

Abstract
A series of 2’-substituted cyclobutyl nucleoside analogs were efficiently prepared by constructing the core cyclobutyl ring using different [2+2] cycloaddition approaches. The triphosphate derivative of a cyclobutyl nucleoside was also synthesized and evaluated against wild-type and mutant HIV reverse transcriptases (RT). Whereas the nucleoside analogs were inactive against HIV-1 in culture, the nucleotide showed good activity not only against wild type and recombinant HIV RT (IC50 = 4.7, 6.9 µM), but also against the M184I and M184V mutants (IC50 = 6.1, 6.9 µM) in cell free assays.
Graphical Abstract
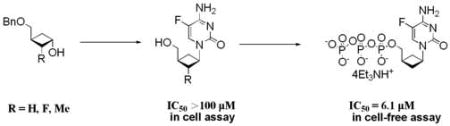
Several 2’-substituted cyclobutyl nucleosides were synthesized and evaluated as anti-HIV agents. Whereas the cyclobutyl nucleosides were not active against HIV in culture, the triphosphate forms were quite active against wild type and mutant forms of HIV reverse transcriptase (RT).
Acquired immune deficiency syndrome (AIDS) has rapidly become one of the major causes of death in the world. It is estimated that over 40 million people have developed human immunodeficiency virus (HIV) infections, which is the causative agent of AIDS.1 In 1985, 3’-azido-3’-deoxythymidine (AZT) was approved as the first synthetic nucleoside to inhibit the replication of HIV. Since then, a number of other synthetic nucleoside analogs have been proven to be effective against HIV. After cellular phosphorylation to the triphosphate form by cellular kinases, the nucleotides are incorporated into a growing strand of viral DNA and cause chain termination due to the absence of the 3’-hydroxyl group.
Despite the enormous success of nucleoside based therapy of HIV infection, there is still no cure for AIDS. One reason is that the prolonged clinical use of nucleoside analogs gives rise to resistant viruses that contain mutations in the RT.2 For example, the M184V/I mutation in HIV-1 RT is associated with high level resistance to lamivudine (3TC) and emtricitabine (FTC). Structural studies suggest that the mechanism of resistance of HIV-1 RT carrying the M184V/I mutation involves steric hindrance, which would prevent further incorporation of nucleoside analogs such as 3TC and FTC in the nucleotide forms.3
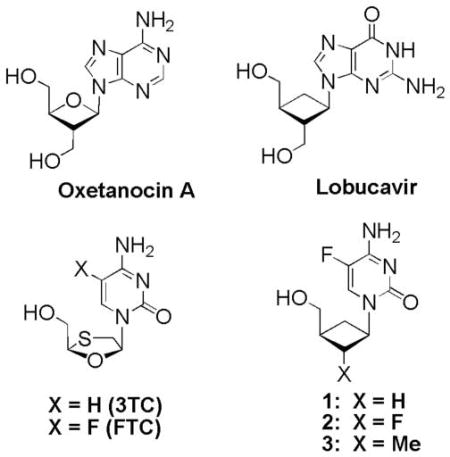
We postulated that the more rigid and smaller cyclobutyl ring would enable the nucleoside analog to fit into the more sterically hindered active site of RT containing the M184V/I mutants. Nucleoside analogs of this type received a great deal of attention several years ago with the discovery of naturally occurring Oxetanocin A, which shows activity against HIV and Lobucavir (Cyclobut-G) with activity against HBV. However, virtually all of the reported derivatives possess the 2’3’-bis-hydroxyl motif that enables them to be phosphorylated and incorporated into growing strands of DNA. We were interested in preparing a series of 2’-substituted cyclobutyl nucleosides that only have the 3’-hydroxymethyl for phosphorylation, but not the 2’-hydroxymethyl motif. We reasoned that these nucleoside analogs could fit into the more sterically hindered active site of M184I/V mutant forms of RT.
In this paper, we report the synthesis and biological evaluation of 2’-substituted cyclobutyl nucleosides as potential anti-HIV agents. To elucidate the mechanism of action of these novel cyclobutyl nucleosides, a triphosphate derivative 21 was also synthesized and evaluated against wild-type and mutant HIV-1 RT.
The 5-fluoro-1-[cis-3-(hydroxymethyl)-cyclobuyl]-cytosine 1 was synthesized using a reported procedure.4 As shown in Scheme 1, reduction of the cyclobutanone 4 with L-Selectride in THF solution gave cis-compound 5, which was converted to trans-(3-benzyloxymethyl)cyclobutanol 6 via Mitsunobu reaction followed by hydrolysis of the resulting ester. Cyclobutanol 6 reacted with N3-benzoyl-5-fluorouracil under Mitsunobu conditions to give nucleoside 7 which was deprotected to give 5-fluorouracil nucleoside 8 in moderate yield. Finally, compound 8 was converted to the corresponding 5-fluorocytosine derivative 1 by the procedure shown in Scheme 2.
Scheme 1.

Scheme 2.

Fluorine as a substituent is isosteric with hydrogen; therefore, it should not affect the spatial disposition of atoms in the molecule. On the other hand, the physicochemical properties of a fluorine atom can, due to its high electron negativity, profoundly affect the electrostatic properties of the bonds around it, thereby influencing both the conformation of the four membered ring and the innate strength of the bonds.
In the synthesis of 2’-fluoro-substituted cyclobutyl nucleosides, we found that electrophilic fluorination with Selectfluor to be the most efficient method to introduce fluorine in the cyclobutanone 4.5 As shown in Scheme 2, the cyclobutanone 4 was first deprotonated by LDA, followed by lithium enolate quenching with TMSCl to afford the silyl enol ether 9.6 This silyl enol ether was then allowed to react with Selecfluor to give a 3:1 mixture of inseparable diastereomers 10. The crude mixture was reduced by L-Selectride followed by chromatography to isolate pure diastereomer 11 in good yield. Mesylation of 11 gave compound 12, which then was coupled with N3-(4-methoxybenzyl)-5-fluorouracil under basic conditions to give 53% of nucleoside 13. Removal of both the p-methoxylbenzyl and benzyl groups by aluminum trichloride afforded compound 14 in 80% yield. The stereochemistry of 14 was confirmed by X-ray crystal structure analysis.7 Conversion of 14 to 5-fluoro-1-[trans-2-fluoro-cis-3-(hydroxylmethyl)-cyclobutyl]cytosine 2 was achieved in the standard 3 step process as shown in Scheme 2.
To compare the electronic properties of the 2’-substituted analogs, the 2’-methylcyclobutyl nucleoside was synthesized and evaluated. As shown in Scheme 3, a keteniminium salt was used as the key intermediate to form the cyclobutanone.8 The keteniminium triflate generated from N,N-dimethylpropionamide in situ reacted with allyl benzyl ether to give the intermediate cyclobutaniminium salt which is hydrolyzed to provide cyclobutanone 15 as the major diastereomer with the methyl group trans to the benzyl group (dr = 9:1). Reduction of the cyclobutanone 15 gave primarily one diastereomer 16 with the methyl group and the hydroxyl group cis to each other as confirmed by 1H NOE analysis. The remainder of the synthesis of 5-fluoro-1-[trans-2’-methyl-cis-3’-hydroxymethyl)cyclobutyl]cytosine 3 is shown in Scheme 2.
Scheme 3.

Although they were not found to be toxic, unfortunately, none of the nucleosides 1, 2 and 3 showed significant anti-HIV activity up to 100 µM in primary human lymphocytes infected with HIV-1 (strain LAI). To elucidate the mechanism of the nucleotides incorporating into the viral DNA chain, the triphosphate of nucleoside 1 was synthesized according to Scheme 4.
Scheme 4.

The monophosphate 20 was generated by the reaction of the nucleoside with phosphorus oxychloride using trimethylphosphate as the solvent. The monophosphate 20 was activated as its morphine phosphoramidate, which was subsequently combined with nucleophilic pyrophosphate to provide triphosphate 21 (Scheme 4).9, 10, 11
Although the previously described anti-HIV studies demonstrated that compound 1 was not active up to 100 µM, its triphosphate 21 exhibited comparable anti-HIV activity to 3TC-TP against recombinant HIV-RT and wild type HIV-RT (IC50 = 4.7, 6.9 µM). Furthermore, the triphosphate showed also very good activity against M184I and M184V mutant RT (IC50 = 6.1, 6.9 µM), which were not inhibited by 3TC-TP. The results suggest that the cyclobutyl nucleotide can be incorporated into the DNA chains of HIV RT and M184M/V mutants to terminate the DNA elongation.
All the biochemical results were obtained according to the assay described by Eriksson et al10 and are shown in Table 1.
Table 1.
Comparison of Inhibition of HIV RT in Cell-Free Assays
| HIV RT* | Inhibition of RT Activity (IC50, µM) | |
|---|---|---|
| 3TC-TP | Compound 21 | |
| Recombinant HIV RT (WT) | 3.0 | 4.7 |
| HIV RT (WT) | 6.5 | 6.9 |
| HIV RT (M184I) | >10 | 6.1 |
| HIV RT (M184V) | >10 | 6.9 |
All the HIV RT used, except the recombinant RT, was obtained from viral lysates from PBM cell infected with respective HIV.
In conclusion, several 2’-substituted cyclobutyl nucleosides were synthesized and evaluated as anti-HIV agents. Although the cyclobutyl nucleosides were not active against HIV, the triphosphate form of one of them was quite active against wild-type and M184V or M184I HIV RT. This suggests that the nucleosides are not being phosphorylated by the cellular kinases. It appears that the cellular kinases cannot recognize the modified nucleoside analogs due to their structural and conformational differences from the natural nucleoside substrates. Our future work will be focused on the synthesis of phosphate prodrugs of various cyclobutyl nucleoside analogs to circumvent the problems with phosphorylation.
Acknowledgments
This work was supported by NIH grants 2R37AI-28731 (DCL), 1R37AI-41890 (RFS), Emory’s CFAR grant 5P30-AI150409, and the Department of Veterans Affairs (RFS). We thank Dr. R. Pai and Kim L. Rapp for performing the RT assays.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Pani A, Loi AG, Mura M, Marceddu T, La Colla P, Marongiu ME. Current Drug Targets-Infectious Disorders. 2002;2:17. doi: 10.2174/1568005024605873. [DOI] [PubMed] [Google Scholar]
- 2.Schuurman R, Nijhuis M, van Leeuwen R, Schipper P, de Jong D, Collis P, Danner SA, Mulder J, Loveday C. J. Infect. Dis. 1995;171:1411. doi: 10.1093/infdis/171.6.1411. [DOI] [PubMed] [Google Scholar]
- 3.Gao HQ, Boyer PL, Sarafianos SG, Arnold E, Hughes SH. J. Mol. Bio. 2000;300:403. doi: 10.1006/jmbi.2000.3823. [DOI] [PubMed] [Google Scholar]
- 4.(a) Kaiwar V, Reese CB, Gray EJ, Neidle S. J. Chem. Soc. Perkin Trans. 1. 1995;18:2281. [Google Scholar]; (b) Frieden M, Giraud M, Reese CB, Song Q. J. Chem. Soc. Perkin Trans. 1. 1998;7:2827. [Google Scholar]; (c) He W, Togo H, Ogawa H, Yokoyama M. Heteroat. Chem. 1997;8:411. [Google Scholar]
- 5.(a) Nyffeler PT, Durón SG, Burkart MD, Vincent SP, Wong C-H. Angew. Chem. Int. Ed. 2005;44:192. doi: 10.1002/anie.200400648. [DOI] [PubMed] [Google Scholar]; (b) Singh RP, Shreeve JM. Acc. Chem. Res. 2004;37:31. doi: 10.1021/ar030043v. [DOI] [PubMed] [Google Scholar]; (c) Stavber S, Jereb M, Zupan M. Synthesis. 2002;17:2609. [Google Scholar]; (d) Stavber G, Zupan M, Jereb M, Stavber S. Org. Lett. 2004;6:4973. doi: 10.1021/ol047867c. [DOI] [PubMed] [Google Scholar]
- 6.Vidal J, Huet F. J. Org. Chem. 1988;53:611. [Google Scholar]
-
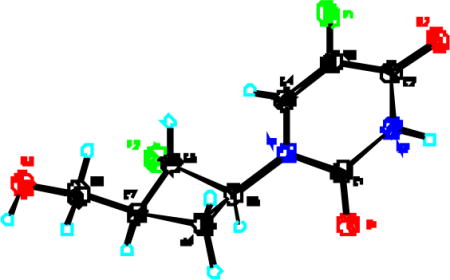
7.X-Ray structure of 5-fluoro-1-[trans-2-fluoro-cis-3-(hydroxymethyl)cyclobutyl]uracil 14 (CCDC 640733):
- 8.Falmagne JB, Schmit C, Escudero J, Vanlierde H, Ghosez L. Org. Synth. 1990;69:199. [Google Scholar]
- 9.(a) Moffatt JG, Khorana HG. Can. J. Chem. 1961;39:649. [Google Scholar]; (b) Moffatt JG. Can. J. Chem. 1964;42:599. [Google Scholar]
- 10.Eriksson BF, Chu CK, Schinazi RF. Antimicrob. Agents. Chemother. 1989;33:1729. doi: 10.1128/aac.33.10.1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Compound 21: 1H NMR (D2O, 400MHz): δ 1.22 (t, J = 7.2Hz, 36H), 2.14 (m, 2H), 2.49 (m, 3H), 3.08 (m, 2H), 3.19 (q, J = 7.2Hz, 24H), 4.64 (m, 1H), 8.12 (d, J = 7.2 Hz, 1H). 31P NMR (D2O, referenced to H3PO4, 162MHz): δ −22.9 (1P), −10.5 (2P).