

Summary
Objective
Childhood Absence Epilepsy (CAE) is a genetic generalized epilepsy syndrome with polygenic inheritance, with genes for GABA receptors and T-type calcium channels implicated in the disorder. Previous studies of T-type calcium channel electrophysiology have shown genetic changes and medications have multiple effects 1–3. The aim of this study was to use an established thalamocortical computer model 4 to determine how T-type calcium channels work in concert with cortical excitability to contribute to pathogenesis and treatment response in CAE.
Methods
The model is comprised of cortical pyramidal, cortical inhibitory, thalamocortical relay, and thalamic reticular single compartment neurons, implemented with Hodgkin-Huxley model ion channels and connected by AMPA, GABAA, and GABAB synapses. Network behavior for different combinations of T-type calcium channel conductance, inactivation time, steady state activation/inactivation shift, and cortical GABAA conductance were simulated.
Results
Decreasing cortical GABAA conductance and increasing T-type calcium channel conductance converted spindle to spike and wave oscillations; smaller changes were required if both were changed in concert. In contrast, left shift of steady state voltage activation/inactivation did not lead to spike and wave oscillations, whereas right shift reduced network propensity for oscillations of any type.
Significance
These results provide a window into mechanisms underlying polygenic inheritance in CAE, as well as a mechanism for treatment effects and failures mediated by these channels. While the model is a simplification of the human thalamocortical network, it serves as a useful starting point for predicting the implications of ion channel electrophysiology in polygenic epilepsy such as CAE.
Keywords: Multiscale Thalamocortical Model, Computer Model of Epilepsy, Polygenic Epilepsy
Introduction
Childhood Absence Epilepsy (CAE) is the most common childhood epilepsy syndrome, and is clinically characterized by frequent, brief (10–20 second) non-convulsive seizures with impairment of consciousness. Electrographically, absence seizures are characterized by generalized 3Hz spike and wave discharges 5. Ethosuximide, valproic acid, and lamotrigine are most commonly used to treat CAE, but there is significant variability in patient response to medication, with even the first line choice ethosuximide showing a 47% short term failure rate 6.
CAE is thought to have a genetic etiology, supported by a 75% concordance rate in monozygotic twins 7. Inheritance is polygenic, with genes for GABA receptors, calcium channels, and non-ion channel genes such as SLC2A1 all implicated in contributing to the disorder 8. Genetic techniques have made it possible to characterize the electrophysiological properties of specific ion channel isoforms in vitro. As animal models have shown increased T-type calcium channel currents associated with an absence phenotype 9, studies have explored the effects of genetic changes 1 and analogues of ethosuximide 2 on T-type calcium channel electrophysiology with the goal of understanding how they may cause or prevent absence seizures.
However, it is often difficult to predict how a change in one channel’s electrophysiology will affect overall cortical network function. For example, one study showed multiple T-type calcium channel variants to have altered inactivation time and steady state voltage activation/inactivation curves predicted to cause increased T-type calcium channel bursting in models, which the authors suggested conferred an increased propensity for absence seizures 1. Another study found that ethosuximide analogues cause decreased channel conductance and a smaller steady state voltage activation/inactivation window, which the authors suggested conferred a decreased propensity for absence seizures 2. In a more recent study of a T-type calcium channel variant associated with absence epilepsy, ethosuximide was found to have an opposite effect on the steady state voltage activation/inactivation window, and also led to decreased inactivation time. These effects were significantly reduced in a T-type calcium channel CACNA1H gene variant (P640L) found in individuals with absence seizures who did not respond to initial therapy with ethosuximide 3.
Given these varied effects, approaches are needed to integrate electrophysiology data into a broader context in order to better understand their significance. Multiscale computer models are a novel approach to exploring how changes in channel electrophysiology due to genetic variations or medication effects influence the network as a whole 10–12. The primary goal of this study was to use an established multiscale thalamocortical model to show how T-type calcium channels work in concert with cortical excitability (mediated by cortical GABAA) to contribute to pathogenesis and treatment response in CAE. The secondary goal was to use these results to predict the clinical implications of the electrophysiology of the P640L T-type calcium channel variant described in a recent NIH funded CAE study.
Methods
A previously established thalamocortical model 4 shows that spindles may be transformed to 3Hz spike and wave discharges with increased cortical excitability, mediated by cortical GABAA currents. As this model incorporates parameters that directly correspond to T-type calcium channel electrophysiology, it is well suited to determine how changes in these parameters affect the transformation of spindle oscillations to spike and wave oscillations in the network. Because of the previously established importance of cortical GABAA currents, we chose to simulate the effects of changes in cortical GABAA paired with changes in each of three different T-type calcium channel electrophysiology parameters: total current, inactivation time, and shift in steady state activation/inactivation voltage.
The model was implemented in the NEURON simulation environment as outlined by Destexhe 4,13,14 (available at http://senselab.med.yale.edu/ModelDB/showModel.cshtml?model=234233), and was run on a HP Envy Laptop with a 2.4GHz Intel i7-4700MQ CPU. The model is summarized graphically (Fig 1). In brief, the thalamocortical model was comprised of four one-dimensional layers of 100 neurons, corresponding to cortical pyramidal (PY) neurons, cortical inhibitory (IN) neurons, thalamic relay (TC) neurons and thalamic reticular nucleus (RE) neurons. Network connections are depicted in Fig 1, and included connections between neighboring RE neurons. Within cortical and thalamic layers, neurons projected to ten adjacent neurons, centered on the presynaptic cell. Connections between thalamus and cortex had greater divergence, projecting to twenty neurons adjacent to the equivalent neuron in the other layer. Each cell was defined by a single compartment with a resting potential determined by a sum of intrinsic and post-synaptic currents. AMPA and GABAA postsynaptic currents were described by a simple two-state kinetic scheme, whereas GABAB postsynaptic currents were defined by a non-linear scheme in which dimers of GABAB receptor activate g-proteins which in turn modulate potassium channel currents. Intrinsic currents were defined as the product of maximal conductance (g), activation and inactivation variables (h,m), and the difference between membrane potential and reversal potential (V,E). Activation and inactivation variables followed the 2 state kinetic scheme described by Hodgkin and Huxley 15. Parameters for each intrinsic current had been previously obtained by fitting the kinetic model to voltage clamp data for each respective cell type. Parameters for synaptic conductance had been estimated from experimental data and refined using a search of parameter space 14. All neurons had sodium (INa) and potassium (IK) currents that are responsible for action potentials; additionally, PY neurons had a slow voltage-dependent K current (Im) responsible for adapting trains of action potentials, TC neurons had a T-type calcium (IT) and hyperpolarization (Ih) current, and RE neurons had an IT current.
Figure 1.

Pictorial representation of the thalamocortical model. The network scale shows the arrangement of neurons (represented as dots) into pyramidal (PY) and inhibitory (IN) layers in the cortex, as well as reticular nucleus (RE) and thalamocortical (TC) layers in the thalamus; synaptic connectivity is also shown. The neuron scale lists the types of channels incorporated into each neuron type. The molecular scale depicts Hodgkin-Huxley functions used to represent each channel, which directly correspond to electrophysiology measurements. The network scale image is adapted from “Mechanisms underlying the synchronizing action of corticothalamic feedback through inhibition of thalamic relay cells.” Destexhe, A., D. Contreras, and M. Steriade, J Neurophysiol, 1998. 79(2): p. 999–1016, adapted with permission.
In this study, we explored the effects of changes to the RE T-type calcium channel IT current paired with changes in cortical excitability. T-type calcium channel maximum conductance (g), steady state voltage shift (shift), and inactivation time (τ0 ) were modeled by inserting variables into the original equations defining RE T-type calcium channel current (1), steady state activation/inactivation (4, 5) and inactivation time (6) as follows:
| (1) |
| (2) |
| (3) |
| (4) |
| (5) |
| (6) |
Increased cortical excitability was modeled by decreasing inhibitory GABAA currents in cortical pyramidal neurons, similar to the paradigm used by Destexhe 4. As previous experiments have measured cloned T-type calcium channel function in isolation (that is, out of the context of specific neural tissue or a genetic profile dictating cortical excitability) 1–3, a wide variety of combinations of cortical GABAA conductance and RE T-type calcium channel function were simulated. T-type calcium channel changes were modeled in the thalamic reticular nucleus specifically, as the CACNA1H gene harboring the P640L variant has been shown to be up-regulated in the RE nucleus and associated with increased T-type calcium channel currents in GAERS rats 16.
Parameters for simulations were selected from Destexhe’s model, starting with baseline values g=0.003 uS, shift=-2mV, and τ0 =28.3 ms. Two parameters were held at baseline, and the other was incrementally increased, then decreased over a series of 15–20 simulations until oscillations were no longer sustained. This process was repeated over a range values for cortical GABAA conductance (0 – 100% of baseline). Across all simulations, oscillations occurred spontaneously every 30–40 seconds due to the spontaneous discharges in TC neurons. Between spontaneous oscillations, additional oscillations could be triggered by a stimulus. For each simulation, we allowed the network to come to a steady state (approximately 10 seconds of simulation time following the start of the last spontaneous oscillation). Then a stimulus of 700 nA for 100ms was applied to a group of five pyramidal neurons. This process was repeated with the same stimulus applied to a more widespread group of twenty pyramidal neurons, a stimulus of −100nA for 100ms applied to five thalamic relay (TC) neurons, and a stimulus of −100nA for 100ms applied to twenty TC neurons. Results of each simulation were displayed using raster plots or heat maps of voltage as a function of time and neuron for each layer, and the duration, average frequency, and overall organization of resulting activity was observed. All figures included in this report were generated using the five pyramidal neuron stimulus, with the exception of Fig 2F which was generated using the twenty TC neuron stimulus.
Figure 2.

Raster plots showing representative simulation results for different oscillation types. Each panel corresponds to a cell layer; within each panel, each row (1–100) shows the activity of a specific neuron over time. Plots of IN layer activity were similar to PY layer activity and so were excluded. A) 8–10 Hz spindle oscillation with alternating thalamic bursts, B) 3–4 Hz spike and wave oscillations with synchronized thalamic bursts, C) transitional pattern with fragments of 8–10 Hz and 3–4 Hz oscillations, and D) disorganized spontaneous RE neuron bursting with no network oscillations. Panels E and F contrast two different mechanisms of spike and wave oscillations. E) 3Hz spike and wave oscillations mediated by T-type calcium channel dependent RE neuron bursting, F) 4–5Hz spike and wave oscillations elicited by a large coordinated stimulus that are mediated by RE tonic bursting alone that occur even in the absence of T-type calcium channel currents. Oscillations in panel E are more suggestive of spike and wave oscillations seen in CAE.
Results
All oscillations following the stimulus could be characterized as one of three patterns: spindle oscillations, spike and wave oscillations, or transitional patterns (Fig 2). Spindle oscillations were characterized by 8–10 Hz well organized discharges in PY neurons and a pattern of alternating discharges in groups of TC neurons (Fig 2A). Spike and wave oscillations were characterized by well-organized 2–4 Hz bursts of discharges from PY neurons, as well as synchronized discharges from all TC neurons and increased bursting in RE neurons (Fig 2B). Between these regions, a transitional region was characterized by variable combinations of these two patterns with frequencies ranging from 4–8Hz within a given oscillations. Regardless of type, oscillations lasted no more than 2–3 seconds due to increased hyperpolarization-activated Ih currents in TC neurons. At upper extremes of RE T-type calcium channel conductance and inactivation time (greater than 300% baseline), disorganized fragments of spike and wave oscillations progressed to absence of oscillation. At lower extremes of RE T-type calcium channel conductance and inactivation time, the absence or shortening of RE bursting leads to failure of initiation or propagation of oscillations in the network.
Increased T-type calcium channel conductance and/or inactivation time worked in concert with increased cortical excitability to convert spindle oscillations to spike and wave discharges (Fig 3A,B). Relatively large regions in parameter space supported spindle oscillations; reducing cortical GABAA conductance by as much as 75% and increasing T-type calcium conductance or inactivation time up to 130% of baseline did not substantially change in the nature of oscillation. However, changes in a single parameter beyond these thresholds led to combinations of spindle and spike and wave oscillations, and further increases led to spike and wave oscillations exclusively. Additionally, smaller changes of both parameters in combination also led to transitional or spike and wave oscillations.
Figure 3.
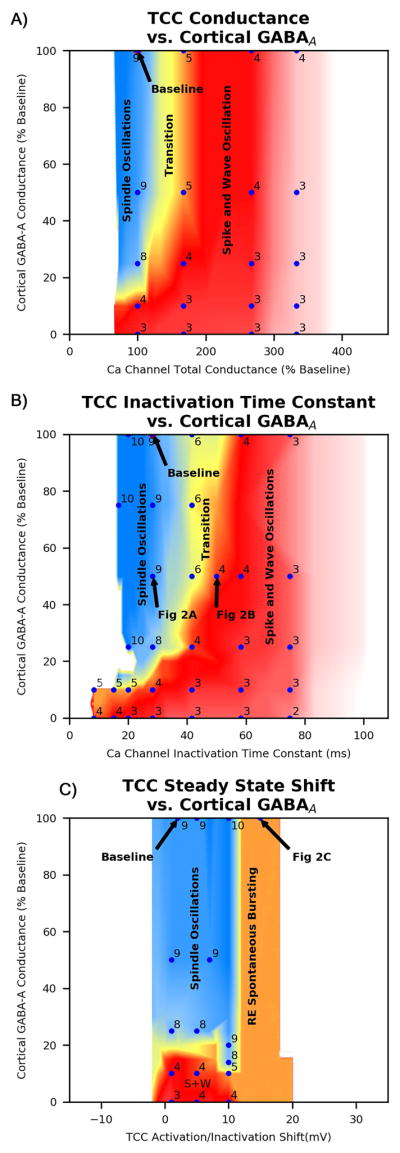
Heat maps showing network oscillation frequency and character for combinations of A) RE T-type calcium channel conductance vs cortical GABAA conductance, B) RE T-type calcium channel Inactivation Time vs cortical GABAA conductance, and C) RE T-type calcium channel Steady State Activation/Inactivation Shift vs cortical GABAA conductance. Blue data points represent individual simulations and are labeled with the overall oscillation frequency; several points also labeled with a figure number that corresponds to that particular simulation. Oscillation type is represented by color: spindle oscillations are blue, transitional oscillations are yellow, spike and wave oscillations are red, disorganized spike and wave oscillations are pink, and absence of oscillation is white.
In contrast, shifts in steady state voltage activation/inactivation curves did not result in spike and wave discharges. Although left shift (activation/inactivation at more negative voltages, reflected by positive values for the shift variable) in RE T-type calcium channel steady state voltage curves did increase T-type calcium bursting and spontaneous firing of RE neurons, it did not lead to coordinated spike and wave discharges. Instead, disorganized RE bursting resulted in fragmented spindle oscillations, a shorter period between spontaneous spindle oscillations, and no oscillation in response to stimulus (Fig 2C, 3C).
Although steady state activation/inactivation shift did not cause spike and wave discharges, right shift in steady state activation/inactivation decreased network propensity for any sort of oscillation. The increase in stimulus threshold required to evoke an oscillation was noted to be proportionate to magnitude of right shift. A right shift of magnitude greater than 4mV (shift = −4) prevented oscillations with small stimulus, a right shift greater than 10mV prevented oscillations with a medium stimulus (10 neurons), and a right shift greater than 16mV prevented oscillations with a large stimulus (20 neurons).
Stimulus size had one other significant effect. In the absence of both cortical GABA-A currents and RE T-type calcium channel currents, widespread TC discharges led to 4–5Hz spike and wave oscillations (albeit not as well formed) despite the absence of RE T-type calcium channel currents (Fig 2F). In this case, spike and wave discharges were initiated and sustained with RE tonic firing alone. This represents a second observed mechanism for diffuse spike and wave oscillations in this model, although these oscillations were less typical of those seen in CAE.
After establishing regions in parameter space that led to different oscillation types, we then used these results to predict clinical implications of the P640L channel variant electrophysiology. In a pharmacogenetic study of 466 individuals with childhood absence epilepsy, this channel variant was shown to be associated with decreased clinical response to ethosuximide; as part of this study, channel electrophysiology was characterized for both wild type calcium channels and P450L variant channels with and without ethosuximide 3. Quantitative changes in channel electrophysiology for each t-type calcium channel parameter were mapped onto parameter space to predict qualitative changes in network function; the original data paired with model predictions are summarized in (Table 1). At a concentration of 3mM ethosuximide there is no change in conductance for either the wild type or the P640L T-type calcium channel; however, there were statistically significant changes in both steady state activation / inactivation shift and inactivation time. Applying simulation results (Fig 3) suggest that right shift in RE T-type calcium channel steady state activation/inactivation does not affect oscillation type, whereas decreased RE T-type calcium channel inactivation time moves the network toward a propensity for spindle oscillations. The region in parameter space for which 3mM ethosuximide is expected to convert spike and wave oscillations to spindle oscillations is shown (Fig 4), and is clearly larger for the wild type T-type calcium channel than the P640L T-type calcium channel. This is consistent with the observation that individuals with absence seizures and the P640L T-type calcium channel variant are less likely to respond to ethosuximide initial therapy 3.
Table 1.
Wild type and P640L Channel Electrophysiology and Predicted Network Effects
| Glauser et al Electrophysiology Results | ||||
|---|---|---|---|---|
| Ethosuximide | 0 mM | 3 mM | ||
| Channel Type | Wild | P640L Variant | Wild | P640L Variant |
| Conductance | 100% | 100% | 98% | 102% |
| Shift of SS Activation / Inactivation Window (mV) | 0 | 2 | −7.5 | −11 |
| Inactivation Time Constant Tau (ms) | 19.8 | 18.1 | 14.2 (5.7ms decrease) | 16.2 (1.9ms decrease) |
| Model Predicted Network Effects | ||||
| Ethosuximide | 0 mM | 3 mM | ||
| Channel Type | Wild | P640L Variant | Wild | P640L Variant |
| Conductance | No effect | No effect | ||
| Shift SS Activation / Inactivation Window | No effect | No effect | ||
| Inactivation Time Constant Tau | No effect | Large decrease in SW (Fig 5A) | Small decrease in SW (Fig 5B) | |
The upper portion of the table summarizes changes in wild type and P640L variant T-Type Calcium Channel Electrophysiology when exposed to ethosuximide as reported by Glauser et al. The lower portion of the table shows the network effect the model predicts for each change in T-type calcium channel parameters. Statistically significant differences in T-type calcium channel parameters from baseline that emerged during electrophysiology experiments are highlighted in bold. Note that although ethosuximide caused a significant right shift in steady state activation/inactivation, the model predicts that this change will not alter overall network behavior. In contrast, the model predicts that decreased inactivation time constant will treat seizure (convert spike and wave oscillations to spindle oscillations) for an area in parameter space. This effect is reduced for the variant calcium channel (see Fig 5).
Figure 4.

Predicted response to ethosuximide for A) individuals with wild type T-type calcium channel and B) P640L variant T-type calcium channel. The white region represents the set of parameters for which the model predicts 3mM ethosuximide will convert spike and wave oscillations to spindle oscillations, corresponding to individuals who would be expected to be adequately treated. Panel B shows a reduced parameter space expected to be treated with the P640L T-type calcium channel, consistent with clinical observations of reduced response to ethosuximide in individuals with this variant.
Discussion
These results provide useful guidelines for predicting clinical implications of changes in T-type calcium channel electrophysiology. On a broader scale, this model sheds light on mechanisms behind pathogenicity and medication effects in CAE. We showed that combinations of changes in T-type calcium channel and GABAA channels can lead to the common endpoint of CAE, consistent with observation of multiple pathogenic mutations in GABAA subunits and T-type calcium channel in animals and humans 8,17. These concepts are illustrated (Fig 5A), showing the effects of different combinations of change in T-type calcium channel and cortical GABAA channels in four hypothetical individuals compared to a baseline individual. In general, the model predicts a substantial tolerance for changes in channel function without an effect on network oscillations, consistent with observations that significant changes in channel function do not necessarily lead to epilepsy 18. Note that although individuals #3 and #4 have the same increase in T-type calcium channel inactivation time, the model predicts that the same T-type calcium channel mutation leads to seizures in only individual #3. Other mechanisms for spike and wave oscillations may not involve RE T-type calcium channels at all; we found a sufficiently large stimulus can lead to spike and wave oscillations in the absence of RE T-type calcium channel currents. This finding is consistent with a recent study showing that CaV3.2/3.3 knockout mice have complete abolition of RE bursting but retain a propensity for absence seizures 19. Taken together, these findings highlight the need to consider patterns of multiple ion channels in order to understand mechanisms behind polygenic epilepsy.
Figure 5.

Examples of pathogenesis of channel variants and treatment. A) Four hypothetical individuals (1–4) with different combinations of genetic variants. #1 and #2 have single variants leading to change in GABAA conductance alone, while #3 and #4 have a combination a shared T-type calcium channel variant increasing inactivation time and a second variant altering GABAA conductance. Note that individuals #1 and #3 are predicted to have seizures, while #2 and #4 are not. B) Treatment responses for three different hypothetical individuals (5–7) predicted to have seizures. #5 shows a treatment response for VPA but not ETX, while #6 shows a response to ETX but not VPA. #7 represents a patient with the P640L variant that blunts response to ETX, leading to treatment failure.
Mechanisms by which antiepileptic drugs prevent absence seizures are illustrated (Fig 5B), where we show three hypothetical individuals with an initial genetic profile at points 5, 6, and 7. A sufficient decrease in T-type calcium channel conductance or inactivation time is predicted to restore spike and wave oscillations to normal spindle oscillations, provided cortical excitability is not too high (Fig 5B #6 horizontal arrow), consistent with a commonly postulated mechanism of action for ethosuximide in absence epilepsy 18. Additionally, right shift of steady state voltage activation/inactivation is predicted to decrease network oscillations, a phenomenon that has been observed in animals 20. Similarly, for mild increases in T-type calcium channel conduction or inactivation time, a decrease in cortical excitability restored spindle oscillations (#5 vertical arrow), explaining the utility of medications with gabaergic properties such as benzodiazepines or valproic acid 6,21.
Conversely, larger changes in these channels may lead to treatment failures. Reducing T-type calcium channel conductance or inactivation time is predicted to have no effect on spike and wave discharges if cortical excitability is significantly increased (Fig 5B #5 horizontal arrow). In this case, the model predicts treatment failure with ethosuximide but success with gabaergic medications. If T-type calcium channel conductance or inactivation time is sufficiently increased, changes in cortical GABAA conductance are predicted to have no effect (Fig 5B #6 vertical arrow), suggesting treatment failure with gabaergic medications but success with ethosuximide. Either medication might be effective with a mild change in both TCCs and GABAA subunits, whereas with a more severe change in both channels might require simultaneous treatment with medications that act on both channels. An alternate mechanism for treatment failure, the blunted reduction of inactivation time by ethosuximide in the P640L T-type calcium channel variant, is illustrated by individual #7.
This study highlights some of the limitations of predicting network-level effects of changes in ion channel function from electrophysiology alone. Even activity at the neuron level may be misleading; several have used computer models to predict changes in thalamic cell bursting due to genetic changes or medications 1,22, with the assumption that increased bursting implies a propensity for seizures. However, we found that this is not universally true; although a left shift in steady state activation/inactivation curves of RE T-type calcium channels increases spontaneous bursting, it does not lead to synchronous bursting necessary for spike and wave oscillations (Fig 2C, 3C). As another example, past studies have focused on conduction block as the primary mechanism by which succinimides act on T-type calcium channels 2, but our results suggest that both conduction and inactivation time play an important role.
Although it does not include developmental properties that correspond with age of onset or offset of CAE, the thalamocortical model utilized incorporates many of the mechanisms thought to underlie absence seizures. Although the mechanisms have long been debated 23, key features are thought to be a thalamocortical loop including the thalamic reticular nucleus 24; transformation of sleep spindles to spike and wave discharges associated with increased thalamic reticular nucleus bursting 25, and an associated increased T-type calcium channel currents 9. Hyperactivity in cortical deep layer pyramidal neurons has also been implicated in the initiation of absence seizures GAERS rats 26. Maheshwari recently outlined a functional framework for absence epilepsy that incorporates four key features: cortical hyperexcitability, decreased feedforward fast phasic inhibition, tonic inhibition (by both GABAB and extra-synaptic GABAA receptors), and deinactivation of T-type calcium channels 17. All of these features are included in the Destexhe model with the exception of tonic inhibition.
A number of other potentially important physiologic features are not included in the model. Consider ion channels: NMDA receptors were excluded from the original model because they did not significantly change behavior of network 4. This is a limitation, as evidence suggests that T-type calcium channels modulate NMDA signaling, resulting in increased glutamate levels and absence seizures in rats 27. Additionally, the channel types represented in this model have multiple isoforms differentially expressed throughout the brain 18, and redundant systems likely modulate the expression of different isoforms and mask the effects of some genetic variants. Maheshwari describes several such changes due to altered feedforward inhibition that have been observed experimentally 17. This model does not incorporate such changes, which may serve to reduce the effects of changes in calcium channels and cortical excitation described in this study.
The model has a number of other functional and structural simplifications. Individual neurons are represented as a single compartment, although this limitation was accounted for (albeit imperfectly) when choosing parameters for the original single compartment RE cell model 28. A recent study implicated copy number variants in a variety of genes that regulate synaptic adhesion, organization, vesicle release and gap junctions in patients with CAE 29; the effects of these variants cannot be directly tested with this model. The anatomic layout is also vastly simplified; this is another significant limitation, as combined fMRI/MEG evidence has pointed to possible focal cortical sources of absence seizures 30. Finally, evidence suggests that metabolic pathways absent from this model could provide additional redundant systems that compensate for channel variants 31. Each of these limitations offers an avenue for future exploration with more complex multiscale models.
This study provides a useful general analysis of interactions of changes in T-type calcium channel and cortical GABAA function. However, there are limitations inherent to this approach. This study analyzed changes in each of 3 T-type calcium channel parameters in isolation; in reality a genetic change or medication modulates these parameters in concert 1,2. Furthermore, in reality genetic changes affect the thalamocortical network in a number of different ways simultaneously. For example, genetic changes in TCCs likely reach beyond the reticular nucleus of the thalamus, as these channels are expressed in other parts of the thalamus as well as the cortex 32. GABAA also is active in a number of different locations including the reticular nucleus 4,33; the effects of gabaergic medications in locations outside of the cortex were not explored in this study. Similarly, medications such as ethosuximide act on a variety of calcium channels isoforms that are expressed throughout the brain, and may act on other channels as well.
While it is not practical to simulate every possible combination of network parameters, an alternative approach is to use a computer model to simulate a specific set of parameters for channel electrophysiology, determined by an individual’s genetic profile. Computer simulation could then be used to determine whether that particular genetic profile could lead to epilepsy, as well as predict response to different anti-epileptic drugs. Obtaining a genetic profile for an individual is already a reality thanks to high-throughput genetic testing 12,34, and efforts are being made to standardize reporting of electrophysiology features of channel variants or medications in order to develop central databases 35. More complex computer models of mouse and human thalamocortical loops have already been created 30,36, and more ambitious projects to develop anatomic models of mouse or human brains are underway 37. This combination of technologies promises to help untangle the mechanisms underlying polygenic epilepsy and lead to novel treatments. They also could lead to a new era of individualized medicine in which multiscale computer models are paired with a patient’s genetic profile to predict that individual’s optimal drug combinations, saving patients from morbidity and mortality due to both uncontrolled seizures and side effects of unnecessary medications.
Key Points.
Increased T-type calcium channel currents act in concert with decreased GABAA currents to cause spike and wave oscillations
Medications acting on these channels abolish spike and wave oscillations, provided they are well matched to changes in channel function
Computer models can be used to predict pathogenicity and medication response for a combination of genetic variants
Acknowledgments
This work was supported in part by a grant from the National Institute of Health (NINDS U01-045911). We wish to thank Donald Gilbert and the rest of the Cincinnati Children’s Neurology Division for their enthusiastic support of this research, as well as for providing helpful feedback on the initial manuscript.
Footnotes
Disclosure of Conflicts of Interest
None of the authors has any conflict of interest to disclose.
Ethical Publication Statement
We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
References
- 1.Vitko I, Chen Y, Arias JM, et al. Functional characterization and neuronal modeling of the effects of childhood absence epilepsy variants of CACNA1H, a T-type calcium channel. J Neurosci. 2005;25:4844–4855. doi: 10.1523/JNEUROSCI.0847-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gomora JC, Daud AN, Weiergraber M, et al. Block of cloned human T-type calcium channels by succinimide antiepileptic drugs. Mol Pharmacol. 2001;60:1121–1132. [PubMed] [Google Scholar]
- 3.Glauser TA, Holland K, O’Brien VP, et al. Pharmacogenetics of Antiepileptic Drug Efficacy in Childhood Absence Epilepsy. Ann Neurol. 2017 doi: 10.1002/ana.24886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Destexhe A. Spike-and-wave oscillations based on the properties of GABAB receptors. J Neurosci. 1998;18:9099–9111. doi: 10.1523/JNEUROSCI.18-21-09099.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tenney JR, Glauser TA. The current state of absence epilepsy: can we have your attention? Epilepsy Curr. 2013;13:135–140. doi: 10.5698/1535-7511-13.3.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Glauser TA, Cnaan A, Shinnar S, et al. Ethosuximide, valproic acid, and lamotrigine in childhood absence epilepsy. N Engl J Med. 2010;362:790–799. doi: 10.1056/NEJMoa0902014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lennox WG. The heredity of epilepsy as told by relatives and twins. J Am Med Assoc. 1951;146:529–536. doi: 10.1001/jama.1951.03670060005002. [DOI] [PubMed] [Google Scholar]
- 8.Yalcin O. Genes and molecular mechanisms involved in the epileptogenesis of idiopathic absence epilepsies. Seizure. 2012;21:79–86. doi: 10.1016/j.seizure.2011.12.002. [DOI] [PubMed] [Google Scholar]
- 9.Tsakiridou E, Bertollini L, de Curtis M, et al. Selective increase in T-type calcium conductance of reticular thalamic neurons in a rat model of absence epilepsy. J Neurosci. 1995;15:3110–3117. doi: 10.1523/JNEUROSCI.15-04-03110.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lytton WW. Computer modelling of epilepsy. Nat Rev Neurosci. 2008;9:626–637. doi: 10.1038/nrn2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Traub RD, Contreras D, Whittington MA. Combined experimental/simulation studies of cellular and network mechanisms of epileptogenesis in vitro and in vivo. J Clin Neurophysiol. 2005;22:330–342. [PubMed] [Google Scholar]
- 12.Klassen T, Davis C, Goldman A, et al. Exome sequencing of ion channel genes reveals complex profiles confounding personal risk assessment in epilepsy. Cell. 2011;145:1036–1048. doi: 10.1016/j.cell.2011.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Destexhe A, Contreras D, Sejnowski TJ, et al. A model of spindle rhythmicity in the isolated thalamic reticular nucleus. J Neurophysiol. 1994;72:803–818. doi: 10.1152/jn.1994.72.2.803. [DOI] [PubMed] [Google Scholar]
- 14.Destexhe A, Contreras D, Steriade M. Mechanisms underlying the synchronizing action of corticothalamic feedback through inhibition of thalamic relay cells. J Neurophysiol. 1998;79:999–1016. doi: 10.1152/jn.1998.79.2.999. [DOI] [PubMed] [Google Scholar]
- 15.Hodgkin AL, Huxley AF. A quantitative description of membrane current and its application to conduction and excitation in nerve. J Physiol. 1952;117:500–544. doi: 10.1113/jphysiol.1952.sp004764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Talley EM, Solorzano G, Depaulis A, et al. Low-voltage-activated calcium channel subunit expression in a genetic model of absence epilepsy in the rat. Brain Res Mol Brain Res. 2000;75:159–165. doi: 10.1016/s0169-328x(99)00307-1. [DOI] [PubMed] [Google Scholar]
- 17.Maheshwari A, Noebels JL. Monogenic models of absence epilepsy: windows into the complex balance between inhibition and excitation in thalamocortical microcircuits. Prog Brain Res. 2014;213:223–252. doi: 10.1016/B978-0-444-63326-2.00012-0. [DOI] [PubMed] [Google Scholar]
- 18.Zamponi GW, Lory P, Perez-Reyes E. Role of voltage-gated calcium channels in epilepsy. Pflugers Arch. 2010;460:395–403. doi: 10.1007/s00424-009-0772-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee SE, Lee J, Latchoumane C, et al. Rebound burst firing in the reticular thalamus is not essential for pharmacological absence seizures in mice. Proc Natl Acad Sci U S A. 2014;111:11828–11833. doi: 10.1073/pnas.1408609111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Steriade M. Sleep oscillations in corticothalamic neuronal networks and their development into self-sustained paroxysmal activity. Rom J Neurol Psychiatry. 1993;31:151–161. [PubMed] [Google Scholar]
- 21.Mikkelsen B, Birket-Smith E, Bradt S, et al. Clonazepam in the treatment of epilepsy. A controlled clinical trial in simple absences, bilateral massive epileptic myoclonus, and atonic seizures. Arch Neurol. 1976;33:322–325. doi: 10.1001/archneur.1976.00500050008002. [DOI] [PubMed] [Google Scholar]
- 22.Lytton WW, Sejnowski TJ. Computer model of ethosuximide’s effect on a thalamic neuron. Ann Neurol. 1992;32:131–139. doi: 10.1002/ana.410320204. [DOI] [PubMed] [Google Scholar]
- 23.Avoli M. A brief history on the oscillating roles of thalamus and cortex in absence seizures. Epilepsia. 2012;53:779–789. doi: 10.1111/j.1528-1167.2012.03421.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fuentealba P, Steriade M. The reticular nucleus revisited: intrinsic and network properties of a thalamic pacemaker. Prog Neurobiol. 2005;75:125–141. doi: 10.1016/j.pneurobio.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 25.Steriade M, Contreras D. Relations between cortical and thalamic cellular events during transition from sleep patterns to paroxysmal activity. J Neurosci. 1995;15:623–642. doi: 10.1523/JNEUROSCI.15-01-00623.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Polack PO, et al. Deep layer somatosensory cortical neurons initiate spike-and-wave discharges in a genetic model of absence seizures. J Neurosci. 2007;27(24):6590–9. doi: 10.1523/JNEUROSCI.0753-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang G, Bochorishvili G, Chen Y, et al. CaV3. 2 calcium channels control NMDA receptor-mediated transmission: a new mechanism for absence epilepsy. Genes Dev. 2015;29:1535–1551. doi: 10.1101/gad.260869.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Destexhe A, Neubig M, Ulrich D, et al. Dendritic low-threshold calcium currents in thalamic relay cells. J Neurosci. 1998;18:3574–3588. doi: 10.1523/JNEUROSCI.18-10-03574.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Addis L, Rosch RE, Valentin A, et al. Analysis of rare copy number variation in absence epilepsies. Neurol Genet. 2016;2:e56. doi: 10.1212/NXG.0000000000000056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tenney JR, Fujiwara H, Horn PS, et al. Low- and high-frequency oscillations reveal distinct absence seizure networks. Ann Neurol. 2014;76:558–567. doi: 10.1002/ana.24231. [DOI] [PubMed] [Google Scholar]
- 31.Neymotin SA, McDougal RA, Bulanova AS, et al. Calcium regulation of HCN channels supports persistent activity in a multiscale model of neocortex. Neuroscience. 2016;316:344–366. doi: 10.1016/j.neuroscience.2015.12.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Destexhe A, Contreras D, Steriade M. LTS cells in cerebral cortex and their role in generating spike-and-wave oscillations. Neurocomputing. 2001;38–40:555–563. [Google Scholar]
- 33.Thomas E, Lytton WW. Computer model of antiepileptic effects mediated by alterations in GABA(A)-mediated inhibition. Neuroreport. 1998;9(4):691–6. doi: 10.1097/00001756-199803090-00024. [DOI] [PubMed] [Google Scholar]
- 34.Chambers C, Jansen LA, Dhamija R. Review of Commercially Available Epilepsy Genetic Panels. J Genet Couns. 2016;25:213–217. doi: 10.1007/s10897-015-9906-9. [DOI] [PubMed] [Google Scholar]
- 35.Hinard V, Britan A, Rougier JS, et al. ICEPO: the ion channel electrophysiology ontology. Database (Oxford) 2016:2016. doi: 10.1093/database/baw017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Traub RD, Contreras D, Cunningham MO, et al. Single-column thalamocortical network model exhibiting gamma oscillations, sleep spindles, and epileptogenic bursts. J Neurophysiol. 2005;93:2194–2232. doi: 10.1152/jn.00983.2004. [DOI] [PubMed] [Google Scholar]
- 37.Tiesinga P, Bakker R, Hill S, et al. Feeding the human brain model. Curr Opin Neurobiol. 2015;32:107–114. doi: 10.1016/j.conb.2015.02.003. [DOI] [PubMed] [Google Scholar]