

Abstract
Dysregulated expression and function of dopamine D2 receptors (D2Rs) are implicated in drug addiction, Parkinson’s disease and schizophrenia. In the current study, we examined whether D2Rs are modulated by regulator of G protein signaling 2 (RGS2), a member of the RGS family that regulates G protein signaling via acceleration of GTPase activity. Using neuroblastoma 2a (N2A) cells, we found that RGS2 was immunoprecipitated by aluminum fluoride-activated Gαi2 proteins. RGS2 siRNA knockdown enhanced membrane [35S] GTPγS binding to activated Gαi/o proteins, augmented inhibition of cAMP accumulation and increased ERK phosphorylation in the presence of a D2/D3R agonist quinpirole when compared to scrambled siRNA treatment. These data suggest that RGS2 is a negative modulator of D2R-mediated Gαi/o signaling. Moreover, RGS2 knockdown slightly increased constitutive D2R internalization and markedly abolished quinpirole-induced D2R internalization assessed by immunocytochemistry. RGS2 knockdown did not compromise agonist-induced β-arrestin membrane recruitment; however, it prevents β-arrestin dissociation from the membrane after prolonged quinpirole treatment during which time β-arrestin moved away from the membrane in control cells. Additionally, confocal microscopy analysis of β-arrestin post-endocytic fate revealed that quinpirole treatment caused β-arrestin to translocate to the early and the recycling endosome in a time-dependent manner in control cells whereas translocation of β-arrestin to these endosomes did not occur in RGS2 knockdown cells. The impaired β-arrestin translocation likely contributed to the abolishment of quinpirole-stimulated D2R internalization in RGS2 knockdown cells.
1. Introduction
Dysfunctional dopamine D2 receptors (D2Rs) are implicated in numerous neurological and psychiatric diseases. Agonists or antagonists of D2Rs have been used for the treatment of Parkinson’s disease and schizophrenia [see review in (Beaulieu and Gainetdinov, 2011)]. Thus, it is important to understand the regulation of D2R function. D2Rs are coupled to inhibitory Gαi/o proteins to produce intracellular signaling (Neve et al., 2004). Activation of Gαi/o proteins by D2R agonists promotes the exchange of GDP to GTP on the Gα subunit and subsequent dissociation of G proteins into Gα and Gβγ subunits, which act on various downstream effectors to produce differential cellular and behavioral responses. For example, the inhibitory Gαi/o subunit couples to adenylyl cyclase to inhibit cAMP production whereas the Gβγ subunit stimulates the MAPK signaling cascade. The extent and duration of D2R signaling is critically controlled by the family of regulators of G protein signaling (RGS) proteins that limit G protein activity (Masuho et al., 2013). All RGS proteins contain a RGS domain which binds directly to the activated Gα subunit to facilitate GTP hydrolysis, thus rapidly terminating G protein signaling and receptor responses (Hepler, 1999; Watson et al., 1996). There are more than 20 subtypes of RGS proteins that are distributed in a brain region- and neuron-dependent manner (Gold et al., 1997; Hooks et al., 2008), suggesting that modulation of GPCR signaling by RGS proteins may be receptor-type and brain region-specific.
The majority of D2Rs are localized on postsynaptic non-dopaminergic neurons in the striatum and play an important role in motor function [see review in (Beaulieu and Gainetdinov, 2011)]. Among the members of the RGS family, RGS4, RGS7 and RGS9 are enriched in striatum (Mancuso et al., 2010; McGinty et al., 2008) and have been shown to directly regulate D2R signaling in heterologous expression systems. For example, RGS9 dose-dependently reduces dopamine-stimulated activation of Gαi/o proteins in HEK293 cells stably expressing D2Rs (Masuho et al., 2013). RGS4 overexpression reduces the ability of quinpirole (a D2R/D3R agonist) to inhibit forskolin-stimulated cAMP production in HEK293 cells (Min et al., 2012). Furthermore, there is compelling evidence that RGS9 controls striatal postsynaptic D2R activity and associated motor function (Kovoor et al., 2005; Rahman et al., 2003). In addition to their enriched expression in striatum, D2Rs are also present on the somas and dendrites of midbrain dopamine neurons (Sesack et al., 1994). These receptors serve as autoreceptors to provide negative feedback inhibition of dopamine transmission in the synapse (Bello et al., 2011; Mercuri et al., 1997). Compared to striatal postsynaptic D2Rs, the regulation of midbrain D2R signaling by RGS proteins has yet to be examined. Given the differential distribution patterns of RGS subtypes in the brain, it is likely that the function of midbrain D2Rs (autoreceptors) is regulated by RGS subtypes other than RGS9 proteins because RGS9 is not expressed in dopaminergic neurons (Mancuso et al., 2010). We and others have shown that RGS2 is highly expressed in the cell bodies of midbrain dopaminergic neurons where D2Rs (autoreceptors) are located (Calipari et al., 2014; Labouebe et al., 2007). However, the functional interaction between RGS2 and D2Rs in neuronal-like cell lines has yet to be investigated. Thus, one purpose of the present study was to examine whether RGS2 regulates D2R-mediated G protein signaling in neuroblastoma 2a (N2A) cells. This study will provide the basis for future interrogation of the functional interaction between these two proteins in midbrain dopaminergic neurons.
The rate of D2R internalization contributes to the duration and strength of receptor signaling in response to agonist stimulation. D2Rs undergo a complex process of internalization which involves D2R phosphorylation and β-arrestin recruitment to the surface receptors. Upon binding to phosphorylated D2Rs, β-arrestin interacts with a set of endocytic proteins, including clathrin and endocytic adaptor protein 2 (AP2), via a conserved motif in the C terminus tail to facilitate its dissociation from the membrane (Kim et al., 2001; Lan et al., 2009; Laporte et al., 2000; Namkung et al., 2009). Recent evidence indicates that RGS proteins not only act as GTPase-activating proteins, but also play an important role in GPCR internalization. For instance, overexpression of RGS9 inhibits dopamine-induced D2R internalization in HEK293 cells (Celver et al., 2010). Thus, the second purpose of this study was to examine whether RGS2 regulated constitutive and agonist-stimulated D2R internalization. Because D2R internalization is primarily mediated by β-arrestin (Kim et al., 2001), we examined the effect of RGS2 knockdown on β-arrestin-mediated D2R internalization. Here we report that RGS2 negatively modulated D2R-mediated Gαi/o protein signaling. Moreover, RGS2 knockdown prevents agonist-induced D2R internalization by disrupting β-arrestin dissociation from the membrane. The present study provides important new information on the functional interactions between RGS2 and D2Rs which may occur in midbrain dopaminergic neurons.
2. Materials and Methods
2.1. Chemicals and antibodies
Geneticin and lipofectamine 2000 were purchased from Life Technologies (Carlsbad, CA). [3H] Raclopride and [35S] GTPγS were purchased from Perkin Elmer (Billerica, MA). The Protease and Phosphatase Inhibitor Cocktails and all other chemicals were purchased from Sigma-Aldrich (St. Louis, MO), unless stated otherwise. The protein A/G-beads and the HRP-conjugated secondary antibodies were purchased from Santa Cruz Biotechnology, Inc (Santa Cruz, CA). All fluorescent secondary antibodies were purchased from Invitrogen/ThermoFisher (Carlsbad, CA).
2.2. Cell culture and transfection
Parental N2A cells obtained from the American Type Culture Collection (Manassas, VA) were cultured in Opti-MEM media supplemented with 10% fetal growth serum and 1% penicillin/streptomycin. N2A cells stably expressing D2Rs (N2A-D2R) were generated by transfecting cells with the short form of human D2R gene (Missouri S&T cDNA Resource Center) using lipofectamine 2000 (Invitrogen) and selected by geneticin (400 μg/ml). The expression of D2Rs was confirmed by the mRNA level and the saturation binding of the D2/D3 antagonist [3H] raclopride (Bmax = 1.64 pmole/mg protein). For transient transfection, parental N2A cells were transfected with FLAG-D2Rs and all experiments were performed 48 hrs after transfection.
2.3. Validation of a RGS2 antibody for Western blot and RGS2 protein knockdown via siRNA
In order to detect the endogenous level of RGS2 protein, we verified the specificity of a commercially available RGS2 antibody (SAB1406388, Sigma-Aldrich). N2A-D2R cells were transiently transfected with 3xHA RGS2 cDNA (Missouri S&T cDNA Resource Center) or empty pcDNA3.1 vector using lipofectamine 2000. Cells were lysed 48 hours after transfection and the protein concentrations were determined using the BCA Protein Assay (Thermo Fisher Scientific). Protein (20 μg) was loaded on 16% Tris-Glycine gels for SDS-polyacrylamide gel (SDS-PAGE) electrophoresis then was transferred to polyvinylidene difluoride membranes. RGS2 proteins were probed by either a rabbit anti-HA tag primary antibody (SAB4300603, Sigma-Aldrich) or a mouse anti-RGS2 antibody (SAB1406388, Sigma-Aldrich) followed by goat anti-rabbit IgG HRP or goat anti-mouse IgG HRP.
Using this validated antibody, we measured the efficiency of endogenous RGS2 protein knockdown via siRNA. Briefly, N2A-D2R cells were transiently transfected with a mouse RGS2 siRNA (Mm01_00052882) or a corresponding scrambled siRNA obtained from Sigma-Aldrich. Cells were lysed in solubilization buffer containing 50 mM Tris-HCl (pH 7.4), 150 mM NaCl and 1% Triton X-100. Western blot was performed to determine RGS2 levels using mouse anti-RGS2 antibody (SAB1406388, Sigma-Aldrich). After incubation with goat anti-mouse secondary, immunoreactive bands were revealed by enhanced chemiluminescent substrate onto X-ray films. Densitometric analysis was conducted using ImageJ (NIH). RGS2 immunoreactive bands were normalized to β-actin (Santa Cruz Biotechnology) and expressed as relative to the control.
2.4. Immunoprecipitation of the RGS2-Gαi2 complex
It has been shown that treatment with aluminum fluoride (AlF4−) can induce the active state conformation of Gα proteins. AlF4− binds to the γ phosphate binding site of the Gα subunit and interacts with GDP, thus mimicking the γ phosphate of GTP and causing the Gα protein to replicate its activated state (Traver et al., 2000; Vincent et al., 1998). We performed immunoprecipitation to examine whether RGS2 coupled to activated Gαi/o proteins in the presence of aluminum fluoride as described previously (Dulin et al., 1999; Tesmer et al., 1997). Briefly, N2A-D2R cells were lysed in lysis buffer (20mM HEPES, 150 mM NaCl, 1% Triton-X, 2mM MgCl2, 1x protease and phosphatase inhibitors) in the presence or absence of AlF4− (NaF 10 mM and AlCl3 100 μM) followed by immunoprecipitation with rabbit anti-Gαi2 antibody (Santa Cruz Biotechnology). Immunoprecipitates were then pulled down by protein A/G beads (Santa Cruz Biotechnology) and immunoblotted for mouse anti-Gαi2 and mouse anti-RGS2 (SAB1406388, Sigma-Aldrich).
2.5. Scintillation proximity assay for quinpirole-induced membrane [35S] GTPγS binding
[35S] GTPγS binding to activated Gαi/o proteins was performed on membranes prepared from the control and RGS2 knockdown cells using the procedure previously described (Blume et al., 2015). Briefly, cells were homogenized in cold hypotonic buffer (20 mM HEPES, pH 7.4, 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 1 mM dithiothreitol and the Protease Inhibitor Cocktail) followed by centrifugation at 1,000 × g for 10 min at 4°C. Supernatants were collected and centrifuged at 40,000 × g for 1 hour at 4°C. The resulting pellet was resuspended in TME buffer (20 mM Tris, 5 mM MgCl2, 1 mM Tris-EDTA, 1 mM DTT, pH 7.4). For [35S] GTPγS binding, membrane (5 μg) was added to the assay buffer (20 mM NaHepes, pH 7.4, 100 mM NaCl, 5 mM MgCl2, and 1 mM dithiothreitol) containing 0.1 nM [35S] GTPγS, 10 μM GDP and quinpirole (1 nM-100 μM) for 1 hr at 30°C. The radioactivity was detected on a Top-Count scintillation counter (PerkinElmer). Non-specific binding was determined in the presence of 10 μM GTPγS and the basal level was in the absence of quinpirole. Specific GTPγS binding was determined by subtracting non-specific activity from the total binding. Data were presented as fold over the baseline. Sigmoidal quinpirole dose-response curves were generated using three-parameter non-linear regression analysis. The values for EC50 and maximal responses (Emax) were extrapolated from the curves.
2.6. cAMP accumulation assay
cAMP accumulation assay was performed using the LANCE Ultra cAMP competitive immunoassay kit (PerkinElmer, Waltham, MA). Briefly, N2A-D2R cells suspended in HBSS buffer (5 mM Hepes, 0.5mM IBMX and 0.1% BSA) were seeded onto 384-well white, opaque OptiPlates (Perkin Elmer). Forskolin (10 μM) and quinpirole (0.1 nM-1 μM) were added to the wells and incubated at room temperature for 30 mins. Next, Eu-cAMP tracer and ULight-anti-cAMP were added to the mixture which was then incubated for 1 hr at room temperature. Plates were read for TR-FRET signal using the Victor3 plate reader (Perkin Elmer) with 340 nm wavelength excitation and 665 nm wavelength emission. The cAMP standard curve was fitted as a sigmoidal 4PL (X, log(agonist)) equation in Prism (GraphPad Software) with the corresponding equation: Y = bottom + (top − bottom)/(1 + 10^[(LogEC50 − X) * Hill slope]). The true cAMP concentration in each sample was extrapolated from the cAMP standard curve with X representing log [cAMP] and Y representing TR-FRET signal emitted at 665 nm. Data were presented as percent of the maximal cAMP accumulation produced by stimulation with forskolin. The values of IC50 were determined.
2.7. ERK phosphorylation
Cells were starved for 2 hrs before treatment with vehicle or quinpirole (1 μM) for 5, 10 and 15 mins followed by solubilization in the buffer containing the Protease and Phosphatase Inhibitor Cocktail. Western blotting was performed to assess the level of phosphorylated ERK (pERK) by rabbit anti-pERK antibody (#9101, Cell Signaling) followed by secondary goat anti-rabbit IgG-HRP. Membranes were then stripped and total ERK (tERK) was probed with mouse anti-ERK (#4696, Cell Signaling) followed by secondary goat anti-mouse IgG-HRP. The level of pERK was normalized to total ERK and represented as relative to its own vehicle treatment.
2.8. Confocal microscopy on constitutive and stimulated-D2R internalization
Surface and intracellular D2Rs were labeled with different fluorophores. The specificity of primary D2R antibodies was validated in parental N2A cells that do not express endogenous D2Rs. Fluorescent images were acquired with a Zeiss LSM 710 laser scanning confocal microscope. Confocal image planes were acquired with a 63X/NA 1.4 PlanNeoFluor oil-immersion objective. Alexa 405, 488 and 594 were excited at 405 nm, 408 nm and 594 nm with a diode, Argon and HeNe laser, respectively. Alexa 633 and 647 signals were also excited at 633 nm with a HeNe laser. Fluorescent channels were acquired sequentially to prevent cross-excitation of laser signal and minimize bleed-through. All images were acquired with identical settings for excitation intensity, detector sensitivity and pinhole to allow intensity comparison between samples. Images for analysis were obtained by taking a Z-axis stack of image planes (1024 ×1024 pixels) with 0.2 μm steps encompassing the entire cell structure and combining image planes into a maximum intensity projection stack. Fluorescent intensity was quantitated for each channel using ImageJ software (NIH). All images were adjusted with Gaussian blur for presentation purposes only. Alexa 405 emission was pseudo colored as green upon acquisition for optimal presentation appearance. At least three independent experiments were performed and 15-50 cells were analyzed for each group from every experiment.
Constitutive D2R internalization was examined using a modified antibody feeding technique as described previously (Bartlett et al., 2005; Xiao et al., 2009). Cells were blocked with 6% normal horse serum in phosphate-buffered saline (PBS) for 30 min at 4°C and surface D2Rs were labeled with mouse anti-D2R (sc-5303, Santa Cruz) for 45 min at 4°C. Cells were then warmed up to 37°C for 5, 15, 30, 60 or 90 min to allow for constitutive internalization of surface D2Rs. Cells were also pretreated with the D2R antagonist raclopride (1 μM) for 30 min to examine whether constitutive D2R internalization could be blocked by raclopride. Internalization was terminated by replacing the media with cold PBS. The remaining primary antibody-bound surface D2Rs were incubated with rabbit anti-mouse Alexa 647 for quantitation of surface D2Rs that were not internalized. Cells were fixed with 4% formaldehyde for 15 min and permeabilized with 0.1% Triton-X for 10 min. Internalized surface D2Rs bound with the primary D2R antibody were labeled with secondary antibody goat anti-mouse Alexa 405 for quantitation of internalized D2Rs. Constitutive D2R internalization was determined as the percent of D2R internalization by calculating the percent of fluorescent intensity of Alexa 647 (internalized) out of the total (Alexa 647 + Alexa 405) (total surface D2Rs before internalization). Data were presented as relative to the level of D2R internalization in control cells after vehicle treatment.
To examine the effect of RGS2 knockdown on quinpirole-stimulated D2R internalization, cells were treated with quinpirole (1 μM) for 5, 15, 30 or 60 min. The amount of internalized D2Rs was assessed using the same method as described for constitutive internalization. To evaluate if D2R internalization was dependent upon G protein activation, clathrin, or ERK activation, cells were incubated with or without pertussis toxin (PTX, 100 nM, overnight), a clathrin inhibitor (Concanavalin A, 250 μg/mL, 1 hr) or an ERK inhibitor (PD0325901, 10 μM, 15 min) at 37°C prior to the treatment with either vehicle or quinpirole (1 μM, 30 min). Immunocytochemistry was performed to label the surface and the intracellular D2Rs as described for the constitutive D2R internalization. Data were presented as relative to the level of D2R internalization in control cells after vehicle treatment.
2.9. β-arrestin translocation and fate
2.9.1. Co-localization of β-arrestin and surface D2Rs
N2A-D2R cells were treated with vehicle or quinpirole (1 μM, 5 or 30 min). Then surface D2Rs were labeled with a mouse anti-D2R antibody (sc-5303, Santa Cruz) at 4°C followed by goat anti-mouse Alexa Fluor 594. Cells were fixed with 4% formaldehyde and permeabilized with 0.1% Triton-X. Cytoplasmic β-arrestin was subsequently labeled with rabbit anti-β-arrestin (ab2914, Abcam) followed by goat anti-rabbit Alexa Fluor 405 antibodies. Colocalization between β-arrestin and surface D2Rs was quantified using Coloc2 software (FIJI) as previously described (Cleghorn et al., 2015) and Pearson R correlation coefficients were generated for individual cells. Pearson R values were averaged across cells for each group and data were presented relative to the level of colocalization between β-arrestin and surface D2Rs in control cells with vehicle treatment.
2.9.2. The post-endocytic fate of β-arrestin
Briefly, N2A-D2R cells were stimulated with vehicle or quinpirole (1μM, 5 or 30 min) followed by fixation, permeabilization and blocking with 6% horse serum in PBS. Cytoplasmic β-arrestin and Rab5 (a marker for the early endosome), transferrin receptors (a marker for the recycling endosome) or LAMP1 (a marker for the late endosome) were probed with the following primary antibodies: rabbit anti-β-arrestin (ab2914, Abcam), mouse anti-Rab5 (sc-46692, Santa Cruz), goat anti-Transferrin (ab166929, Abcam), and mouse anti-LAMP1 (sc-17768, Santa Cruz). Then cells were incubated with rabbit anti-mouse Alexa 405, mouse anti-rabbit Alexa 555 and donkey anti-goat Alexa 633, respectively. Colocalization of β-arrestin with Rab5, transferrin receptors or LAMP1 was calculated using Coloc2 software (FIJI). Data are presented relative to the level of colocalization between β-arrestin and each endocytic marker in control cells with vehicle treatment.
2.10. Statistical Analysis
Graph Pad Prism 5 (La Jolla, CA, USA) was used for statistical analysis. All data are presented as mean ± SEM. A two-tailed unpaired Student’s t-test analysis was performed for comparisons between two groups. A two-way analysis of variance (ANOVA) was used followed by Bonferroni post hoc analysis for multiple comparisons. A value of p≤0.05 was considered statistically significant.
3. Results
3.1. Validation of a RGS2 antibody and verification of RGS2 knockdown efficiency by Western blotting
We verified the specificity of a commercially available RGS2 antibody. Shown in Fig.1, 3xHA-RGS2 proteins were detected by both a HA antibody and a RGS2 antibody in N2A cells transfected with 3xHA-RGS2 cDNA. The notable weak expression of the exogenous RGS2 proteins was likely due to the high level of endogenous RGS2 proteins. Using this validated antibody, we examined the knockdown efficiency of RGS2 proteins by RGS2 siRNA. There was a 60.9±1.2% (p<0.01) reduction in the level of endogenous RGS2 proteins in knockdown cells when compared to the control cells transfected with a corresponding scrambled siRNA (Fig. 2).
Fig. 1.
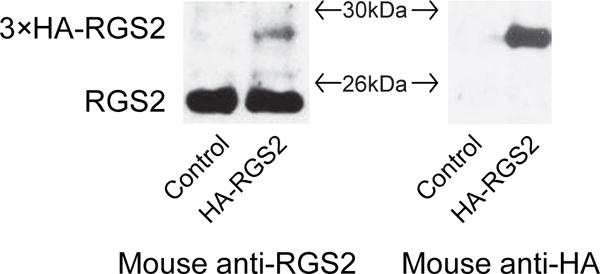
Verification of a commercially available RGS2 antibody. Parental N2A cells were transfected with 3xHA-RGS2 or pcDNA3.1 empty vector. Cells were lysed and Western blotting was performed to measure endogenous RGS2 and exogenous HA-RGS2 protein levels. (A) A mouse anti-RGS2 antibody (SAB1406388, Sigma-Aldrich) was used to detect endogenous RGS2 (~24KDa) and exogenous 3xHA-RGS2 (~27KDa). HA-RGS2 was not detected in control cells. (B) A mouse anti-HA antibody was applied to detect HA-RGS2 protein. There was a pronounced band (~27KDa) in cells transfected with 3xHA-RGS2 and no band was detected in control cells.
Fig. 2.
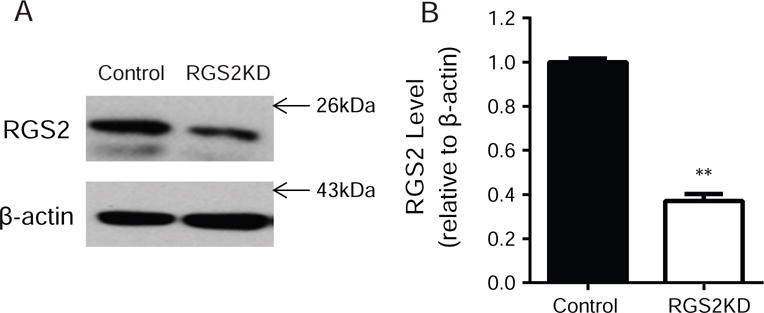
Verification of RGS2 protein knockdown via siRNA. N2A-D2R cells were transfected with RGS2 siRNA or corresponding scrambled siRNA. RGS2 protein levels were measured 48 hrs after transfection by Western blotting. (A) Representative Western blot for endogenous RGS2 protein level in control and RGS2 knockdown (RGS2KD) cells probed by mouse anti-RGS2 antibody (SAB1406388, Sigma-Aldrich). (B) Quantification of RGS2 protein levels. RGS2 siRNA knockdown significantly reduced the endogenous RGS2 protein level when compared to scrambled siRNA treated control cells (**P<0.01 vs. control, N=3).
3.2. Aluminum fluoride promoted the association of RGS2 with Gαi2
RGS proteins are known to interact with the high-affinity, activated state of Gα proteins. To test the interaction between RGS2 and activated Gαi/o proteins, cells were treated with or without aluminum fluoride, which can interact with GDP while bound to Gα and mimic the γ-phosphate of GTP. Our previous study shows that midbrain D2Rs are primarily coupled to the Gαi2 subtype for receptor activation and signaling (Calipari et al., 2014). Thus, we investigated if RGS2 specifically interacted with the activated form of Gαi2 by performing immunoprecipitation. In the absence of AlF4−, there was no detectable amount of RGS2 that was immunoprecipitated by Gαi2. In contrast, the presence of AlF4− (AlCl3 and NaF) caused a significant coupling between RGS2 and Gαi2 (Fig. 3A), suggesting that RGS2 interacts with activated Gαi2 in N2A cells.
Fig. 3.
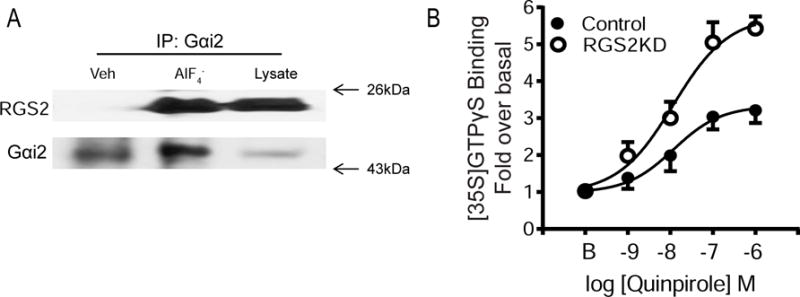
RGS2 interacts with activated Gαi proteins and modulates Gαi/o signaling. (A) Immunoprecipitation of activated Gαi2 and RGS2. Cells were lysed in Triton X-containing buffer (see Materials and Methods) in the presence or absence of AlF4− (NaF 10mM and AlCl3 100 μM) for 1 hr. Cell lysates were immunoprecipitated overnight with rabbit anti-Gαi2 antibody. Immunoprecipitates or whole cell lysates were immunoblotted with mouse anti-RGS2 and rabbit anti-Gαi2 antibodies. (B) RGS2 knockdown increases quinpirole-stimulated [35S] GTPγS binding. Cell membranes (5 μg) were incubated with [35S] GTPγS (0.1 nM), GDP (10 μM) and quinpirole (1 nM – 10 μM) for 1 hr at 30°C. Non-specific binding was determined in the presence of cold GTPγS (10 μM). Quinpirole stimulated a dose-dependent increase in [35S] GTPγS binding in both control and RGS2 knockdown (RGS2KD) cells. However, the Emax value was significantly greater in RGS2 knockdown cells (**P<0.01, N=3). There was no significant difference in the basal binding or the EC50 values between the two groups.
3.3. RGS2 negatively modulated D2R-mediated G protein signaling
To determine whether RGS2 regulates D2R-mediated G protein signaling, we first examined D2R-stimulated Gαi/o protein activation using [35S] GTPγS binding on membranes prepared from the control and RGS2 knockdown cells. There was no difference in the basal binding between the two groups (control: 79.3±3.4 fmole/mg protein; knockdown: 85.7±4.2 fmole/mg protein). Moreover, quinpirole stimulated a dose-dependent increase in [35S] GTPγS binding in both control and RGS2 knockdown cells and was blocked by a D2R antagonist raclopride (1 μM). However, the Emax value was significantly greater in RGS2 knockdown cells (5.54±0.33) than in control cells (3.47±0.31) (Fig. 3B; N=3, p<0.05). There was no significant group difference in the values of EC50 (57.05±22.80 nM and 41.21±20.48 nM for control and knockdown cells, respectively).
To determine the effect of RGS2 knockdown on downstream Gα-mediated signaling, we measured the ability of D2Rs to inhibit forskolin-stimulated cAMP accumulation. There was no difference in forskolin-stimulated cAMP accumulation between control (4.90±0.89 nM) and RGS2 knockdown cells (5.01±0.77 nM). However, the inhibition of cAMP accumulation by quinpirole was greater in RGS2 knockdown cells (IC50=0.89±0.02 nM) than in the controls (IC50=2.37±0.42 nM) (Fig 4A, N=5, p<0.05).
Fig. 4.
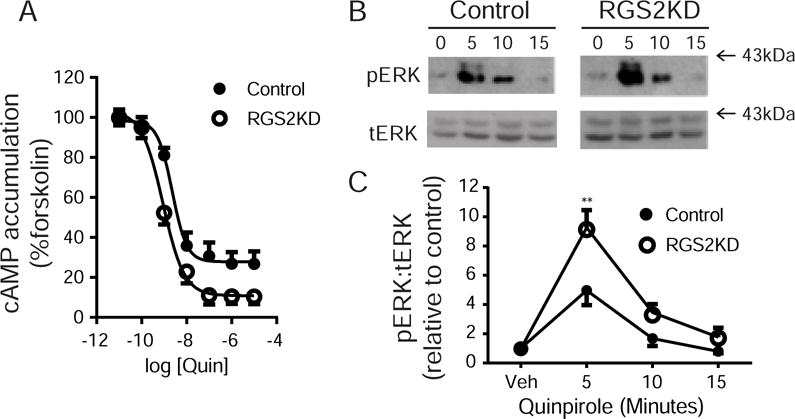
RGS2 knockdown increases quinpirole-stimulated cAMP accumulation and ERK phosphorylation. (A) Measurement of cAMP accumulation. Cells were harvested and cAMP accumulation was measured using the LANCE Ultra cAMP immunoassay kit as described in Materials and Methods. Control and RGS2 knockdown cells were stimulated with forskolin (10 μM) and increasing doses of quinpirole (0.1 nM-10 μM) for 30 mins. The inhibition of cAMP accumulation by quinpirole was significantly greater in RGS2 knockdown cells (IC50=0.89±0.02 nM) compared to control cells (IC50=2.37±0.42 nM) (**P< 0.01, N=5). Data were presented as percent response of forskolin treatment. (B) Representative blots of phosphorylated ERK (pERK) and total ERK (tERK) for control and RGS2 knockdown (RGS2KD) cells. (C) Quantification of pERK and tERK levels. Quinpirole stimulated ERK phosphorylation in a time-dependent manner for both control and knockdown cells. However, the pERK level was significantly greater after 5 min of quinpirole stimulation in RGS2 knockdown cells compared to control cells (**P< 0.01, a two-way ANOVA with Bonferroni post hoc test, N=4).
Finally, we examined the effect of RGS2 knockdown on quinpirole-stimulated ERK phosphorylation. There was no significant difference in the basal level of phosphorylated ERK between groups. However, a two-way ANOVA revealed significant main effects of group, F(1,24) = 16.22, p<0.01; time, F(3,24) = 42.29, p<0.01; and interaction, F(3,24) = 4.50, p<0.05. The overall phosphorylated ERK level was greater in RGS2 knockdown cells than in the control cells. Bonferroni post hoc analysis revealed a significantly higher level of phosphorylated ERK after 5 min of quinpirole stimulation in RGS2 knockdown cells compared to control cells (Figs. 4B-C, N=4, p<0.01).
3.4. RGS2 knockdown slightly increased constitutive D2R internalization but markedly attenuated quinpirole-stimulated D2R internalization
Constitutive D2R internalization was examined using a modified antibody feeding technique to directly label internalized D2Rs as illustrated in Fig. 5A. Data were expressed as a ratio of internalized D2Rs vs. the total surface D2Rs labeled prior to internalization. A two-way ANOVA indicated significant main effects of groups, F(1,530) = 13.13, p<0.01, and time, F(5,530) = 21.07, p<0.01. Constitutive internalization occurred in a time-dependent manner in both control and RGS2 knockdown cells (Figs. 5B-5C; n=55-70 cells/group). RGS2 knockdown produced a modest but significant overall increase in constitutive D2R internalization when compared to the scrambled siRNA treated control cells (Fig. 5C). Further, D2R internalization was not altered by treatment with raclopride, suggesting that constitutive D2R internalization is ligand-independent (Fig. 5C).
Fig. 5.
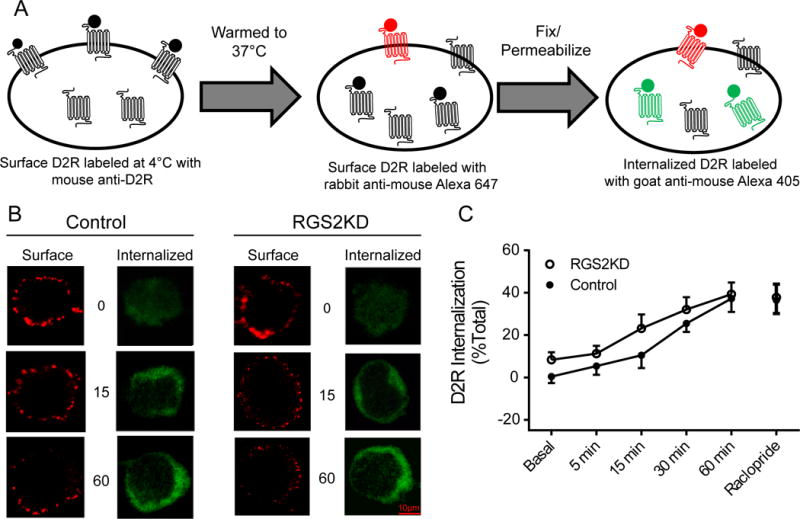
RGS2 knockdown slightly enhances constitutive D2R internalization. (A) A schematic diagram of surface and intracellular D2R labeling for measurement of constitutive D2R internalization. Surface D2Rs were saturated with mouse anti-D2R. Then cells were warmed to 37°C to allow constitutive internalization for the indicated time points. At the end of each time point, remaining primary antibody-bound surface D2Rs that did not internalize were labeled with goat anti-mouse Alexa 647 (red). D2Rs that were internalized were identified by labeling with goat anti-mouse Alexa 405 (blue, pseudo-colored to green). (B) Representative confocal images of surface and internalized D2Rs in control and RGS2 knockdown (RGS2KD) cells. Scale bar, 10 μm. (C) Quantification of constitutive D2R internalization. Internalized D2Rs were represented as percent of total D2Rs. Constitutive internalization occurred in a time-dependent manner in both control and RGS2 knockdown cells (P<0.01, a two-way ANOVA, n=55-70 cells/group). RGS2 knockdown produced a small but significant overall increase in constitutive D2R internalization when compared to control cells. Pre-treatment with raclopride (1μM, 30 min) did not inhibit constitutive D2R internalization. Experiments were replicated three times. Data are expressed as relative to the vehicle treatment in the control cells.
Quinpirole-stimulated D2R internalization was measured by the procedure illustrated in Fig. 6A. A two-way ANOVA revealed that there was a significant main effect of group, F(1,499)=47.86, p<0.01, time, F(4,499) = 15.12, and interaction, F(4,499) = 5.119, p<0.01. RGS2 knockdown abolished quinpirole-stimulated D2R internalization. Bonferroni post hoc analysis revealed that quinpirole induced a time-dependent increase in D2R internalization in both control and RGS2 knockdown cells (Figs. 6B-C). For control cells, D2R internalization reached a steady state 30 min after quinpirole treatment when 61.7± 1.8% of the surface D2Rs internalized. In contrast, only 16.34±1.6% of the surface D2Rs were internalized in RGS2 knockdown cells at this same time point. Notably, overnight pertussis toxin treatment (PTX, 100 nM) abolished quinpirole-stimulated D2R internalization in control cells (Fig. 6D), suggesting that D2R internalization in N2A cells is dependent upon agonist-induced G protein activation.
Fig. 6.
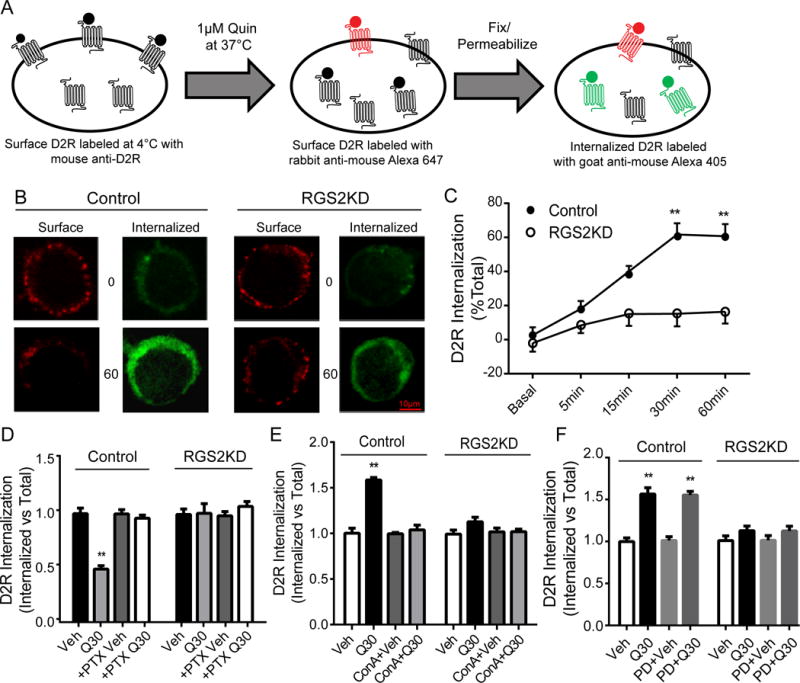
RGS2 knockdown abolishes quinpirole-stimulated D2R internalization. (A) A schematic diagram of surface and intracellular D2R labeling for measurement of quinpirole stimulated D2R internalization. Quinpirole-stimulated D2R internalization was measured by immunocytochemistry as described for constitutive D2R internalization and stimulated with quinpirole (1 μM) at 37°C for the indicated time points. Remaining primary antibody-bound surface D2Rs that did not internalize after quinpirole treatment were labeled with goat anti-mouse Alexa 647 (red) and internalized D2Rs were identified by labeling with goat anti-mouse Alexa 405 (blue, pseudo-colored to green). (B) Representative confocal images of surface and internalized D2Rs in control and RGS2 knockdown (RGS2KD) cells at various time points. Scale bar, 10 μm. (C) Quinpirole induced a time-dependent increase in D2R internalization in both control and RGS2 knockdown cells (P<0.01, a two-way ANOVA, n=55-70 cells/group); however, D2R internalization in RGS2 knockdown cells was significantly inhibited at all the indicated time points (**P<0.01 vs. control, a two-way ANOVA with Bonferroni post hoc test). Moreover, D2R internalization induced by quinpirole treatment (1μM, 30 min, Q30) was abolished by pre-treatment with (D) pertussis toxin (PTX, 100 nM, overnight, n=40-60 cells/group) or (E) a clathrin inhibitor Concanavalin A (ConA, 250 μg/mL, 1 hr, n=45-55 cells/group). (F) Inhibition of ERK activation with an ERK inhibitor PD0325901 (PD, 1μM, 30 min, n=45-60 cells/group) had no effect on D2R internalization. Experiments were replicated three times. Data are expressed as relative to their own vehicle treatment.
3.5. D2R internalization is clathrin- but not ERK-dependent
D2R internalization has been shown to be clathrin-dependent in non-neuronal cell lines (e.g. HEK293 and COS-7) (Kabbani et al., 2004; Kim et al., 2001). To confirm that D2R internalization involves clathrin in N2A cells, we treated cells with the clathrin inhibitor, concanavalin A (ConA, 250 μg/mL, 1 hr) before quinpirole treatment (1 μM, 30 min). ConA blocked quinpirole-stimulated D2R internalization in control cells and had no effect in RGS2 knockdown cells (Fig. 6E). Moreover, pre-treatment with the dynamin inhibitor, dynasore, also blocked quinpirole-stimulated D2R internalization in control cells and did not alter quinpirole-stimulated D2R internalization in RGS2 knockdown cells (see S1 in Supplemental Materials), thus confirming that D2R internalization is also dynamin-dependent. Furthermore, we assessed whether D2R internalization was dependent on ERK signaling. Pretreatment with an ERK inhibitor PD0325901 (10 μM, 15 min) had no effect on quinpirole-stimulated D2R internalization in both control and RGS2 knockdown cells (Fig. 6F), suggesting that the effect of RGS2 knockdown on D2R internalization is not mediated by downstream intracellular ERK signaling.
3.6. RGS2 knockdown disrupted β-arrestin dissociation from the membrane
Because D2R internalization is both clathrin- and β-arrestin-dependent, we further investigated whether disruption of D2R internalization by RGS2 knockdown was due to impaired translocation of β-arrestin. We examined quinpirole-stimulated β-arrestin translocation by quantitating the colocalization between β-arrestin and surface D2Rs. There was no difference in the colocalization between β-arrestin and surface D2Rs at the basal state in control and RGS2 knockdown cells (Figs. 7A-B). However, a two-way ANOVA revealed a significant interaction, F(2,326)=11.04, p<0.01 and significant main effects of group, F(1,326)=16.05, p<0.01, and treatment time, F(2,326)=56.26,p<0.01. Short-term (5 min) quinpirole treatment increased colocalization by 25.2±2.1% and 27.8±3.1% in control and RGS2 knockdown cells, respectively. Prolonged quinpirole treatment (30 min) caused a significant reduction (21.5±1.9%) in the colocalization in control cells whereas there was no change in RGS2 knockdown cells (Figs. 7A-B). We further performed segmentation quantification of β-arrestin translocation (see Fig. S2 in Supplemental Materials) and confirmed the observation that β-arrestin did not dissociate from the membrane after 30 min of quinpirole treatment in RGS2 knockdown cells.
Fig. 7.
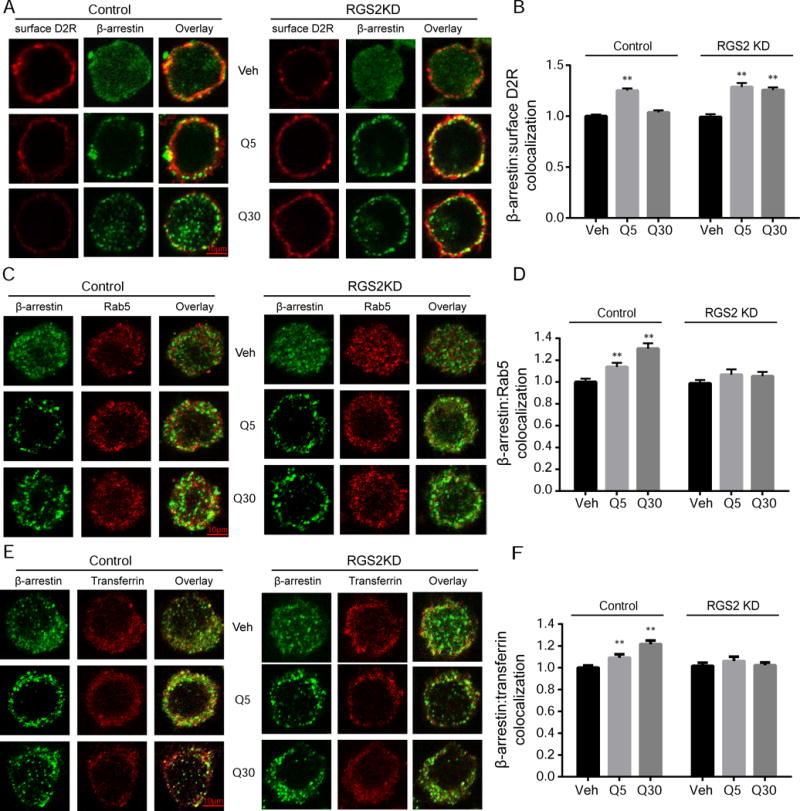
RGS2 knockdown impaired β-arrestin dissociation from the membrane. Surface D2Rs (red) and cytoplasmic β-arrestin (green) were labeled with different fluorophores. (A) Representative confocal images of surface D2Rs and β-arrestin in control and RGS2 knockdown (RGS2KD) cells after treatment with vehicle (Veh) or quinpirole (1 μM) for 5 min (Q5) or 30 min (Q30). Scale bars, 10 μm. (B) Quantification of β-arrestin and surface D2R colocalization. β-arrestin colocalization with surface D2Rs significantly increased after 5 min quinpirole treatment in both control and RGS2 knockdown cells (**P<0.01 vs. vehicle, a two-way ANOVA with Bonferroni post hoc test, n=50-55 cells/group). Prolonged quinpirole treatment (30 min) reduced colocalization between β-arrestin and surface D2Rs in control cells whereas β-arrestin and surface D2R colocalization remained persistently elevated in RGS2 knockdown cells (**P<0.01 vs. vehicle, a two-way ANOVA with Bonferroni post hoc test). Experiments were replicated three times. Data are expressed as relative to the control. (C) Representative confocal images of β-arrestin (green) and Rab5 (red) in control and RGS2 knockdown cells after treatment with vehicle (Veh) or quinpirole (1 μM) for 5 min (Q5) or 30 min (Q30). Scale bars, 10 μm. (D) Quantification of β-arrestin and Rab5 colocalization. Quinpirole stimulated β-arrestin and Rab5 colocalization in a time-dependent manner in the control cells (** P<0.01 vs. vehicle, a two-way ANOVA with Bonferroni post hoc test, n=45-60 cells/group) but not in RGS2 knockdown cells. (E) Representative confocal images of β-arrestin (green) and transferrin (red) in the control and RGS2 knockdown cells after treatment with vehicle (Veh) or quinpirole (1 μM) for 5 min (Q5) or 30 min (Q30). Scale bars, 10 μm. (F) Quantification of β-arrestin and transferrin colocalization. Quinpirole stimulated β-arrestin and transferrin colocalization in a time-dependent manner in the control cells (** P<0.01 vs. vehicle, a two-way ANOVA with Bonferroni post hoc test, n=45-60 cells/group) but not in RGS2 knockdown cells.
3.7. RGS2 knockdown inhibited translocation of β-arrestin to early and recycling endosomes
Immunocytochemistry was used to evaluate the post-endocytic fate of β-arrestin by determining the colocalization between β-arrestin and endosomal markers. Quinpirole-induced translocation of β-arrestin into early endosomes was examined by confocal microscopy analysis of β-arrestin and Rab5 colocalization. A two-way ANOVA showed a significant interaction, F(2,204)=5.220, p<0.01, and significant effects of group, F(1, 204)=12.03, p<0.01 and treatment time, F(2,204)=11.94, p<0.01. Bonferroni post hoc analysis revealed that β-arrestin and Rab5 colocalization increased in a time-dependent manner following quinpirole stimulation in control cells (Figs. 7C-D, n=45-60 cells/group, p<0.01) and no significant change was observed in RGS2 knockdown cells.
Next, the localization of β-arrestin in the recycling endosome was examined by the colocalization between β-arrestin and transferrin. A two-way ANOVA revealed a significant interaction, F(2,306)=6.921, p<0.01, and significant effects of group, F(1, 306)=7.857, p<0.01 and treatment time, F(2,306)=7.272, p<0.01. Bonferroni post hoc analysis revealed that colocalization between β-arrestin and transferrin increased in a time-dependent manner after stimulation with quinpirole in control cells (Figs. 7E-F, n=45-60 cells/group, p<0.01) and there was no significant change in RGS2 knockdown cells. Moreover, quinpirole stimulation had no effect on β-arrestin and LAMP1 colocalization in either control or RGS2 knockdown cells (see Fig. S3 in Supplemental Materials). Our observation of persistent association of β-arrestin and D2Rs was further confirmed by immunoprecipitation of FLAG-D2Rs and β-arrestin (see Fig. 4 in Supplemental Materials). Quinpirole (1μM, 30 min) caused an enhanced coupling between these two proteins in RGS2 knockdown cells compared to the vehicle treatment. However, the same treatment with quinpirole in the control cells did not induce any change in the coupling between D2R and arrestin.
4. Discussion
In the current study, we show for the first time that RGS2 negatively modulates D2R-mediated Gαi/o protein signaling in neuroblastoma N2A cells. Moreover, RGS2 critically regulates agonist-stimulated D2R internalization. RGS2 knockdown abolished quinpirole-stimulated D2R internalization and impaired β-arrestin dissociation from the membrane. These data suggest that RGS2 plays an integral role in modulation of both D2R activity and trafficking.
A significant finding of the present study is that RGS2 is a negative modulator of D2R-mediated Gαi/o signaling. It has been shown that D2R signaling can be mediated by several RGS proteins including RGS4, RGS7 and RGS9 (Druey and Kehrl, 1997; Masuho et al., 2013). The present study adds another member of the RGS family to the D2R signaling network. Here, we show that RGS2 interacts with activated Gαi2 proteins. RGS2 knockdown robustly enhanced quinpirole-stimulated Gαi/o protein activation, attenuated cAMP accumulation and increased ERK phosphorylation. Our observation contrasts with a previous report that RGS2 overexpression had no influence on D2R-mediated Gαi/o signaling in HEK293 cells (Min et al., 2012). This discrepancy is likely attributed to differences in cell lines (HEK293 vs. neuroblastoma N2A cells) and technical approaches (overexpression vs. knockdown) used in these experiments. A potential ceiling effect may contribute to no change in D2R signaling by RGS2 overexpression in HEK293 cells. It is worth noting that RGS2 was initially shown to be a selective and potent negative regulator of Gαq signaling using purified Gα subunits and RGS proteins in a single-turnover GTPase assay (Heximer et al., 1997). However, recent evidence indicates that RGS2 can also regulate Gαi/o signaling in certain types of cultured cells and animal tissues. For example, RGS2 was identified as a primary terminator of β2-adrenergic receptor-mediated Gαi signaling (Chakir et al., 2011). Our study provides additional evidence of a non-Gαq linked receptor system that is regulated by RGS2 proteins in neuroblastoma N2A cells.
The second finding of the present study is that RGS2 regulates constitutive and stimulated D2R internalization. Emerging evidence indicates that RGS proteins, in addition to their GTPase acceleration activity, can regulate GPCR trafficking. For example, RGS4 overexpression in HEK293 cells attenuated agonist-induced internalization of δ-opioid receptors (Leontiadis et al., 2009; Papakonstantinou et al., 2015). RGS14 knockdown prevented agonist-induced reduction in surface μ-opioid receptors in mouse brain tissue (Rodriguez-Munoz et al., 2007). Moreover, RGS9 overexpression in HEK293 cells blocked dopamine-induced reduction of surface D2Rs (Celver et al., 2010). These data suggest that RGS proteins may potentially act as a scaffold to regulate GPCR translocation between specific subcellular compartments. Our study also revealed a trafficking role of RGS2 in modulation of constitutive and quinpirole-stimulated D2R internalization in N2A cells. In control N2A cells, D2Rs undergo constitutive internalization with a maximal 30% of surface D2Rs internalized. Our observation is consistent with previous studies showing approximately 25% of surface D2Rs constitutively internalize after 30 minutes in HEK293 cells (Vickery and Von Zastrow, 1999; Kabbani et al., 2004). Blockade of D2Rs did not prevent constitutive D2R internalization, suggesting that constitutive D2R internalization does not depend on the intracellular signaling events. This observation is in agreement with the observation in HEK293 cells (Vickery and von Zastrow, 1999). RGS2 knockdown slightly increased constitutive D2R internalization but markedly reduced agonist-stimulated D2R internalization in N2A cells. Our finding is contradictory to a recent report showing that RGS2 overexpression in HEK293 cells had no effect on agonist-induced reduction of surface D2R levels (Min et al., 2012). A major difference in experimental approaches (knockdown vs. overexpression) may contribute to the disparate outcomes. When endogenous RGS2 proteins are abundantly expressed, overexpression may not be effective due to a potential ceiling effect. As a matter of fact, overexpression of RGS2 proteins in N2A cells did not alter D2R internalization in our hands, which is likely due to the high level of endogenous RGS2 expressed in N2A cells.
Lastly, our study suggests that RGS2 is critically involved in β-arrestin-mediated D2R internalization. It is well established that agonist-induced D2R internalization is primarily mediated by β-arrestin. Upon G protein activation, D2Rs are phosphorylated by G protein-coupled receptor kinases, which are recruited by the released Gβγ subunit, allowing for binding of β-arrestin to the receptor and subsequent internalization of the receptors into clathrin-coated vesicles (Beaulieu and Gainetdinov, 2011). Our data show that PTX pretreatment inhibits quinpirole-induced internalization, suggesting that D2R internalization in N2A cells is controlled by the canonical processes involving the G protein activation. This observation agrees with the report that disruption of D2R coupling to Gαi/o proteins by treatment with PTX completely abolishes agonist-induced D2R internalization in HEK293 cells (Celver et al., 2010). Moreover, D2R internalization requires recruitment of β-arrestin to the receptor, subsequent binding to endocytic proteins, such as clathrin and AP2, followed by dissociation from the membrane for internalization (Kim et al., 2001). In control N2A cells, we observed β-arrestin recruitment to and subsequent dissociation from the membrane following treatment with D2R agonist that is in agreement with the literature (Cho et al., 2010). However, it appears that RGS2 knockdown does not compromise β-arrestin recruitment to the membrane but prevents its dissociation from the membrane after prolonged D2R stimulation. The persistent association of β-arrestin with surface D2Rs in RGS2 knockdown cells assessed by confocal microscopy was further confirmed by no change in the association between β-arrestin and each marker for the early endosome (Rab5), the recycling endosomal (transferrin) or the lysosomes (LAMP1) across a time course of quinpirole treatment. In contrast, β-arrestin translocated to the early endosome and recycling endosome in a time-dependent manner in control N2A cells, which are in agreement with the reports that β-arrestin traffics from the membrane to the early endosome and recycling endosome after prolonged stimulation of various types of GPCRs (Tulipano et al., 2004; Wagener et al., 2009). Lastly, our immunoprecipitation data provide additional evidence that β-arrestin and D2Rs are persistently associated in RGS2 knockdown cells after prolonged quinpirole stimulation although we are aware that one caveat of this approach is that it measures the association between β-arrestin and total D2Rs, not the surface D2Rs.
Collectively, our data suggest that RGS2 is critical in regulation of agonist-induced β-arrestin translocation. RGS2 knockdown impaired β-arrestin dissociation from the membrane after prolonged stimulation, which may explain the abolishment of agonist-stimulated D2R internalization in the current study. One potential explanation for these observations is that RGS2 knockdown may attenuate D2R phosphorylation which could lead to a weak association between D2Rs and β-arrestin and subsequent disruption of D2R internalization. It has been shown that D2R phosphorylation is critical in β-arrestin recruitment and binding to the receptors (Cho et al., 2010). Alternatively, RGS2 may directly interact with D2Rs and/or β-arrestin to regulate β-arrestin-mediated D2R internalization. It has been reported that RGS2 physically binds to α1-adrenergic and M1 muscarinic receptors (Bernstein et al., 2004; Hague et al., 2005). Additionally, β-arrestin was shown to bind directly to the DEP domain of RGS9 (Zheng et al., 2011), which is required for RGS9-mediated D2R internalization in HEK293 cells (Celver et al., 2010). Therefore, future studies are necessary to examine whether RGS2 directly interacts with β-arrestin and/or D2Rs to regulate D2R trafficking. Because RGS2 and dopamine D2 receptors (autoreceptors) are co-expressed in the midbrain dopaminergic neurons (Calipari et al., 2014; Han et al., 2006), this study suggests a potential regulation of D2 autoreceptors by RGS2 proteins in the brain.
Supplementary Material
Acknowledgments
This work was supported by NIH grants (DA0006634, DA03690, T32-AA007565, AA023999 and AA014445) and a pilot grant from the Center for Molecular Communication and Signaling at Wake Forest University. The authors would like to thank for the Microscopic Imaging Core Facility in the Department of Biology at Wake Forest University.
Footnotes
Author Contributions:
Participated in research design: Luessen, Chen, McCool, Howlett
Conducted experiments: Luessen, Hinshaw, Sun
Performed data analysis: Luessen, Hinshaw, Sun, Marrs
Wrote or contributed to the writing of the manuscript: Luessen, Chen, McCool
References
- Bartlett SE, Enquist J, Hopf FW, Lee JH, Gladher F, Kharazia V, Waldhoer M, Mailliard WS, Armstrong R, Bonci A, Whistler JL. Dopamine responsiveness is regulated by targeted sorting of D2 receptors. Proc Natl Acad Sci U S A. 2005;102:11521–11526. doi: 10.1073/pnas.0502418102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaulieu JM, Gainetdinov RR. The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol Rev. 2011;63:182–217. doi: 10.1124/pr.110.002642. [DOI] [PubMed] [Google Scholar]
- Bello EP, Mateo Y, Gelman DM, Noain D, Shin JH, Low MJ, Alvarez VA, Lovinger DM, Rubinstein M. Cocaine supersensitivity and enhanced motivation for reward in mice lacking dopamine D2 autoreceptors. Nat Neurosci. 2011;14:1033–1038. doi: 10.1038/nn.2862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein LS, Ramineni S, Hague C, Cladman W, Chidiac P, Levey AI, Hepler JR. RGS2 binds directly and selectively to the M1 muscarinic acetylcholine receptor third intracellular loop to modulate Gq/11alpha signaling. J Biol Chem. 2004;279:21248–21256. doi: 10.1074/jbc.M312407200. [DOI] [PubMed] [Google Scholar]
- Blume LC, Eldeeb K, Bass CE, Selley DE, Howlett AC. Cannabinoid receptor interacting protein (CRIP1a) attenuates CB1R signaling in neuronal cells. Cellular signalling. 2015;27:716–726. doi: 10.1016/j.cellsig.2014.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calipari ES, Sun H, Eldeeb K, Luessen DJ, Feng X, Howlett AC, Jones SR, Chen R. Amphetamine Self-Administration Attenuates Dopamine D2 Autoreceptor Function. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology. 2014 doi: 10.1038/npp.2014.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celver J, Sharma M, Kovoor A. RGS9-2 mediates specific inhibition of agonist-induced internalization of D2-dopamine receptors. J Neurochem. 2010;114:739–749. doi: 10.1111/j.1471-4159.2010.06805.x. [DOI] [PubMed] [Google Scholar]
- Chakir K, Zhu W, Tsang S, Woo AY, Yang D, Wang X, Zeng X, Rhee MH, Mende U, Koitabashi N, Takimoto E, Blumer KJ, Lakatta EG, Kass DA, Xiao RP. RGS2 is a primary terminator of beta(2)-adrenergic receptor-mediated G(i) signaling. J Mol Cell Cardiol. 2011;50:1000–1007. doi: 10.1016/j.yjmcc.2011.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho D, Zheng M, Min C, Ma L, Kurose H, Park JH, Kim KM. Agonist-induced endocytosis and receptor phosphorylation mediate resensitization of dopamine D(2) receptors. Mol Endocrinol. 2010;24:574–586. doi: 10.1210/me.2009-0369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleghorn WM, Branch KM, Kook S, Arnette C, Bulus N, Zent R, Kaverina I, Gurevich EV, Weaver AM, Gurevich VV. Arrestins regulate cell spreading and motility via focal adhesion dynamics. Mol Biol Cell. 2015;26:622–635. doi: 10.1091/mbc.E14-02-0740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Druey KM, Kehrl JH. Inhibition of regulator of G protein signaling function by two mutant RGS4 proteins. Proc Natl Acad Sci U S A. 1997;94:12851–12856. doi: 10.1073/pnas.94.24.12851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulin NO, Sorokin A, Reed E, Elliott S, Kehrl JH, Dunn MJ. RGS3 inhibits G protein-mediated signaling via translocation to the membrane and binding to Galpha11. Mol Cell Biol. 1999;19:714–723. doi: 10.1128/mcb.19.1.714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold SJ, Ni YG, Dohlman HG, Nestler EJ. Regulators of G-protein signaling (RGS) proteins: region-specific expression of nine subtypes in rat brain. J Neurosci. 1997;17:8024–8037. doi: 10.1523/JNEUROSCI.17-20-08024.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hague C, Bernstein LS, Ramineni S, Chen Z, Minneman KP, Hepler JR. Selective inhibition of alpha1A-adrenergic receptor signaling by RGS2 association with the receptor third intracellular loop. J Biol Chem. 2005;280:27289–27295. doi: 10.1074/jbc.M502365200. [DOI] [PubMed] [Google Scholar]
- Han J, Mark MD, Li X, Xie M, Waka S, Rettig J, Herlitze S. RGS2 determines short-term synaptic plasticity in hippocampal neurons by regulating Gi/o-mediated inhibition of presynaptic Ca2+ channels. Neuron. 2006;51:575–586. doi: 10.1016/j.neuron.2006.07.012. [DOI] [PubMed] [Google Scholar]
- Hepler JR. Emerging roles for RGS proteins in cell signalling. Trends Pharmacol Sci. 1999;20:376–382. doi: 10.1016/s0165-6147(99)01369-3. [DOI] [PubMed] [Google Scholar]
- Heximer SP, Watson N, Linder ME, Blumer KJ, Hepler JR. RGS2/G0S8 is a selective inhibitor of Gqalpha function. Proc Natl Acad Sci U S A. 1997;94:14389–14393. doi: 10.1073/pnas.94.26.14389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooks SB, Martemyanov K, Zachariou V. A role of RGS proteins in drug addiction. Biochem Pharmacol. 2008;75:76–84. doi: 10.1016/j.bcp.2007.07.045. [DOI] [PubMed] [Google Scholar]
- Kabbani N, Jeromin A, Levenson R. Dynamin-2 associates with the dopamine receptor signalplex and regulates internalization of activated D2 receptors. Cellular signalling. 2004;16:497–503. doi: 10.1016/j.cellsig.2003.09.011. [DOI] [PubMed] [Google Scholar]
- Kim KM, Valenzano KJ, Robinson SR, Yao WD, Barak LS, Caron MG. Differential regulation of the dopamine D2 and D3 receptors by G protein-coupled receptor kinases and beta-arrestins. J Biol Chem. 2001;276:37409–37414. doi: 10.1074/jbc.M106728200. [DOI] [PubMed] [Google Scholar]
- Kovoor A, Seyffarth P, Ebert J, Barghshoon S, Chen CK, Schwarz S, Axelrod JD, Cheyette BN, Simon MI, Lester HA, Schwarz J. D2 dopamine receptors colocalize regulator of G-protein signaling 9-2 (RGS9-2) via the RGS9 DEP domain, and RGS9 knock-out mice develop dyskinesias associated with dopamine pathways. J Neurosci. 2005;25:2157–2165. doi: 10.1523/JNEUROSCI.2840-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labouebe G, Lomazzi M, Cruz HG, Creton C, Lujan R, Li M, Yanagawa Y, Obata K, Watanabe M, Wickman K, Boyer SB, Slesinger PA, Luscher C. RGS2 modulates coupling between GABAB receptors and GIRK channels in dopamine neurons of the ventral tegmental area. Nat Neurosci. 2007;10:1559–1568. doi: 10.1038/nn2006. [DOI] [PubMed] [Google Scholar]
- Lan H, Liu Y, Bell MI, Gurevich VV, Neve KA. A dopamine D2 receptor mutant capable of G protein-mediated signaling but deficient in arrestin binding. Mol Pharmacol. 2009;75:113–123. doi: 10.1124/mol.108.050534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laporte SA, Oakley RH, Holt JA, Barak LS, Caron MG. The interaction of beta-arrestin with the AP-2 adaptor is required for the clustering of beta 2-adrenergic receptor into clathrin-coated pits. J Biol Chem. 2000;275:23120–23126. doi: 10.1074/jbc.M002581200. [DOI] [PubMed] [Google Scholar]
- Leontiadis LJ, Papakonstantinou MP, Georgoussi Z. Regulator of G protein signaling 4 confers selectivity to specific G proteins to modulate mu- and delta-opioid receptor signaling. Cellular signalling. 2009;21:1218–1228. doi: 10.1016/j.cellsig.2009.03.013. [DOI] [PubMed] [Google Scholar]
- Mancuso JJ, Qian Y, Long C, Wu GY, Wensel TG. Distribution of RGS9-2 in neurons of the mouse striatum. J Neurochem. 2010;112:651–661. doi: 10.1111/j.1471-4159.2009.06488.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuho I, Xie K, Martemyanov KA. Macromolecular composition dictates receptor and G protein selectivity of regulator of G protein signaling (RGS) 7 and 9-2 protein complexes in living cells. J Biol Chem. 2013;288:25129–25142. doi: 10.1074/jbc.M113.462283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGinty JF, Shi XD, Schwendt M, Saylor A, Toda S. Regulation of psychostimulant-induced signaling and gene expression in the striatum. J Neurochem. 2008;104:1440–1449. doi: 10.1111/j.1471-4159.2008.05240.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercuri NB, Saiardi A, Bonci A, Picetti R, Calabresi P, Bernardi G, Borrelli E. Loss of autoreceptor function in dopaminergic neurons from dopamine D2 receptor deficient mice. Neuroscience. 1997;79:323–327. doi: 10.1016/s0306-4522(97)00135-8. [DOI] [PubMed] [Google Scholar]
- Min C, Cheong SY, Cheong SJ, Kim M, Cho DI, Kim KM. RGS4 exerts inhibitory activities on the signaling of dopamine D2 receptor and D3 receptor through the N-terminal region. Pharmacol Res. 2012;65:213–220. doi: 10.1016/j.phrs.2011.08.008. [DOI] [PubMed] [Google Scholar]
- Namkung Y, Dipace C, Javitch JA, Sibley DR. G protein-coupled receptor kinase-mediated phosphorylation regulates post-endocytic trafficking of the D2 dopamine receptor. J Biol Chem. 2009;284:15038–15051. doi: 10.1074/jbc.M900388200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neve KA, Seamans JK, Trantham-Davidson H. Dopamine receptor signaling. J Recept Signal Transduct Res. 2004;24:165–205. doi: 10.1081/rrs-200029981. [DOI] [PubMed] [Google Scholar]
- Papakonstantinou MP, Karoussiotis C, Georgoussi Z. RGS2 and RGS4 proteins: New modulators of the kappa-opioid receptor signaling. Cellular signalling. 2015;27:104–114. doi: 10.1016/j.cellsig.2014.09.023. [DOI] [PubMed] [Google Scholar]
- Rahman Z, Schwarz J, Gold SJ, Zachariou V, Wein MN, Choi KH, Kovoor A, Chen CK, DiLeone RJ, Schwarz SC, Selley DE, Sim-Selley LJ, Barrot M, Luedtke RR, Self D, Neve RL, Lester HA, Simon MI, Nestler EJ. RGS9 modulates dopamine signaling in the basal ganglia. Neuron. 2003;38:941–952. doi: 10.1016/s0896-6273(03)00321-0. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Munoz M, de la Torre-Madrid E, Gaitan G, Sanchez-Blazquez P, Garzon J. RGS14 prevents morphine from internalizing Mu-opioid receptors in periaqueductal gray neurons. Cellular signalling. 2007;19:2558–2571. doi: 10.1016/j.cellsig.2007.08.003. [DOI] [PubMed] [Google Scholar]
- Sesack SR, Aoki C, Pickel VM. Ultrastructural localization of D2 receptor-like immunoreactivity in midbrain dopamine neurons and their striatal targets. J Neurosci. 1994;14:88–106. doi: 10.1523/JNEUROSCI.14-01-00088.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesmer JJ, Berman DM, Gilman AG, Sprang SR. Structure of RGS4 bound to AlF4–activated G(i alpha1): stabilization of the transition state for GTP hydrolysis. Cell. 1997;89:251–261. doi: 10.1016/s0092-8674(00)80204-4. [DOI] [PubMed] [Google Scholar]
- Traver S, Bidot C, Spassky N, Baltauss T, De Tand MF, Thomas JL, Zalc B, Janoueix-Lerosey I, Gunzburg JD. RGS14 is a novel Rap effector that preferentially regulates the GTPase activity of galphao. Biochem J. 2000;350(Pt 1):19–29. [PMC free article] [PubMed] [Google Scholar]
- Tulipano G, Stumm R, Pfeiffer M, Kreienkamp HJ, Hollt V, Schulz S. Differential beta-arrestin trafficking and endosomal sorting of somatostatin receptor subtypes. J Biol Chem. 2004;279:21374–21382. doi: 10.1074/jbc.M313522200. [DOI] [PubMed] [Google Scholar]
- Vickery RG, von Zastrow M. Distinct dynamin-dependent and -independent mechanisms target structurally homologous dopamine receptors to different endocytic membranes. J Cell Biol. 1999;144:31–43. doi: 10.1083/jcb.144.1.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent S, Brouns M, Hart MJ, Settleman J. Evidence for distinct mechanisms of transition state stabilization of GTPases by fluoride. Proc Natl Acad Sci U S A. 1998;95:2210–2215. doi: 10.1073/pnas.95.5.2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagener BM, Marjon NA, Revankar CM, Prossnitz ER. Adaptor protein-2 interaction with arrestin regulates GPCR recycling and apoptosis. Traffic. 2009;10:1286–1300. doi: 10.1111/j.1600-0854.2009.00957.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson N, Linder ME, Druey KM, Kehrl JH, Blumer KJ. RGS family members: GTPase-activating proteins for heterotrimeric G-protein alpha-subunits. Nature. 1996;383:172–175. doi: 10.1038/383172a0. [DOI] [PubMed] [Google Scholar]
- Xiao MF, Xu JC, Tereshchenko Y, Novak D, Schachner M, Kleene R. Neural cell adhesion molecule modulates dopaminergic signaling and behavior by regulating dopamine D2 receptor internalization. J Neurosci. 2009;29:14752–14763. doi: 10.1523/JNEUROSCI.4860-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng M, Cheong SY, Min C, Jin M, Cho DI, Kim KM. beta-arrestin2 plays permissive roles in the inhibitory activities of RGS9-2 on G protein-coupled receptors by maintaining RGS9-2 in the open conformation. Mol Cell Biol. 2011;31:4887–4901. doi: 10.1128/MCB.05690-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.