

Abstract
The therapeutic value of numerous small molecules hinges on their ability to permeate the plasma membrane. This is particularly true for tenofovir (TFV), adefovir, and other antiviral nucleosides that demonstrate potent antiviral activity but poor bioavailability. Using TFV as a model substrate, we hybridized two disparate prodrug strategies to afford novel reduction-sensitive lipid conjugates of TFV that exhibit subnanomolar activity toward HIV-1 and are stable in human plasma for more than 24 h with a therapeutic index approaching 30000. These compounds significantly rival the clinically approved formulation of TFV and revitalize the potential of disulfide-bearing prodrugs which have seen limited in vitro and in vivo success since their debut over 20 years ago. We further demonstrate the utility of these conjugates as a tool to indirectly probe the enzymatic hydrolysis of phosphonomonoesters that may further advance the development of other prodrug strategies for nucleosides, peptides, and beyond.
Graphical Abstract
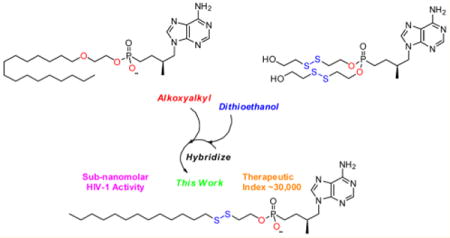
INTRODUCTION
Tenofovir (TFV) is an acyclic nucleoside that demonstrates broad spectrum antiviral activity against human immunodeficiency virus (HIV),1–3 hepatitis B virus (HBV),4,5 and herpes simplex type-2 virus (HSV-2).6 TFV structurally resembles 2′,3′-dideoxyadenosine, which lacks the requisite 3′ hydroxyl moiety necessary for DNA polymerization and triggers obligate chain termination upon incorporation of tenofovir diphosphate (TFVdpp) into the growing viral DNA strand. A common structural feature among acyclic nucleosides (tenofovir, adefovir, cidofovir) is a metabolically stable phosphonate linkage that permanently affixes the phosphonate to the acyclic sugar linker and nucleobase. This serves to prevent undesirable chemical and enzymatic hydrolysis and bypasses the initial phosphorylation to the monophosphate, which is the kinetic bottleneck during the conversion of conventional nucleosides to their active triphosphate.7 Unfortunately, the persistent nature of the phosphonate moiety plagues TFV and other acyclic nucleosides with significant dianionic character at physiological pH that restricts diffusion across the plasma membrane, resulting in rapid renal clearance and depreciated bioavailability and antiviral activity in vivo. When orally administered to mice,8 the bioavailability of TFV is approximately 2% and that of adefovir has been reported to be <1% in monkeys9 and 8–11% in rats.10 These undesirable properties can be ameliorated by masking the anionic phosphonic acid with various prodrugs that alter the pharmacokinetic profile of the parent nucleoside, enhance cellular permeability, and improve bioavailability. Several eclectic prodrug strategies have been developed for this purpose and are extensively reviewed elsewhere.11,12 The clinically approved prodrug formulation of TFV is tenofovir disoproxil fumarate (TDF), which is manufactured by Gilead Sciences. TDF features two isopropyloxymethyl carbonate masking units esterified to the phosphonate that relies on an esterase-activated cleavage mechanism to liberate TFV following successful delivery to the target tissue as shown in Scheme 1.13 The installation of two isopropyl carbonate esters increases the oral bioavailability of TFV to 25%, enhances tissue distribution, and improves biological stability.8 However, the ubiquitous distribution of esterases renders a significant fraction of TDF susceptible to premature hydrolysis, resulting in systemic exposure to TFV, a known nephrotoxin.14,15 Continuous administration of TDF has been reported to induce lactic acidosis, Fanconi syndrome, acute renal failure, and bone loss.16,17 It is therefore desirable to exploit alternative prodrug strategies to enhance intracellular delivery and reduce systemic exposure of tenofovir. To date, two lead candidates have surfaced in clinical trials: tenofovir-alafenamide (TAF)18,19 and hexadecyloxypropyl-tenofovir (1)20 illustrated in Figure 1. TAF is an isopropylalaninyl phenyl ester that requires two disparate enzymes for prodrug release: carboxyesterase and cathepsin A.21 The latter enzyme, cathepsin A, is a serine protease localized almost exclusively to lysosomal endosomes and ensures selective intracellular delivery of TFV. Similarly, 1 also relies on the catalytic activity of an intracellular hydrolase, phospholipase C and/or sphingomylenase, to liberate TFV within the cytosol.20,22 TAF is currently in phase III clinical trials and demonstrates little to no nephrotoxicity and more potent antiviral activity than TDF at 1/10 the dose.18 On the other hand, 1 has made little progression through the clinical trial pipeline since the completion of phase I in 2011. Preliminary data disclosed on Chimerix’s website23 reveal 1 is well-tolerated and achieves significant concentrations of TFVdpp up to 1 week after a single 400 mg dose, suggesting the potential for a convenient, once-a-week dosing regimen.
Scheme 1.
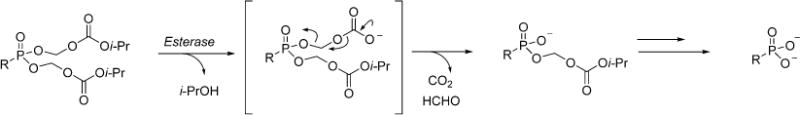
Decomposition of Disoproxil Fumarate Prodrugs in Vivo
Figure 1.
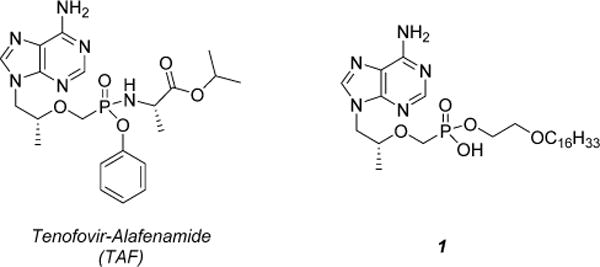
Prodrug conjugates of tenofovir in the clinical trial pipeline to compete with TDF.
Other lipid prodrug strategies have also been developed to enhance nucleoside delivery. Mitra and collaborators reported biotinylated lipid conjugates of acyclovir that demonstrate profound cellular targeting and uptake in MDCK-MDR1 and Caco-2 cell lines, resulting in enhanced antiviral activity.24–26 Although the exact mechanism of release remains to be elucidated, these structures also feature an ester moiety that may be subject to premature plasma hydrolysis, which is less than ideal for the treatment of chronic systemic infections.
We sought to design disulfide-linked lipid conjugates similar to 1 that readily permeate the plasma membrane and rapidly releases the nucleoside within the target cell without the need for enzymatic activation. In contrast to other prodrug strategies, our design relies on the reduction-sensitive nature of the S–S bond that selectively bows to the universal glutathione redox potential that exists at the plasma membrane between the extra- and intracellular milieu.27,28 Glutathione, often referred to as GSH, is a tripeptide typically stockpiled in millimolar concentrations (2–10 mM) within the cytosol that participates in a multitude of biological functions to promote the reduction of disulfides, detoxification of xenobiotics,29,30 mediate immunoregulation,31 and curb oxidative stress32 among others. GSH is seldom found in the extracellular space and thus creates a gradient that fosters intracellular reduction of disulfides and simultaneously promotes an oxidative environment to support S–S formation in the surrounding media.
The literature is rife with examples that harness this system for drug delivery and biomedical applications, however, the field has primarily focused on nanoparticles,33–36 copolymers,37,38 and other relatively complex macromolecular39–41 or inorganic structures42–44 as delivery vehicles. In an attempt to extend this technology to a useful prodrug for nucleosides, Gosselin and collaborators previously examined dithioethanol (DTE) conjugates to mediate the delivery of adefovir, AZT, and 3′-deoxyuridine (ddU) to HIV-1 infected MT-4, CEM-SS, and CEM-TK− cell lines in vitro.45,46 A proposed cleavage mechanism for these masking groups is illustrated in Scheme 2. Reduction of 2 releases β-mercaptoethanol and metastable intermediate 3 that spontaneously collapses on the thioethanol linker to generate thiirane and the free nucleoside 4 when R = H. Of note, S-acylthioethyl (SATE) prodrugs also pass through common intermediate 3 following hydrolysis of an S-acyl moiety via nonspecific carboxyesterases to liberate the target nucleoside.47 Conjugation of bis(DTE) to adefovir increases the HIV-1 activity of the parent nucleoside by 10-fold in CEM-TK− cells and confers exceptional stability (t1/2 > 24 h at pH 2, 7.4, culture medium, and human gastric juice). However, these conjugates rapidly degrade in human serum (t1/2 < 5 min), which limits their clinical utility. The apparent instability of the disulfide linkage in serum and the presumed toxicity of thiirane have stalled efforts to advance this technology forward. Herein, we hybridized the DTE approach with the alkoxyalkyloxypropyl prodrug strategy parlayed for 1 to afford novel reduction-sensitive lipid conjugates of tenofovir shown in Figure 2 that exhibit subnanomolar activity toward HIV-1 and are stable in human plasma for more than 24 h with a therapeutic index near 30000.
Scheme 2.
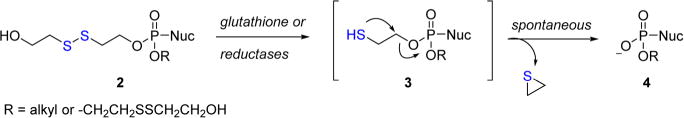
Mechanism of DTE Prodrug Cleavage in Vivo
Figure 2.
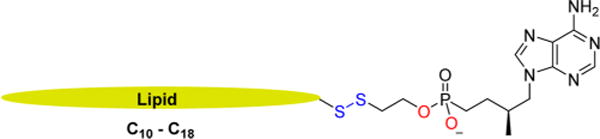
Generalized representation of current prodrug strategy.
RESULTS AND DISCUSSION
Chemistry
Our original synthesis toward TFV disulfide conjugates 10 (Scheme 3) began with commercially available TFV (CombiBlocks) that was converted to the bis-chloridate with excess oxalyl chloride and dimethylformamide (DMF) in dichloromethane (DCM). When catalytic DMF was used, the reaction stalled due to the presence of the C-6 amino moiety on the purine ring system, which depletes the catalytic Vilsmeyer chlorinating agent and becomes concomitantly protected as N-formimidine. This was resolved by adding 1.2 equiv of DMF to rapidly afford the formimidine protected bis-chloridate (6) that was subsequently treated with excess β-mercaptoethanol to generate a mixture of bis- and mono- adducts of 7 that were not isolated. When 7 stirred with aqueous saturated sodium bicarbonate for 30 min, the complexity of the mixture reduced to a single species whose m/z ratio was consistent with 8. Acid-mediated hydrolysis of 8 afforded 9 in moderate yield whose structural assignment was unambiguously characterized by HRMS, 1H, 13C, and 31P NMR. The key step of the synthesis involved the construction of the critical disulfide linkage between 9 and hexadecanethiol. Unfortunately, this endeavor proved unsuccessful with a variety of oxidizing agents including iodine, oxone, O2, and H2O2. Cyclization of the mercaptoethanol linker was observed when 9 reacted with iodine, and unidentifiable byproducts or no appreciable reaction occurred with the latter oxidants. This reaction sequence was thus abandoned and an alternative synthesis of was sought. The new reaction sequence is presented in Scheme 4 where β-mercaptoethanol was preoxidized with a handful of aliphatic thiols (C10–C18) to produce the corresponding disulfide-bridged linkers 11a–e in moderate yields of 34–45%. These compounds were crystalline solids with sharp melting points (11a is an oil) that could be cleanly purified by silica gel column chromatography. Alcohols 11a–e were subsequently coupled to TFV using the same DMF/oxalyl chloride methodology introduced in Scheme 3 to afford monoesters 12a–e in yields of 12–49% after hydrolysis of the remaining chloridate with water and deprotection of formimidine in warm ethanol.
Scheme 3.
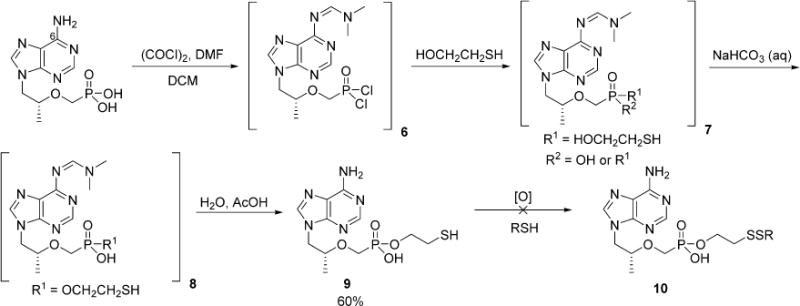
Initial Synthesis of Monoester Disulfide Conjugates of TFV
Scheme 4.
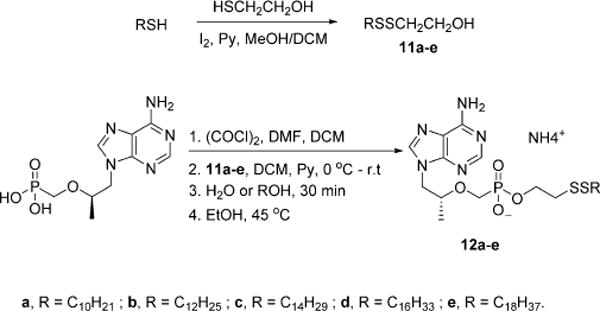
Synthesis of Disulfide Lipid Phosphonomonoesters of TFV
Despite the installation of a greasy hydrocarbon tail, the exposed phosphonic acid moiety retained significant polarity that initially plagued normal phase chromatographic purification of 12a–e on silica gel. Relatively polar solvent gradients of DCM/MeOH/NH4OH (80:20:0.1) failed to move compounds on aluminum-backed silica TLC plates, and increasing the percentage of methanol compromised the integrity of the silica and promoted streaking. However, when the concentration of ammonium hydroxide was raised from 0.1 to 1–3% (v/v), appreciable movement was observed by TLC and these conditions were successfully applied to column chromatography to furnish 12a–e as their ammonium salts as determined by elemental analysis.
With monoesters 12a–e in hand, the corresponding bis-disulfides 13a–e shown in Figure 3a were synthesized by employing the same protocol outlined in Scheme 4 using two equivalents of 11a–e during coupling to TFV instead of one. All five transformations occurred as predicated to afford 13a–e in 30–40% yield after purification without incident. Finally, a series of mixed phosphonodiesters (14–22) were synthesized and are displayed in Figure 3b. Scheme 4 is again referenced for the synthesis of these compounds using 11d as the coupling lipid and various alcohols to quench the remaining phosphonochloridate in step 3 rather than water. Note that 14–22 were prepared as an inconsequential mixture of diastereomers and were not separated.
Figure 3.
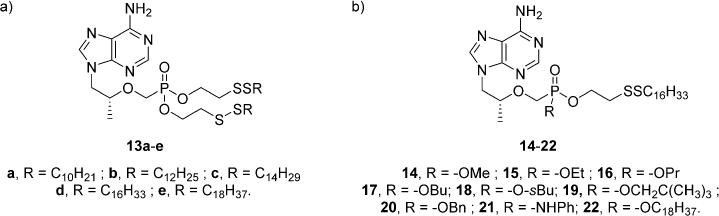
(a) Bis-disulfide conjugates of TFV. (b) Mixed phosphonoesters of TFV.
All compounds were evaluated against HIV-infected PBMCs and antiviral activity was assessed by measuring reverse transcriptase in cell supernatants (see Experimental Section). Compounds 12a–e, 13a–e, and 14 were assayed against HBV, and qPCR was used to quantify viral DNA following incubation in HepG2.2.15 cells after 6 days. Table 1 details the antiviral activity of conjugates 12a–e with modifications to the lipid tail. These compounds resemble lysogenic phospholipids characterized by an anionic phosphate headgroup and a single aliphatic tail. This structural motif confers an inverted conical-like shape that facilitates translocation between the inner and outer leaflets of the plasma membrane and procures detergent-like properties that disrupts lipid bilayers.48,49 As shown in Table 1, compound 12a possesses the shortest lipid (C10) and is 18-fold less potent than TDF (85 vs 4.6 nM) with a relatively poor therapeutic index (TI) of 590 at the EC50. Extending the length of the alkyl chain by two carbon atoms results in 12b whose HIV-1 activity (3.0 nM) is comparable to TDF and 7-fold more active than 1 (20 nM). Maximum antiviral activity was obtained for conjugates 12c–e, with alkyl chain lengths ranging from 14 to 18 carbon atoms, respectively. 12c–e exhibit subnanomolar EC50 values of ∼0.5 nM that outrivals TDF and 1 and 12d boasts a TI50 that exceeds 20000, whereas that of 12c approaches 30000. This broad therapeutic window is sustained even at the EC90 for both 12c and 12d and is nearly an order of magnitude wider than the TI90 of TDF (11000 vs 1300; data not shown). The observed correlation between chain length and antiviral activity corresponds to Hostetler’s work, who found that ∼20 atoms is the ideal linker length necessary to achieve maximum potency for alkyloxypropyl conjugates of cidofovir.22 Note that compounds 12c–e possess linker lengths of 19, 20, and 21 atoms, respectively, when the mercaptoethanol bridge is taken into account. Table 1 also reveals that increasing chain length is associated with a concomitant increase in cytotoxicity, a phenomenon that has been documented for a variety of surfactants in numerous aquatic organisms.50
Table 1.
HIV-1 and HBV Activity of Lysogenic Phospholipids 9 and 12a–e Compared to TDF, 1, and DTE–TFVa
| HIV-1 | HBV | |||||
|---|---|---|---|---|---|---|
|
|
||||||
| compd | EC50b (PBMCs) | CC50c (PBMCs) | TI (CC50/EC50) | EC50b (HepG2) | CC50c (HepG2) | TI (CC50/EC50) |
| TDF | 0.0046 | 43.7 | 9500 | 0.34 | 64.5 | 190 |
| TFV | 0.319 | >100 | >300 | |||
| 9 | 18.6 | >100.0 | >5.38 | 41.9 | >100.0 | >2.4 |
| DTE–TFV | 1.61 | >100.0 | >62.1 | 11.6 | >100.0 | >8.6 |
| 12a | 0.085 | >50.0 | >590 | >50.0 | >50.0 | >1 |
| 12b | 0.0030 | 25.0 | 8300 | 1.67 | >50.0 | >30 |
| 12c | 0.00050 | 14.0 | 28000 | 0.444 | >25 | >56 |
| 12d | 0.00065 | 14.3 | 22000 | 0.020 | >25 | >1200 |
| 1 | 0.02 | >100.0 | >5000 | |||
| 12e | 0.00060 | 6.36 | 11000 | 0.505 | >50 | >50 |
All data represent an average of triplicate experiments.
EC50, effective concentration (in μM) required to inhibit HIV-1 or HBV by 50%.
CC50, effective concentration (in μM) required to reduce the viability of uninfected cells by 50%.
With respect to HBV activity, all conjugates in Table 1 demonstrated rather unremarkable activity with the exception of 12d, whose potency (20 nM) and TI50 (1250) bested that of TDF, which further supports the advantage of our lipids over carbonate prodrug strategies.
In addition to 12a–e, DTE–TFV and 9 were also assessed in this assay and were found to be 5- and 58-fold less active than TFV against HIV-1, respectively. The dramatic potency loss observed for 9 is interesting given that this compound is predicated to readily undergo intramolecular cyclization (Scheme 2) to release the bound nucleoside in vivo.11,46 We suspect that the β-mercaptoethanol linker resists cleavage following intracellular delivery and obstructs the phosphorylation of the parent nucleoside to the active diphosphate. Additional experiments are currently being conducted by our laboratory to shed light on these potential mechanistic nuances.
In contrast to 12a–e, 13a–e resemble conventional phospholipids with two aliphatic tails that assume a cylindrical shape and do not readily traverse the plasma membrane on their own accord.51 We therefore speculated that 13a–e would exhibit compromised antiviral activity when compared to their lysogenic brethren. Indeed, all phosphonodiesters in Table 2 proved to be significantly less potent than 12a–e. Conjugates 13a and 13e achieved potencies comparable to TFV against HIV-1, whereas 13c was the only compound to demonstrate a 2-fold increase in activity when compared to the parent nucleoside. All compounds were relatively inactive against HBV relative to TDF. If it is assumed that reduction is facile upon entry and that 12a–e and 13a–e interact with the same cellular machinery following cleavage of the linker, then the results presented in Table 2 strongly implicate membrane trans-location as the rate-limiting step governing the EC50 value of 13a–e. It is therefore interesting that 13a,c,e and TFV all have strikingly similar HIV-1 activity despite their disparate alkyl chain lengths and, in the case of TFV, no chain at all. Previous work has demonstrated that phospholipid translocation depends on several factors such as lipid chain length, composition of the polar headgroup, and the presence of various ATP-dependent enzymes (flippase, floppase, scram-blase, aminophospholipid translocase, etc.) to affect this locomotion.51–53 In the context of this work, the exact mechanisms (whether active or passive) responsible for the diffusion of TFV and 13a–e through the membrane remain to be determined, however, it may be tentatively concluded that these forces exhibit a nonobvious degree of selectivity that facilitates the transport of lipids 13a,c,e with similar efficiencies in lieu of conjugates 13b and 13d.
Table 2.
HIV-1 and HBV Activity of Bis-disulfide Conjugates 13a–ea
| HIV-1 | HBV | |||||
|---|---|---|---|---|---|---|
|
|
||||||
| compd | EC50b (PBMCs) | CC50c (PBMCs) | TI (CC50/EC50) | EC50b (HepG2) | CC50c (HepG2) | TI (CC50/EC50) |
| TDF | 0.0046 | 43.7 | 9500 | 0.34 | 29.2 | 86 |
| 13a | 0.349 | >100.0 | >287 | >100 | >100.0 | >1 |
| 13b | 1.18 | >100.0 | >85.0 | >100 | >100.0 | >1 |
| 13c | 0.189 | >100.0 | >529 | >100 | >100.0 | >1 |
| 13d | 6.32 | >25 | 4.0 | >25 | >25 | >1 |
| 13e | 0.331 | >25 | >76 | >25 | >25 | >1 |
All data represent an average of triplicate experiments.
EC50, effective concentration (in μM) required to inhibit HIV-1 or HBV by 50%.
CC50, effective concentration (in μM) required to reduce the viability of uninfected cells by 50%.
Stability Studies
To be considered clinical candidates for oral delivery, conjugates 12b–e must demonstrate sufficient hydrolytic, nucleophilic, and plasma stability. Despite the dismal reported plasma stability profile of bis(DTE)-conjugates (t1/2 < 5 min),45 we reasoned 12b–e could form micelles in solution and protect the delicate S–S linkage from reductases circulating in human serum.54 Compounds 12c and 12d were selected as model compounds and subjected to various media including human serum, PBS buffer (pH 7.4), Dulbecco’s Modified Eagle Medium (DMEM), and carbonate/bicarbonate buffer (pH 9). Aliquots of each sample were analyzed by LC-MS at varying times points over the course of 2 h with a final time point at 24 h to assess decomposition. This data is presented in graphical and tabular format within the Supporting Information.
To our delight, both 12c and 12d demonstrated robust stability in human plasma with a half-life of >24 h. In contrast, DTE–TFV readily degraded in the presence of human plasma (t1/2 < 34 min), which is in agreement with previous findings. These results lend credence to the idea that micelle formation enhances the plasma stability of 12c and 12d relative to DTE–TFV whose truncated, polar tail precludes sturdy micelle formation and renders DTE–TFV susceptible to reductases or other mechanisms of decomposition.
We were curious how 12c,d would fare in the presence of base (pH 9), PBS (pH 7.4), and upon exposure to nucleophilic media (DMEM). Unfortunately, the hydrophobic nature of our prodrug strategy skewed the stability data of 12c,d under these conditions (see Supporting Information). Preliminary experiments with 12d and DTE–TFV in PBS initially revealed that 12d rapidly decomposed in PBS solution with t1/2 < 20 min (data not shown) while DTE–TFV boasted a half-life of 3.5 h. Poor PBS stability was also observed for other alkoxyalkyl conjugates in PBS but not for less hydrophobic conjugates. In light of these observations, we suspected that chemical decomposition was not the operative mechanism behind this phenomenon in PBS but rather adsorption of the lipid to the glass surface was responsible for abstracting analyte from the solution. To test this hypothesis, we assessed the stability of hexadecyloxypropyl 2′-deoxy-2′-fluorouridine, a nonlabile alkoxyalkyl lipid conjugate of 2′-deoxy-2′-fluorouridine in PBS buffer using Pyrex, Kimax, and silanized glassware. These experiments revealed that both Pyrex and silanized glass encourage substantial lipid adsorption and give rise to a rapid (but artificial) decomposition profile for lipid-bound nucleosides (data not shown). Interestingly, Kimax glassware did not produce this effect despite its near identical composition to Pyrex. This prompted us to reassess the PBS stability of our disulfide lipid conjugates in Kimax glassware. Note that all stability experiments were performed in Kimax glassware using hexadecyloxypropyl 2′-deoxy-2′-fluorouridine as an internal standard. With these conditions, both 12c and d exhibited similar stabilities at pH 9, PBS, and DMEM. Unfortunately, neither 12c nor 12d exhibit a half-life of >2 h in these media, which is in stark contrast to their stability in human serum (>24 h). We contend that the precipitation of 12d and 12c from the media is responsible for the poor half-life of these compounds in PBS, carbonate buffer, and DMEM. This is consistent with the observation that the C14 chain of 12c procures a near 2-fold stability increase over the C16 linker of 12d in all examined media (with the exception of human serum). It is plausible that the proteins present in human serum provide additional van der Waals contacts to facilitate dissolution. Taken together, the inherent plasma stability of 12c,d illuminates the possibility for selective intracellular delivery that may reduce systemic exposure of TFV or other bound cargo.
As a Tool to Probe Enzymatic Cleavage
A subset of prodrug strategies rely on the presence of specific hydrolases such as phospholipase C or cathepsin A to sever the P–O or P–N linkage between prodrug and nucleoside. Unfortunately, little is known about the substrate specificity of these enzymes. Seminal reports by Kelly et al. concluded 5′-nucleotide phosphodiesterase preferentially cleaves aryl esters over aliphatic and benzyl derivatives,55,56 however, a systematic analysis of various ester derivatives has not been examined in PBMCs. We saw this as an opportunity to indirectly probe the enzymatic machinery responsible for the hydrolysis of TFV phosphonomonoesters in PBMCs using our reduction-sensitive lipids as a delivery vehicle. Conjugates 14–22 were prepared using hexadecyldisulfanylethanol (HDE) as the lipid of choice. We hypothesized that HDE would ensure thorough cellular permeation and relatively rapid intracellular cleavage to expose the corresponding phosphonomonoester which would then be subsequently hydrolyzed in a rate-limiting step to TFV (Scheme 5). The rate of hydrolysis is thus dependent on the nature of the alkyl or aryl ester and therefore governs the HIV-1 activity of 14–22.
Scheme 5.
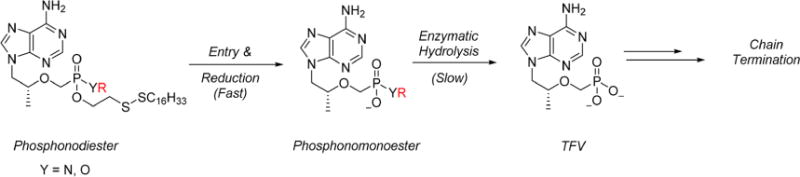
Predicated Decomposition Pathway for Phosphonodiesters 14–22
As expected, none of these compounds (14–22) exhibited antiviral activity comparable to 12c–e, presumably due to the additional cleavage step (Table 3). Successively increasing the chain length of the alkyl ester from methyl to n-Bu resulted in significant potency loss with 13 being the most active (26 nM) of the linear series (14–16, 17, and 22). When 14 is omitted, HIV-1 activity concomitantly increased with chain length from ethyl to n-Bu.
Table 3.
HIV-1 Activity of Lipid Conjugates 14–22 in PBMCsa
| compd | R– | EC50b (PBMCs) | CC50c (PBMCs) | TI (CC50/EC50) |
|---|---|---|---|---|
| TDF | NA | 0.0046 | 43.7 | 9500 |
| TFV | NA | 0.319 | >100.0 | >300 |
| 14 | MeO– | 0.026 | 38.2 | 1500 |
| 15 | EtO– | 0.349 | 36.2 | 104 |
| 16 | PrO– | 0.216 | 10.6 | 49.0 |
| 17 | n-BuO– | 0.19 | 27.9 | 150 |
| 18 | s-BuO– | 0.049 | 23.7 | 480 |
| 19 | C(CH3)3CH2O– | 0.14 | >100.0 | >700 |
| 20 | PhCH2O– | 0.018 | 41.1 | 2300 |
| 21 | PhNH– | 0.005 | 34.2 | 7000 |
| 22 | C8H17O– | 0.911 | >50.0 | >50 |
All data represent an average of triplicate experiments.
EC50, effective concentration (in μM) required to inhibit HIV-1 by 50%.
CC50, effective concentration (in μM) required to reduce the viability of uninfected cells by 50%.
Serafinowska and co-workers investigated the in vivo cleavage for a series of dialkyl and dibenzyl prodrugs of 9-[2-(phosphonomethoxy)ethoxy]adenine (PEEA).57 After oral administration to mice, the concentration of short chain dialkyl esters was relatively unchanged and little of the corresponding monoester was detected. As the chain length increased from methyl to n-Bu, the concentration of the monoester metabolite also increased, however, the monoesters failed to appreciably convert to free PEEA and accumulated in the blood. Our data suggests that cleavage of methyl monoester 14 in human PBMCs is superior to the corresponding ethyl, propyl, and n-butyl esters (15, 16, and 17, respectively) and that conversion to TFV increases with chain length for this series. Note that the antiviral activity of 14–22 directly corresponds to [TFV] and thus provides an indirect readout for the concentration of the free nucleoside. A rigorous comparison between these studies warrants caution due to significant differences in experimental design, method of quantitation, and the system under investigation (rat vs human PBMCs), nonetheless, the apparent dichotomy raises the possibility that the extent of alkoxyalkyl prodrug cleavage depends not only on the identity of the alkylmonoester but is equally dependent on the identity of the parent nucleoside.
Unfortunately, the effect of chain length could only be studied to a certain extent with our lipid delivery system. Longer alkyl chains impose significant phospholipid character with the HDE linker and become “stuck” in the plasma membrane, a phenomenon observed with conjugates 13a–e. The activity of compound 22 (0.911 μM) is likely plagued by the same phenomenon and obscures the relationship between the EC50 and enzymatic cleavage of the octyl ester. Fortunately, the remaining esters in Table 3 are sufficiently smaller than the HDE linker and do not exhibit this behavior.
Conjugates 17, 18, and 19 suggest that a moderate increase in steric encumbrance around the phosphonate center correlates with an increase in antiviral activity. s-Bu ester 18 is superior to its n-Bu analogue 17 by nearly 4-fold. The neopentyl derivative 19 had unimpressive activity of 0.14 μM although the corresponding n-pentyl ester was not synthesized for comparison.
Aryl phosphonoamidate 21 and benzyl ester 20 are the most potent compounds in Table 3 (5 and 18 nM, respectively), which is consistent with the notion that aryl and benzyl esters are superior substrates over their alkyl counterparts.
CONCLUSIONS
Our results demonstrate that disulfide-bearing lipid conjugates of tenofovir significantly improve the potency of the parent nucleoside by over 600-fold and that these conjugates readily permeate the cell membrane, are stable in human plasma for >24 h, and have significantly higher therapeutic indices than TDF against both HIV and HBV at the EC50 and EC90. Although we do not provide any direct evidence here, we believe this system relies on the presence of glutathione to facilitate reduction and does not require distal enzymatic activation for release. Further experiments are being conducted by our laboratory. This strategy has the potential for selective intracellular delivery and enhanced permeability to viral sanctuaries such as the gut-associated lymphatic tissue, central nervous system, and reproductive organs.
EXPERIMENTAL SECTION
Chemical Synthesis
All reagents were obtained from commercial suppliers and used without further purification. Reaction progress was monitored by either thin layer chromatography (TLC) using precoated aluminum-backed silica gel plates (60 F254 Merk, article 5554) or liquid chromatography–mass spectrometry (LC-MS) on an Agilent Technologies 6100 quadrupole instrument equipped with UV detection at 254 and 210 nm and a Varian C8 analytical column. Hanes reagent and UV detection at 254 nm were the preferred visualization agents for TLC. LC-MS analysis was performed using a stepwise H2O/MeOH gradient, with the % MeOH increasing from 75 to 95% over the course of 3 min unless otherwise specified. Flash column chromatography was conducted using CombiFlash Rf 200 (Telendyne-Isco) automated flash chromatography system with hand-packed RediSep columns. Evaporation of solvents was carried out on a rotary evaporator under reduced pressure and under ultrahigh vacuum (UHV) where appropriate. 1H NMR and 13C NMR spectra were recorded at ambient temperature on a Varian 400 spectrometer. 31P spectra were recorded at ambient temperature on either a Mercury 300 or Varian 400 spectrometer. Unless otherwise specified, all NMR spectra were obtained in deuterated chloroform (CDCl3) and referenced to the residual solvent peak. Chemical shifts are given in δ values, and coupling constants are reported in hertz (Hz). Coupling constants are not reported in the 13C spectrum of diastereomeric mixtures. Melting points were determined on a MelTemp melting apparatus and are uncorrected. High resolution mass-spectra (HRMS) were acquired on a VG 70-S Nier Johnson or JEOL mass spectrometer. Elemental analyses were performed by Atlantic Microlabs (Norcross, GA) for C, H, N analysis and are in agreement with the proposed structures with purity ≥95%.
General Procedure A
To a stirring solution of dry tenofovir (0.05 g, 0.174 mmol) in anhydrous DCM (6 mL) and N,N-dimethylformamide (0.016 mL, 0.209 mmol) was gradually added excess oxalyl chloride (0.075 mL, 0.870 mmol). The mixture stirred exposed to atmosphere for 1 h at room temperature until a colorless, transparent solution was observed and no starting material coated the walls of the vessel. The solvent and excess oxalyl chloride was evaporated under reduced pressure to produce a pale-yellow foam which was redissolved in anhydrous DCM (5 mL) and placed under argon. The vessel was equipped with a magnetic stir bar and chilled to 0 °C. Then a solution of 2-(hexadecyldisulfanyl)ethanol (0.070 g, 0.209 mmol) and pyridine (0.084 mL, 1.045 mmol) in anhydrous DCM was slowly added dropwise. After stirring for 10 min at this temperature, the mixture was naturally warmed to room temperature and stirred for an hour.
General Procedure B
To a stirring solution of dry tenofovir (0.1 g, 0.348 mmol) in anhydrous DCM (6 mL) and N,N-dimethylformamide (0.032 mL, 0.418 mmol) was gradually added excess oxalyl chloride (0.149 mL, 1.741 mmol). The mixture was stirred at rt exposed to air for 15 min or until complete dissolution of starting material was observed. The solvent and excess oxalyl chloride was evaporated under reduced pressure, and the resulting residue was placed under argon and redissolved in anhydrous DCM (5 mL) to afford a clear colorless solution. The mixture was then chilled to 0 °C.
2-((2-Hydroxyethyl)disulfanyl)ethyl Hydrogen ((((R)-1-(6-Amino-9H-purin-9-yl)propan-2-yl)oxy)methyl)phosphonate (DTE–TFV)
To a stirring solution of dry tenofovir (0.3 g, 1.045 mmol) in anhydrous DCM (100 mL) and N,N-dimethylformamide (0.121 mL, 1.567 mmol) was gradually added excess oxalyl dichloride (0.537 mL, 6.27 mmol) at room temperature. The mixture stirred for 1 h then cooled to 0 °C and quenched with excess 2-mercaptoethanol (0.735 mL, 10.45 mmol). The mixture was naturally warmed to room temperature and stirred for an additional hour. Progress was monitored by LC-MS (H2O/MeOH gradient, 50–95% MeOH, 3 min). Upon completion, aqueous HCl and methanol was added. The solution was stirred at room temperature (pH = 1) overnight. The pH was then gradually raised to 5 using a saturated solution of aqueous sodium bicarbonate and the mixture stirred for 2 h at room temperature. The organic solvents were removed under reduced pressure, and the resulting aqueous liquid was stirred overnight exposed to atmosphere. The resulting solid was then purified on a C18 reverse phase column using a H2O/MeOH gradient (1% formic acid) (10–20% MeOH) to afford the title compound (75 mg, 0.160 mmol, 15.29% yield) as a white solid. 1H NMR (400 MHz, D2O) δ 8.39 (s, 1H), 8.38 (s, 1H), 8.19 (s, 2H), 4.44 (dd, J = 14.7, 2.9 Hz, 1H), 4.26 (dd, J = 14.7, 7.8 Hz, 1H), 4.04–3.90 (m, 1H), 3.89–3.78 (m, 2H), 3.73 (dt, J = 11.7, 7.5 Hz, 3H), 3.47 (dd, J = 13.3, 9.8 Hz, 1H), 2.80–2.65 (m, 4H), 1.20 (d, J = 6.3 Hz, 3H). 13C NMR (101 MHz, D2O) δ 165.73, 149.74, 148.51, 145.54, 144.37, 117.55, 75.48 (d, J = 12.7 Hz), 62.83 (d, J = 159.4 Hz), 62.49 (d, J = 5.5 Hz), 59.03, 48.49, 39.80, 38.47 (d, J = 6.2 Hz), 15.54. 31P NMR (D2O) δ: 17.89. HRMS (ESI) m/z calculated for C13H23O5N5PS2 [M]+ 424.08727, found 424.08777. Anal. Calculated for C13H22N5O5PS2: C, 36.87; H, 5.24; N, 16.54. Found: C, 36.58; H, 5.24; N, 16.43. Melting point: 130–140 °C. Solid is amorphous.
2-Mercaptoethyl Hydrogen ((((R)-1-(6-Amino-9H-purin-9-yl)-propan-2yl)oxy)methyl)phosphonate (9)
To a stirring solution of dry tenofovir (1 g, 3.48 mmol) in anhydrous DCM (100 mL) and N,N-dimethylformamide (0.537 mL, 6.96 mmol) was gradually added excess oxalyl chloride (1.19 mL, 13.93 mmol) at room temperature and stirred for 20 min. The mixture was then cooled to 0 °C and quenched with excess 2-mercaptoethanol (2.451 mL, 34.8 mmol). The mixture stirred for 1 h at this temperature. Aqueous HCl was then added (3 mL), followed by 15 mL of methanol (pH = 1). The mixture stirred at room temperature overnight, then the pH was gradually raised to 5 using saturated aqueous sodium bicarbonate. The organic solvents were evaporated under reduced pressure at 30 °C and the resulting residue purified on a C18 reverse phase column using a H2O/MeOH gradient (isocratic 10% MeOH) to afford the title compound 2-mercaptoethyl hydrogen ((((R)-1-(6-amino-9H-purin-9-yl)propan-2-yl)oxy)methyl)phosphonate (0.6043 g, 1.740 mmol, 50.0% yield) as a white solid. 1H NMR (400 MHz, D2O) δ 8.38 (s, 1H), 8.37 (s, 1H), 4.44 (dd, J = 14.7, 3.1 Hz, 1H), 4.26 (dd, J = 14.7, 7.7 Hz, 1H), 4.04–3.92 (m, 1H), 3.77–3.66 (m, 3H), 3.48 (dd, J = 13.4, 9.6 Hz, 1H), 2.53 (td, J = 6.4, 4.2 Hz, 2H), 1.19 (d, J = 6.3 Hz, 3H). 13C NMR (101 MHz, D2O) δ 149.73, 148.53, 145.45, 144.35, 117.52, 75.55 (d, J = 12.5 Hz), 66.10 (d, J = 5.6 Hz), 62.92 (d, J = 159.5 Hz), 48.37, 24.40 (d, J = 6.5 Hz), 15.58. 31P NMR (162 MHz, D2O) δ 20.77. HRMS (ESI) m/z calculated for C11H19O4N5PS [M + H]+ 348.08899, found 348.08929. Anal. Calculated for C11H20N5O5PS (as a monohydrate): C, 36.16; H, 5.52; N, 19.17. Found: C, 35.73; H, 5.53; N, 19.07. Melting point: decomposes at 100 °C.
2-(Decyldisulfanyl)ethanol (11a)
To a stirring solution of decane-1-thiol (6.07 mL, 28.7 mmol) and 2-mercaptoethanol (2.019 mL, 28.7 mmol) in DCM/MeOH (75:25, 100 mL) was added pyridine (4.99 mL, 57.4 mmol) followed by the gradual addition of diiodine (7.28 g, 28.7 mmol). The solution stirred for 5 h at room temperature then the solvents were evaporated under reduced pressure and the resulting solid was dissolved in EtOAc and washed with brine. The organic layer was collected and dried over anhydrous sodium sulfate. Then the mixture was filtered, concentrated, and purified on a silica column using a hexanes/EtOAc gradient (0–12% EtOAc) to afford the title compound 2-(decyldisulfanyl)ethanol (3.10 g, 12.38 mmol, 43.2% yield) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 3.90 (dd, J = 11.9, 5.9 Hz, 2H), 2.85 (dd, J = 7.0, 4.6 Hz, 2H), 2.75–2.66 (m, 2H), 2.11 (t, J = 6.3 Hz, 1H), 1.75–1.62 (m, 2H), 1.58–1.19 (m, 14H), 0.88 (t, J = 6.9 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 60.26, 41.12, 39.02, 31.86, 29.52, 29.47, 29.28, 29.19, 29.11, 28.49, 22.66, 14.11. HRMS (ESI) m/z calculated for C12H26OS2Na [M + Na]+ 273.13173, found 273.13120.
2-(Dodecyldisulfanyl)ethanol (11b)
To a stirring solution of dodecane-1-thiol (5.92 mL, 24.70 mmol) and 2-mercaptoethanol (1.739 mL, 24.70 mmol) DCM/MeOH (75:25, 250 mL) was added pyridine (4.29 mL, 49.4 mmol) followed by the gradual addition of diiodine (6.27 g, 24.70 mmol). The solution stirred for 5 h at room temperature, then the solvents were evaporated under reduced pressure and the resulting solid was dissolved in EtOAc and washed with brine. The organic layer was collected and dried over anhydrous sodium sulfate. Then, the mixture was filtered, concentrated, and purified on a silica column using a hexanes/EtOAc gradient (0–15% EtOAc) to afford the title compound 2-(dodecyldisulfanyl)ethanol (3.072 g, 11.03 mmol, 44.6% yield) as a waxy solid. 1H NMR (400 MHz, CDCl3) δ 3.90 (dd, J = 11.8, 6.0 Hz, 2H), 2.85 (t, J = 5.8 Hz, 2H), 2.75–2.67 (m, 2H), 2.04 (t, J = 6.3 Hz, 1H), 1.77–1.58 (m, 2H), 1.47–1.16 (m, 18H), 0.89 (t, J = 6.9 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 60.26, 41.13, 39.02, 31.89, 29.62, 29.61, 29.56, 29.47, 29.33, 29.20, 29.11, 28.50, 22.67, 14.12. HRMS (ESI) m/z calculated for C14H30OS2Na [M + Na]+ 301.16303, found 301.16255. Melting point: 30–32 °C.
2-(Tetradecyldisulfanyl)ethanol (11c)
To a stirring solution of tetradecane-1-thiol (5.91 mL, 21.70 mmol) and 2-mercaptoethanol (1.527 mL, 21.70 mmol) in DCM/MeOH (75:25, 100 mL) was added pyridine (3.77 mL, 43.4 mmol) followed by the gradual addition of diiodine (5.51 g, 21.70 mmol). The solution stirred for 5 h at room temperature then the solvents were evaporated under reduced pressure and the resulting solid was dissolved in EtOAc and washed with brine. The organic layer was collected and dried over anhydrous sodium sulfate. Then, the mixture was filtered, concentrated, and purified on a silica column using a hexanes/EtOAc gradient (0–12% EtOAc) to afford the title compound 2-(tetradecyldisulfanyl)ethanol (2.941 g, 9.59 mmol, 44.2% yield) as a white foam. 1H NMR (400 MHz, CDCl3) δ 3.90 (dd, J = 11.9, 6.0 Hz, 2H), 2.85 (t, J = 5.8 Hz, 2H), 2.75–2.66 (m, 2H), 2.10 (t, J = 6.3 Hz, 1H), 1.74–1.62 (m, 2H), 1.47–1.06 (m, 22H), 0.88 (t, J = 6.9 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 60.26, 41.12, 39.02, 31.90, 29.67, 29.65, 29.63, 29.62, 29.57, 29.48, 29.34, 29.20, 29.11, 28.50, 22.68, 14.12. HRMS (ESI) m/z calculated for C16H34OS2Na [M + Na]+ 329.19433, found 329.19369. Melting point: 41–42 °C.
2-(Hexadecyldisulfanyl)ethanol (11d)
To a stirring solution of hexadecane-1-thiol (8.74 mL, 28.4 mmol) and 2-mercaptoethanol (2 mL, 28.4 mmol) in MeOH/DCM (50:50, 200 mL) was added pyridine (4.94 mL, 56.8 mmol) followed by the gradual added diiodine (7.21 g, 28.4 mmol) until the color of the solution remained brown. The solution stirred for 2 h at room temperature then the resulting suspension was filtered and the supernatant collected. The solvents were evaporated under reduced pressure, and the resulting solid was washed with water and extracted into DCM. The organic layer was concentrated and the residue purified on a silica column using hexanes/EtOAc (0–8%) gradient to afford the title compound 2-(hexadecyldisulfanyl)ethanol (3.98 g, 11.89 mmol, 41.9% yield) as a fluffy white powder. 1H NMR (400 MHz, CDCl3) δ 3.90 (q, J = 5.6 Hz, 2H), 2.95–2.80 (m, 2H), 2.77–2.63 (m, 2H), 2.01 (t, J = 6.0 Hz, 1H), 1.69 (dt, J = 14.9, 7.3 Hz, 2H), 1.46–1.17 (m, 26H), 0.89 (t, J = 6.9 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 60.29, 41.16, 39.05, 31.92, 29.69 (4), 29.66, 29.63, 29.58, 29.49, 29.36, 29.21, 29.13, 28.51, 22.69, 14.13. HRMS (ESI) m/z calculated for C18H38OS2Na [M + Na]+: 357.22563, found 357.22533. Melting point: 50–51 °C.
2-(Octadecyldisulfanyl)ethanol (11e)
To a stirring solution of octadecane-1-thiol (5.93 mL, 17.45 mmol) and 2-mercaptoethanol (1.224 mL, 17.45 mmol) in DCM/MeOH (75:25, 100 mL) was added pyridine (3.03 mL, 34.9 mmol) followed by the gradual addition of diiodine (4.43 g, 17.45 mmol) and stirred for 5 h at room temperature. Then, the suspension was filtered and the supernatant collected. The solvents were evaporated under reduced pressure and the resulting solid was dissolved in EtOAc and washed with brine. The organic layer was collected, dried over anhydrous sodium sulfate, and filtered. The crude was concentrated and purified on a silica column using a hexanes/EtOAc gradient (0–15% EtOAc) to afford the title compound 2-(octadecyldisulfanyl)ethanol (2.168 g, 5.98 mmol, 34.3% yield) as a white powder. 1H NMR (400 MHz, CDCl3) δ 3.90 (dd, J = 11.8, 5.9 Hz, 2H), 2.85 (t, J = 5.8 Hz, 2H), 2.75–2.68 (m, 2H), 2.03 (t, J = 6.2 Hz, 1H), 1.74–1.63 (m, 2H), 1.45–1.17 (m, 30H), 0.89 (t, J = 6.9 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 60.26, 41.13, 39.02, 31.91, 29.68 (3), 29.66 (2), 29.64 (2), 29.63, 29.57, 29.48, 29.35, 29.20, 29.11, 28.50, 22.68, 14.12. HRMS (ESI) m/z calculated for C20H42OS2Na [M + Na]+ 385.25693, found 385.25638. Melting point: 57–58 °C.
2-(Decyldisulfanyl)ethyl Hydrogen ((((R)-1-(6-Amino-9H-purin-9-yl)propan-2-yl)oxy)methyl)phosphonate (12a)
Following general procedure B, a mixture of 2-(decyldisulfanyl)ethanol (0.105 g, 0.418 mmol) and pyridine (0.168 mL, 2.089 mmol) in anhydrous DCM was slowly added dropwise to the reaction vessel. The mixture stirred at this temperature for 15 min, then naturally warmed to room temperature and stirred for 3 h. Then, aqueous HCl (1.2 M, 0.5 mL) was added and the mixture continued stirring for an additional 30 min. Methanol was added until the aqueous/organic interface disappeared and the reaction mixture stirred at room temperature overnight. Then, the solution was diluted with DCM, dried over anhydrous magnesium sulfate, filtered, and dried under UHV. The crude was dissolved in minimal DCM and purified on a silica column using a DCM/DCM:MeOH:NH4OH (80:20:1) gradient (0–60%) to afford the title compound 2-(decyldisulfanyl)ethyl hydrogen ((((R)-1-(6-amino-9H-purin-9-yl)propan-2-yl)oxy)methyl)phosphonate (88.9 mg, 0.171 mmol, 49.1% yield) as an off-white solid. 1H NMR (400 MHz, CD3OD) δ 8.36 (s, 1H), 8.24 (s, 1H), 4.42 (dd, J = 14.4, 3.1 Hz, 1H), 4.27 (dd, J = 14.4, 6.7 Hz, 1H), 4.08–4.00 (m, 2H), 3.95 (ddd, J = 9.5, 6.3, 3.1 Hz, 1H), 3.76 (dd, J = 12.8, 9.4 Hz, 1H), 3.53 (dd, J = 12.8, 10.0 Hz, 1H), 2.89–2.76 (m, 2H), 2.69 (dd, J = 15.7, 8.5 Hz, 2H), 1.65 (dt, J = 14.7, 7.2 Hz, 2H), 1.44–1.24 (m, 14H), 1.21 (d, J = 6.2 Hz, 3H), 0.92 (t, J = 6.9 Hz, 3H). Carbon referenced to CD3OD (49.0 ppm). 13C NMR (100 MHz, CD3OD) δ 157.37, 153.33, 151.71, 145.21, 120.41, 77.86 (d, J = 12.9 Hz), 66.37 (d, J = 160.4 Hz), 65.23 (d, J = 5.6 Hz), 67.17, 65.57, 65.26, 65.20, 49.99, 41.23 (d, J = 5.9 Hz), 40.81, 33.87, 31.47, 31.26, 31.17, 31.00, 30.32, 24.55, 17.78, 15.31. 31P NMR (162 MHz, CD3OD) δ 16.86. HRMS (ESI) m/z calculated for C21H39N5O4PS2 [M + H]+ 520.21756, found 520.21788. Melting point: 155–160 °C. Solid is amorphous. Anal. Calculated for C21H43N6O5PS2 (as a monohydrate ammonium salt): C, 45.47; H, 7.81; N, 15.15. Found: C, 45.56; H, 7.53; N, 15.10.
2-(Dodecyldisulfanyl)ethyl Hydrogen ((((R)-1-(6-Amino-9H-purin-9-yl)propan-2-yl)oxy)methyl)phosphonate (12b)
Following general procedure B, a mixture of 2-(dodecyldisulfanyl)ethanol (0.116 g, 0.418 mmol) and pyridine (0.168 mL, 2.089 mmol) in anhydrous DCM was slowly added dropwise to the reaction mixture. The solution stirred at this temperature for 15 min, then naturally warmed to room temperature and stirred for 3 h. Then, aqueous HCl (1.2 M, 0.5 mL) was added and the mixture continued stirring for an additional 30 min. Methanol was added until the aqueous/organic interface disappeared and the reaction mixture stirred overnight. Then, the solution was diluted with DCM and dried over anhydrous magnesium sulfate. The mixture was filtered and the supernatant collected, concentrated, and purified on a silica column using a DCM/DCM:MeOH:NH4OH (80:20:1) gradient (0–70%) to afford the title compound 2-(dodecyldisulfanyl)ethyl hydrogen ((((R)-1-(6-amino-9H-purin-9-yl)propan-2-yl)oxy)methyl)phosphonate (40.8 mg, 0.074 mmol, 21.39% yield) as a white solid. 1H NMR (399 MHz, CD3OD) δ 8.32 (s, 1H), 8.21 (s, 1H), 4.39 (dd, J = 14.4, 3.1 Hz, 1H), 4.23 (dd, J = 14.4, 6.8 Hz, 1H), 4.06–3.96 (m, 2H), 3.91 (td, J = 6.5, 3.1 Hz, 1H), 3.73 (dd, J = 12.8, 9.4 Hz, 1H), 3.50 (dd, J = 12.8, 10.1 Hz, 1H), 2.88–2.72 (m, 2H), 2.65 (dd, J = 14.8, 7.6 Hz, 2H), 1.67–1.55 (m, 2H), 1.41–1.22 (m, 18H), 1.18 (d, J = 6.2 Hz, 3H), 0.89 (t, J = 6.9 Hz, 3H). Carbon referenced to CD3OD signal (49.00 ppm). 13C NMR (100 MHz, CD3OD) δ 156.75, 152.82, 150.89, 144.30, 119.57, 76.98 (d, J = 12.9 Hz), 65.54 (d, J = 160.7 Hz), 64.39 (d, J = 5.6 Hz), 49.12, 40.39 (d, J = 5.9 Hz), 39.97, 33.04, 30.74, 30.72, 30.68, 30.62, 30.44, 30.32, 30.15, 29.48, 23.71, 16.89, 14.44. 31P NMR (162 MHz, CD3OD) δ 16.96. HRMS (ESI) m/z calculated for C23H43N5O4PS2 [M + H]+ 548.24886, found 548.24906. Anal. Calculated for C23H45N6O4PS2 (as an ammonium salt): C,48.92; H, 8.03; N, 14.88. Found: C, 48.51; H, 7.96; N, 14.06. Melting point: 165–170 °C.
2-(Tetradecyldisulfanyl)ethyl Hydrogen ((((R)-1-(6-Amino-9H-purin-9-yl)propan-2-yl)oxy)methyl)phosphonate (12c)
Following general procedure B, a mixture of 2-(tetradecyldisulfanyl)ethanol (0.128 g, 0.418 mmol) and pyridine (0.168 mL, 2.089 mmol) in anhydrous DCM was slowly added dropwise to the solution. The mixture stirred at this temperature for 15 min then naturally warmed to room temperature and stirred for 3 h. Then, aqueous HCl (1.2 M, 0.5 mL) was added and the mixture continued stirring for an additional 30 min. Methanol was added until the aqueous/organic interface disappeared and the reaction mixture stirred overnight. Then, the solution was diluted with DCM, dried over anhydrous magnesium sulfate, filtered, and dried under UHV. The crude was dissolved in minimal DCM and purified on a 4 g silica column using a DCM/DCM:MeOH:NH4OH (80:20:1) gradient (0–65%) to afford the title compound 2-(tetradecyldisulfanyl)ethyl hydrogen ((((R)-1-(6-amino-9H-purin-9-yl)propan-2-yl)oxy)methyl)phosphonate (27.5 mg, 0.048 mmol, 13.72% yield) as a white foam. Proton spectrum referenced to CD3OD (3.31 ppm). 1H NMR (399 MHz, CD3OD/CDCl3) δ 8.33 (s, 1H), 8.21 (s, 1H), 4.39 (dd, J = 14.4, 3.0 Hz, 1H), 4.22 (dd, J = 14.4, 6.8 Hz, 1H), 4.03 (q, J = 6.9 Hz, 2H), 3.90 (td, J = 6.5, 3.0 Hz, 1H), 3.75 (dd, J = 12.8, 9.4 Hz, 1H), 3.50 (dd, J = 12.7, 10.0 Hz, 1H), 2.88–2.73 (m, 2H), 2.65 (dd, J = 14.0, 6.7 Hz, 2H), 1.68–1.56 (m, 2H), 1.41–1.21 (m, 24H), 1.18 (d, J = 6.2 Hz, 3H), 0.88 (t, J = 6.9 Hz, 3H). Carbon spectrum referenced to CD3OD (49.0 ppm). 13C NMR (100 MHz, CD3OD/CDCl3) δ 155.79, 151.60, 150.52, 144.35, 119.31, 76.68 (d, J = 12.7 Hz), 65.26 (d, J = 160.0 Hz), 64.13 (d, J = 5.7 Hz), 40.16 (d, J = 6.0 Hz), 39.81, 32.82, 30.53, 30.51, 30.46, 30.40, 30.22, 30.12, 29.96, 29.31, 23.50, 16.84, 14.40. 31P NMR (162 MHz, CD3OD/CDCl3) δ 16.83. HRMS (ESI) m/z calculated for C25H47N5O4PS2 [M + H]+ 576.28016, found 576.28038. Anal. Calculated for C25H50N6O5PS2 (as a monohydrate ammonium salt): C, 49.24; H, 8.26; N, 13.78. Found: C, 49.33; H, 8.19; N, 13.55. Melting point: 149–154 °C.
2-(Hexadecyldisulfanyl)ethyl Hydrogen ((((R)-1-(6-Amino-9H-purin-9-yl)propan-2-yl)oxy)methyl)phosphonate (12d)
Following general procedure A, the reaction was quenched with excess water and stirred for 30 min at room temperature. Then, aqueous HCl and methanol was added and the reaction mixture stirred at room temperature overnight. Complete hydrolysis of formimidine was confirmed by LC-MS (isocratic 95% MeOH, 5% H2O, 7 min). The mixture was washed with brine and the crude extracted into DCM. The organic layer was collected, concentrated, and purified on a silica column using a DCM/DCM:MeOH:NH4OH (90:10:0.1) gradient (0–65%) to afford the title compound 2-(hexadecyldisulfanyl)ethyl hydrogen ((((R)-1-(6-amino-9H-purin-9-yl)propan-2-yl)oxy)methyl)-phosphonate (29.8 mg, 0.049 mmol, 28.3% yield) as a white solid. 1H NMR (400 MHz, CD3OD) δ 8.32 (s, 1H), 8.20 (s, 1H), 4.43–4.33 (m, 1H), 4.23 (dd, J = 14.4, 6.7 Hz, 1H), 4.00 (qd, J = 7.0, 1.7 Hz, 2H), 3.90 (pd, J = 6.3, 3.1 Hz, 1H), 3.73 (dd, J = 12.8, 9.4 Hz, 1H), 3.49 (dd, J = 12.7, 10.1 Hz, 1H), 2.86–2.73 (m, 2H), 2.64 (dd, J = 7.7, 6.8 Hz, 2H), 1.68–1.55 (m, 2H), 1.39–1.25 (m, 26H), 1.17 (d, J = 6.2 Hz, 3H), 0.94–0.85 (m, 3H). 13C NMR (101 MHz, CD3OD) δ 155.36, 151.48, 149.41, 142.91, 139.91, 118.10, 75.52 (d, J = 13.1 Hz), 64.05 (d, J = 160.2 Hz), 62.96 (d, J = 5.7 Hz), 38.87 (d, J = 6.2 Hz), 38.46, 31.65, 29.38, 29.35, 29.34, 29.28, 29.23, 29.06, 28.93, 28.71, 28.06, 22.32, 15.41, 13.04. 31P NMR (162 MHz, CD3OD) δ 16.87. HRMS (ESI) m/z calculated for C27H51O4N5PS2 [M + H]+ 604.31146, found 604.31149. Anal. Calculated for C27H53O5N6PS2 (as a monohydrate ammonium salt): C, 50.92; H, 8.39; N, 13.20. Found: C, 51.45; H, 8.42; N, 12.79. Melting point: 138–142 °C.
2-(Octadecyldisulfanyl)ethyl Hydrogen ((((R)-1-(6-Amino-9H-purin-9-yl)propan-2-yl)oxy)methyl)phosphonate (12e)
Following general procedure B, a mixture of 2-(octadecyldisulfanyl)ethanol (0.152 g, 0.418 mmol) and pyridine (0.168 mL, 2.089 mmol) in anhydrous DCM was slowly added dropwise to the solution. The mixture stirred at this temperature for 15 min, then naturally warmed to room temperature and stirred for 3 h. Then, aqueous HCl (1.2 M, 0.5 mL) was added and the mixture continued stirring for an additional 30 min. Methanol was added until the aqueous/organic interface disappeared and the reaction mixture stirred overnight. Then, the solution was diluted with DCM, dried over anhydrous magnesium sulfate, filtered, and dried under UHV. The crude was dissolved in minimal DCM and purified on a silica column using a DCM/DCM:MeOH:NH4OH (80:20:1) gradient (0–70%) to afford the title compound 2-(octadecyldisulfanyl)ethyl hydrogen ((((R)-1-(6-amino-9H-purin-9-yl)propan-2-yl)oxy)methyl)phosphonate (54.5 mg, 0.086 mmol, 24.77% yield) as an off-white solid. Proton referenced to residual TMS. 1H NMR (399 MHz, CD3OD/CDCl3) δ 8.23 (s, 1H), 8.12 (s, 1H), 4.30 (dd, J = 14.4, 2.9 Hz, 1H), 4.13 (dd, J = 14.4, 6.7 Hz, 1H), 3.93 (q, J = 6.9 Hz, 2H), 3.81 (td, J = 6.3, 3.0 Hz, 1H), 3.65 (dd, J = 12.7, 9.4 Hz, 1H), 3.40 (dd, J = 12.6, 10.1 Hz, 1H), 2.72 (td, J = 6.8, 2.9 Hz, 2H), 2.56 (dd, J = 14.3, 6.9 Hz, 2H), 1.59–1.45 (m, 2H), 1.28–1.13 (m, 30H), 1.09 (d, J = 6.2 Hz, 3H), 0.79 (t, J = 6.6 Hz, 3H). Carbon referenced to CD3OD signal (49.00 ppm). 13C NMR (100 MHz, CD3OD/CDCl3) δ 156.35, 152.48, 150.60, 144.07, 119.35, 76.73 (d, J = 12.7 Hz), 65.30 (d, J = 159.9 Hz), 64.14 (d, J = 5.6 Hz), 48.95, 40.15 (d, J = 6.8 Hz), 39.80, 32.82, 30.53, 30.46, 30.40, 30.22, 30.12, 29.96, 29.31, 23.50, 16.84, 14.41. 31P NMR (162 MHz, CD3OD/CDCl3) δ 16.76. HRMS (ESI) m/z calculated for C29H55N5O4PS2 32.34276, found 632.34318. Anal. Calculated for C29H59N6O5PS2 (as a monohydrate ammonium salt): C, 52.23; H, 8.92; N, 12.60. Found: C, 52.48; H, 8.88; N, 12.66. Melting point: 154–158 °C.
2-(Decyldisulfanyl)ethyl (2-(Decylsulfinothioyl)ethyl) ((((R)-1-(6-Amino-9H-purin-9-yl)propan-2-yl)oxy)methyl)phosphonate (13a)
Following general procedure B, a mixture of 2-(decyldisulfanyl)ethanol (0.192 g, 0.766 mmol) and pyridine (0.168 mL, 2.089 mmol) in anhydrous DCM was slowly added dropwise to the solution. The mixture stirred at this temperature for 15 min, then naturally warmed to room temperature and stirred for 3 h. Then, 1.2 M HCl (0.1 mL) was added and stirred for 30 min. Methanol was added until the organic and aqueous layers coalesced and the mixture stirred at room temperature overnight. The mixture was diluted with DCM, dried over anhydrous magnesium sulfate, and filtered. The supernatant was collected, concentrated, and purified on a silica column using a DCM/MeOH gradient (0–3% MeOH) to afford the title compound 2-(decyldisulfanyl)ethyl (2-(decylsulfinothioyl)ethyl) ((((R)-1-(6-amino-9H-purin-9-yl)propan-2-yl)oxy)methyl)phosphonate (97.7 mg, 0.130 mmol, 37.3% yield) as a viscous amorphous solid. 1H NMR (400 MHz, CDCl3) δ 8.33 (s, 1H), 7.96 (s, 1H), 6.10 (s, 2H), 4.35 (dd, J = 14.4, 3.1 Hz, 1H), 4.26 (ddt, J = 14.7, 11.5, 5.2 Hz, 4H), 4.13 (dd, J = 14.4, 7.6 Hz, 1H), 3.98–3.92 (m, 1H), 3.89 (dd, J = 13.7, 8.9 Hz, 1H), 3.64 (dd, J = 13.7, 9.5 Hz, 1H), 2.86 (dt, J = 16.6, 6.7 Hz, 4H), 2.70–2.63 (m, 4H), 1.69–1.57 (m, 4H), 1.41–1.10 (m, 31H), 0.86 (t, J = 6.8 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 155.55, 152.89, 150.07, 141.73, 119.19, 76.46 (d, J = 11.9 Hz), 64.23 (apparent t, J = 6.6 Hz), 62.65 (d, J = 168.5 Hz), 48.14, 39.07, 39.06, 38.38 (d, J = 5.7 Hz), 38.34 (d, J = 5.8 Hz), 31.85, 29.51, 29.48, 29.27, 29.20, 29.09, 28.47, 22.64, 16.54, 14.10. 31P NMR (162 MHz, CDCl3) δ 21.40. HRMS (ESI) m/z calculated for C33H63N5O4PS4 [M + H]+ 752.34950, found 752.35098. Anal. Calculated for C33H62N5O4PS4: C, 52.70; H, 8.31; N, 9.31. Found: C, 52.80; H, 8.46; N, 9.29.
2-(Dodecyldisulfanyl)ethyl (2-(Dodecylsulfinothioyl)ethyl) ((((R)-1-(6-Amino-9H-purin-9-yl)propan-2-yl)oxy)methyl)phosphonate (13b)
Following general procedure B, a mixture of 2-(dodecyldisulfanyl)ethanol (0.213 g, 0.766 mmol) and pyridine (0.168 mL, 2.089 mmol) in anhydrous DCM was slowly added dropwise to the solution. The mixture stirred at this temperature for 15 min, then naturally warmed to room temperature and stirred for 3 h. Then 1.2 M HCl (0.1 mL) was added and stirred for 30 min. Methanol was added until the organic and aqueous layers coalesced and the mixture stirred at room temperature overnight. The mixture was diluted with DCM, dried over anhydrous magnesium sulfate, and filtered. The supernatant was collected, concentrated, and purified on a silica column using a DCM/MeOH gradient (0–3% MeOH) to afford the title compound 2-(dodecyldisulfanyl)ethyl (2-(dodecylsulfinothioyl)ethyl) ((((R)-1-(6-amino-9H-purin-9-yl)-propan-2-yl)oxy)methyl)phosphonate (86.8 mg, 0.107 mmol, 30.8% yield) as an opaque, waxy solid. 1H NMR (400 MHz, CDCl3) δ 8.35 (s, 1H), 7.96 (s, 1H), 5.90 (s, 2H), 4.36 (dd, J = 14.4, 3.1 Hz, 1H), 4.33–4.21 (m, 4H), 4.14 (dd, J = 14.5, 7.6 Hz, 1H), 4.00–3.93 (m, 1H), 3.90 (dd, J = 13.7, 8.9 Hz, 1H), 3.64 (dd, J = 13.7, 9.5 Hz, 1H), 2.87 (dt, J = 17.8, 6.7 Hz, 4H), 2.72–2.62 (m, 4H), 1.70–1.59 (m, 4H), 1.44–1.11 (m, 39H), 0.87 (t, J = 6.9 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 155.67, 152.86, 150.02, 141.65, 119.15, 76.46 (d, J = 12.0 Hz), 64.25 (d, J = 5.4 Hz), 64.19 (d, J = 6.4 Hz), 62.65 (d, J = 168.5 Hz), 48.11, 39.04, 39.03, 38.37 (d, J = 3.4 Hz), 38.31 (d, J = 3.4 Hz), 31.86, 29.60, 29.58, 29.55, 29.47, 29.30, 29.20, 29.08, 28.47, 22.64, 16.53, 14.10. 31P NMR (162 MHz, CDCl3) δ 21.39. HRMS (ESI) m/z calculated for C37H71N5O4PS4 [M + H]+ 808.41210, found 808.41335. Anal. Calculated for C37H70N5O4PS4: C, 54.98; H, 8.73; N, 8.67. Found: C, 54.31; H, 8.97; N, 8.43. Melting point: 41–42 °C.
2-(Tetradecyldisulfanyl)ethyl (2-(Tetradecylsulfinothioyl)ethyl) ((((R)-1-(6-Amino-9H-purin-9-yl)propan-2-yl)oxy)methyl)-phosphonate (13c)
Following general procedure B, a mixture of 2-(tetradecyldisulfanyl)ethanol (0.128 g, 0.418 mmol) and pyridine (0.224 mL, 2.79 mmol) in anhydrous DCM was slowly added dropwise to the solution. The mixture stirred at this temperature for 15 min, then naturally warmed to room temperature and stirred for 3 h. Water (2 mL) was then added to the reaction mixture and stirred for an additional 30 min. The solvents were evaporated under reduced pressure, and the resulting residue was dissolved in ethanol (6 mL) and heated to 45 °C overnight to facilitate cleavage of formimidine. The solvent was again evaporated under reduced pressure, and the residue was purified on a silica column using a DCM/MeOH gradient (0–3% MeOH) to afford the title compound 2-(tetradecyldisulfanyl)-ethyl (2-(tetradecylsulfinothioyl)ethyl) ((((R)-1-(6-amino-9H-purin-9-yl)propan-2-yl)oxy)methyl)phosphonate (38.9 mg, 0.045 mmol, 12.93% yield) as a white foam. 1H NMR (400 MHz, CDCl3) δ 8.31 (s, 1H), 7.98 (s, 1H), 6.40 (s, 2H), 4.41–4.16 (m, 5H), 4.12 (dd, J = 14.4, 7.6 Hz, 1H), 4.00–3.81 (m, 2H), 3.62 (dd, J = 13.7, 9.5 Hz, 1H), 2.85 (dt, J = 16.7, 6.7 Hz, 4H), 2.70–2.60 (m, 4H), 1.70–1.54 (m, 4H), 1.42–1.13 (m, 47H), 0.91–0.77 (m, 6H). 13C NMR (101 MHz, CDCl3) δ 154.95, 151.72, 149.94, 142.13, 119.05, 76.39 (d, J = 11.7 Hz), 64.29 (d, J = 12.8 Hz), 64.23, 62.62 (d, J = 168.7 Hz), 48.22, 39.06, 39.05, 38.37 (d, J = 5.9 Hz), 38.34 (d, J = 5.8 Hz), 31.90, 29.67, 29.66, 29.63, 29.59, 29.51, 29.34, 29.23, 29.11, 28.50, 22.67, 16.52, 14.12. 31P NMR (162 MHz, CDCl3) δ 21.2. HRMS (ESI) m/z calculated for C41H79N5O4PS4 [M + H]+ 864.47470, found 864.47511. Anal. Calculated for C41H78N5O4PS4: C, 57.00; H, 9.10; N, 8.10. Found: C, 56.00; H, 9.23; N, 7.46. Melting point: 29–31 °C.
2-(Hexadecyldisulfanyl)ethyl (2-(Hexadecylsulfinothioyl)ethyl) ((((R)-1-(6-Amino-9H-purin-9-yl)propan-2-yl)oxy)methyl)-phosphonate (13d)
Following general procedure B, a mixture of 2-(hexadecyldisulfanyl)ethanol (0.256 g, 0.766 mmol) and pyridine (0.168 mL, 2.089 mmol) in anhydrous DCM was slowly added dropwise to the solution. The mixture stirred at this temperature for 15 min, then naturally warmed to room temperature and stirred for 3 h. Then 1.2 M HCl (0.1 mL) was added and stirred for 30 min. Methanol was added until the organic and aqueous layers coalesced and the mixture stirred at room temperature overnight. The mixture was diluted with DCM, dried over anhydrous magnesium sulfate, and filtered. The supernatant was collected, concentrated, and purified on a 4 g silica column using a DCM/MeOH gradient to afford the title compound 2-(hexadecyldisulfanyl)ethyl (2-(hexadecylsulfinothioyl)-ethyl) ((((R)-1-(6-amino-9H-purin-9-yl)propan-2-yl)oxy)methyl)-phosphonate (98.6 mg, 0.107 mmol, 30.8% yield) as a white solid. 1H NMR (400 MHz, CDCl3) δ 8.36 (s, 1H), 8.06 (s, 1H), 6.07 (s, 2H), 4.37 (dd, J = 14.4, 3.0 Hz, 1H), 4.33–4.22 (m, 4H), 4.16 (dd, J = 14.4, 7.6 Hz, 1H), 3.99–3.93 (m, 1H), 3.90 (dd, J = 13.7, 9.0 Hz, 1H), 3.65 (dd, J = 13.7, 9.5 Hz, 1H), 2.88 (dt, J = 17.6, 6.7 Hz, 4H), 2.73–2.63 (m, 4H), 1.70–1.59 (m, 4H), 1.46–1.14 (m, 55H), 0.87 (t, J = 6.9 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 155.46, 152.99, 150.02, 142.08, 118.71, 76.34 (d, J = 12.0 Hz), 64.29 (d, J = 4.6 Hz), 64.23 (d, J = 4.6 Hz), 62.64 (d, J = 168.7 Hz), 48.23, 39.03, 39.02, 38.38 (d, J = 2.4 Hz), 38.32 (d, J = 2.5 Hz), 31.89, 29.67, 29.63, 29.59, 29.50, 29.33, 29.23, 29.10, 28.49, 22.66, 16.49, 14.11. 31P NMR (162 MHz, CDCl3) δ 21.36. HRMS (ESI) m/z calculated for C45H87N5O4PS4 [M + H]+ 920.53730, found 920.53987. Anal. Calculated for C45H86N5O4PS4: C, 58.72; H, 9.42; N, 7.61. Found: C, 58.05; H, 9.50; N, 7.44. Melting point: 39–42 °C.
2-(Octadecyldisulfanyl)ethyl (2-(Octadecylsulfinothioyl)ethyl) ((((R)-1-(6-Amino-9H-purin-9-yl)propan-2-yl)oxy)methyl)-phosphonate (13e)
Following general procedure B, a mixture of 2-(octadecyldisulfanyl)ethanol (0.278 g, 0.766 mmol) and pyridine (0.168 mL, 2.089 mmol) in anhydrous DCM was slowly added dropwise to the solution. The mixture stirred at this temperature for 15 min, then naturally warmed to room temperature and stirred for 3 h. Then 1.2 M HCl (0.1 mL) was added and stirred for 30 min. Methanol was added until the organic and aqueous layers coalesced and the mixture stirred at room temperature overnight. The mixture was diluted with DCM, dried over anhydrous magnesium sulfate, and filtered. The supernatant was collected, concentrated, and purified on a silica column using a DCM/MeOH gradient (0–3% MeOH) to afford the title compound 2-(octadecyldisulfanyl)ethyl (2-(octadecylsulfinothioyl)ethyl) ((((R)-1-(6-amino-9H-purin-9-yl)-propan-2-yl)oxy)methyl)phosphonate (114.4 mg, 0.117 mmol, 33.6% yield) as a white solid. 1H NMR (400 MHz, CDCl3) δ 8.35 (s, 1H), 7.97 (s, 1H), 5.86 (s, 2H), 4.36 (dd, J = 14.4, 3.1 Hz, 1H), 4.27 (ddt, J = 16.3, 14.7, 5.2 Hz, 5H), 4.14 (dd, J = 14.4, 7.7 Hz, 1H), 3.99–3.93 (m, 1H), 3.90 (dd, J = 13.7, 8.9 Hz, 1H), 3.65 (dd, J = 13.7, 9.5 Hz, 1H), 2.87 (dt, J = 18.1, 6.7 Hz, 4H), 2.72–2.63 (m, 4H), 1.70–1.59 (m, 4H), 1.45–1.16 (m, 63H), 0.87 (t, J = 6.9 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 155.64, 152.83, 150.03, 141.68, 119.18, 76.46 (d, J = 11.9 Hz), 64.25 (d, J = 5.8 Hz), 64.19 (d, J = 5.7 Hz), 62.65 (d, J = 168.4 Hz), 48.13, 39.04, 39.03, 38.36 (d, J = 5.8 Hz), 38.32 (d, J = 5.7 Hz), 31.88, 29.67, 29.62, 29.58, 29.50, 29.33, 29.22, 29.09, 28.48, 22.65, 16.53, 14.10. 31P NMR (162 MHz, CDCl3) δ 21.40. HRMS (ESI) m/z calculated for C49H95N5O4PS4 [M + H]+ 976.59990, found 976.60338. Anal. Calculated for C49H94N5O4PS4: C, 60.27; H, 9.70; N, 7.17. Found: C, 59.99; H, 9.82; N, 7.14. Melting point: 56–57 °C.
2-(Hexadecyldisulfanyl)ethyl Methyl (((1-(6-Amino-9H-purin-9-yl)propan-2-yl)oxy)methyl)phosphonate (14)
Following general procedure A, the reaction was quenched with excess methanol and stirred for 30 min at room temperature. Then aqueous HCl (1.2 M) was added and the mixture stirred at room temperature overnight. Additional MeOH was added until the organic and aqueous phases coalesced. Reaction progress was monitored by LC-MS (isocratic 95% MeOH, 5% H2O, 7 min). The crude product was washed with brine and extracted into DCM. The organic layer was collected, concentrated, and purified on a silica column using a DCM/DCM:MeOH:NH4OH (90:10:0.1) gradient (0–26%) to afford the title compound 2-(hexadecyldisulfanyl)ethyl methyl (((1-(6-amino-9H-purin-9-yl)propan-2-yl)oxy)methyl)phosphonate (38 mg, 0.062 mmol, 35.3% yield) as a white solid. 1H NMR (400 MHz, CDCl3) δ 8.34 (s, 1H), 7.96 (s, 1H), 6.08 (s, 2H), 4.36 (dd, J = 14.4, 3.0 Hz, 1H), 4.30–4.20 (m, 2H), 4.13 (dd, J = 14.5, 7.8 Hz, 1H), 3.96–3.84 (m, 2H), 3.71 (dd, J = 23.5, 10.8 Hz, 3H), 3.62 (dd, J = 13.7, 9.0 Hz, 1H), 2.86 (ddd, J = 13.3, 12.4, 6.7 Hz, 2H), 2.72–2.63 (m, 2H), 1.69–1.59 (m, 2H), 1.33–1.22 (m, 29H), 0.87 (t, J = 6.8 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 155.35, 152.68, 150.06, 141.83, 119.15, δ 76.50 (d, J = 11.9 Hz), 76.47 (d, J = 11.7 Hz), 64.27 (d, J = 6.3 Hz), 64.16 (d, J = 6.4 Hz), 62.34 (d, J = 168.7 Hz), 52.92 (d, J = 6.5 Hz), 52.81 (d, J = 6.8 Hz), 48.22, 48.20, 41.23, 39.07, 39.05, 38.42, 31.90, 29.68, 29.66, 29.64, 29.58, 29.52, 29.50, 29.34, 29.24, 29.22, 29.13, 29.10, 28.51, 28.48, 22.68, 16.53 (d, J = 5.4 Hz), 14.12. 31P NMR (162 MHz, CDCl3) δ 22.16, 22.09. HRMS (ESI) m/z calculated for C28H53O4N5PS2 [M + H]+ 618.32711, found 618.32597. Anal. Calculated for C28H52N5O4PS2: C, 54.43; H, 8.48; N, 11.34. Found: C, 54.20; H, 8.56; N, 11.08. Melting Point: 48–50 °C.
(2-(Hexadecyldisulfanyl)ethyl) ((((R)-1-(6-Amino-9H-purin-9-yl)-propan-2-yl)oxy)methyl)phosphonate (15)
Following general procedure A, the reaction was quenched with excess EtOH and stirred at room temperature for 30 min. Formimidine hydrolysis was mediated by the addition of excess 2-aminoethanol. The solution stirred at room temperature until complete cleavage of formimidine was observed by LC-MS (isocratic 95% MeOH, 5% H2O, 7 min). The solvent was evaporated under reduced pressure, and the resulting residue was purified on a silica column using a DCM/DCM:MeOH:NH4OH (90:10:0.1) gradient (0–26%) to afford the title compound which required further purification on a C18 reverse phase column using a H2O/MeOH gradient (50–95% MeOH) to produce ethyl (2-(hexadecyldisulfanyl)ethyl) ((((R)-1-(6-amino-9H-purin-9-yl)propan-2-yl)oxy)methyl)phosphonate (14.5 mg, 0.023 mmol, 13.18% yield) as an amorphous solid. 1H NMR (400 MHz, CDCl3) δ 8.35 (s, 1H), 7.98 (s, 1H), 5.86 (s, 2H), 4.37 (dd, J = 14.4, 2.9 Hz, 1H), 4.26 (ddd, J = 21.3, 10.7, 4.3 Hz, 2H), 4.19–3.97 (m, 4H), 3.94 (dd, J = 7.8, 3.2 Hz, 1H), 3.88 (dd, J = 13.6, 8.9 Hz, 1H), 3.67–3.57 (m, 1H), 2.87 (dt, J = 19.2, 6.7 Hz, 2H), 2.68 (td, J = 7.2, 5.7 Hz, 2H), 1.65 (td, J = 14.5, 7.2 Hz, 2H), 1.49–1.14 (m, 31H), 0.88 (t, J = 6.7 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 155.31, 152.79, 141.84, 76.44, 76.32, 64.14, 64.08, 64.04, 63.97, 63.97, 63.51, 62.69, 62.62, 62.56, 61.83, 48.19, 39.07, 39.05, 38.46, 38.43, 38.40, 38.37, 31.90, 29.67, 29.65, 29.63, 29.58, 29.49, 29.34, 29.22, 29.10, 29.10, 28.48, 22.67, 16.53, 16.48, 16.41, 16.36, 14.11. 31P NMR (162 MHz, CDCl3) δ 21.91, 21.86. HRMS (ESI) m/z calculated for C29H54N5O4PS2 [M + H]+: 632.34276, found 632.34174. Anal. Calculated for C29H54N5O4PS2: C, 55.12; H, 8.61; N, 11.08. Found: C, 55.48; H, 8.37; N, 10.44.
2-(Hexadecyldisulfanyl)ethyl Propyl ((((R)-1-(6-Amino-9H-purin-9-yl)propan-2-yl)oxy)methyl)phosphonate (16)
Following general procedure A, the reaction was quenched with excess propanol and stirred at room temperature for 30 min. Then aqueous acetic acid was added and the reaction mixture stirred at room temperature overnight. Complete hydrolysis of formimidine was confirmed by LC-MS (isocratic 95% MeOH, 5% H2O, 7 min). The mixture was washed with brine and the crude extracted into DCM. The organic layer was collected, concentrated, and purified on a silica column using a DCM/DCM:MeOH:NH4OH (90:10:0.1) gradient (0–17%) to afford the title compound 2-(hexadecyldisulfanyl)ethyl propyl ((((R)-1-(6-amino-9H-purin-9-yl)propan-2-yl)oxy)methyl)phosphonate (46 mg, 0.071 mmol, 40.9% yield) as an amorphous solid. 1H NMR (400 MHz, CDCl3) δ 8.33 (s, 1H), 7.96 (d, J = 1.2 Hz, 1H), 6.08 (s, 3H), 4.35 (dd, J = 14.4, 3.0 Hz, 1H), 4.30–4.19 (m, 2H), 4.13 (ddd, J = 14.4, 7.6, 2.2 Hz, 1H), 4.03–3.83 (m, 4H), 3.62 (ddd, J = 13.6, 9.6, 2.5 Hz, 1H), 2.86 (dt, J = 16.2, 6.7 Hz, 2H), 2.71–2.59 (m, 2H), 1.73–1.56 (m, 4H), 1.44–1.04 (m, 28H), 0.89 (ddd, J = 13.7, 11.7, 7.2 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 155.52, 152.87, 150.08, 141.73, 119.16, 76.44, 76.42, 76.32, 76.31, 68.09, 68.03, 67.97, 64.15, 64.09, 64.06, 64.00, 63.46, 61.79, 48.17, 48.14, 39.07, 39.05, 38.48, 38.45, 38.42, 38.39, 31.88, 29.66, 29.64, 29.62, 29.57, 29.48, 29.33, 29.21, 29.09, 28.48, 23.87, 23.83, 23.81, 23.77, 22.66, 16.52, 16.50, 14.11, 9.96, 9.92. 31P NMR (121 MHz, CDCl3) δ 21.92, 21.87. HRMS (ESI) m/z calculated for C30H57O4N5PS2 [M + H]+ 646.35841, found 646.35757. Anal. Calculated for C30H56N5O4PS2: C, 55.79; H, 8.74; N, 10.84. Found: C, 54.97; H, 8.78; N, 10.07.
Butyl (2-(Hexadecyldisulfanyl)ethyl) ((((R)-1-(6-Amino-9H-purin-9-yl)propan-2-yl)oxy)methyl)phosphonate (17)
Following general procedure A, the reaction was quenched with excess n-butanol and stirred at room temperature for 30 min. Then aqueous acetic acid and methanol were added to remove the formimidine moiety. After stirring at room temperature overnight, complete cleavage was observed by LC-MS (isocratic 95% MeOH, 5% H2O, 7 min). The mixture was washed with brine, extracted into DCM, concentrated, and purified on a silica column using a DCM/DCM:MeOH:NH4OH (90:10:0.1) gradient (0–26%) to afford the title compound butyl (2-(hexadecyldisulfanyl)ethyl) ((((R)-1-(6-amino-9H-purin-9-yl)propan-2-yl)oxy)methyl)phosphonate (61.6 mg, 0.093 mmol, 53.6% yield) as an amorphous solid. 1H NMR (400 MHz, CDCl3) δ 8.34 (s, 1H), 7.96 (d, J = 1.4 Hz, 1H), 5.99 (s, 2H), 4.35 (dd, J = 14.4, 3.0 Hz, 1H), 4.29–4.19 (m, 2H), 4.13 (ddd, J = 14.5, 7.6, 2.8 Hz, 1H), 4.10–3.91 (m, 4H), 3.87 (dd, J = 13.6, 9.0 Hz, 1H), 3.62 (ddd, J = 13.6, 9.5, 3.0 Hz, 1H), 2.87 (dt, J = 15.2, 6.7 Hz, 2H), 2.71–2.61 (m, 2H), 1.68–1.54 (m, 4H), 1.36–1.17 (m, 30H), 0.96–0.81 (m, 6H). 13C NMR (101 MHz, CDCl3) δ 155.47, 152.85, 150.07, 150.05, 141.74, 119.12, 77.33, 77.02, 76.70, 76.44, 76.42, 76.31, 66.34, 64.14, 64.08, 64.01, 63.46, 61.78, 48.16, 48.13, 39.06, 39.05, 38.48, 38.45, 38.42, 38.39, 32.47, 32.43, 32.41, 32.38, 31.88, 29.65, 29.64, 29.62, 29.56, 29.48, 29.32, 29.21, 29.09, 28.47, 22.66, 18.63, 18.59, 16.52, 16.49, 14.10, 13.57, 13.56. 31P NMR (121 MHz, CDCl3) δ 21.14, 21.11. HRMS (ESI) m/z calculated for C31H59O4N5PS2 [M + H]+ 660.37406, found 660.37489. dr: 1:1 by 31P NMR. Anal. Calculated for C31H59N5O4PS2: C, 56.42; H, 8.86; N, 10.61. Found: C, 55.63; H, 8.87; N, 10.06.
2-(Hexadecyldisulfanyl)ethyl Isobutyl ((((R)-1-(6-Amino-9H-purin-9-yl)propan-2-yl)oxy)methyl)phosphonate (18)
Following general procedure A, the reaction was quenched with excess isobutanol and stirred at room temperature for 30 min. Formimidine hydrolysis was mediated by the addition of excess 2-aminoethanol. The solution stirred at room temperature until complete cleavage of formimidine was observed by LC-MS (isocratic 95% MeOH, 5% H2O, 7 min). Upon completion, the crude was washed with brine, extracted into DCM, concentrated, and purified on a silica column using a DCM/DCM:MeOH:NH4OH (90:10:0.1) gradient (0–33%) to afford the title compound 2-(hexadecyldisulfanyl)ethyl isobutyl ((((R)-1-(6-amino-9H-purin-9-yl)propan-2-yl)oxy)methyl)phosphonate (8.1 mg, 0.012 mmol, 7.05% yield) as an amorphous solid. 1H NMR (400 MHz, CDCl3) δ 8.35 (s, 1H), 7.97 (s, 1H), 5.74 (s, 2H), 4.37 (dd, J = 14.4, 3.0 Hz, 1H), 4.31–4.20 (m, 2H), 4.15 (dd, J = 14.4, 7.6 Hz, 1H), 3.98–3.74 (m, 4H), 3.64 (ddd, J = 13.6, 9.5, 1.4 Hz, 1H), 2.94–2.82 (m, 2H), 2.73–2.64 (m, 2H), 1.90 (dtd, J = 20.1, 13.4, 6.7 Hz, 1H), 1.69–1.58 (m, 2H), 1.39–1.21 (m, 28H), 0.98–0.81 (m, 9H). 13C NMR (101 MHz, CDCl3) δ 155.30, 152.87, 141.80, 119.14, 76.39, 76.37, 76.27, 76.25, 72.44, 72.38, 72.31, 64.19, 64.13, 64.09, 64.02, 63.38, 61.70, 48.21, 48.17, 39.09, 39.07, 38.49, 38.46, 38.44, 38.40, 31.90, 29.67, 29.63, 29.58, 29.50, 29.34, 29.22, 29.17, 29.15, 29.11, 28.49, 22.67, 18.62, 18.58, 16.51, 14.12. 31P NMR (121 MHz, CDCl3) δ 21.06, 21.03. HRMS (ESI) m/z calculated for C31H58O4N5PS2 [M + H]+ 660.37406, found 660.37445. Anal. Calculated for C31H57N5O4PS2: C, 56.42; H, 8.86; N, 10.61. Found: C, 56.65; H, 8.95; N, 10.25.
2-(Hexadecyldisulfanyl)ethyl Neopentyl ((((R)-1-(6-Amino-9H-purin-9-yl)propan-2-yl)oxy)methyl)phosphonate (19)
Following general procedure A, the reaction was quenched with excess neopentanol and stirred at room temperature for 30 min. Formimidine hydrolysis was mediated by the addition of methanolic ammonia (7N). The solution stirred at room temperature until complete cleavage of formimidine was observed by LC-MS (isocratic 95% MeOH, 5% H2O, 7 min). Upon completion, the solvents were evaporated under reduced pressure and the crude was purified on a silica column using a DCM/DCM:MeOH:NH4OH (90:10:0.1) gradient (0–26%) to afford the title compound 2-(hexadecyldisulfanyl)ethyl neopentyl ((((R)-1-(6-amino-9H-purin-9-yl)propan-2-yl)oxy)methyl)phosphonate (51.2 mg, 0.076 mmol, 43.6% yield) as an amorphous solid. 1H NMR (400 MHz, CDCl3) δ 8.31 (s, 1H), 7.98 (s, 1H), 6.33 (s, 2H), 4.36 (dd, J = 14.4, 2.8 Hz, 1H), 4.32–4.20 (m, 2H), 4.15 (dd, J = 14.5, 7.4 Hz, 1H), 3.95 (dd, J = 6.0, 4.6 Hz, 1H), 3.88 (dd, J = 13.6, 9.2 Hz, 1H), 3.75–3.55 (m, 3H), 2.87 (dt, J = 13.5, 6.7 Hz, 2H), 2.73–2.58 (m, 2H), 1.62 (dd, J = 10.7, 6.8 Hz, 2H), 1.36–1.11 (m, 29H), 0.94–0.75 (m, 12H). 13C NMR (101 MHz, CDCl3) δ 155.43, 152.45, 149.94, 149.93, 141.74, 118.84, 118.82, 76.30, 76.24, 76.18, 76.13, 75.73, 75.71, 75.66, 75.64, 64.22, 64.15, 64.09, 63.29, 61.61, 48.20, 48.16, 39.05, 39.04, 38.48, 38.45, 38.42, 38.39, 32.08, 32.03, 32.02, 31.97, 31.88, 29.65, 29.64, 29.61, 29.56, 29.47, 29.32, 29.20, 29.08, 28.47, 25.94, 25.90, 22.65, 16.48, 16.45, 14.10. 31P NMR (162 MHz, CDCl3) δ 20.95, 20.93. HRMS (ESI) m/z calculated for C32H61O4N5PS2 [M + H]+ 674.38971, found 674.38928. dr: 1.2:1 by 31P NMR. Anal. Calculated for C32H60N5O4PS2: C, 57.03; H, 8.97; N, 10.39. Found: C, 57.11; H,8.92; N, 10.11.
Benzyl (2-(Hexadecyldisulfanyl)ethyl) ((((R)-1-(6-Amino-9H-purin-9-yl)propan-2-yl)oxy)methyl)phosphonate (20)
Following general procedure A, the reaction was quenched with excess benzyl alcohol and stirred at room temperature for 30 min. Formimidine hydrolysis was mediated by the addition of methanolic ammonia (7N). The solution stirred at room temperature until complete cleavage of formimidine was observed by LC-MS (isocratic 95% MeOH, 5% H2O, 7 min). Upon completion, the crude was washed with brine, extracted into DCM, concentrated, and purified on a silica column using a DCM/DCM:MeOH:NH4OH (90:10:0.1) gradient (0–33%) to afford the title compound benzyl (2-(hexadecyldisulfanyl)ethyl) ((((R)-1-(6-amino-9H-purin-9-yl)propan-2-yl)oxy)methyl)phosphonate (64.6 mg, 0.093 mmol, 53.5% yield) as an amorphous solid. 1H NMR (400 MHz, CDCl3) δ 8.34 (s, 1H), 7.94 (s, 1H), 7.34 (ddd, J = 16.8, 7.7, 6.2 Hz, 5H), 6.31 (s, 2H), 5.15–5.01 (m, 2H), 4.33–4.07 (m, 4H), 3.95–3.78 (m, 2H), 3.65–3.50 (m, 1H), 2.82 (dt, J = 13.4, 6.5 Hz, 2H), 2.72–2.60 (m, 2H), 1.68–1.58 (m, 2H), 1.45–1.09 (m, 28H), 0.88 (t, J = 6.5 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 155.64, 155.63, 152.87, 150.00, 141.68, 135.90, 135.84, 135.78, 128.58, 128.57, 128.07, 119.07, 76.45, 76.42, 76.33, 76.30, 68.07, 68.01, 64.13, 64.09, 64.07, 64.03, 63.69, 62.01, 48.07, 39.04, 38.34, 38.31, 38.28, 38.25, 31.88, 29.65, 29.61, 29.56, 29.48, 29.32, 29.20, 29.09, 29.06, 28.48, 28.46, 22.65, 16.53, 16.43, 14.11. 31P NMR (121 MHz, CDCl3) δ 21.65 (2). HRMS (ESI) m/z calculated for C34H56O4N5PS2 [M + H]+ 694.35841, found 694.35784. Anal. Calculated for C34H55N5O4PS2: C, 58.85; H, 8.13; N, 10.09. Found: C, 58.50; H, 8.08; N, 9.69.
2-(Hexadecyldisulfanyl)ethyl P-((((R)-1-(6-Amino-9H-purin-9-yl)-propan-2-yl)oxy)methyl)-N-phenylphosphonamidate (21)
Following general procedure A, the reaction was quenched with excess aniline and stirred at room temperature for 30 min. Then formimidine hydrolysis was mediated by the addition of excess 2-aminoethanol. The solution stirred at room temperature until complete cleavage of formimidine was observed by LC-MS (isocratic 95% MeOH, 5% H2O, 7 min). The solvent was evaporated under reduced pressure, and the resulting residue was purified on a silica column using a DCM/DCM:MeOH:NH4OH (90:10:0.1) gradient (0–26%) to afford the title compound which required further purification on a C18 reverse phase column using a H2O/MeOH gradient (50–95% MeOH) to produce unadulterated 2-(hexadecyldisulfanyl)ethyl P-((((R)-1-(6-amino-9H-purin-9-yl)propan-2-yl)oxy)methyl)-N-phenylphosphonamidate (39.6 mg, 0.058 mmol, 33.5% yield) as a white foam. 1H NMR (400 MHz, CDCl3) δ 8.29 (s, 1H), 8.27 (s, 1H), 7.95 (s, 1H), 7.91 (s, 1H), 7.18 (dt, J = 13.5, 7.8 Hz, 2H), 6.93 (ddd, J = 18.7, 15.8, 7.9 Hz, 3H), 6.69 (s, 2H), 4.44–4.15 (m, 4H), 4.12–3.89 (m, 3H), 3.83–3.67 (m, 2H), 2.93–2.84 (m, 2H), 2.62 (t, J = 7.4 Hz, 2H), 1.64–1.56 (m, 2H), 1.34–1.21 (m, 22H), 1.19 (d, J = 6.2 Hz, 2H), 1.12 (d, J = 6.2 Hz, 2H), 0.88 (t, J = 6.8 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 155.57, 152.48, 152.45, 149.96, 149.81, 141.85, 139.44, 139.25, 129.45, 129.38, 122.23, 118.86, 118.80, 118.12, 118.06, 118.00, 117.94, 76.48, 76.36, 76.11, 76.00, 63.23, 63.21, 62.56, 62.50, 61.68, 61.66, 48.33, 48.22, 38.95, 38.28, 38.22, 31.89, 29.67, 29.65, 29.63, 29.57, 29.48, 29.33, 29.20, 29.07, 28.46, 22.67, 16.40, 16.22, 14.12. 31P NMR (121 MHz, CDCl3) δ 21.83, 21.76. HRMS (ESI) m/z calculated for C33H56O3N6PS2 [M + H]+ 679.35874, found 679.35910. dr: 1.5:1 by 31P NMR. Anal. Calculated for C33H56N5O4PS2: C, 58.38; H, 8.17; N, 12.38. Found: C, 57.69; H,7.81; N, 11.58.
2-(Hexadecyldisulfanyl)ethyl Octyl ((((R)-1-(6-Amino-9H-purin-9-yl)propan-2-yl)oxy)methyl)phosphonate (22)
Following general procedure A, the reaction was quenched with excess 1-octanol and stirred for an additional 2 h. After stirring, the solvent was evaporated under reduced pressure and the residue redissolved in ethanol. The mixture was heated to 45 °C and stirred overnight. Then the mixture was washed with brine and the crude extracted into DCM. The organic layer was collected, concentrated, and purified on a silica column using a DCM/DCM:MeOH:NH4OH (90:10:0.1) gradient (0–18%) to afford the title compound 2-(hexadecyldisulfanyl)ethyl octyl ((((R)-1-(6-amino-9H-purin-9-yl)propan-2-yl)oxy)methyl)phosphonate (75.2 mg, 0.105 mmol, 60.3% yield) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 8.35 (s, 1H), 7.97 (s, 1H), 5.85 (s, 2H), 4.36 (dd, J = 14.4, 3.0 Hz, 1H), 4.30–4.19 (m, 2H), 4.14 (ddd, J = 14.4, 7.6, 2.4 Hz, 1H), 4.08–3.83 (m, 4H), 3.62 (ddd, J = 13.5, 9.5, 3.6 Hz, 1H), 2.94–2.81 (m, 2H), 2.73–2.63 (m, 2H), 1.72–1.51 (m, 4H), 1.41–1.10 (m, 39H), 0.93–0.71 (m, 6H). 13C NMR (101 MHz, CDCl3) δ 155.40, 155.39, 152.88, 150.13, 150.11, 141.79, 119.21, 119.20, 76.44, 76.33, 66.71, 66.65, 66.58, 64.15, 64.09, 64.06, 63.99, 63.50, 61.82, 48.17, 48.13, 39.08, 39.07, 38.49, 38.45, 38.43, 38.40, 31.89, 31.74, 30.53, 30.49, 30.47, 30.43, 29.67, 29.63, 29.58, 29.50, 29.34, 29.23, 29.16, 29.15, 29.10, 29.09, 29.07, 28.49, 25.41, 25.37, 22.67, 22.61, 16.54, 16.52, 14.11, 14.08. 31P NMR (162 MHz, CDCl3) δ 21.09, 21.07. dr: 1.2:1. HRMS (ESI) m/z calcd for C35H67O4N5PS2 [M + H]+ 716.43666, found 716.43556. Anal. Calculated for C35H66O4N5PS2: C, 58.71; H, 9.29; N, 9.78. Found: C, 58.53; H, 9.35; N, 9.52.
Supplementary Material
Acknowledgments
We acknowledge Manohar Saindane for transporting numerous samples from Emory to the EIDD for analysis.
ABBREVIATIONS USED
- DCM
dichloromethane
- DMF
dimethylformamide
- TFV
tenofovir
- TDF
tenofovir disoproxil fumarate
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmed-chem.6b00428.
Assay details, stability studies, and associated spectra for all synthesized compounds (PDF) Molecular formula strings (CSV)
Author Contributions
Kyle Giesler drafted the manuscript and performed the chemical synthesis. Jose Marengo conducted stability studies and provided edits to the final manuscript. All authors have given approval to the final version of the manuscript.
Notes
The authors declare no competing financial interest.
References
- 1.Gallant JE, DeJesus E, Arribas JR, Pozniak AL, Gazzard B, Campo RE, Lu B, McColl D, Chuck S, Enejosa J, Toole JJ, Cheng AK. Tenofovir Df, Emtricitabine, and Efavirenz Vs. Zidovudine, Lamivudine, and Efavirenz for Hiv. N Engl J Med. 2006;354:251–260. doi: 10.1056/NEJMoa051871. [DOI] [PubMed] [Google Scholar]
- 2.Fung HB, Stone EA, Piacenti FJ. Tenofovir Disoproxil Fumarate: A Nucleotide Reverse Transcriptase Inhibitor for the Treatment of Hiv Infection. Clin Ther. 2002;24:1515–1548. doi: 10.1016/s0149-2918(02)80058-3. [DOI] [PubMed] [Google Scholar]
- 3.De Clercq E. Clinical Potential of the Acyclic Nucleoside Phosphonates Cidofovir, Adefovir, and Tenofovir in Treatment of DNA Virus and Retrovirus Infections. Clin Microbiol Rev. 2003;16:569–596. doi: 10.1128/CMR.16.4.569-596.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ying C, De Clercq E, Nicholson W, Furman P, Neyts J. Inhibition of the Replication of the DNA Polymerase M550v Mutation Variant of Human Hepatitis B Virus by Adefovir, Tenofovir, L-Fmau, Dapd, Penciclovir and Lobucavir. J Viral Hepat. 2000;7:161–165. doi: 10.1046/j.1365-2893.2000.00210.x. [DOI] [PubMed] [Google Scholar]
- 5.Delaney WEt, Ray AS, Yang H, Qi X, Xiong S, Zhu Y, Miller MD. Intracellular Metabolism and in Vitro Activity of Tenofovir against Hepatitis B Virus. Antimicrob Agents Chemother. 2006;50:2471–2477. doi: 10.1128/AAC.00138-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Andrei G, Lisco A, Vanpouille C, Introini A, Balestra E, van den Oord J, Cihlar T, Perno CF, Snoeck R, Margolis L, Balzarini J. Topical Tenofovir, a Microbicide Effective against Hiv, Inhibits Herpes Simplex Virus-2 Replication. Cell Host Microbe. 2011;10:379–389. doi: 10.1016/j.chom.2011.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.De Clercq E, Holy A. Acyclic Nucleoside Phosphonates: A Key Class of Antiviral Drugs. Nat Rev Drug Discovery. 2005;4:928–940. doi: 10.1038/nrd1877. [DOI] [PubMed] [Google Scholar]
- 8.Kearney BP, Flaherty JF, Shah J. Tenofovir Disoproxil Fumarate - Clinical Pharmacology and Pharmacokinetics. Clin Pharmacokinet. 2004;43:595–612. doi: 10.2165/00003088-200443090-00003. [DOI] [PubMed] [Google Scholar]
- 9.Balzarini J, Naesens L, Slachmuylders J, Niphuis H, Rosenberg I, Holy A, Schellekens H, De Clercq E. 9-(2-Phosphonylmethoxyethyl)Adenine (Pmea) Effectively Inhibits Retrovirus Replication in Vitro and Simian Immunodeficiency Virus Infection in Rhesus Monkeys. AIDS. 1991;5:21–28. doi: 10.1097/00002030-199101000-00003. [DOI] [PubMed] [Google Scholar]
- 10.Starrett JE, Jr, Tortolani DR, Russell J, Hitchcock MJM, Whiterock V, Martin JC, Mansuri MM. Synthesis, Oral Bioavailability Determination, and in Vitro Evaluation of Prodrugs of the Antiviral Agent 9-[2-(Phosphonomethoxy)Ethyl]Adenine (Pmea) J Med Chem. 1994;37:1857–1864. doi: 10.1021/jm00038a015. [DOI] [PubMed] [Google Scholar]
- 11.Pradere U, Garnier-Amblard EC, Coats SJ, Amblard F, Schinazi RF. Synthesis of Nucleoside Phosphate and Phosphonate Prodrugs. Chem Rev. 2014;114:9154–9218. doi: 10.1021/cr5002035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ray AS, Hostetler KY. Application of Kinase Bypass Strategies to Nucleoside Antivirals. Antiviral Res. 2011;92:277–291. doi: 10.1016/j.antiviral.2011.08.015. [DOI] [PubMed] [Google Scholar]
- 13.Hecker SJ, Erion MD. Prodrugs of Phosphates and Phosphonates. J Med Chem. 2008;51:2328–2345. doi: 10.1021/jm701260b. [DOI] [PubMed] [Google Scholar]
- 14.Karras A, Lafaurie M, Furco A, Bourgarit A, Droz D, Sereni D, Legendre C, Martinez F, Molina JM. Tenofovir-Related Nephrotoxicity in Human Immunodeficiency Virus-Infected Patients: Three Cases of Renal Failure, Fanconi Syndrome, and Nephrogenic Diabetes Insipidus. Clin Infect Dis. 2003;36:1070–1073. doi: 10.1086/368314. [DOI] [PubMed] [Google Scholar]
- 15.Coca S, Perazella MA. Rapid Communication: Acute Renal Failure Associated with Tenofovir: Evidence of Drug-Induced Nephrotoxicity. Am J Med Sci. 2002;324:342–344. doi: 10.1097/00000441-200212000-00011. [DOI] [PubMed] [Google Scholar]
- 16.Lewis W, Day BJ, Copeland WC. Mitochondrial Toxicity of Nrti Antiviral Drugs: An Integrated Cellular Perspective. Nat Rev Drug Discovery. 2003;2:812–822. doi: 10.1038/nrd1201. [DOI] [PubMed] [Google Scholar]
- 17.Herlitz LC, Mohan S, Stokes MB, Radhakrishnan J, D’Agati VD, Markowitz GS. Tenofovir Nephrotoxicity: Acute Tubular Necrosis with Distinctive Clinical, Pathological, and Mitochondrial Abnormalities. Kidney Int. 2010;78:1171–1177. doi: 10.1038/ki.2010.318. [DOI] [PubMed] [Google Scholar]
- 18.Ruane PJ, DeJesus E, Berger D, Markowitz M, Bredeek UF, Callebaut C, Zhong LJ, Ramanathan S, Rhee MS, Fordyce MW, Yale K. Antiviral Activity, Safety, and Pharmacokinetics/Pharmacodynamics of Tenofovir Alafenamide as 10-Day Monotherapy in Hiv-1-Positive Adults. JAIDS, J Acquired Immune Defic Syndr. 2013;63:449–455. doi: 10.1097/QAI.0b013e3182965d45. [DOI] [PubMed] [Google Scholar]
- 19.Markowitz M, Zolopa A, Squires K, Ruane P, Coakley D, Kearney B, Zhong LJ, Wulfsohn M, Miller MD, Lee WA. Phase I/Ii Study of the Pharmacokinetics, Safety and Antiretroviral Activity of Tenofovir Alafenamide, a New Prodrug of the Hiv Reverse Transcriptase Inhibitor Tenofovir, in Hiv-Infected Adults. J Antimicrob Chemother. 2014;69:1362–1369. doi: 10.1093/jac/dkt532. [DOI] [PubMed] [Google Scholar]
- 20.Painter GR, Almond MR, Trost LC, Lampert BM, Neyts J, De Clercq E, Korba BE, Aldern KA, Beadle JR, Hostetler KY. Evaluation of Hexadecyloxypropyl-9-R-[2-(Phosphonomethoxy)Propyl]-Adenine, Cmx157, as a Potential Treatment for Human Immunodeficiency Virus Type 1 and Hepatitis B Virus Infections (Vol 51, Pg 3505, 2007) Antimicrob Agents Chemother. 2007;51:4538–4538. doi: 10.1128/AAC.00460-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Birkus G, Kutty N, He GX, Mulato A, Lee W, McDermott M, Cihlar T. Activation of 9-[(R)-2-[[(S)-[[(S)-1-(Isopropoxycarbonyl)Ethyl]Amino] Phenoxyphosphinyl]-Methoxy]-Propyl]Adenine (Gs-7340) and Other Tenofovir Phosphonoamidate Prodrugs by Human Proteases. Mol Pharmacol. 2008;74:92–100. doi: 10.1124/mol.108.045526. [DOI] [PubMed] [Google Scholar]
- 22.Hostetler KY. Alkoxyalkyl Prodrugs of Acyclic Nucleoside Phosphonates Enhance Oral Antiviral Activity and Reduce Toxicity: Current State of the Art. Antiviral Res. 2009;82:A84–98. doi: 10.1016/j.antiviral.2009.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chimerix’s Antiviral Cmx157 Demonstrates Positive Phase 1 Results with Favorable Pharmacokinetics, Safety, and Tolerability. Chimerix; Durham, NC: Dec 13, 2010. http://ir.chimerix.com/releasedetail.cfm?releaseid=752310. [Google Scholar]
- 24.Vadlapudi AD, Vadlapatla RK, Mitra AK. Sodium Dependent Multivitamin Transporter (Smvt): A Potential Target for Drug Delivery. Curr Drug Targets. 2012;13:994–1003. doi: 10.2174/138945012800675650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vadlapudi AD, Vadlapatla RK, Kwatra D, Earla R, Samanta SK, Pal D, Mitra AK. Targeted Lipid Based Drug Conjugates: A Novel Strategy for Drug Delivery. Int J Pharm. 2012;434:315–324. doi: 10.1016/j.ijpharm.2012.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vadlapudi AD, Vadlapatla RK, Earla R, Sirimulla S, Bailey JB, Pal D, Mitra AK. Novel Biotinylated Lipid Prodrugs of Acyclovir for the Treatment of Herpetic Keratitis (Hk): Transporter Recognition, Tissue Stability and Antiviral Activity. Pharm Res. 2013;30:2063–2076. doi: 10.1007/s11095-013-1059-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schafer FQ, Buettner GR. Redox Environment of the Cell as Viewed through the Redox State of the Glutathione Disulfide/Glutathione Couple. Free Radical Biol Med. 2001;30:1191–1212. doi: 10.1016/s0891-5849(01)00480-4. [DOI] [PubMed] [Google Scholar]
- 28.Gilbert HF. [2] Thiol/Disulfide Exchange Equilibria and Disulfidebond Stability. Methods Enzymol. 1995;251:8–28. doi: 10.1016/0076-6879(95)51107-5. [DOI] [PubMed] [Google Scholar]
- 29.Coles B, Ketterer B, Hinson JA. The Role of Glutathione and Glutathione Transferases in Chemical Carcinogenesis. Crit Rev Biochem Mol Biol. 1990;25:47–70. doi: 10.3109/10409239009090605. [DOI] [PubMed] [Google Scholar]
- 30.Pastore A, Federici G, Bertini E, Piemonte F. Analysis of Glutathione: Implication in Redox and Detoxification. Clin Chim Acta. 2003;333:19–39. doi: 10.1016/s0009-8981(03)00200-6. [DOI] [PubMed] [Google Scholar]
- 31.Droge W, Schulze-Osthoff K, Mihm S, Galter D, Schenk H, Eck HP, Roth S, Gmunder H. Functions of Glutathione and Glutathione Disulfide in Immunology and Immunopathology. FASEB J. 1994;8:1131–1138. [PubMed] [Google Scholar]
- 32.Schulz JB, Lindenau J, Seyfried J, Dichgans J. Glutathione, Oxidative Stress and Neurodegeneration. Eur J Biochem. 2000;267:4904–4911. doi: 10.1046/j.1432-1327.2000.01595.x. [DOI] [PubMed] [Google Scholar]
- 33.Wang YC, Wang F, Sun TM, Wang J. Redox-Responsive Nanoparticles from the Single Disulfide Bond-Bridged Block Copolymer as Drug Carriers for Overcoming Multidrug Resistance in Cancer Cells. Bioconjugate Chem. 2011;22:1939–1945. doi: 10.1021/bc200139n. [DOI] [PubMed] [Google Scholar]
- 34.Kam NW, Liu Z, Dai H. Functionalization of Carbon Nanotubes Via Cleavable Disulfide Bonds for Efficient Intracellular Delivery of Sirna and Potent Gene Silencing. J Am Chem Soc. 2005;127:12492–12493. doi: 10.1021/ja053962k. [DOI] [PubMed] [Google Scholar]
- 35.Abdullah-Al-Nahain, Lee H, Lee YS, Lee KD, Park SY. Development of Disulfide Core-Crosslinked Pluronic Nanoparticles as an Effective Anticancer-Drug-Delivery System. Macromol Biosci. 2011;11:1264–1271. doi: 10.1002/mabi.201100083. [DOI] [PubMed] [Google Scholar]
- 36.Hirosue S, Kourtis IC, van der Vlies AJ, Hubbell JA, Swartz MA. Antigen Delivery to Dendritic Cells by Poly(Propylene Sulfide) Nanoparticles with Disulfide Conjugated Peptides: Cross-Presentation and T Cell Activation. Vaccine. 2010;28:7897–7906. doi: 10.1016/j.vaccine.2010.09.077. [DOI] [PubMed] [Google Scholar]
- 37.Cerritelli S, Velluto D, Hubbell JA. Peg-Ss-Pps: Reduction-Sensitive Disulfide Block Copolymer Vesicles for Intracellular Drug Delivery. Biomacromolecules. 2007;8:1966–1972. doi: 10.1021/bm070085x. [DOI] [PubMed] [Google Scholar]
- 38.Koo AN, Lee HJ, Kim SE, Chang JH, Park C, Kim C, Park JH, Lee SC. Disulfide-Cross-Linked Peg-Poly(Amino Acid)S Copolymer Micelles for Glutathione-Mediated Intracellular Drug Delivery. Chem Commun. 2008:6570–6572. doi: 10.1039/b815918a. [DOI] [PubMed] [Google Scholar]
- 39.Kurtoglu YE, Navath RS, Wang B, Kannan S, Romero R, Kannan RM. Poly(Amidoamine) Dendrimer-Drug Conjugates with Disulfide Linkages for Intracellular Drug Delivery. Biomaterials. 2009;30:2112–2121. doi: 10.1016/j.biomaterials.2008.12.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McKenzie DL, Smiley E, Kwok KY, Rice KG. Low Molecular Weight Disulfide Cross-Linking Peptides as Nonviral Gene Delivery Carriers. Bioconjugate Chem. 2000;11:901–909. doi: 10.1021/bc000056i. [DOI] [PubMed] [Google Scholar]
- 41.Tang FX, Hughes JA. Introduction of a Disulfide Bond into a Cationic Lipid Enhances Transgene Expression of Plasmid DNA. Biochem Biophys Res Commun. 1998;242:141–145. doi: 10.1006/bbrc.1997.7923. [DOI] [PubMed] [Google Scholar]
- 42.Brown SD, Nativo P, Smith JA, Stirling D, Edwards PR, Venugopal B, Flint DJ, Plumb JA, Graham D, Wheate NJ. Gold Nanoparticles for the Improved Anticancer Drug Delivery of the Active Component of Oxaliplatin. J Am Chem Soc. 2010;132:4678–4684. doi: 10.1021/ja908117a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ghosh P, Han G, De M, Kim CK, Rotello VM. Gold Nanoparticles in Delivery Applications. Adv Drug Delivery Rev. 2008;60:1307–1315. doi: 10.1016/j.addr.2008.03.016. [DOI] [PubMed] [Google Scholar]
- 44.Slowing II, Vivero-Escoto JL, Wu CW, Lin VSY. Mesoporous Silica Nanoparticles as Controlled Release Drug Delivery and Gene Transfection Carriers. Adv Drug Delivery Rev. 2008;60:1278–1288. doi: 10.1016/j.addr.2008.03.012. [DOI] [PubMed] [Google Scholar]
- 45.Benzaria S, Pelicano H, Johnson R, Maury G, Imbach JL, Aubertin AM, Obert G, Gosselin G. Synthesis, in Vitro Antiviral Evaluation, and Stability Studies of Bis(S-Acyl-2-Thioethyl) Ester Derivatives of 9-[2-(Phosphonomethoxy)Ethyl] Adenine (Pmea) as Potential Pmea Prodrugs with Improved Oral Bioavailability. J Med Chem. 1996;39:4958–4965. doi: 10.1021/jm960289o. [DOI] [PubMed] [Google Scholar]
- 46.Gosselin G, Imbach JL, Perigaud C. Biologically Active Phosphotriester-Type Compounds. US 6555676 B2. U.S. Patent. 2002
- 47.Shafiee M, Deferme S, Villard AL, Egron D, Gosselin G, Imbach JL, Lioux T, Pompon A, Varray S, Aubertin AM, Van Den Mooter G, Kinget R, Perigaud C, Augustijns P. New Bis(Sate) Prodrug of Azt 5′-Monophosphate: In Vitro Anti-Hiv Activity, Stability, and Potential Oral Absorption. J Pharm Sci. 2001;90:448–463. doi: 10.1002/1520-6017(200104)90:4<448::aid-jps1003>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 48.Raggers RJ, Pomorski T, Holthuis JCM, Kalin N, van Meer G. Lipid Traffic: The Abc of Transbilayer Movement. Traffic. 2000;1:226–234. doi: 10.1034/j.1600-0854.2000.010305.x. [DOI] [PubMed] [Google Scholar]
- 49.Contreras FX, Sanchez-Magraner L, Alonso A, Goni FM. Transbilayer (Flip-Flop) Lipid Motion and Lipid Scrambling in Membranes. FEBS Lett. 2010;584:1779–1786. doi: 10.1016/j.febslet.2009.12.049. [DOI] [PubMed] [Google Scholar]
- 50.Showell M. Handbook of Detergents: Formulation. Taylor & Francis; London: 2005. [Google Scholar]
- 51.Boon JM, Smith BD. Chemical Control of Phospholipid Distribution across Bilayer Membranes. Med Res Rev. 2002;22:251–281. doi: 10.1002/med.10009. [DOI] [PubMed] [Google Scholar]
- 52.Bevers EM, Comfurius P, Dekkers DW, Zwaal RF. Lipid Translocation across the Plasma Membrane of Mammalian Cells. Biochim Biophys Acta, Mol Cell Biol Lipids. 1999;1439:317–330. doi: 10.1016/s1388-1981(99)00110-9. [DOI] [PubMed] [Google Scholar]
- 53.Daleke DL. Regulation of Transbilayer Plasma Membrane Phospholipid Asymmetry. J Lipid Res. 2003;44:233–242. doi: 10.1194/jlr.R200019-JLR200. [DOI] [PubMed] [Google Scholar]
- 54.Oumzil K, Khiati S, Grinstaff MW, Barthelemy P. Reduction-Triggered Delivery Using Nucleoside-Lipid Based Carriers Possessing a Cleavable Peg Coating. J Controlled Release. 2011;151:123–130. doi: 10.1016/j.jconrel.2011.02.008. [DOI] [PubMed] [Google Scholar]
- 55.Kelly SJ, Butler LG. Enzymic-Hydrolysis of Phosphonate Esters - Reaction-Mechanism of Intestinal 5′-Nucleotide Phosphodiesterase. Biochemistry. 1977;16:1102–1104. doi: 10.1021/bi00625a011. [DOI] [PubMed] [Google Scholar]
- 56.Kelly SJ, Butler LG. Enzymic Hydrolysis of Phosphonate Esters. Biochem Biophys Res Commun. 1975;66:316–321. doi: 10.1016/s0006-291x(75)80330-5. [DOI] [PubMed] [Google Scholar]
- 57.Serafinowska HT, Ashton RJ, Bailey S, Harnden MR, Jackson SM, Sutton D. Synthesis and in Vivo Evaluation of Prodrugs of 9-[2-(Phosphonomethoxy)Ethoxy]Adenine. J Med Chem. 1995;38:1372–1379. doi: 10.1021/jm00008a015. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.