

Abstract
This chapter describes detailed methods used for laser capture microdissection (LCM) of discrete subpopulations of cells. Topics covered include preparing tissue blocks, cryostat sectioning, processing slides, performing the LCM, and purification of RNA from LCM samples. Notes describe the fine points of each operation, which can often mean the difference between success and failure.
Keywords: Laser capture, Laser capture microdissection, Tissue purification, Small sample analysis, Gene expression profiling
1. Introduction
For many research projects the purity of the starting biological sample being investigated is of the utmost importance. For example, in studies of the molecular events driving kidney development, there is great power in being able to divide the entire kidney into discrete subcomponents. The higher the resolution of the analysis the better one is able to assign molecular processes that emerge to specific compartments and cell types. In particular, a microarray-based analysis of the total developing kidney might identify expression of genes involved in Wnt signaling. But the results would not determine which cell types are sending signals, and which are receiving them. On the other hand, an analysis of gene expression patterns in the multiple individual distinct compartments of the developing kidney would identify precise ligand and receptor expression patterns for each element (1). This clearly yields a much more useful set of data. Indeed, there are many situations in the study of development and disease where there is great advantage to be gained through the analysis of small regions of a sample. For example, one the first applications of laser capture technology was in the study of tumors, which can exhibit significant morphological heterogeneity (2).
Laser capture microdissection (LCM) allows the purification of discrete subregions of a sample, in some cases even down to single cell resolution. There are multiple variations of the methodology. In its earliest form, a transparent thermoplastic film is placed over a tissue section and a near infrared (IR) (3) wavelength laser is used to heat the region above the cells of interest. This melts the film onto the cells, creating an adhesion that allows the cells to be purified as the film is removed (4). Other versions use a fine ultraviolet (UV) wavelength laser beam to cut out and isolate the region of interest from a tissue section (5–7). This chapter focuses on a combination system made by Applied Biosystems/Arcturus, which uses both IR and UV lasers for microdissection, although most of the principles discussed would equally apply to any commercial LCM product.
The ArcturusXT and Arcturus Veritas instruments combine IR/UV lasers and can be used with special polyethylene naphthalate (PEN) plastic membrane slides. In one preferred format the PEN membrane covers a glass slide, with the tissue sections then placed on top of the membrane. A UV laser is used to cut around the cells of interest, through both the membrane and the tissue (Fig. 1). The chief advantage of this system is that the region to be purified is not directly attached to the glass slide, thereby facilitating easy removal. An overlying thermoplastic membrane is melted onto the region to be captured using the IR laser, and then when this membrane is lifted from the slide it carries with it both the isolated cells and their underlying PEN membrane. This strategy can achieve very reproducible LCM purification of selected regions.
Fig. 1.
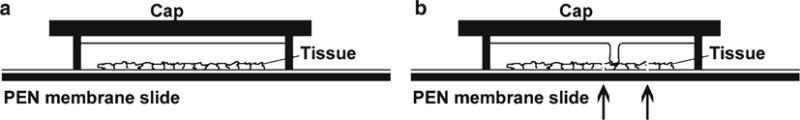
Laser capture principle. (a) Polyethylene naphthalate (PEN) membrane slide is shown with tissue on the membrane. A Cap is shown over the tissue, suspending a thermoplastic membrane above the cells of interest. (b) The arrows point to positions where the ultraviolet laser has been used to cut through the PEN membrane and the overlying cells at the border of the region of interest. In addition, the overlying membrane has been heated with the IR laser to create a melt spot with the cells to be captured.
One important advantage of LCM is the excellent preservation of the in vivo properties of the cells purified. The tissue of interest is rapidly removed and frozen during the embedding process. This contrasts with competing procedures, such as fluorescent activated cell sorting (FACS), which requires cell dissociation procedures that can sometimes result in significant perturbation of gene expression patterns (3).
2. Materials
2.1. Equipment
Laser capture machine, Arcturus XT or Arcturus Veritas (Applied Biosystems), including inverted microscope. Similar machines are available from Zeiss, Leica, and other suppliers.
Cryostat. Microm HM520 (Thermo Scientific), or equivalent.
−80°C freezer.
2.2. Supplies
PEN membrane glass slides (Arcturus, LCM0522).
Poly-L-lysine.
2-Methylbutane.
Acetone.
CapSure HS LCM Caps (Arcturus, LCM0214).
OCT (Sakura Finetek Corp., 4583).
Liquid nitrogen.
Xylene.
100% Ethanol.
Qiagen RNeasy Micro RNA purification kit (Qiagen).
Fluorescein labeled peanut agglutinin (PNA) (Vector lab).
Mayer’s hematoxylin.
Cryomolds.
Eosin Y solution.
Scott’s Tap Water Substitute Blueing Solution.
Qiagen RNeasy Micro Kit (Qiagen).
Ovation Pico WTA System (Nugen, 3300-12).
WT-Ovation One-Direct Amplification (Nugen, 3500).
3. Methods
3.1. Preparing Tissue Blocks
Rapidly dissect out tissue of interest, such as embryonic kidneys, and store briefly in ice-cold PBS (see Note 1).
Process through OCT only as many kidneys, or other tissues, as you plan to freeze in one block at a time (see Note 2).
Place kidneys in precooled OCT in a 60 mm plate cooled with ice.
Quickly mix kidneys in with OCT using a sterile pipette tip, then transfer carefully to a new 60-mm plate with cold OCT, quickly mix again, and place in a mold with ice-cold OCT covering the bottom. Transfers can be made with a pipetman and a 1-ml pipette with the end enlarged by slicing the tip off with a razor.
Cover kidneys with additional OCT and position kidneys, or other tissues of interest, near each other in the mold, in a central position, not too near the top.
Immediately freeze in 2-methylbutane in a pyrex beaker resting in liquid nitrogen. The 2-methylbutane should be frozen solid. Hold the tinfoil mold with forceps to keep vertical and gently move in a circular motion against the surface of the 2-methylbutane to improve thermal contact (see Note 3).
When the OCT is completely frozen, place the mold in dry ice and then store for long term in either a −80°C or a liquid nitrogen freezer.
3.2. Cryostat Sectioning
Throughout this procedure be very careful not to cut yourself on the sharp blades used in the cryostat. Wear gloves throughout to reduce RNase contamination.
Place specimen block in the chamber for 5–10 min to temperature equilibrate. Remove tissue OCT block from the mold. Place chuck that has been at room temperature in chamber and let cool a minute, but not too much. Place OCT on chuck and let cool a minute, but not freeze, and then place tissue OCT block on chuck, and let freeze in position. One can place additional OCT around the base and spread with gloved finger to help hold the block in place.
The temperature of the cryostat is critically important. Set to −15°C chamber and −15°C specimen (see Note 4).
Begin sectioning. Use the trim setting of 40–60 μm to remove most excess OCT, until tissue is visible. When close to the tissue of interest change to 7–10 μm sections (see Note 5).
Collect sections on membrane slides (see Note 6). Collect 5–10 sections per slide. It is important that the sections are placed in the central region of the slide, as the LCM machine cannot work on sections near edges. It is also important, however, to space the sections so that the Cap can be placed on each section without overlapping another one (see Note 7). Try to work fast, as the RNA in one section can be degrading while the other sections are being collected.
Freeze the slides quickly with dry ice and store at −80°C.
Clean up the cryostat. Remove dirty blade, brushes, OCT, etc.
3.3. Processing Slides
A limitation of LCM is the relatively poor histology of cryostat sections. This is particularly true when no additional staining procedure is used. Nevertheless, in some cases the structure of interest is so well demarcated that no special stains are necessary. One example would be the glomerulus of the kidney. A good general rule is to use as small a number of processing steps as possible. The more steps, the more opportunity for the RNA to diffuse out of the sample and the greater the likelihood of RNA degradation. Another general rule is the colder, the better, as this also reduces RNAse activity. Also, the less exposure to water, the better, since RNAs dissolve in water, causing losses from the tissue section, and ribonucleases require water. Therefore, the more the sample is maintained in a dehydrated state, the better the RNA recovery and the better the resulting RNA integrity.
With these recommendations in mind we present three variations of a protocol for processing slides for LCM. The first is a very straightforward procedure, with a minimum of steps, for use when very obvious structures are to be isolated. The second procedure adds lectin staining which can greatly assist the identification of more subtle structures. The third procedure incorporates hematoxylin and eosin staining, which can provide even more detailed structural definition.
Remove the slide from −80°C and immediately place on a room temperature metal surface, such as a slide warmer with the heat turned off. After a few seconds, move the slide to a new position, again at room temperature, to promote gentle warming to room temperature. Let the slide sit 2–3 min.
Fix the sample by placing in an ice-cold 1:1 solution of acetone and 75% ethanol for 2 min.
To dissolve OCT transfer to room temperature 70% ethanol, with gentle dipping of the slide for 1–2 min.
Transfer to fresh 70% ethanol, with gentle dipping, for 15–30 s. If no staining is required then go straight to 95% ethanol, step 7, for lectin staining go to step 5, and for hematoxylin and eosin staining go to step 6.
For lectin staining place the slide on an ice-cold metal block and flood the surface sections with lectin staining solution. PNA lectin provides a nice pan epithelial stain that can be very useful for distinguishing structures. Each lectin will need to be optimized for concentration and staining time to achieve the best signal to noise ratio. For PNA a good starting point is 5 μl/ml of 1/10 PBS diluted with autoclaved water. Stain for 6 min. Then rinse in ice-cold 1/10 PBS, 2 × 10 s with gentle dipping, and then 1 × 3 min. Gently dip in 70% ethanol and continue with dehydration series, step 7.
For LCM compatible H&E staining (8) take slides from 70% ethanol (step 4) and successively dip in water: 10 s, Mayer’s hematoxylin: 15 s, water: 10 s, Scott’s Tap Water Substitute: 10 s, 70% ethanol: 10 s (optional), eosin: 3–10 s, and then go to dehydration series, step 7.
Gently dip in 95% ethanol: 10 s, and repeat in fresh 95% ethanol: 10 s.
Further dehydrate with gentle dipping 2× in 100% ethanol: 45 s each.
Xylene with gentle dipping 2 × 1.5 min.
Air-dry in vertical position. Then, proceed immediately to LCM.
3.4. LCM Procedure
The LCM should be carried out as quickly as possible to minimize RNA degradation. The rate of RNA degradation is tissue specific and can be tested simply by allowing slides processed for LCM to sit at room temperature for variable time periods and then purifying the RNA from the sections on the slides and determining levels of RNA integrity on an Agilent Bioanalyzer. The processed slides are dehydrated, which significantly reduces RNAse activity. We have found that for embryonic kidneys RNA loss is minimal after 30 min and acceptable for up to 1 h. For other tissues with higher levels of endogenous RNAse, however, RNA degradation can occur much more quickly.
The details below are specific to the Arcturus Veritas machine, but the principles also apply to other platforms.
Turn the LCM machine on, activate the Veritas software, insert username and password, and click start a new session.
Load slides and caps, fill in relevant information on pop-up windows, and click OK.
Roadmap low power magnification images appear that show where the tissue sections on the slides are located. Double-click on a section to provide a higher power image of the region of interest.
Use the mouse to place a Cap over the region of interest. Ideally, the tissue should be centrally located and not in contact with the support struts of the Cap (see Note 7).
Drag a region without tissue to the center of the field. Double-click the mouse to fi re the IR laser. Adjust the aim by left clicking the mouse and using “capture laser is here” function.
Adjust the power and duration of the IR laser to achieve an optimal melt spot when the laser is fi red. A blurred circle that cannot be focused indicates that the Cap plastic was not melted sufficiently to extend down to the PEN slide. In this case, the power and/or duration of the laser pulse needs to be turned higher (Fig. 2). An ideal melt spot is a sharp black line circle with a clear center. This shows that the Cap plastic was heated sufficiently to melt, descend to the PEN slide membrane and form a region where the Cap and slide plastics melted together. It is very important that an appropriate melt spot is formed (see Note 8).
Return to the tissue region of interest and from the menu bar activate the UV cutting laser. Adjust the aim using the mouse activated drop down menu function (cutting laser is here).
The power of the UV laser needs to be properly adjusted from the window on the left. A “low” power setting of 3–5 is typically appropriate (see Note 9).
Click “capture image” to get a photograph of the tissue section before LCM. It is generally advisable to capture images before, after using the UV cutting laser, and after separating the sample of interest from the slide, to thoroughly document the tissue sample that was taken.
Use the mouse to draw a line around the tissue of interest.
Activate the UV cutting laser, which will then cut along the line drawn with the mouse (Fig. 3).
Use the menu bar to return to the IR capture laser. Click the hand. Use manual setting. Automatic setting will trigger a large number of melt spots, creating “plastic sandwich” problems (see Note 10).
Double-click the mouse to create a melt spot on the tissue of interest (see Note 10). One variant of the LCM procedure just uses the IR capture laser to melt plastic onto the cells of interest. No UV cutting laser is used. Then when the Cap is removed the cells that are attached to the melted plastic are lifted from the surface. An advantage of this method is that since no cutting laser is used there is no UV laser damage to cells. This method, therefore, is sometimes useful for the capture of very small samples, with only a few cells, where the UV laser would of necessity be cutting very near all of the cells of interest. Another advantage is that PEN membrane slides, which again are expensive, are not required. A major disadvantage, however, is that the cells must be pulled off of the underlying surface, and from the flanking cells, to which they are attached. It is often challenging to achieve good “lifting” of the cells of interest.
Move the field of view by double-clicking on a region of the roadmap image where there is no tissue, and use the mouse activated “place cap in center of field” function to move the Cap to this position. The image should now show the captured tissue attached to the Cap. (If not, then see Note 11.)
Take photographs of the Cap with the captured material as well as the tissue with the region of interest removed.
Fig. 2.
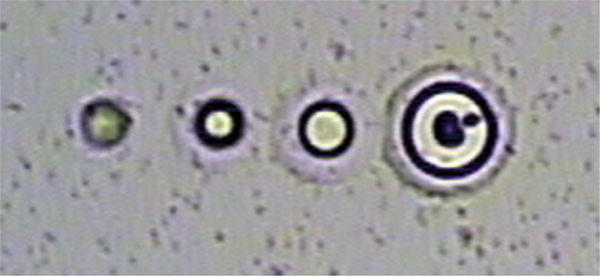
Ideal infrared (IR) laser melt spots. A series of spots generated with the IR laser are shown. The blurred spot on the far left indicates insufficient heating to melt the thermoplastic membrane to create a genuine melt spot. The membrane remains suspended above the sample. The next spot is small, but would work well for capturing minute samples of just one or a few cells. The third spot is generally ideal for excellent attachment of the thermoplastic membrane to the sample tissue. The far right spot is quite large, but could be effective for collecting large samples, where maximal contact is useful.
Fig. 3.
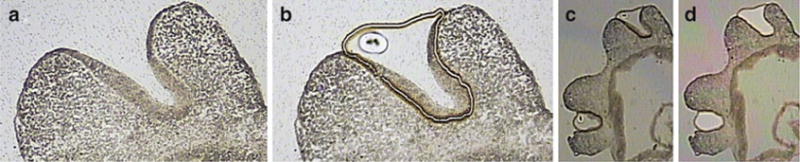
Laser capture microdissection example. (a) A cryostat section showing E10.5 embryonic developing craniofacial region. (b) The ultraviolet cutting laser was used to cut around the cells of the olfactory pit. The infrared laser was used to generate a large melt spot, showing melting of the thermoplastic membrane of the Cap to the membrane of the PEN slide. (c) A lower magnification image showing both olfactory pits. (d) An “after” image showing both olfactory pits cleanly removed from the tissue section, and now residing on the Cap.
3.5. Processing Caps
The laser capture tissue now residing on the Cap can be used for transcription profiling, via RNA-Seq or microarrays, or for DNA analysis or proteomics. For example, RNA can be purified using the Zymo ZR RNA MicroPrep kit, and target amplifications for microarray analysis can be performed using Nugen products designed for extremely small samples, such as Ovation Pico WTA System (for samples with just a few nanograms of total RNA), or WT-Ovation One-Direct Amplification (for samples as small as a single cell).
Acknowledgments
This work was supported by NIH grants RC4DK090891, UO1DK070251, and UO1DE020049. We thank Lauren Kadel and Andrew S. Potter for technical assistance with laser capture.
Footnotes
Process samples as quickly as possible. The more time that passes before freezing, the greater the chance of RNA degradation.
The OCT is hyperosmotic and tends to absorb the water from the tissue it touches, which can in particular ruin the edges of the tissue of interest. Nevertheless, be sure to carefully rinse the PBS away because it can interfere with the sectioning. Leaving the sample in OCT for too long a period, however, can seriously distort tissue morphology.
As you move the mold against the surface a melted region of 2-methylbutane will appear. Keep the mold in this melted region, moving it to improve thermal contact and to prevent freezing of the mold into the 2-methylbutane as it continues to cool and refreeze.
If the sample is still too brittle, with flaking and shattering during sectioning, then try warming the specimen and chamber temperatures a degree or two.
The membrane slides are quite expensive, so use regular slides until tissue is clearly visible, and then switch to membranes slides.
We have found that embryonic kidney sections do not adhere well to the membranes of PEN slides. To prevent loss of sections during later processing steps it is necessary to pretreat the slides by dipping in 1/10 dilution of poly lysine, and then air drying in a vertical position.
The Caps hold the thermoplastic membrane immediately above the sample section. The Cap rests on struts arranged in a circle around the region of interest on the PEN membrane slide. If these struts rest on tissue then the tissue can stick to them, and end up contaminating the sample. This material is informally referred to as “Cap crap”. The best way to avoid Cap crap is to have sections that do not make unintended contact with the Caps. Small sections that can be contained within the strut-encircled region are ideal. Also, it is important to have flat sections, without wrinkles or folds that might extend vertically far enough to contact the membrane of the Cap. Contaminants can be in part addressed using an ablation laser. This is a high intensity laser that is fi red at the unwanted material. In our experience, however, the ablation laser will often explode the contaminating material into many small pieces that then are scattered on the Cap. If possible, it is best to simply avoid the contaminating material in the first place.
If the melt spot is too small then there might not be sufficient contact for the Cap to lift the UV cut tissue section and underlying PEN membrane from the slide. On the other hand, if the melt spot is too large then it might cover too wide a region and reduce the resolution of the LCM, and/or create “plastic sandwich” problems, as described in Note 10. In some cases, it seems impossible to actually create a melt spot. Instead, only a blurred circle is formed, suggesting that the plastic has not been able to melt all the way down to the tissue section. This can be the result of the cap not lying flat on the PEN membrane slide. In some cases, the struts are on a thickened region of tissue, perhaps a folded part of tissue section, or a part of tissue section that is elevated after coming loose from the membrane. In this case, try moving the Cap to a new region of the slide, where the tissue section is not protruding.
If the UV power setting is too low then the PEN membrane and tissue are not completely cut and the sample cannot be separated, or “lifted”, from the slide. On the other hand, if the power setting is too high, then it can cause damage of flanking tissue. If large swaths of tissue are being collected then UV damage of the relatively small edge regions may be acceptable. On the other hand, if a small region of only a few cells is being collected, or a single cell layer, then the UV power setting must be kept to a minimum to reduce damage. This is an empirical trial and error process, finding the minimal setting that allows good sample “lifts”.
It is often advisable to locate the melt spot near an edge of the tissue region to be captured. If the melt spot region is too extensive then this can result in a “plastic sandwich”. The captured tissue is trapped between the PEN slide membrane and the melted Cap membrane. This enclosed tissue is difficult to access, which can result in dramatically reduced yields of RNA.
Sometimes the sample fails to lift from the PEN slide with the Cap. This can be caused by incomplete cutting with the UV laser, or ineffective melting with the IR laser. One can repeat the cutting with the UV laser. If additional cuts still do not release the sample then the UV laser power may need to be increased. One can also improve the lifting by increasing the melted contact surface with the IR laser. In some cases, the first lift attempt will only remove the cells from the melt spot region, but not carry with it the underlying membrane. In this case it is advisable to remelt to the spot where the cells have been removed. The plastic-to-plastic melt creates a firmer attachment than plastic to cells.
References
- 1.Brunskill EW, Aronow BJ, Georgas K, et al. Atlas of gene expression in the developing kidney at microanatomic resolution. Dev Cell. 2008;15:781–791. doi: 10.1016/j.devcel.2008.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Curran S, McKay JA, McLeod HL, Murray GI. Laser capture microscopy. Mol Pathol. 2000;53:64–68. doi: 10.1136/mp.53.2.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Geho DH, Bandle RW, Clair T, Liotta LA. Physiological mechanisms of tumor-cell invasion and migration. Physiology (Bethesda) 2005;20:194–200. doi: 10.1152/physiol.00009.2005. [DOI] [PubMed] [Google Scholar]
- 4.Emmert-Buck MR, Bonner RF, Smith PD, et al. Laser capture microdissection. Science. 1996;274:998–1001. doi: 10.1126/science.274.5289.998. [DOI] [PubMed] [Google Scholar]
- 5.Kolble K. The LEICA microdissection system: design and applications. J Mol Med. 2000;78:B24–B25. [PubMed] [Google Scholar]
- 6.Micke P, Ostman A, Lundeberg J, Ponten F. Laser-assisted cell microdissection using the PALM system. Methods Mol Biol. 2005;293:151–166. doi: 10.1385/1-59259-853-6:151. [DOI] [PubMed] [Google Scholar]
- 7.Schermelleh L, Thalhammer S, Heckl W, et al. Laser microdissection and laser pressure catapulting for the generation of chromosome-specific paint probes. Biotechniques. 1999;27:362–367. doi: 10.2144/99272rr04. [DOI] [PubMed] [Google Scholar]
- 8.Espina V, Wulfkuhle JD, Calvert VS, et al. Laser-capture microdissection. Nat Protoc. 2006;1:586–603. doi: 10.1038/nprot.2006.85. [DOI] [PubMed] [Google Scholar]