

Abstract
A novel series of CXCR4 antagonists with substituted piperazines as benzimidazole replacements is described. These compounds showed micromolar to nanomolar potency in CXCR4-mediated functional and HIV assays, namely inhibition of X4 HIV-1IIIB virus in MAGI-CCR5/CXCR4 cells and inhibition of SDF-1 induced calcium release in Chem-1 cells. Preliminary SAR investigations led to the identification of a series of N-aryl piperazines as the most potent compounds. Results show SAR that indicates type and position of the aromatic ring, as well as type of linker and stereochemistry are significant for activity. Profiling of several lead compounds showed that one (49b) reduced susceptibility towards CYP450 and hERG, and the best overall profile when considering both SDF-1 and HIV potencies (6–20 nM).
Keywords: Piperazine, G-protein coupled receptor, CXC chemokine receptor 4, CXCR4 antagonists, HIV
The CXC chemokine receptor 4 (CXCR4) is an alpha G-protein coupled chemokine receptor that is widely expressed on most hematopoietic cells, predominantly leukocytes including neutrophils, monocytes, and macrophages.1 As the specific ligand for CXCR4, CXCL12 (SDF-1, stromal cell-derived factor-1) plays a vital role in hematopoietic stem cell (HSC) homing to the bone marrow and HSC quiescence,2–4 making the CXCR4/CXCL12 axis key to many essential physiological activities such as homeostatic regulation of leukocyte trafficking, hematopoiesis and embryonic development. 5,6 It has also been shown that CXCR4 assists the entry of HIV into CD4+ T cells as a co-receptor with CD4.7,8 Not surprisingly, the discovery of the involvement of CXCR4 in HIV infection triggered tremendous research efforts in the development of CXCR4 antagonists, primarily as potential HIV entry inhibitors.9 The compound AMD 3100 (1, Perlixafor™, Chart 1), a CXCR4 antagonist, first entered clinical trials as an anti-T-tropic HIV agent, but was later approved by the FDA in a different therapeutic setting as supportive treatment for autologous stem cell transplantation due to its efficacy in mobilizing HSC from bone marrow to the peripheral blood stream.10,11 This serendipity dramatically broadened the breadth and scope of CXCR4 research, leading to the discovery of various CXCR4 antagonists both peptide-based12 and small molecules.13 Recently, the CXCR4/CXCL12 axis has been linked to more than 23 types of cancer including ovarian, prostate, oesophageal, melanoma, neuroblastoma and renal cell carcinoma, where the receptor/ligand interaction is a key factor in cancer metastasis, angiogenesis, and tumor growth via direct and indirect mechanisms. 14–19 Furthermore, the involvement of CXCR4/CXCL12 in various, yet essential signaling pathways and physiological activities has made it an attractive target for the development of novel treatments in the areas of HSC mobilization during autologous transplantation, T-tropic HIV infection, metastatic tumors, tissue regeneration, inflammation and WHIM syndrome.20
Chart 1.

Selected small molecule CXCR4 antagonists.
Efforts in the search for CXCR4 antagonists has resulted in several small molecules being discovered (Chart 1, 2–7), with one of these (2, AMD11070) entering human clinical trials.1,19,21–23 Further work revealed other analogs of note including a 11070 congener (GSK812397, 3), IT1t (4), and AMD3100 homologues WZ811 (5) and MSX-122 (6).24–27 Recently, we reported a series of TIQ analogs represented by TIQ-15 (7), where the benzimidazole portion was replaced by a tetrahydro-isoquinoline ring.28 Considering the complexity of the many roles of CXCR4, further research is still greatly needed for the discovery of CXCR4 antagonists that have increased affinity with the ability to target CXCR4 differentially, while showing validated drug-like properties including bioavailability, pharmacokinetics and pharmacodynamics. Inspired by previous work in our group and others,24,26–28 we herein describe a series of novel CXCR4 small molecule antagonists 8 consisting of chiral N-aryl piperazines as replacements for the benzimidazole ring. As a complementary study to our previous research on CXCR4 antagonists, which contain a fused N-heterocycle and aromatic ring system (7) and those done on AMD11070 (2), current efforts involved the incorporation of a piperazine moiety linked with an aromatic ring with various linkers as the replacement of the 1,2,3,4-tetrahydroisoquinoline28 and benzimidazole ring systems.22 The goals were to improve the ADMET properties of these molecules as piperazines have shown privileged pharmacological merits in drug development, particularly within GPCR ligands.29,30
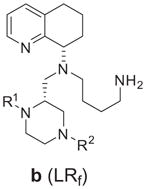
Our synthetic efforts focused on providing a versatile route that would allow substitution at both piperazine nitrogen atoms, while allowing access to both stereoisomers on the piperazine ring. Although the anti-diastereomer/R-stereoisomer is preferred in the TIQ series (7), we were not certain how this would affect activity in this series. The general synthesis of the piperazines (8) is shown in Scheme 1. Syntheses commenced from readily available racemic N-Boc piperazine carboxylic acid 9. A number of divergent routes could be utilized with this starting material to access different derivatives. For example: route one involves simple conversion to the methyl ester (10); route two consisted of conversion to the alcohol followed by TBS protection of the alcohol to give 11; and route three consists of simultaneous di-benzylation of both the carboxylic acid and piperazine nitrogen moieties to give compound 12. The other intermediates (10,11) were then utilized to make various N-piperazine building blocks (13–17). The methyl ester 10 was converted to N-phenyl sulfonamide, phenyl (via Buchwald–Hartwig) and carboxybenzyl (CBZ) derivatives 15–17 followed by reduction to the corresponding alcohols (21–23) and oxidation to give piperazine aldehydes 27–29. The silyl ether 11 was acylated to give benzoyl and furanoyl compounds 13 and 14, followed by deprotection (19, 20) and oxidation to give piperazine aldehydes 25 and 26. Finally, N-benzyl derivative 12 could be converted to the aldehyde 24 by either reduction to the alcohol (18) and oxidation or directly by di-isobutyl aluminum hydride reduction. The first of two consecutive reductive aminations with the piperazine aldehydes 24–29 with chiral S-THQ 30 gave intermediates 31–36 that were now diastereomerically differentiated (syn and anti). In the case of the N-benzyl derivative 31, these diastereomers were readily separable by chromatography. An X-ray structure determination on suitable crystals of the lower Rf isomer of 31 revealed this to be the syn-isomer (31b, Supplementary material).
Scheme 1.

Reagents. (i) MeOH, EDCI, THF, DCM; (ii) BH3-SMe2, THF; (iii) TBSCl, NEt3, DCM; (iv) BnBr, K2CO3, DMF; (v) PhCOCl or PhSO2Cl or 2-furyl-COCl or CbzCl, NEt3, DCM; (vi) PhBr, Pd2dba3, DavePhos, Cs2CO3, 1,4-dioxane; (vii) NaBH4, CaCl2, THF, EtOH; (viii) Bu4NF, THF; (ix) Dess–Martin, NaHCO3, DCM; (x) (COCl)2, DMSO, NEt3, DCM; (xi) 30, NaBH(OAc)3, DCM; (xii) 37, NaBH(OAc)3, DCM, AcOH; (xiii) H2, Pd(C), MeOH; (xiv) Cl-SO2-2-naphthyl or Cl-SO2-2-thiophenyl or Cl-SO2-(4Cl)Ph, NEt3, DCM; (xv) PhNCO, DCM; (xvi) TFA, DCM.
This stereochemistry observation for 31b was consistent with compounds in the TIQ series (7) where the upper and lower Rf stereoisomers corresponded to anti and syn diastereomers, respectively. Most of the other isomers (32–36) were separable at the next step upon subsequently attaching the Boc protected butyl amine side chain (37). Attachment of this side chain was accomplished by reacting the secondary amines 31–36 with derivative 37 to give the di-Boc protected compounds 38–43. As mentioned before, the N-benzyl isomers (31) were separated prior to this step and then carried through individually. The N-acyl and sulfonyl compounds (39–41, and 43) were separated into their anti and syn diastereomers at this stage. The phenyl derivative 42 was inseparable and carried forward as a mixture. All these intermediates were N-Boc deprotected by treatment with trifluoro acetic acid to give the corresponding final compounds (48–53a/b, Table 1) as free amines. The phenyl derivative 52 was inseparable at either stage and tested as a diastereomeric mixture.
Table 1.
| Compound number |

|
|
MAGI HIV1IIIB IC50a,b (μM) | SDF-1 Ca2+ Flux CEM-1 IC50a (μM) | |
|---|---|---|---|---|---|
| R1 | R2 | (*)-R/S | |||
| 68a | H | H | R | 0.31 | 0.255 |
| 68b | H | H | S | 1.1 | 1.2 |
| 52a/b | H | Ph | Mix. (1:1) | 0.72 | 0.13 |
| 48a | H | CH2Ph | R | 0.15 | 0.23 |
| 48b | H | CH2Ph | S | 0.03 | 0.19 |
| 49a | H | C(O)Ph | S | 0.06 | 0.002 |
| 49b | H | C(O)Ph | R | 0.02 | 0.006 |
| 69a | C(O)Ph | H | R | >100 | — |
| 69b | C(O)Ph | H | S | >100 | — |
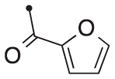
| 50a/b | H |
|
Mix. (2:1) | 0.21 | — |
| 50b | H | R | 0.15 | 0.035 | |
| 53a | H | CO2CH2Ph | S | 0.34 | 0.018 |
| 53b | H | CO2CH2Ph | R | 1.35 | — |
| 51a | H | SO2Ph | S | 0.05 | 0.002 |
| 51b | H | SO2Ph | R | 0.02 | 0.023 |
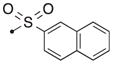
| 54a | H |
|
S | 4.6 | 0.071 |
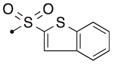
| 55a | H |
|
S | 3.44 | 0.039 |
| 56a | H |
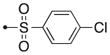
|
S | 0.05 | 0.002 |
| 57a | H | C(O)NHPh | S | 0.35 | 21 |
All assays were performed in duplicate.
The cytotoxicities (TC50’s) for all compounds were greater than 10 μM.
A modular modification on Scheme 1 utilized a late stage intermediate, the upper Rf isomer of the N-Boc/N-Cbz differentially protected piperazine compound 43a, to make several variations on the bottom nitrogen (54a–57a, Scheme 1). This was accomplished by first deprotecting the Cbz group by hydrogenation followed by treatment with the appropriate electrophile (ArSO2Cl or PhNCO) to give the di-Boc compounds (44a–47a). Subsequent Boc deprotection with trifluoroacetic acid gave the corresponding final compounds.
Alternatively, the two derivatives, one with no piperazine substituents (68) and the other with a N-benzoyl group transposed to the top piperazine nitrogen (69) were synthesized from another commercially available N-Boc protected piperazine carboxylic acid (58, Scheme 2). Compound 58 was first protected by Cbz-Cl (59), followed by reduction (60) and Parikh–Doering oxidation to furnish aldehyde 61. Similar to Scheme 1, intermediate 61 was sequentially reacted with tetrahydro-quinoline 30 and Boc/Cbz-protected amino butyraldehydes 37/63 to give differentially protected derivatives 64 and 65. These were followed by hydrogenolysis removal of Cbz on the top piperazine nitrogen to give 66, followed by reaction with benzoyl chloride for compound 67. TFA-facilitated Boc deprotection of 67 provided the final product 69. Alternatively, the bis-Cbz derivative 65 was Boc-deprotected and then hydrogenated to give 68. In these cases, the stereoisomers were separated at compounds 65 and 67. Since the stereochemistry of intermediate 31b was determined and confirmed by X-ray crystal structure (Scheme 1, Supplementary material) the stereochemical assignments of all other compounds were deduced from this observation and the lower Rf compound was assigned as the syn diastereomer (Table 1, URf (a) and LRf (b)). The labeling of the compounds by the R,S-nomenclature system is based on priority rankings of the substituents around the chiral center on the piperazine ring, which changed from compound to compound. This assignment along with the Rf properties is listed along with the compound structure in Table 1.
Scheme 2.

Reagents: (i) CbzCl, NEt3, 1,4-dioxane; (ii) BH3-SMe2, THF, 0 °C; (iii) SO3-pyridine, NEt3, DCM, DMSO; (iv) 30, NaBH(OAc)3, 1,2-DCE; (v) 37 or 63, NaBH(OAc)3, AcOH, DCM; (vi) H2, Pd(C), EtOH; (vii) BzCOCl, NEt3, DCM; (viii) TFA, DCM.
We wanted to determine the ability of these compounds to block CXCR4 signaling as well as HIV entry. The activities of these compounds against CXCR4 were evaluated by a combination of two assays: (1) the viral attachment assay with HIV-1IIIB in CCR5/ CXCR4-expressing HeLa-CD4-LTR-β-gal (MAGI) cells measuring the compound’s ability to block potential viral entry;31,32 (2) inhibition of CXCL12 induced calcium (Ca2+) flux/release in Chem-1 cells showing the compound’s potential to initiate the receptor’s G-protein function.33 The results of these two assays for the piperazine compounds is shown in Table 1 and indicate stereochemistry, as well as inclusion and location of the aromatic substitution on the piperazine ring are of central importance to potency in both assays. Some of the compounds with substituents at the lower nitrogen atom (48–57a) showed moderate to potent activities in blocking viral entry with IC50 values below 100 nM in one or both assays, showing that the aromatic substituent contributed to a 10–100 fold increase in activity. This is demonstrated by comparing these compounds to the ones with an N–H at this position (63a/b). The compound with no linkage to the phenyl ring (52) gave no significant contribution to activity, but introduction of a linker with and without heteroatoms, such as oxygen and sulfur, to the aromatic group did slightly improve the activity especially when the linker length was one atom (48, 49, 51 compared to 52). Furthermore, the linkage between the piperazine ring and nature of the aromatic group played a significant role as hetero cycles, multiple rings (50, 54a, 55a), carbamate and urea linkages (53, 57a) resulted in a drop off in activity of 10-fold or more. The only variation that seemed tolerable in addition to benzyl, benzamide and phenyl sulfonamide linkages-groups (48, 49, 51a/b) was para substitution on the aromatic ring (p-chlorophenyl, 56a), albeit with no increase in activity though. When the group was placed on the top piperazine nitrogen (64) all activity is eliminated confirming our choice in focusing on the bottom nitrogen of the piperazine ring. Marginal differences were observed when comparing stereoisomers but this difference in the most potent compounds (49a/b and 51a/b) was only 2–3 fold. This is significant when compared to the TIQ scaffold (7) where a 100-fold difference was observed between R and S stereoisomers.
Generally speaking, most compounds behaved slightly better as CXCR4 ligands in blocking G-protein-mediated calcium flux. All compounds showed CXCR4 antagonist behavior with varying potencies (Table 1, 1μM down to 2 nM). In contrast to the MAGI-HIV results, almost all aromatic groups increased potency, and most had IC50 values below 100 nM in the calcium flux assay. An example of this difference is seen in comparing sulfonamide groups on the upper Rf isomer (51a, 54a, 55a, 56a) where the calcium flux potency range is about 30-fold (2–71 nM), while the HIV assay range is about 100-fold (50–4600 nM). This smaller difference in calcium flux potency also applies to linker atoms and stereochemistry supporting the general trend. This trend is not observed in the TIQ scaffold where ring position and stereochemistry contribute to a 100-fold or more in potency differences. Furthermore, compound 48b shows a unique property when compared to the rest showing a preference of blocking HIV versus SDF-1 effects (HIV selectivity = 0.19/0.03 = 6 fold) compared to 49b (HIV selectivity = 0.006/0.02 = 0.3 fold). This effect is further supported by the observation that compound 48b has a less potent ability to displace 125I-SDF-1 than TIQ-15 (IC50 = 2920 nM for 48b vs 112 nM for 7). This discrepancy in activity might become the gateway to find more selective CXCR4 based HIV entry blockers.
To better understand the SAR of our efforts we engaged in a molecular modeling study using the existing CXCR4 X-ray crystal structures.34 As we have previously published studies using both the small molecule and peptide crystal structures with AMD11070, we utilized these findings to focus our efforts here.35 We selected the most potent diastereomeric pair with the benzamide (49a and 49b) for docking studies with the CXCR4:CVX15 grid, which we hypothesize will adopt ‘peptide-mimetic’ binding poses. Docking these isomers and optimizing the resulting grids resulted in unique and different docking poses (Fig. 1). In the case of the upper Rf (49a) diastereomer, the molecule sits in the receptor and makes hydrogen bond interactions with three residues and is depicted in Figure 1A as follows: (i) the butyl amine side chain forms a salt bridge with aspartic acid residue 97 (Asp97); (ii) the top piperazine N–H forms a salt bridge with glutamic acid residue 288 (Glu288); and (iii) the benzoyl carbonyl forms a H-bond interaction with the side chain of arginine 188 (Arg188). The lower Rf diastereomer (49b) forms similar interactions but with a few differences and is depicted in Figure 1B as follows: (i) the butyl amine side chain forms a salt bridge with aspartic acid 187 (asp187); and (ii) the piperazine N–H forms a salt bridge with glutamic acid residue 288 (Glu288). To compare and contrast the two seemingly significant and different poses for these isomers (Fig. 1A and B), it can be seen that the two molecules occupy similar positions in the receptor with regards to the piperazine ring, as both involve interaction of the piperazine N–H with the glutamic acid residue Glu288. However, there are notable differences. One is that the carbonyl of the benzoyl group on 49a picks up a van der Waals interaction with the side chain of Arg188, while 49b does not. The other main difference is that the butyl amine side chains interact with different aspartic acid residues (Asp97 for 49a vs Asp 187 for 49b) showing a fundamentally different positioning for the butyl amine and THQ substituents. These two poses are consistent with findings of the crystal structure determinations with the CVX15 peptide and IT1t with CXCR4, which include the following residues: (i) for IT1t—Asp97, Asp187, and Glu288; and (ii) for CVX15—Asp187, Arg188. Given these parameters it seems both isomers 49a and 49b are hybrid binders in that they interact with residues identified in both ligand–crystal structure complexes.
Figure 1.

Docking poses of diastereomeric piperazines 49a and 49b in the CVX15:CXCR4 crystal structure showing critical amino acids and key hydrogen bonds between protein and ligand (green dotted lines). (A) Compound 49a. (B) Compound 49b.
Our final efforts in this series involved accessing the drug-like characteristics of our best leads to identify and compare to other series of compounds. We selected the two compounds 49b and 51b since they had the best selection of potency in both assays and contained different aryl groups on the piperazine ring (benzamide vs sulfonamide). We decided upon a small but significant subset of ADMET assays, consisting of CYP450 inhibition, and hERG channel binding. The assays consisted of measuring the effect of these compounds and the corresponding results are shown in Table 2 along with previous reported values for compounds 2 and 7. The findings are supportive of the piperazine addition as a better drug-like fragment than the benzimidazole on AMD11070 (2) in regards to the CYP450 activities. First, when comparing the CYP450 inhibition for the three isozymes (3A4, 2D6, and 2C19), both 49b and 51b show marked improvement compared to 2, with the benzamide 49b having the lowest response and similar to TIQ-15 (7).28,36,37 The sulfonamide (51b) had slightly higher inhibition of 2D6 in comparison but both had low responses against 3A4 and 2C19. Furthermore, when comparing these three series (benzimidazole, TIQ, and piperazine), one can conclude that the benzimidazole moiety results in substantial CYP450 inhibition supporting our efforts to alter this section of the molecule to improve ADMET properties. When tested against the hERG channel, both compounds showed beneficial effects in comparison to TIQ-15 (7) in an absolute sense. In these cases, both compounds show less than 50% at 1 μM. Whereas, compound 49b has the best value (55% at 10 μM), the results must be compared to on-target potencies to provide a fair comparison. In the case of 49b, the anti-viral potency is slightly higher than 7 (PBMC-HIV-1IIIB IC50 = 88 nMvs 24 nM for 7) by about 3-fold while the hERG activity is about 10-fold different (55% at 10 μM vs 50% at 1 μM). Therefore, the benzamide 49b may hold a slight advantage in the therapeutic index in comparison to compound 7 with more potent anti-viral activity. A pharmacokinetic (PK) study of 49b may provide further evidence of drug-like potential.
Table 2.
CYP450 and hERG parameters for 49b, 51b, 2, and 7
| Compound | CYP450 (% @ 1 μM)a | hERGb (%) | |||
|---|---|---|---|---|---|
|
|
|
||||
| 2C19 | 2D6 | 3A4 | 1 μM | 10 μM | |
| 49b | 2 | 19 | 0 | 18 | 55 |
| 51b | 2 | 41 | 0 | 37 | 79 |
| 2 | 20 | 100 | 35 | — | — |
| 7 | 0 | 8 | 6 | 50 | 93 |
Isolated human enzymes and fluorometric substrates.
Displacement of 3H-dofetilide in HEK293 cells.
In conclusion, we have presented the discovery of a novel series of N-aryl piperazine based CXCR4 antagonists (8). These compounds showed potent effects against blocking the functional responses of SDF-1 and blockade of HIV entry in assays measuring these effects. Preliminary SAR provided an optimal linker between the piperazine nitrogen and aromatic group as being a carbonyl or sulfonamide. Furthermore, our initial efforts in aryl SAR show some tolerance but also a limitation on the size and type of group. Finally, assessment of drug-like properties show two potential lead compounds (49b and 51b) where a reduction in CYP activity is observed as well as reduction in hERG potential. Our results also show that to fully ascertain the ability to deliver these agents orally, more experiments are needed. The optimization of these compounds as both anti-HIV entry agents and CXCR4 antagonists will be reported in due course.
Supplementary Material
Acknowledgments
A.R.P. would like to thank the National Science Foundation – United States for funding.
Footnotes
Supplementary data (procedures and characterization data for the compounds in Table 1, Schemes 1 and 2, as well as the X-ray crystal structure of 31b) associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.bmcl.2015.04.036.
References and notes
- 1.Peled A, Wald O, Burger J. Expert Opin Invest Drugs. 2012;21:341. doi: 10.1517/13543784.2012.656197. [DOI] [PubMed] [Google Scholar]
- 2.Lapidot T, Petit I. Exp Hematol. 2002;30:973. doi: 10.1016/s0301-472x(02)00883-4. [DOI] [PubMed] [Google Scholar]
- 3.Sugiyama T, Kohara H, Noda M, Nagasawa T. Immunity. 2006;25:977. doi: 10.1016/j.immuni.2006.10.016. [DOI] [PubMed] [Google Scholar]
- 4.Tzeng Y, Li H, Kang YL, Chen WC, Chen WC. Blood. 2011;117:429. doi: 10.1182/blood-2010-01-266833. [DOI] [PubMed] [Google Scholar]
- 5.Tachibana K, Hirota S, Iizasa H, Yoshia H, Kawabata K, Kataoka Y, Kitamura Y, Matsushima K, Yoshida N, Nishikawa S, Kishimoto T, Nagasawa T. Nature. 1998;393:591. doi: 10.1038/31261. [DOI] [PubMed] [Google Scholar]
- 6.Zou Y, Kottman AH, Kuroda M, Taniuchi I, Littman DR. Nature. 1998;393:595. doi: 10.1038/31269. [DOI] [PubMed] [Google Scholar]
- 7.Nagasawa T, Tachibana K, Kawabata K. Adv Immunol. 1999;71:211. doi: 10.1016/s0065-2776(08)60403-4. [DOI] [PubMed] [Google Scholar]
- 8.Oberlin E, Amara A, Bachelerie F, Bessia C, Virelizier JL, Arenzana-Seisdedos F, Schwartz O, Heard JM, Clark-Lewis I, Legler DF, Loetscher M, Baggiollni M, Moser B. Nature. 1996;382:833. doi: 10.1038/382833a0. [DOI] [PubMed] [Google Scholar]
- 9.Murakami T, Yamamoto N. Future Microbiol. 2010;5:1025. doi: 10.2217/fmb.10.67. [DOI] [PubMed] [Google Scholar]
- 10.De Clercq E. Nat Rev Drug Disc. 2003;2:581. doi: 10.1038/nrd1134. [DOI] [PubMed] [Google Scholar]
- 11.Dipersio JF, Yasothan U, Kirkpatrick D. Nat Rev Drug Disc. 2009;8:105. doi: 10.1038/nrd2819. [DOI] [PubMed] [Google Scholar]
- 12.Hassan S, Buchanan M, Jahan K, Aguilar-Mahecha A, Gaboury L, Muller WJ, Alsawafi Y, Mourskaia AA, Siegel PM, Salvucci O, Basic M. Int J Cancer. 2011;129:225. doi: 10.1002/ijc.25665. [DOI] [PubMed] [Google Scholar]
- 13.Debnath B, Xu S, Grande F, Garofalo A, Neamati N. Theranostics. 2013;3:47. doi: 10.7150/thno.5376. http://dx.doi.org/10.7150/thno.5376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Balkwell F. Nat Rev Cancer. 2004;4:540. doi: 10.1038/nrc1388. [DOI] [PubMed] [Google Scholar]
- 15.Zlotnik A. J Pathol. 2008;215:211. doi: 10.1002/path.2350. [DOI] [PubMed] [Google Scholar]
- 16.Teicher BA, Fricker SP. Clin Cancer Res. 2010;16:2927. doi: 10.1158/1078-0432.CCR-09-2329. [DOI] [PubMed] [Google Scholar]
- 17.Burger JA, Peled A. Leukemia. 2009;23:43. doi: 10.1038/leu.2008.299. [DOI] [PubMed] [Google Scholar]
- 18.Domanska UM, Kruizinga RC, Nagengast WB, Timmer-Bosscha H, Huls G, de Vries EGE, Walenkamp AME. Eur J Cancer. 2013;49:219. doi: 10.1016/j.ejca.2012.05.005. [DOI] [PubMed] [Google Scholar]
- 19.Wilson LJ, Liotta DC. Drug Dev Res. 2011;72:598. [Google Scholar]
- 20.Tamamura H, Tsutsumi H, Nomura W, Tanaka T, Fujii N. Expert Opin Drug Discov. 2008;3:1155. doi: 10.1517/17460441.3.10.1155. [DOI] [PubMed] [Google Scholar]
- 21.Choi WT, Duggineni S, Xu Y, Huang Z, An J. J Med Chem. 2012;55:977. doi: 10.1021/jm200568c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Skerlj RT, Bridger GJ, Kaller A, McEachem EJ, Crawford JB, Zhou Y, Atsma B, Langille J, Nan S, Veale D, Wilson T, Harwig C, Hatse S, Princen K, DeClercq E, Schols D. J Med Chem. 2010;53:3376. doi: 10.1021/jm100073m. [DOI] [PubMed] [Google Scholar]
- 23.Stone ND, Dunaway SB, Flexner C, Tierney C, Calandra GB, Becker S, Cao YJ, Wiggins IP, Conley J, MacFarland RT, Park JG, Lalama C, Snyder S, Kallungal B, Klingman KL, Hendrix CW. Antimicrob Agents Chemother. 2007;51:2351. doi: 10.1128/AAC.00013-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jenkinson S, Thomson M, McCoy D, Edelstein M, Danehower S, Lawrence W, Wheelan P, Spaltenstein A, Gudmundsson K. Antimicrob Agents Chemother. 2010;54:817. doi: 10.1128/AAC.01293-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thoma G, Streiff MB, Kovarik J, Glickman F, Wagner T, Beerli C, Zerwes HG. J Med Chem. 2008;51:7915. doi: 10.1021/jm801065q. [DOI] [PubMed] [Google Scholar]
- 26.Zhan W, Liang Z, Zhu A, Kurtkaya S, Shim H, Snyder JP, Liotta DC. J Med Chem. 2007;50:5655. doi: 10.1021/jm070679i. [DOI] [PubMed] [Google Scholar]
- 27.Liang Z, Zhan W, Zhu A, Yoon Y, Lin S, Sasaki M, Klapproth JMA, Yang H, Grossniklaus HE, Xu J, Rojas M, Voll RJ, Goodman MM, Arrendale RF, Liu J, Yun CC, Snyder JP, Liotta DC, Shim H. PLoS One. 2012;7:e34038. doi: 10.1371/journal.pone.0034038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Truax VM, Zhao H, Katzman BM, Prosser AR, Alcaraz AA, Saindane MT, Howard RB, Culver D, Arrendale RF, Gruddanti PR, Evers TJ, Natchus MG, Snyder JP, Liotta DC, Wilson LJ. ACS Med Chem Lett. 2013;4:1025. doi: 10.1021/ml400183q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Patel RV, Park SW. Mini Rev Med Chem. 2013;13:1579. doi: 10.2174/13895575113139990073. [DOI] [PubMed] [Google Scholar]
- 30.Meher CP, Rao AM, Omar Md. AJPSR. 2013;3:43. [Google Scholar]
- 31.Kimpton J, Emerman M. J Virol. 1992;66:2232. doi: 10.1128/jvi.66.4.2232-2239.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lackman-Smith C, Osterling C, Luckenbaugh K, Mankowski M, Snyder B, Lewis G, Paull J, Profy A, Ptak RG, Buckheit RW, Watson KM, Cummins JE, Sanders-Beer BE. Antimicrob Agents Chemother. 2008;52:1768. doi: 10.1128/AAC.01328-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.GPCR Profiler Assay. Eurofins/Panlabs Discovery Services; St. Charles, MO: [Google Scholar]
- 34.Wu B, Chien EYT, Mol CD, Fenalti G, Liu W, Katritch V, Abagyan R, Brooun A, Wells P, Bi FC, Hamel DJ, Kuhn P, Handel TM, Cherezov V, Stevens RC. Science. 2010;330:1066. doi: 10.1126/science.1194396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cox BD, Prosser AR, Katzman BM, Alcaraz AA, Liotta DC, Wilson LJ, Snyder JP. ChemBioChem. 2014;15:1614. doi: 10.1002/cbic.201402056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nyunt M, Becker S, MacFarland RT, Chee P, Scarborough R, Everts S, Calandra GB, Hendrix CW. J Acquir Immune Defic Syndr. 2008;47:559. doi: 10.1097/QAI.0b013e3181627566. [DOI] [PubMed] [Google Scholar]
- 37.Cao YJ, Flexner CW, Dunaway S, Park JG, Klingman K, Wiggins I, Conley J, Radebaugh C, Kashuba AD, MacFarland RT, Becker S, Hendrix CW. Antimicrob Agents Chemother. 2008;52:1630. doi: 10.1128/AAC.01460-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.