviral.2011.08.015. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.