

Abstract
Factor XI (FXI) is the zymogen of a plasma protease, factor XIa (FXIa), that contributes to thrombin generation during blood coagulation by proteolytic activation of several coagulation factors, most notably factor IX (FIX). FXI is a homolog of prekallikrein (PK), a component of the plasma kallikrein-kinin system. While sharing structural and functional features with PK, FXI has undergone adaptive changes that allow it to contribute to blood coagulation. Here we review current understanding of the biology and enzymology of FXI, with an emphasis on structural features of the protein as they relate to protease function.
Keywords: Factor XI, Factor XIa, Prekallikrein, Factor XIIa, Factor IX
Introduction
Factor XI (FXI) is the zymogen of the coagulation protease factor XIa (FXIa) [1,2]. In humans, the protein circulates in plasma at a concentration of ~30 nM (15-45 nM) almost entirely as a non-covalent complex with the glycoprotein high molecular weight kininogen (HK) [3–5]. FXI is unique among coagulation proteases in that it is a dimer of identical 80 kDa subunits [6–9]. It was first identified by Robert Rosenthal and his colleagues in 1953 as a plasma constituent missing in members of a family with a mild trauma-induced bleeding disorder, and was originally called plasma thromboplastin antecedent (PTA) [10,11]. In the past, the autosomal bleeding disorder associated with FXI deficiency has been referred to as Rosenthal syndrome, PTA deficiency or hemophilia C (to distinguish it from deficiencies of factor VIII [hemophilia A] and factor IX [hemophilia B].
In the original cascade/waterfall hypotheses of coagulation, FXI is converted to FXIa by the protease factor XIIa (FXIIa) [12,13]. FXIa then promotes clotting by converting factor IX (FIX) to the protease FIXaβ. However, the absence of abnormal bleeding associated with deficiency of factor XII (FXII, the precursor of FXIIa) indicates this is not an important mechanism for stopping bleeding at a wound site (hemostasis) [14]. Furthermore, the mild bleeding disorder associated with FXI deficiency suggests FXI serves a supportive role in hemostasis, rather than a major role in initiating clot formation [15–17]. The diagram in Figure 1 is a model depicting relationships between FXI and other plasma components that we currently use in our laboratories [18,19]. Here, FXI is a bridge between tissue factor-initiated thrombin generation (Left Panel) and the kallikrein-kinin system (KKS), a group of proteins involved in several host-defense and homeostatic processes (Right Panel) [20]. FXI can support thrombin generation independent of the KKS, can function as a substrate and protease within the KKS, or serve as a bidirectional interface that permits the two systems to influence each other. The role of FXI as it relates to these systems has been covered in several recent reviews [16,19–22]. Here, we discuss current understanding of FXI protein structure, molecular biology, and enzymology.
Figure 1. Factor XI, thrombin generation, and the kallikrein-kinin system (KKS).

Proteolytic reactions required for thrombin generation at an injury site are shown in the gray box on the left, while proteolytic reactions involving the KKS are in the box on the right. Protease zymogens are indicated in black, and their active forms by a lower case “a”. Cofactors are in red. Requirements for calcium ions (Ca2+) or phospholipid (PL) are indicated. Thrombin generation is initiated by the factor VIIa/tissue factor (TF) complex, which activates factors (F) X and IX. Activated FX (FXa) converts prothrombin to thrombin. Thrombin generated early in coagulation converts FXI to FXIa, which sustains thrombin production through FIX activation. Note that FXI activation during thrombin generation does not require FXIIa. In the KKS, artificial or abnormal surfaces facilitate FXII autoactivation. FXIIa converts prekallikrein (PK) to α-kallikrein, which activates additional FXII and cleaves high molecular–weight kininogen (HK), liberating bradykinin (BK). The KKS can promote thrombin generation through FXIIa-mediated activation of FXI. There is evidence that FXIa, in turn, can activate FXII. In plasma, PK and FXI circulate as complexes with HK, which may serve as a cofactor for PK and FXI activation. FXI may act as a bidirectional bridge, allowing the thrombin generation system and KKS to influence each other. Image modified from Bane et al [18] and Gailani et al [19] with permission.
Evolutionary History
Homologs for components of the thrombin generation mechanism shown in the left panel of Figure 1 are found in all but the most primitive vertebrate organisms [23–25]. Genes for KKS proteins (FXII, the protease precursor prekallikrein [PK] and the cofactor HK) are not present in the genomes of zebrafish or pufferfish, but are found in amphibians, reptiles and mammals, suggesting an adaptation to terrestrial environments [24–26]. The gene for FXI arose through duplication of the PK gene early in mammalian evolution or in a reptilian ancestor of mammals [26]. Originally, it was thought that the genome of the primitive egg-laying mammal (monotreme) the duck-billed platypus had a single gene for a PK-like protein [28], however, subsequent work identified sequences corresponding to separate PK and FXI genes in this species (M. Ponczek, personal communication).
cDNAs for PK [27,28] and FXI [7,8] encode proteins with four 90 to 91 amino acid repeats called apple domains (or PAN domains for Plasminogen-Apple-Nematode) that are homologs of the N-terminal domains of plasminogen and hepatocyte growth factor [29]. While PAN domains are present in genes from a variety of organisms, no other proteins with four apple domains in the configuration seen in PK and FXI have been identified. Both proteins contain a C-terminal trypsin-like catalytic (protease) domain. The diagram in Figure 2 shows the amino acid sequence, disulfide bonds, and domain organization of human FXI as described by McMullen and co-workers [8]. PK is similarly organized [28]. The homology with PK has facilitated studying FXI structure-function relationships.
Figure 2. Human Factor XI amino acid sequence, disulfide bond structure, and domain organization.

Factor XI (FXI) is comprised of four apple domains (A1 to A4) and a trypsin-like Catalytic Domain. In this diagram, each amino acid is represented by a circle, with cysteine residues indicated in yellow. Conversion of FXI to FXIa involves cleavage after Arg369 (arrow). Residues identified as having specific functional importance are indicated by various colors. The catalytic triad required for proteolytic activity is indicated in magenta (His413, Asp462, Ser557). Resides involved in interactions with thrombin (orange - Glu66, Lys83, Gln84), HK (pink – Lys103, Gly104, Asn106, Tyr107, Asn108, His143, Leu148, and Leu163), factor IX (green - Ile183, Arg184, Asp185), the platelet GP1b receptor (grey - Ser248, Arg250, Lys255, Phe260, and Gln263), and polyanions (red – ABS1 Arg250, Lys252, Lys253, and Lys255; ABS2 Lys529, Arg530, and Arg531) are shown. The Abbreviations ABS1 and ABS2 stand for Anion binding Site 1 and 2, respectively. Residues in blue (Leu284, Glue287, Ile290, Tyr329 and Lys331) are components of the interface between the two subunits of the FXI dimer, with Cys321 forming a disulfide bond between the dimer subunits. Black diamonds indicate locations of putative N-linked glycosylation sites. After McMullen et al. [8], with permission.
Factor XI Synthesis
The genes for FXI and PK are located within ~5 kilobases of each other on the distal end of the long arm of chromosome 4 (4q35) in humans, and are organized similarly (15 exons, exon 2 signal peptide, exons 3-10 apple domains, exon 11-15 protease domain) [30–32]. Like other coagulation protease precursors, plasma FXI is synthesized primarily in hepatocytes [33–35]. Normal expression of FXI is controlled by the transcription factor hepatocyte nuclear factor-4α (HNF-4α) [32]. In humans, FXI is also expressed in the Islets of Langerhans in the pancreas, and in renal tubule cells [36]. Expression in mice appears to be largely confined to the liver [35].
There is inconsistent literature regarding whether or not human platelets contain and/or express FXI, and the form the protein takes [37,38]. Several groups identified FXI mRNA identical to that found in liver in human platelets and several megakaryocytic cell lines [39-41]. Recently, Zucker et al. presented data showing FXI pre-mRNA in human platelets that is spliced to a mature form on platelet activation [41]. The activated platelet then translates the message, producing a protein that migrates similarly to plasma FXI on SDS-PAGE and is recognized by antibodies to human FXI. A similar process has been reported for platelet expression of interleukin 1β, tissue factor, and cyclooxygenase 2 [42]. The relative importance of FXI synthesized or carried by platelets to that of plasma FXI is not known.
Factor XI Structure
Papagrigoriou et al. reported a structure for zymogen human FXI in 2006 [9]. Each apple domain consists of seven β-strands running antiparallel to each other that support a single α-helix (Fig. 3A). Three internal disulfide bonds constrain the apple structure. The apple domains (A1 to A4 from the N-terminus) form a planar disk-like structure ~60 Å wide, with A1 and A2 running anti-parallel to A3 and A4, separated by a 180° turn in the polypeptide chain (Fig. 3B). The apple domain disk has been likened to a “saucer” on which the protease domain (the cup) rests (Fig. 3C). The identical subunits of the FXI dimer interface through the A4 domains, with Cys321 forming an interchain disulfide bond (Fig. 3D). The apple domain disks are inclined at a 70° angle relative to each other creating an inverted V-shape. The two A2 domains are separated furthest from each other, while the A3 and A1 domains on one subunit are located close to the A1 and A3 domains, respectively, of the other subunit.
Figure 3. Factor XI zymogen structure.

(A) Ribbon diagram of the isolated FXI apple 1 domain from the crystal structure of the full length FXI zymogen (pdb:2F83). The α-helix is indicated in red and the β-sheet in blue. Disulfide bonds are in yellow. (B) Topology diagrams for the first, second, third, and fourth apple domains (A1, A2, A3, and A4) are shown in grey, blue, orange, and yellow, respectively. (C) Ribbon diagram and schematic of the FXI monomer with the catalytic domain (CD) colored maroon and the activation loop cleavage site residues Arg369-Val370 colored green. Apple domains (numbered 1 to 4) are indicated by the colors described in panel B. (D) Ribbon diagram and schematic of the FXI dimer with the A4 domains of each subunit forming the dimer interface. The Cys321-Cys321 bond at the top of the diagram covalently connects the subunits. From Papagrigoriou et al. [9].
Each FXI subunit is converted to an active protease by cleavage after Arg369 (Fig. 2) [1,7,8]. While a crystal structure is not available for FXIa, studies using low-resolution electron microscopy and small angle X-ray scattering reveal large-scale conformational changes accompany conversion of FXI to FXIa [43]. As discussed below, FXIa activation of FIX requires FIX to bind to an exosite on the FXIa A3 domain that is not available in FXI [44]. This implies a significant conformational change on activation that unmasks the FIX binding site. This is simulated in the images in Figure 4A to 4C. The interaction is discussed in more detail in the section on Factor XIa Activation of Factor IX.
Figure 4. Model of conversion of FXI to FXIa and the FXI dimer interface.
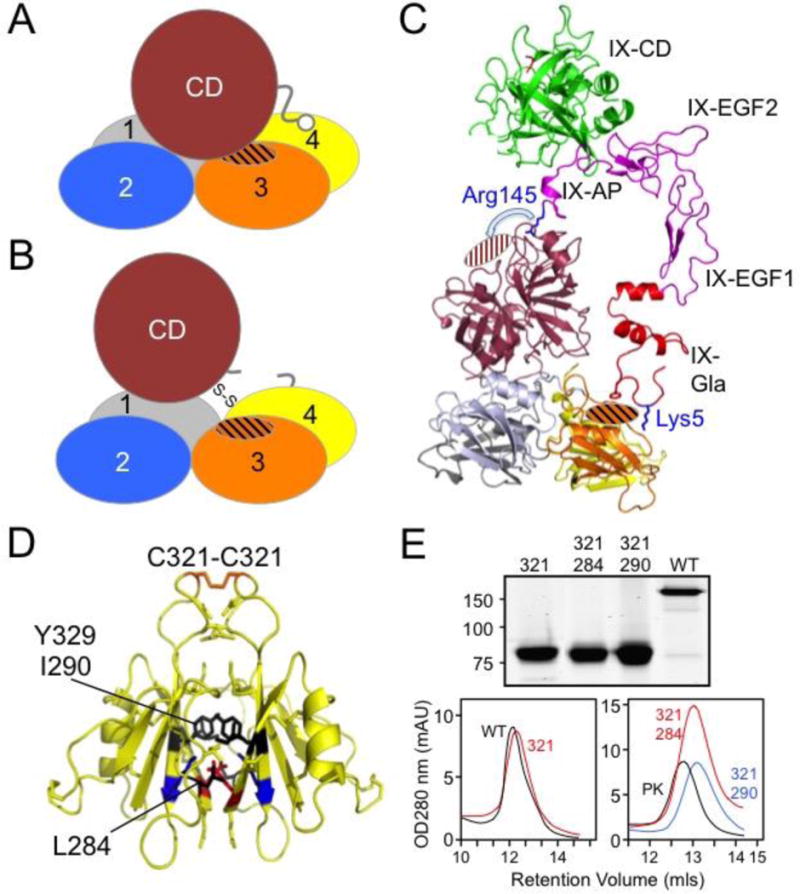
(A) Cartoon of a FXI subunit depicting the catalytic domain (CD) covering a FIX-binding site (cross-hatched area) on the A3 domain. Apple domains are numbered 1 to 4. (B) Cartoon of a FXIa subunit showing the predicted conformational change accompanying cleavage of the Arg369-Val370 bond that results in movement of the catalytic domain relative to the apple domain disk, and unmasking of the FIX binding site. (C) Topological diagram showing the FXI structure with the catalytic domain (maroon) displaced to reveal the FIX binding exosite in the A3 domain (orange cross-hatch). The FIXa crystal structure (pdb:1PFX) is shown as a ribbon diagram to illustrate the scale comparison of a vitamin-K dependent coagulation factor and FXI. The FIX Gla domain (red) including residue Lys5 can contact the FXIa A3 domain exosite, while the EGF domains (magenta) project upwards to position the FIX activation peptide (AP) residue Arg145 in the area of the FXIa S1 pocket (maroon cross-hatched). The FIX protease is colored green. (D) FXI dimer interface. Shown are the diagrams of two FXI A4 domains forming the dimer interface. The Cys321-Cys321 interchain disulfide bond is shown at the top in orange. Hydrophobic residues Leu284, Ile290, and Tyr329 are shown in black, and a salt bridge is formed between Lys331 (blue) and Glu287 (red). From Papagrigoriou et al. [9]. (E) FXI monomers. The top panel is a Coomassie Blue stained SDS-PAG showing non-reduced samples of wild type FXI (WT) and FXI species with Cy321 changed to serine (321). 284 and 290 indicate proteins that have also had Leu284 or Ile290 changed to alanine. The bottom images are elution profiles from size exclusion columns demonstrating the replacement of Cys321 alone elutes with FXI dimer (WT), while proteins with replacement of Cys321 and either Leu284 or Ile390 elute with the monomer prekallikrein (PK). From Geng et al. [53].
The Factor XI Dimer
The A4 domains of the two subunits in the FXI dimer form an 886 Å2 interface, with the β-sheets packed against each other (Fig. 4D) [9,47]. Cys321 [7], located on a loop projecting away from the main body of A4, forms an interchain disulfide bond [45]. Leu284, Ile290 and Tyr329 form the hydrophobic core of the interface [9,46,47], and salt bridges between Lys331 and Glu287 of opposite subunits contribute to dimer stability [9]. Replacing Cys321 does not disrupt the FXI dimer, showing the importance of non-covalent interactions (Fig. 4E) [45–51]. The structure of FXI and mutagenesis data demonstrate the central role of the A4 domain in dimer formation, however, A4 is not the only apple domain driving dimerization. In PK, a monomeric protein, Cys321 forms an intra-domain disulfide bond with Cys326, a residue not present in FXI [30]. Removal of PK Cys326 to leave Cys321 unpaired does not promote PK dimerization [50]. However, FXI in which the A4 domain is replaced with a PK A4 domain lacking Cys326 (leaving Cys321 unpaired) readily dimerizes despite the absence of the FXI A4 domain [50]. Here the A3 and A2 domains, while perhaps not directly contributing to the interface, are somehow involved in promoting dimerization in a manner not made obvious by the available crystal structure.
The FXI dimer is conserved across species, implying functional importance [51]. Critical components of the A4 interface, and an unpaired Cys321, are present in all species for which mRNA sequences are available, with the exception of the rabbit, which has histidine replacing Cys321 [52]. However, gel filtration studies show that rabbit FXI, like recombinant human FXI lacking Cys321, is a non-covalently associated homodimer. A function for the FXI dimer is not clearly established. Monomeric forms of FXI can be generated by replacing Cys321 and either Leu298 or Ile290 with alanine (Fig. 4E) [46,47,53,54]. These proteins display activity similar to dimeric FXI when used to supplement FXI-deficient plasma in activated partial thromboplastin time (aPTT) clotting assays. In our hands they are also similar to dimeric FXI/XIa in terms of ability to undergo autoactivation and activation by thrombin, and to activate FIX [53,55-57]. Unlike dimeric FXI, they fail to reconstitute FXI-deficient mice in thrombosis models, consistent with an important role for the dimer in vivo [53]. At low FXIIa concentrations, FXI monomer is activated more slowly than FXI dimer [46,53]. This may explain the loss of the prothrombotic effect in mice, but would not appear relevant for the proposed role of FXI in hemostasis. We are investigating the possibility that one FXI subunit is required for interaction with a surface such as a platelet, while the other activated subunit engages FIX [57]. Such interactions, while not required for protease function in static assays, may be important for protease function in flowing blood.
Factor XI Post-Translational Modifications
Glycosylation accounts for ~5% of the molecular mass of human FXI (Fig. 5A) [58]. Each subunit has five consensus sites for N-glycosylation (Fig. 2, N72, N108, N335, N432, and N473) [7,8]. Using mass spectrometry, Faid et al. showed that N72, N108, N432, and N473 are glycosylated on >90% of subunits. [59], while N335 is not glycosylated. N335 is located near the dimer interface, and glycosylation at this location would probably interfere with dimerization. A noncanonical site (N145) is glycosylated in ~5% of subunits [59]. We replaced Asn residues at the putative N-glycosylation sites in FXI with alanine and, with the exception of residue 335, noted reductions in apparent molecular mass on SDS-PAGE consistent with loss of a glycosylation site (Fig. 5B, D. Sun, M-f Sun and D Gailani, unpublished data). The glycosylation mutants have normal enzymatic activity in plasma, and activity of plasma FXI is not altered by deglycosylation (D. Sun, M-f Sun and D Gailani, unpublished data). However, glycosylation may play an important role in protein stability in plasma in vivo. FXI plasma levels are often low in patients with defects in N-linked glycosylation [60,61]. We observed that the half-lives of recombinant FXI proteins infused into FXI-deficient mice vary widely depending on the cell line in which they were expressed (Q Cheng, M-f Sun, D Gailani, unpublished observation). The differences in the FXI preparations is likely due to variation in post-translational glycosylation, which may vary substantially between cell lines.
Figure 5. Factor XI glycosylation sites and anion binding sites.
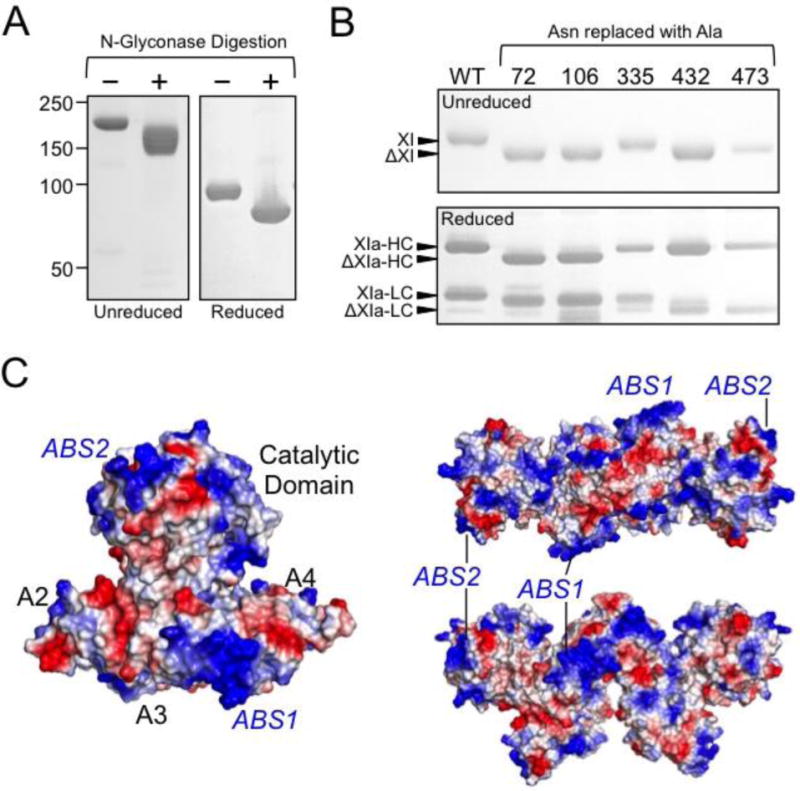
(A) SDS-PAGE of human plasma derived FXI incubated in the absence (−) or presence (+) of N-glycosidase to remove N-linked carbohydrate moieties. Positions of molecular mass standards are shown on the left of the figure. (B) SDS-PAGE (top panel – unreduced, bottom panel – reduced) of human recombinant wild type FXI (WT) or FXI in which asparagine residues have been replaced with alanine (indicated at the top). Positions of glycosylated FXI and the heavy chain (HC) and light chain (LC) of FXIa are indicated at the left of the images, as well as expected positions of FXI, HC, and LC lacking a single N-linked glycosylation site. Note that FXI with alanine replacing N335 appears similar to FXI-WT. (C) Charged surface representation of FXI zymogen monomer (left) and two views of the FXI dimer (right) showing positions of FXI anion binding sites 1 and 2 (ABS1 and ABS2). Blue indicates positively charged regions, and red negatively charged regions of the molecule.
FXI Anion-Binding
Human FXI contains two clusters of basic amino acids, one on the A3 domain starting at Arg250 (R-I-K-K-S-K) [62,63] and the other on the protease domain 170-loop starting at Lys529 (K-R-Y-R) [64], that interact with polyanions such as heparin [62–64], polyphosphate [65–67] and nucleic acids [68] (Fig. 2 & Fig. 5C). We refer to these clusters as anion binding exosite (ABS) 1 and 2, respectively. ABS1 and ABS2 were first recognized as sites required for normal expression of heparin cofactor activity during FXIa inhibition by antithrombin [62–64], and were subsequently shown to mediate binding to polyphosphate chains that enhance FXI activation by thrombin and FXIIa, and promoting FXI autoactivation [65,66]. On a polyanion, FXI/XIa and an inhibitor (antithrombin) or activator (thrombin or FXIIa) bind in proximity to each other to achieve maximum rates of inhibition/activation by a “template” mechanism characterized by bell-shaped dependences on polyanion concentration.
PK lacks an ABS on its A3 domain. While it has basic residues on its 170-loop that correspond to those of FXI ABS2 [29,30], the site does not appear to be required for polyanion binding [68]. PK binds more weakly than FXI to polyanions (Ivanov and Gailani, unpublished observation), even though polyanions enhance PK/kallikrein mediated reactions [68-71]. For PK, normal activation and activity require HK, which facilitates PK binding to the polyanion [20,68]. In contrast, HK has an inhibitory effect on FXI activation in the presence of polyanions, and cannot substitute for ABS1 and ABS2 to support polyanion-enhanced FXI activation [68]. These data suggest a fundamental difference in the manner in which FXI and PK interact with polyanions, and that HK may have an inhibitory regulatory role in FXI activation.
The FXI interaction with HK
Almost all FXI (3,4) and 75-90% of PK (4,72) circulates in complex with HK, a highly glycosylated 120-kDa 644 amino acid plasma protein that is organized into 6 domains (D1 to D6) [20,73]. The C-terminal D6 domain binds FXI and PK, with at least 58 amino acids (HK residues 556-613) required for normal FXI binding, and a 31 amino acid segment required for PK binding (74). The physiologic importance of the PK-HK interaction seems clear as HK cleavage by α-kallikrein (activated PK) liberates the vasoactive peptide bradykinin from HK [20,21,73], and HK enhances PK activation by FXIIa [20,68,73,75,76]. The importance of the FXI-HK interaction is less clear. Although HK may facilitate FXI binding to surfaces such as platelets [77-80] and endothelial cells [81], we observed that HK tends to inhibit FXI activation in the presence of DNA or polyphosphate [68].
Studies with FXI/PK chimeras, individual apple domains, and peptide sequences derived from apple domains indicate the FXI A2 domain is required for HK binding [82,83]. Domains A1 and A4 may also contribute [82,83]. A major advance in understanding the FXI-HK interaction came with the determination of the crystal structure for FXI in complex with an eight amino acid peptide derived from the HK D6 domain called HKP [84]. The central region of D6 containing the sequence Asn-Pro-Ile-Ser-Asp-Phe-Pro-Asp (residues 583-590) binds to a pocket in the A2 domain β-sheet (Figure 6A & 6B). The HK Asp-Phe-Pro motif is a major binding determinant, with Asp587 forming a key salt bridge with FXI Lys103, and Phe588 inserting into a hydrophobic pocket in FXI A2
Figure 6. The Factor XI interaction with high molecular weight kininogen.

(A) Cartoon diagram of the crystal structure of the apple domain disk of FXI in complex with a synthetic peptide spanning HK amino acids 582-593 (pdb:5I25). FXI apple domains are colored grey, blue, orange and yellow for A1 to A4 domains respectively. The HK peptide (HKP-magenta) is shown as a stick figure. (B) Diagram of the FXI A2 domain (white) with the HKP peptide shown as a stick diagram in magenta. Electrostatic and hydrogen bonding interactions are shown as purple dotted lines. FXI residues interacting with HKP are shown as sticks in green. On the left is a charged surface representation of the A2 domain (blue-positive, red-negative) bound to HKP (magenta). (C) Space-filling model of FXI dimer with the catalytic domains in maroon and activation loop residues Arg369-Val370 in green. Colors for the apple domains are as in Figure 3. Shown is the location of FXI-bound HKP (magenta) relative to the FXI dimer axis. (D) A 90 degree rotated view of the FXI dimer in panel C. (E) Size-exclusion chromatography (Superdex 200) profile of UV (280 nm) absorbance versus retention volume (ml) for FXI (red), HK (blue), or mixtures of FXI and HK at 1:0.25 (magenta); 1:2 (grey); 1:4 (brown-dashed) molar ratios. FXI elutes close to the expected molecular weight of the dimer (~200 kDa) and the FXI-HK complex elutes at ~400 kDa. (F) Schematic diagram of FXI interacting with HK. From Wong et al. [84].
Figure 6C & 6D shows the two HKP peptides binding to the A2 domains of the FXI dimer on the face of the apple domain disks opposite the protease domains. In Figure 6C the top (northern) hemisphere contains the FXI protease domain (maroon), activation loop (green), and A4 domain (yellow), while the bottom (southern) hemisphere contains the A2 domains (light blue) with the HK-D6 peptides (purple). The A3 domains (orange) and A1 domains (grey) lie within the FXI equatorial region. The HK peptides face the dimer axis in an arrangement that would effectively localize HK in the A2 domain binding pockets in the same plane at a distance of 45Å from each other (Figure 6D). The FXI dimer interface is at an oblique angle so that bound HK peptides are oriented towards each other. The data suggest a FXI dimer binds up to two HK molecules. This is supported by gel filtration experiments, which reveal a 1:2 molar ratio of FXI and HK resulting in a single ~400kDa complex (Figure 6E). Edman degradation analysis confirms an equimolar interaction between FXI subunits and HK polypeptides (i.e. two HK binding to each FXI dimer) [84]. HK has been reported to be a disulfide-linked circular monomer (85) or a dimer [86,87] by SDS-PAGE, however, gel filtration studies consistently indicate the protein is only a dimer [3,84,87]. Given this, it is possible that each FXI dimer actually associates with one HK dimer, as illustrated in Figure 6F.
FXI interaction with laminin
Laminins are large heterotrimeric proteins that are required for normal formation of extracellular matrices. Laminins 411 and 511 are abundant constituents of the extracellular matrix of blood vessels and arteries. FXI binds to human recombinant laminin heterotrimers 111, 411 and 511 in a concentration-dependent manner with Kd values of 11.1nM, 9.2nM, 8.3nM, respectively [84], and to laminin 111 purified from the native basement membrane of a murine sarcoma cell line (Kd 18.8nM) [84]. It does not bind to triple helical collagens [84]. Laminins are now recognized as important contributors to thrombus formation, and their interactions with platelet integrins is well-characterized [88]. Laminin provides a surface that can promote plasma coagulation and thrombus formation through FXII and FXI [89]. HK and FXII have both been reported to bind laminin [90,91]. It is notable that the FXIa substrate FIX binds to basement membrane through a well-characterized interaction with collagen IV [92,93]. The interaction between FXI and laminin may be a mechanism for co-localization of FXIa and its substrate FIX to promote thrombin generation on the extracellular matrix after blood vessel injury.
Conversion of FXI to FXIa
Each FXI subunit may be converted to its active form by several forms of thrombin (α-thrombin, β-thrombin, γ-thrombin, and meizothrombin) [94–97], factor XIIa [20,53,71], or by FXIa (autoactivation) in the presence of polyanions [65,66,68,95]. Regardless of the activating protease, FXI activation requires cleavage of the Arg369-Ile370 bond [7,8] (Fig. 2), creating a ~50 kDa heavy chain that contains the apple domains and a 30 kDa light chain that is the catalytic domain (Fig. 7A). The heavy and light chains are connected by a Cys362-Cys482 disulfide bond (Fig. 2). The designation “FXIa” is used to describe FXI with both subunits cleaved after Arg369. However, when FXI is activated by thrombin or FXIIa the first species formed has only one activated subunit [55,56]. We refer to this form as 1/2-FXIa. On non-reducing SDS-PAGE FXI, 1/2-FXIa, and FXIa migrate at slightly different rates relative to each other (Fig. 7B) [55]. 1/2-FXIa activates FIX by the same mechanism as FXIa with both subunits activated (discussed below). In vitro, conversion of FXI to 1/2-FXIa is considerably more rapid than conversion of 1/2-FXIa to FXIa, suggesting 1/2-FXIa may be a physiologically important species of activated FXI.
Figure 7. Factor XI Conversion to Factor XIa.

(A) Reducing SDS-PAG of FXI and FXIa showing the relative migrations of the 80 kDa FXI subunit (XI) and the heavy chain (HC) and light chain (LC) of FXIa. Positions of molecular mass standards are shown on the left. (B) Non-reducing SDS-PAGE of a time course (hours indicated at the top) of FXI activation by thrombin. Conversion of FXI to FXI (a species with two activated subunits) proceeds through an intermediate (1/2-FXIa) with one activated subunit. The three species (FXI, 1/2-FXIa, and FXIa) migrate slightly differently on non-reducing SDS-PAGE, allowing them to be identified.
In aPTT assays FXIIa activates FXI [2,14]. In mouse and primate models, this reaction appears to contribute to thrombosis [51,71,98,99], however, it is unlikely to be required for hemostasis given the absence of a bleeding diathesis in FXII-deficient individuals [2,14]. Studies comparing FXI dimers and monomers suggest there may be two types of binding interactions between FXIIa and FXI; a high affinity interaction operating by a trans-mechanism in which FXIIa binds to one subunit of the FXI dimer while activating the other subunit, and a lower-affinity cis-activation in which FXIIa binds to and activates the same FXI subunit [46,53]. The FXI residues involved in FXIIa binding are not known. The interaction may involve more than one apple domain. While studies using peptide mimicry point to a binding site on the A4 domain [100], an antibody to A2 selectively interferes with FXI activation by factor XIIa [51].
While clinical observations [14] and some pre-clinical work with gene-deleted mice [101,102] support the premise that FXI contributes to hemostasis in a FXIIa-independent manner, the identity of the protease(s) that activate FXI in vivo are not established. Forms of thrombin are leading candidates. The X-Pro-Arg sequences preceding the FXI activation cleavage site in most mammals are typical thrombin cleavage sites, and differ from sequences found in PK [97]. FXI activation by thrombin is enhanced by polyphosphate [53,65,66], nucleic acids [68,103–105], dextran sulfate [95] and various glycosaminoglycans [106,107] through a template mechanism as described above. Thrombin anion binding exosite II facilitates thrombin binding to polyanions during FXI activation, but anion binding exosite I does not appear to be required for FXI activation [97]. There is limited data regarding a thrombin-binding site on FXI. A1 domain residues Glu66, Lys83, and Gln84 (Fig. 2) have been implicated [9,108,109]. These residues cluster near the interface with the A4 domain, and are in proximity to the activation loop containing the Arg369-Ile370 cleavage site.
The structures of the thrombin and FXIIa protease domains are quite different. Thrombin uses its positively charged anion binding exosites to interact with a variety of substrates [110], while the FXIIa protease domain has a net negative charge and exosites that may bind FXI have not been identified [111,112]. FXI has an overall net positive charge, and charge complementarity with FXIIa may explain why it can activate FXI in the absence of polyanions, while thrombin mediated-FXI activation is enhanced to a much greater degree by polyanions such as polyphosphate [53].
Factor XIa Activation of Factor IX
Conversion of FIX to FIXaβ is a calcium-dependent process catalyzed by factor VIIa (FVIIa) in the presence of tissue factor and phosphatidylserine-rich phospholipid [113,114], or by FXIa in a phospholipid-independent reaction [14,55,115,116]. Regardless of the activating protease, FIX is cleaved first after Arg145 forming the intermediate FIXα, then after Arg180 to form FIXaβ (Fig. 8A) [14,55,116]. FIXα accumulates during FIX activation by FVIIa (Fig. 8B, top), but not during activation by FXIa (Fig. 8B, bottom) [115,116]. This is not accounted for by FXIa’s dimeric structure, as FIXα does not accumulate during activation by FXIa monomer [46] or 1/2-FXIa [55].
Figure 8. Factor XIa Activation of Factor IX.

(A) Schematic diagrams showing conversion of factor IX to the intermediate factor IXα by cleavage after Arg145, and then to factor IXaβ by cleavage after Arg180. Factor IX contains an N-terminal calcium-binding Gla-domain, two epidermal growth factor (EGF) domains, an activation peptide (blue) and a catalytic domain (CD-red). (B) Western blots of time courses of FIX activation by FVIIa/TF (top) or FXIa (bottom). Gels were run under reducing conditions. Positions of FIX zymogen (IX), the large fragment of FIXα, the light chain of FIXα and FIXaβ (LC) and the catalytic domains (CD) of FIXaβ are indicated on the right. (C) Western blots of time courses of FIX activation by FXIa (top) and FXIa with the PKA3 domain (bottom). Note the different time scales in the two panels. (D) Kinetic parameters for cleavage of FIX by FXIa or FXIa-PKA3 based on full-progress experimental traces analyzed with KinTek software (KinTek Explorer Version 2.5). Km and kcat for activation were calculated from individual rate constants for conversion of FIX to FIXα (reaction 1) and FIXα to FIXaβ (reaction 2). Catalytic efficiencies (kcat/Km) are shown for both reactions. (E) Charged surface representations of the FXI (left) and PK (right) A3 domains. Blue indicates positive charge, and red indicates negative charge. Note that the hydrophobic pocket on the surface of FXI A3 is not present in PK A3. (F) Structure of FXI showing the relationship of the A3 domain (green) and catalytic domain (grey) in the zymogen. Arg184 and the adjacent hydrophobic pocket 1 are covered by the catalytic domain. Arg184 forms salt bridges (dotted lines) with Asp488 and Asn566 in the zymogen. From [116,117].
FIX binds to an exosite on the FXIa A3 domain [117-120] followed by engagement at the protease active site and cleavage of the Arg145-Ala146 bond [116]. This facilitates subsequent cleavage of the Arg180-Val181 bond. The catalytic efficiency for the second cleavage is 7-fold greater than for the first, explaining the minimal FIX αaccumulation (Fig. 8C & 8D). Replacing the FXIa A3 domain with the PK A3 domain (FXIa/PKA3) reduces the catalytic efficiency for cleavage of both FIX bonds, with a greater effect on cleavage after Arg180, leading to FIXα accumulation (Fig. 8C & 8D). The same “defect” is observed when FIX is activated by isolated FXIa catalytic domain (no A3 domain), or during FIX activation by FXIa in the absence of Ca2+ ions. Thus, FIX activation requires FIX and FIXα to bind to the FXIa A3 domain in a Ca2+-dependent manner prior to catalysis.
FIX does not bind zymogen FXI [121], indicating binding sites form or are unmasked by conformational changes accompanying conversion to FXIa. Replacing Ile183, Arg184 and Asp185 in the N-terminus of the A3 domain (Fig. 2) with alanine residues causes a defect in FIX activation similar to that observed with FXIa/PKA3 [117,119]. These residues form a charged ridge adjacent to a hydrophobic pocket on the A3 domain surface facing the catalytic domain, a feature not found in PK (Fig. 8E) [117]. FXI Arg184 forms salt bridges with residues Asp488 and Asn566 on the protease domain (Fig. 8F), anchoring the protease domain over the putative FIX binding site (Fig. 8F) [9]. We suspect that the highly conserved Arg184 serves as a latch that is released to expose the FIX binding site on conversion to FXIa.
FIX binding to FXIa requires the FIX Gla domain [116,121]. Mutagenesis studies indicate that residues in the phospholipid-binding Ω-loop of the Gla-domain (residues 4 to 11) are required for FXIa binding [117] (Fig. 9A & 9B). The Ω-loops of vitamin K-dependent proteases mediate binding to phospholipid membranes through charged and hydrophobic interactions (Fig. 9C) [2]. The topology of the putative FIX binding site on FXIa A3 suggests an interaction involving charged and hydrophobic components. Using surface plasmon resonance, we observed that FXIa, but not FXIa/PKA3, competes with phosphatidyl-serine-rich surfaces for FIX binding (Messer, Bajaj, Geng and Gailani, unpublished observation), supporting the conclusion that the FIX-Gla domain engages the FXIa A3 domain. The data suggest that the A3 domain performs a role in FIX activation similar to that of phospholipid during FIX activation by FVIIa, with both interactions contribute to Km for their respective reactions.
Figure 9. Factor IX with factor VII sequence in the Gla-domain.

(A) Primary sequences of the human factor IX (FIX) and factor VII (FVII) Gla-domains. The numbering system shown is for FIX. The symbol γ indicates positions of γ-carboxyglutamic acid residues. Underlined sequences were changed from FIX sequence to FVII sequence to generate chimeras C1, C2, C3 and C4. The amino acids changed in each chimera are highlighted by the gray boxes. (B) Top - stained SDS-polyacrylamide gel of purified plasma FIX (pIX), recombinant wild type FIX (WT), FIX/FVII chimeras (C1, C2, C3, and C4), and FIX in which the Gla-domain from residues 1 to 46 are changed to FVII sequence (FIX/VII-Gla - abbreviated VIIGla). Bottom - FXIa (2 nM) was incubated with 250 nM FIXWT (○), C1 (●), C2 (□), C3 (■), C4 (△) and FIX/VII-Gla (▲) (250 nM) in TBS with calcium, as described under methods. Shown are concentrations of FIXa▲ at various times as determined by densitometry of SDS-polyacrylamide gels. (E) Topological diagram of the human FIX Gla-domain, with residues in the Ω-loop (4–11) that differ from the corresponding region of the human FVII Gla-domain highlighted (magenta). Positions of certain Gla residues are indicated by the symbol ‘γ’. Calcium ions in the vicinity of the Ω-loop are represented by green spheres. The image is derived from a structure for a complex between the human FIX Gla-domain and the antibody 10C12 [142]. Figures were prepared with PYMOL (The PyMOL Molecular Graphics System, Version 1.5.0.4). From Geng et al. [117].
Other FXIa substrates
FXIa activates several components of the plasma thrombin generation mechanism other than FIX. Factor X (FX), a FIX homolog, is a major substrate for FIXaβ and FVIIa/tissue factor [2]. We showed that FXIa also activates FX [122]. The reaction is orders of magnitude less efficient than activation of FIX by FXIa, is not Ca2+-dependent, and does not require the FXIa A3 domain. Whelihan et al. reported that FXIa cleaves factors V and VIII (FV and FVIII) to forms that have a few percent of the cofactor activity of the standard active forms FVa and FVIIIa [123]. We showed that FV activation by FXIa requires the FXIa A3 domain to proceed optimally [112], and Choi et al. reported that polyphosphate accelerates the reaction [124]. One or more of these reactions may occur in vivo. Mice lacking both FXI and FIX are more resistant to injury-induced thrombosis than are mice lacking only one of the proteins [122], indicating FXIa is capable of bypassing FIX to activate other clotting factors. The capacity to activate FV and/or FX may allow FXIa to influence thrombin generation by essentially jumping the gap created by FIX deficiency. The relevance of these findings to hemostasis warrants further investigation in animal models.
FXIa activates components of the KKS. Like its homolog α-kallikrein, FXIa activates FXII [18,126] and cleaves HK to release bradykinin [95,126], although neither reaction is as efficient as the α-kallikrein-mediated reactions. The kallikrein-like properties of FXIa may explain an observation made in septic mice. FXI-deficient mice have lower mortality than wild type mice with infection/inflammation induced by cecal ligation and puncture [18]. Surprisingly, FXI deficiency did not cause appreciable changes in activation of coagulation. Instead, FXI-deficient mice had a milder cytokine response, with reduced consumption of FXII and PK, indicating blunted activation of the KKS. Infusing polyphosphate into mice induces KKS activation. FXI-deficient mice receiving polyphosphate had reduced FXII and PK activation compared with wild type mice [18]. The findings support the hypothesis that FXI/XIa is not only a substrate for the KKS, but contributes to KKS activation, perhaps by activating FXII.
FXIa may promote clot formation/stability through proteolytic inactivation of regulators of coagulation. Puy et al. reported that FXIa cleaves tissue factor pathway inhibitor (TFPI) between the Kunitz 1 and 2 domains, reducing the capacity of this key protein to regulate tissue factor-initiated coagulation [127]. TFPI inhibition by FXIa is enhanced by polyphosphate chains of the length typically released by activated platelets [128]. FXIa also cleaves ADAMTS13, the von Willebrand factor-cleaving metaolproteinase, removing the C-terminal CUB domains [129]. The result is a form of ADAMTS13 with increased activity toward small peptide substrates, but that interacts poorly with von Willebrand factor.
FXI interactions with platelets
FXI lacks the Gla domain that facilitates binding of vitamin K-dependent coagulation proteases to phospholipid surfaces such as membranes of activated platelets or damaged tissues [7–9]. Nevertheless, there is evidence that FXI binds to platelets, and that FXI contributes to platelet activation/accumulation in flowing blood exposed to thrombogenic surfaces. When human blood is perfused over collagen, platelets bind and aggregate in a manner supported by thrombin and fibrin formation [130-132]. Adding an anti-FXI antibody prevents fibrin formation and reduces platelet aggregate size. Anti-FXI antibodies produce a similar effect in a primate model in which thrombus-inducing collagen-coated grafts are introduced into the circulation [51,130,133].
Greengard et al. detected ~1500 FXI binding sites per activated platelet [134], with subsequent work demonstrating binding to the GP1b receptor [135]. FXI competes with von Willebrand factor, but not thrombin, for GP1bα binding [135]. Kossmann et al. recently reported that FXI localized to platelets promotes coagulation and inflammation in mice in a GP1b-dependent manner [136]. In vitro, optimal platelet binding involves residues in the FXI A3 domain (Fig. 2) [135,137], leucine-rich repeats at the N-terminus of GP1bα [135,138], and HK and Zn2+ ions [135,137,138]. The FXI A3 residues involved, Ser248, Arg250, Lys255, Phe260 and Gln263 overlap with elements of ABS1 (Fig. 2). There are ~25,000 GP1b complexes per platelet, a value 10 times greater than the number of FXI binding sites, suggesting FXI binds to a subset of GP1b. White-Adams et al. showed that FXI binds to the platelet ApoER2′ receptor [139]. There are ~2000 copies of ApoER2′ per platelet, and they co-localize with GP1b. This suggests FXI binding may involve a complex of the two receptors. Preliminary data indicate the binding site for ApoER2′ on FXI is distinct from that of GP1b (McCarty and Gailani, unpublished observation).
There is evidence indicating that activated platelets support FXI activation, but the history of work in this area is complicated. A 1998 report indicating that activated platelets enhance FXI activation by thrombin at a rate 2- to 5-fold higher than dextran sulfate [80] was subsequently retracted because the results proved difficult to reproduce [140]. However, Choi et al. reported in 2011 that activated platelets and platelet releasates do enhance FXI activation by thrombin [65]. Key to this process is polyphosphate secreted by the platelets. In our experience, the effects of polyphosphate on FXI activation are inhibited in certain types of buffers, possibly explaining variation across experiments. Verhoef et al. recently reported that platelet polyphosphate chains condense into divalent cation-dependent membrane-associated particles that are considerably larger than the individual polyphosphate chains released from platelets [141]. These particles may serve as binding sites for FXI and FXII, and may be more efficient at promoting protease activation than short-chain (60-100 unit) polyphosphate species in solution.
Summary
FXI is structurally distinct from the vitamin K-dependent proteases that form the core of the vertebrate coagulation mechanism (Fig. 1). FXI retains structural features and activities of its parent molecule PK, and the similarities between the proteins have been invaluable in efforts to characterize unique properties of FXI. Since the duplication event that created separate PK and FXI genes, FXI has undergone important structural changes that are required for its procoagulant activities. These include adoption of a homodimeric conformation, formation of a binding site for the Gla-domain of FIX, and development of ABSs that promote activation. Changes to the activation cleavage site permitting activation by thrombin were also likely instrumental in facilitating the switch from KKS component to coagulation protease. Less clear from a pathophysiologic standpoint are the importance of FXI’s conserved interaction with HK, its ability to bind to platelet receptors, and the capacity of FXIa to neutralize regulatory proteins such as TFPI and ADAMTS13. The reason FXI must be a dimer to be functional in vivo remains a major unanswered question. Furthermore, the major species of FXI (FXIa or 1/2-FXIa) generated during coagulation remains to be determined. Progress on these questions will be important for fully understanding the roles of FXI in coagulation and inflammation, and will better inform translational efforts to develop therapeutic strategies targeting this protein.
HIGHLIGHTS.
Factor XI (FXI) is a homolog of the plasma contact factor prekallikrein
FXI can function as a contact protein or as coagulation protein
FXI also serves as a bridge between contact activation and thrombin generation
FXI is a homodimeric protein, an unusual configuration for a serine protease
FXI has undergone adaptive changes that make it a potent activator of factor IX
Acknowledgments
This work was supported by awards HL58837 and HL81326 (D.G.) and (HL130018) IMV from the National Institutes of Health, National Heart, Lung, and Blood Institute; and Programme Grant RG/12/9/29775 and Project Grant PG/09/025/27136 (J.E.) from the British Heart Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
BIBLIOGRAPHY
- 1.Emsley J, McEwan PA, Gailani D. Structure and function of factor XI. Blood. 2010;115:2569–2577. doi: 10.1182/blood-2009-09-199182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brummel-Ziedens K, Mann KG. Molecular basis of blood coagulation. In: Hoffman H, Benz EJ, Silverstein LS, Heslop HE, Weitz JI, Anastasi J, editors. Hematology, Basic Principles and Practice. 6th. Philadelphia, PA: Elsevier-Saunders; 2013. pp. 1821–1841. [Google Scholar]
- 3.Thompson RE, Mandle R, Jr, Kaplan AP. Association of factor XI and high molecular weight kininogen in human plasma. J Clin Invest. 1977;60:1376–1380. doi: 10.1172/JCI108898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thompson RE, Mandle R, Jr, Kaplan AP. Studies of binding of prekallikrein and Factor XI to high molecular weight kininogen and its light chain. Proc Natl Acad Sci U S A. 1979;76:4862–4866. doi: 10.1073/pnas.76.10.4862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Renné T, Gailani D, Meijers JC, Müller-Esterl W. Characterization of the H-kininogen-binding site on factor XI: a comparison of factor XI and plasma prekallikrein. J Biol Chem. 2002;277:4892–4899. doi: 10.1074/jbc.M105221200. [DOI] [PubMed] [Google Scholar]
- 6.Bouma BN, Griffin JH. Human blood coagulation factor XI. Purification, properties, and mechanism of activation by activated factor XII. J Biol Chem. 1977;252:6432–6437. [PubMed] [Google Scholar]
- 7.K, Chung DW, Hendrickson LE, Davie EW. Amino acid sequence of human factor XI, a blood coagulation factor with four tandem repeats that are highly homologous with plasma prekallikrein. Biochemistry. 1986;25:2417–2424. doi: 10.1021/bi00357a018. [DOI] [PubMed] [Google Scholar]
- 8.McMullen BA, Fujikawa K, Davie EW. Location of the disulfide bonds in human coagulation factor XI: the presence of tandem apple domains. Biochemistry. 1991;30:2056–2060. doi: 10.1021/bi00222a008. [DOI] [PubMed] [Google Scholar]
- 9.Papagrigoriou E, McEwan PA, Walsh PN, Emsley J. Crystal structure of the factor XI zymogen reveals a pathway for transactivation. Nat Struct Mol Biol. 2006;13:557–558. doi: 10.1038/nsmb1095. [DOI] [PubMed] [Google Scholar]
- 10.Rosenthal RL, Dreskin OH, Rosenthal N. New hemophilia-like disease caused by deficiency of a third plasma thromboplastin factor. Proc Soc Exp Biol Med. 1953;82:171–174. doi: 10.3181/00379727-82-20057. [DOI] [PubMed] [Google Scholar]
- 11.Rosenthal RL. Hemophilia and hemophilia-like diseases caused by deficiencies in plasma thromboplastin factors: anti-hemophilic globulin (AHG), plasma thromboplastin component (PTC) and plasma thromboplastin antecedent (PTA) Am J Med. 1954;17:57–69. doi: 10.1016/0002-9343(54)90144-8. [DOI] [PubMed] [Google Scholar]
- 12.Macfarlane RG. An enzyme cascade in the blood clotting mechanism, and its function as a biochemical amplifier. Nature. 1964;202:498–499. doi: 10.1038/202498a0. [DOI] [PubMed] [Google Scholar]
- 13.Davie EW, Ratnoff OD. Waterfall sequence for intrinsic blood clotting. Science. 1964;145:1310–1312. doi: 10.1126/science.145.3638.1310. [DOI] [PubMed] [Google Scholar]
- 14.Gailani D, Neff AT. Rare coagulation factor deficiencies. In: Hoffman H, Benz EJ, Silverstein LS, Heslop HE, Weitz JI, Anastasi J, editors. Hematology, Basic Principles and Practice. 6th. Philadelphia, PA: Elsevier-Saunders; 2013. pp. 1971–1986. [Google Scholar]
- 15.James P, Salomon O, Mikovic D, Peyvandi F. Rare bleeding disorders - bleeding assessment tools, laboratory aspects and phenotype and therapy of FXI deficiency. Haemophilia. 2014;20(Suppl 4):71–75. doi: 10.1111/hae.12402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Puy C, Rigg RA, McCarty OJ. The hemostatic role of factor XI. Thromb Res. 2016;141(Suppl 2):S8–S11. doi: 10.1016/S0049-3848(16)30354-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wheeler AP, Gailani D. Why factor XI deficiency is a clinical concern. Expert Rev Hematol. 2016;9:629–637. doi: 10.1080/17474086.2016.1191944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bane CE, Jr, Ivanov I, Matafonov A, Boyd KL, Cheng Q, Sherwood ER, Tucker EI, Smiley ST, McCarty OJ, Gruber A, Gailani D. Factor XI Deficiency Alters the Cytokine Response and Activation of Contact Proteases during Polymicrobial Sepsis in Mice. PLoS One. 2016;11:e0152968. doi: 10.1371/journal.pone.0152968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gailani D, Gruber A. Factor XI as a therapeutic target. Arterioscler Thromb Vasc Biol. 2016;36:1316–1322. doi: 10.1161/ATVBAHA.116.306925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schmaier AH. The contact activation and kallikrein/kinin systems: pathophysiologic and physiologic activities. J Thromb Haemost. 2016;14:28–39. doi: 10.1111/jth.13194. [DOI] [PubMed] [Google Scholar]
- 21.Foley JH, Conway EM. Cross Talk Pathways Between Coagulation and Inflammation. Circ Res. 2016;118:1392–1408. doi: 10.1161/CIRCRESAHA.116.306853. [DOI] [PubMed] [Google Scholar]
- 22.Weitz JI, Fredenburgh JC. Factors XI and XII as Targets for New Anticoagulants. Front Med (Lausanne) 2017;4:19. doi: 10.3389/fmed.2017.00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jiang Y, Doolittle RF. The evolution of vertebrate blood coagulation as viewed from a comparison of puffer fish and sea squirt genomes. Proc Natl Acad Sci U S A. 2003;100:7527–7532. doi: 10.1073/pnas.0932632100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Doolittle RF. Step-by-step evolution of vertebrate blood coagulation. Cold Spring Harb Symp Quant Biol. 2009;74:35–40. doi: 10.1101/sqb.2009.74.001. [DOI] [PubMed] [Google Scholar]
- 25.Doolittle RF. The evolution of vertebrate clotting. University Science Books; 2013. [Google Scholar]
- 26.Ponczek MB, Gailani D, Doolittle RF. Evolution of the contact phase of vertebrate blood coagulation. J Thromb Haemost. 2008;6:1876–1883. doi: 10.1111/j.1538-7836.2008.03143.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chung DW, Fujikawa K, McMullen BA, Davie EW. Human plasma prekallikrein, a zymogen to a serine protease that contains four tandem repeats. Biochemistry. 1986;25:2410–2417. doi: 10.1021/bi00357a017. [DOI] [PubMed] [Google Scholar]
- 28.McMullen BA, Fujikawa K, Davie EW. Location of the disulfide bonds in human plasma prekallikrein: the presence of four novel apple domains in the amino-terminal portion of the molecule. Biochemistry. 1991;30:2050–2056. doi: 10.1021/bi00222a007. [DOI] [PubMed] [Google Scholar]
- 29.Tordai H, Banyai L, Patthy L. The PAN module: the N-terminal domains of plasminogen and hepatocyte growth factor are homologous with the apple domains of the prekallikrein family and with a novel domain found in numerous nematode proteins. FEBS Lett. 1999;461:63–67. doi: 10.1016/s0014-5793(99)01416-7. [DOI] [PubMed] [Google Scholar]
- 30.Asakai R, Davie EW, Chung DW. Organization of the gene for human factor XI. Biochemistry. 1987;26:7221–7228. doi: 10.1021/bi00397a004. [DOI] [PubMed] [Google Scholar]
- 31.Yu H, Anderson PJ, Freedman BI, Rich SS, Bowden DW. Genomic structure of the human plasma prekallikrein gene, identification of allelic variants, and analysis in end-stage renal disease. Genomics. 2000;69:225–234. doi: 10.1006/geno.2000.6330. [DOI] [PubMed] [Google Scholar]
- 32.Tarumi T, Kravtsov DV, Zhao M, Williams SM, Gailani D. Cloning and characterization of the human factor XI gene promoter: transcription factor hepatocyte nuclear factor 4alpha (HNF-4alpha) is required for hepatocyte-specific expression of factor XI. J Biol Chem. 2002;277:18510–18516. doi: 10.1074/jbc.M201886200. [DOI] [PubMed] [Google Scholar]
- 33.Walker IR, Milner RA, Johnston MA, Rand CA, Neame PB, Hirsh J. Factor XI and factor XII are low in subjects with liver disease. Dig Dis Sci. 1983;28:967–970. doi: 10.1007/BF01311723. [DOI] [PubMed] [Google Scholar]
- 34.Dzik WH, Arkin CF, Jenkins RL. Transfer of congenital factor XI deficiency from a donor to a recipient by liver transplantation. N Engl J Med. 1987;316:1217–1218. [PubMed] [Google Scholar]
- 35.Gailani D, Sun MF, Sun Y. A comparison of murine and human factor XI. Blood. 1997;90:1055–1064. [PubMed] [Google Scholar]
- 36.Cheng Q, Kantz J, Poffenberger G, Powers AC, Gailani D. Factor XI protein in human pancreas and kidney. Thromb Haemost. 2008;100:158–160. doi: 10.1160/TH08-04-0260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gailani D, Zivelin A, Sinha D, Walsh PN. Do platelets synthesize factor XI? J Thromb Haemost. 2004;2:1709–1712. doi: 10.1111/j.1538-7836.2004.00935.x. [DOI] [PubMed] [Google Scholar]
- 38.Hsu TC, Shore SK, Seshsmma T, Bagasra O, Walsh PN. Molecular cloning of platelet factor XI, an alternative splicing product of the plasma factor XI gene. J Biol Chem. 1998;273:13787–13793. doi: 10.1074/jbc.273.22.13787. [DOI] [PubMed] [Google Scholar]
- 39.Martincic D, Kravtsov V, Gailani D. Factor XI messenger RNA in human platelets. Blood. 1999;94:3397–3404. [PubMed] [Google Scholar]
- 40.Podmore A, Smith M, Savidge G, Alhaq A. Real-time quantitative PCR analysis of factor XI mRNA variants in human platelets. J Thromb Haemost. 2004;2:1713–1719. doi: 10.1111/j.1538-7836.2004.00924.x. [DOI] [PubMed] [Google Scholar]
- 41.Zucker M, Hauschner H, Seligsohn U, Rosenberg N. Platelet factor XI: intracellular localization and mRNA splicing following platelet activation. Blood Cells Mol Dis. doi: 10.1016/j.bcmd.2017.04.006. (in press) [DOI] [PubMed] [Google Scholar]
- 42.Rondina MT, Weyrich AS. Regulation of the genetic code by platelets. J Thromb Haemost. 2015;13(Suppl 1):S26–32. doi: 10.1111/jth.12965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Samuel D, Cheng H, Riley PW, Canutescu AA, Nagaswami C, Weisel JW, Bu Z, Walsh PN, Roder H. Solution structure of the A4 domain of factor XI sheds light on the mechanism of zymogen activation. Proc Natl Acad Sci U S A. 2007;104:15693–15698. doi: 10.1073/pnas.0703080104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gailani D, Geng Y, Verhamme I, Sun MF, Bajaj SP, Messer A, Emsley J. The mechanism underlying activation of factor IX by factor XIa. Thromb Res. 2014;133(Suppl 1):S48–51. doi: 10.1016/j.thromres.2014.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Meijers JC, Mulvihill ER, Davie EW, Chung DW. Apple four in human blood coagulation factor XI mediates dimer formation. Biochemistry. 1992;31:4680–4684. doi: 10.1021/bi00134a021. [DOI] [PubMed] [Google Scholar]
- 46.Wu W, Sinha D, Shikov S, Yip CK, Walz T, Billings PC, Lear JD, Walsh PN. Factor XI homodimer structure is essential for normal proteolytic activation by factor XIIa, thrombin, and factor XIa. J Biol Chem. 2008;283:18655–18664. doi: 10.1074/jbc.M802275200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zucker M, Zivelin A, Landau M, Rosenberg N, Seligsohn U. Three residues at the interface of factor XI (FXI) monomers augment covalent dimerization of FXI. J Thromb Haemost. 2009;7:970–975. doi: 10.1111/j.1538-7836.2009.03353.x. [DOI] [PubMed] [Google Scholar]
- 48.Riley PW, Cheng H, Samuel D, Roder H, Walsh PN. Dimer dissociation and unfolding mechanism of coagulation factor XI apple 4 domain: spectroscopic and mutational analysis. J Mol Biol. 2007;367:558–573. doi: 10.1016/j.jmb.2006.12.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dorfman R, Walsh PN. Noncovalent interactions of the Apple 4 domain that mediate coagulation factor XI homodimerization. J Biol Chem. 2001;276:6429–6438. doi: 10.1074/jbc.M010340200. [DOI] [PubMed] [Google Scholar]
- 50.Cheng Q, Sun MF, Kravtsov DV, Aktimur A, Gailani D. Factor XI apple domains and protein dimerization. J Thromb Haemost. 2003;1:2340–2347. doi: 10.1046/j.1538-7836.2003.00418.x. [DOI] [PubMed] [Google Scholar]
- 51.Cheng Q, Tucker EI, Pine MS, Sisler I, Matafonov A, Sun MF, White-Adams TC, Smith SA, Hanson SR, McCarty OJ, Renné T, Gruber A, Gailani D. A role for factor XIIa-mediated factor XI activation in thrombus formation in vivo. Blood. 2010;116:3981–3989. doi: 10.1182/blood-2010-02-270918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sinha D, Marcinkiewicz M, Gailani D, Walsh PN. Molecular cloning and biochemical characterization of rabbit factor XI. Biochem J. 2002;367(Pt 1):49–56. doi: 10.1042/BJ20020232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Geng Y, Verhamme IM, Smith SB, Sun MF, Matafonov A, Cheng Q, Smith SA, Morrissey JH, Gailani D. The dimeric structure of factor XI and zymogen activation. Blood. 2013;121:3962–3969. doi: 10.1182/blood-2012-12-473629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sinha D, Marcinkiewicz M, Lear JD, Walsh PN. Factor XIa dimer in the activation of factor IX. Biochemistry. 2005;44:10416–10422. doi: 10.1021/bi050361x. [DOI] [PubMed] [Google Scholar]
- 55.Smith SB, Verhamme IM, Sun MF, Bock PE, Gailani D. Characterization of Novel Forms of Coagulation Factor XIa: independence of factor XIa subunits in factor IX activation. J Biol Chem. 2008;283:6696–6705. doi: 10.1074/jbc.M707234200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gailani D, Smith SB. Structural and functional features of factor XI. J Thromb Haemost. 2009;7(suppl 1):75–78. doi: 10.1111/j.1538-7836.2009.03414.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gailani D, Ho D, Sun MF, Cheng Q, Walsh PN. Model for a factor IX activation complex on blood platelets: dimeric conformation of factor XIa is essential. Blood. 2001;97:3117–3122. doi: 10.1182/blood.v97.10.3117. [DOI] [PubMed] [Google Scholar]
- 58.Kurachi K, Davie EW. Activation of human factor XI (plasma thromboplastin antecedent) by factor XIIa (activated Hageman factor) Biochemistry. 1977;16:5831–5839. doi: 10.1021/bi00645a030. [DOI] [PubMed] [Google Scholar]
- 59.Faid V, Denguir N, Chapuis V, Bihoreau N, Chevreux G. Site-specific N-glycosylation analysis of human factor XI: identificiation of a noncanonical NXC glycosite. Proteomics. 2014;14:2460–2470. doi: 10.1002/pmic.201400038. [DOI] [PubMed] [Google Scholar]
- 60.Young G, Driscoll MC. Coagulation abnormalities in the carbohydrate-deficient glycoprotein syndrome: case report and review of the literature. Am J Hematol. 1999;60:66–69. doi: 10.1002/(sici)1096-8652(199901)60:1<66::aid-ajh11>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 61.Calvo PL, Pagliardini S, Baldi M, Pucci A, Sturiale L, Garozzo D, Vinciguerra T, Barbera C, Jaeken J. Long-standing mild hypertransaminasaemia caused by congenital disorder of glycosylation (CDG) type IIx. J Inherit Metab Dis. 2008;31(Suppl 2):S437–440. doi: 10.1007/s10545-008-1004-9. [DOI] [PubMed] [Google Scholar]
- 62.Ho DH, Badellino K, Baglia FA, Walsh PN. A binding site for heparin in the apple 3 domain of factor XI. J Biol Chem. 1998;273:16382–16390. doi: 10.1074/jbc.273.26.16382. [DOI] [PubMed] [Google Scholar]
- 63.Zhao M, Abdel-Razek T, Sun MF, Gailani D. Characterization of a heparin binding site on the heavy chain of factor XI. J Biol Chem. 1998;273:31153–31159. doi: 10.1074/jbc.273.47.31153. [DOI] [PubMed] [Google Scholar]
- 64.Yang L, Sun MF, Gailani D, Rezaie AR. Characterization of a heparin-binding site on the catalytic domain of factor XIa: mechanism of heparin acceleration of factor XIa inhibition by the serpins antithrombin and C1-inhibitor. Biochemistry. 2009;48:1517–1524. doi: 10.1021/bi802298r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Choi SH, Smith SA, Morrissey JH. Polyphosphate is a cofactor for the activation of factor XI by thrombin. Blood. 2011;118:6963–6970. doi: 10.1182/blood-2011-07-368811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Geng Y, Verhamme IM, Smith SA, Cheng Q, Sun M, Sheehan JP, Morrissey JH, Gailani D. Factor XI anion-binding sites are required for productive interactions with polyphosphate. J Thromb Haemost. 2013;11:2020–2028. doi: 10.1111/jth.12414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Morrissey JH, Smith SA. Polyphosphate as modulator of hemostasis, thrombosis, and inflammation. J Thromb Haemost. 2015;13(Suppl 1):S92–97. doi: 10.1111/jth.12896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ivanov I, Shakhawat R, Sun MF, Dickeson SK, Puy C, McCarty OJ, Gruber A, Matafonov A, Gailani D. Nucleic acids as cofactors for factor XI and prekallikrein activation: Different roles for high-molecular-weight kininogen. Thromb Haemost. 2017;117:671–681. doi: 10.1160/TH16-09-0691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ivanov I, Matafonov A, Sun MF, Cheng Q, Dickeson SK, Verhamme IM, Emsley J, Gailani D. Proteolytic properties of single-chain factor XII: a mechanism for triggering contact activation. Blood. 2017;129:1527–1537. doi: 10.1182/blood-2016-10-744110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Naudin C, Burillo E, Blankenberg S, Butler L, Renné T. Factor XII contact activation. Semin Thromb Hemost. doi: 10.1055/s-0036-1598003. (in press) [DOI] [PubMed] [Google Scholar]
- 71.Müller F, Mutch NJ, Schenk WA, Smith SA, Esterl L, Spronk HM, Schmidbauer S, Gahl WA, Morrissey JH, Renné T. Platelet polyphosphates are proinflammatory and procoagulant mediators in vivo. Cell. 2009;139:1143–1156. doi: 10.1016/j.cell.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mandle RJ, Colman RW, Kaplan AP. Identification of prekallikrein and high-molecular-weight kininogen as a complex in human plasma. Proc Natl Acad Sci U S A. 1976;73:4179–4183. doi: 10.1073/pnas.73.11.4179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sainz IM, Pixley RA, Colman RW. Fifty years of research on the plasma kallikrein-kinin system: from protein structure and function to cell biology and in-vivo pathophysiology. Thromb Haemost. 2007;98:77–83. [PubMed] [Google Scholar]
- 74.Tait JF, Fujikawa K. Primary structure requirements for the binding of human high molecular weight kininogen to plasma prekallikrein and factor XI. J Biol Chem. 1987;262:11651–11656. [PubMed] [Google Scholar]
- 75.Wiggins RC, Bouma BN, Cochrane CG, Griffin JH. Role of high-molecular-weight kininogen in surface-binding and activation of coagulation Factor XI and prekallikrein. Proc Natl Acad Sci U S A. 1977;74:4636–4640. doi: 10.1073/pnas.74.10.4636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.van Iwaarden F, Bouma BN. Role of high molecular weight kininogen in contact activation. Semin Thromb Hemost. 1987;13:15–24. doi: 10.1055/s-2007-1003472. [DOI] [PubMed] [Google Scholar]
- 77.Ho DH, Baglia FA, Walsh PN. Factor XI binding to activated platelets is mediated by residues R(250), K(255), F(260), and Q(263) within the apple 3 domain. Biochemistry. 2000;39:316–323. doi: 10.1021/bi991851q. [DOI] [PubMed] [Google Scholar]
- 78.Baglia FA, Gailani D, López JA, Walsh PN. Identification of a binding site for glycoprotein Ibalpha in the Apple 3 domain of factor XI. J Biol Chem. 2004;279:45470–45476. doi: 10.1074/jbc.M406727200. [DOI] [PubMed] [Google Scholar]
- 79.Baglia FA, Shrimpton CN, Emsley J, Kitagawa K, Ruggeri ZM, López JA, Walsh PN. Factor XI interacts with the leucine-rich repeats of glycoprotein Ibalpha on the activated platelet. J Biol Chem. 2004;279:49323–49329. doi: 10.1074/jbc.M407889200. [DOI] [PubMed] [Google Scholar]
- 80.Baglia FA, Walsh PN. Prothrombin is a cofactor for the binding of factor XI to the platelet surface and for platelet-mediated factor XI activation by thrombin. Biochemistry. 1998;37:2271–2281. doi: 10.1021/bi972113+. [DOI] [PubMed] [Google Scholar]
- 81.Shariat-Madar Z, Mahdi F, Schmaier AH. Factor XI assembly and activation on human umbilical vein endothelial cells in culture. Thromb Haemost. 2001;85:544–551. [PubMed] [Google Scholar]
- 82.Renné T, Gailani D, Meijers JC, Müller-Esterl W. Characterization of the H-kininogen-binding site on factor XI: a comparison of factor XI and plasma prekallikrein. J Biol Chem. 2002;277:4892–4899. doi: 10.1074/jbc.M105221200. [DOI] [PubMed] [Google Scholar]
- 83.Renné T, Sugiyama A, Gailani D, Jahnen-Dechent W, Walter U, Müller-Esterl W. Fine mapping of the H-kininogen binding site in plasma prekallikrein apple domain 2. Int Immunopharmacol. 2002;2:1867–1873. doi: 10.1016/s1567-5769(02)00170-4. [DOI] [PubMed] [Google Scholar]
- 84.Wong SS, Østergaard S, Hall G, Li C, Williams PM, Stennicke H, Emsley J. A novel DFP tripeptide motif interacts with the coagulation factor XI apple 2 domain. Blood. 2016;127:2915–2923. doi: 10.1182/blood-2015-10-676122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Weisel JW, Nagaswami C, Woodhead JL, DeLa Cadena RA, Page JD, Colman RW. The shape of high molecular weight kininogen. Organization into structural domains, changes with activation, and interactions with prekallikrein, as determined by electron microscopy. J Biol Chem. 1994;269:10100–10106. [PubMed] [Google Scholar]
- 86.Higashiyama S, Ohkubo I, Ishiguro H, Kunimatsu M, Sawaki K, Sasaki M. Human high molecular weight kininogen as a thiol proteinase inhibitor: presence of the entire inhibition capacity in the native form of heavy chain. Biochemistry. 1986;25:1669–1675. doi: 10.1021/bi00355a034. [DOI] [PubMed] [Google Scholar]
- 87.Baba SP, Zehra S, Bano B. Purification and characterization of kininogens from sheep plasma. Protein J. 2005;24:95–102. doi: 10.1007/s10930-004-1516-6. [DOI] [PubMed] [Google Scholar]
- 88.Schaff M, Tang C, Maurer E, Bourdon C, Receveur N, Eckly A, Hechler B, Arnold C, de Arcangelis A, Nieswandt B, Denis CV, Lefebvre O, Georges-Labouesse E, Gachet C, Lanza F, Mangin PH. Integrin α6β1 is the main receptor for vascular laminins and plays a role in platelet adhesion, activation, and arterial thrombosis. Circulation. 2013;128:541–552. doi: 10.1161/CIRCULATIONAHA.112.000799. [DOI] [PubMed] [Google Scholar]
- 89.White-Adams TC, Berny MA, Patel IA, Tucker EI, Gailani D, Gruber A, McCarty OJ. Laminin promotes coagulation and thrombus formation in a factor XII-dependent manner. J Thromb Haemost. 2010;8:1295–1301. doi: 10.1111/j.1538-7836.2010.03850.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Schousboe I. Endothelial cells express a matrix protein which binds activated factor XII in a zinc-independent manner. Thromb Haemost. 2006;95:312–319. doi: 10.1160/TH05-06-0458. [DOI] [PubMed] [Google Scholar]
- 91.Schousboe I, Nystrøm B. High molecular weight kininogen binds to laminin--characterization and kinetic analysis. FEBS J. 2009;276:5228–5238. doi: 10.1111/j.1742-4658.2009.07218.x. [DOI] [PubMed] [Google Scholar]
- 92.Wolberg AS, Stafford DW, Erie DA. Human factor IX binds to specific sites on the collagenous domain of collagen IV. J Biol Chem. 1997;272:16717–16720. doi: 10.1074/jbc.272.27.16717. [DOI] [PubMed] [Google Scholar]
- 93.Gui T, Reheman A, Ni H, Gross PL, Yin F, Monroe D, Monahan PE, Stafford DW. Abnormal hemostasis in a knock-in mouse carrying a variant of factor IX with impaired binding to collagen type IV. J Thromb Haemost. 2009;7:1843–1851. doi: 10.1111/j.1538-7836.2009.03545.x. [DOI] [PubMed] [Google Scholar]
- 94.Naito K, Fujikawa K. Activation of human blood coagulation factor XI independent of factor XII: factor XI is activated by thrombin and factor XIa in the presence of negatively charged surfaces. J Biol Chem. 1991;266:7353–7358. [PubMed] [Google Scholar]
- 95.Gailani D, Broze GJ., Jr Factor XI activation in a revised model of blood coagulation. Science. 1991;253:909–912. doi: 10.1126/science.1652157. [DOI] [PubMed] [Google Scholar]
- 96.von dem Borne PA, Mosnier LO, Tans G, Meijers JC, Bouma BN. Factor XI activation by meizothrombin: stimulation by phospholipid vesicles containing both phosphatidylserine and phosphatidylethanolamine. Thromb Haemost. 1997;78:834–839. [PubMed] [Google Scholar]
- 97.Matafonov A, Sarilla S, Sun MF, Sheehan JP, Serebrov V, Verhamme IM, Gailani D. Activation of factor XI by products of prothrombin activation. Blood. 2011;118:437–445. doi: 10.1182/blood-2010-10-312983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kleinschnitz C, Stoll G, Bendszus M, Schuh K, Pauer HU, Burfeind P, Renné C, Gailani D, Nieswandt B, Renné T. Targeting coagulation factor XII provides protection from pathological thrombosis in cerebral ischemia without interfering with hemostasis. J Exp Med. 2006;203:513–518. doi: 10.1084/jem.20052458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Matafonov A, Leung PY, Gailani AE, Grach SL, Puy C, Cheng Q, Sun MF, McCarty OJ, Tucker EI, Kataoka H, Renné T, Morrissey JH, Gruber A, Gailani D. Factor XII inhibition reduces thrombus formation in a primate thrombosis model. Blood. 2014;123:1739–1746. doi: 10.1182/blood-2013-04-499111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Baglia FA, Jameson BA, Walsh PN. Identification and characterization of a binding site for factor XIIa in the Apple 4 domain of coagulation factor XI. J Biol Chem. 1993;268:3838–3844. [PubMed] [Google Scholar]
- 101.Spronk HMH, Wilhelm S, Heemskerk H, et al. Feedback activation of factor XI by thrombin is essential for haemostasis in vivo. J Thromb Haemost. 2009;7(suppl 2) Abstract PL-TU-003. [Google Scholar]
- 102.Jamsa A, Spronk HMH, Gailani D, Mackman N, Renne T. A critical role of factor XI feedback activation for placental hemostasis. RPTH. 2017;1(Suppl. 1):108. [Google Scholar]
- 103.Gould TJ, Vu TT, Swystun LL, Dwivedi DJ, Mai SH, Weitz JI, Liaw PC. Neutrophil extracellular traps promote thrombin generation through platelet-dependent and platelet-independent mechanisms. Arterioscler Thromb Vasc Biol. 2014;34:1977–1984. doi: 10.1161/ATVBAHA.114.304114. [DOI] [PubMed] [Google Scholar]
- 104.Vu TT, Leslie BA, Stafford AR, Zhou J, Fredenburgh JC, Weitz JI. Histidine-rich glycoprotein binds DNA and RNA and attenuates their capacity to activate the intrinsic coagulation pathway. Thromb Haemost. 2016;115:89–98. doi: 10.1160/TH15-04-0336. [DOI] [PubMed] [Google Scholar]
- 105.Noubouossie DF, Whelihan MF, Yu YB, Sparkenbaugh E, Pawlinski R, Monroe DM, Key NS. In vitro activation of coagulation by human neutrophil DNA and histone proteins but not neutrophil extracellular traps. Blood. 2017;129:1021–1029. doi: 10.1182/blood-2016-06-722298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Gailani D, Broze GJ., Jr Effects of glycosaminoglycans on factor XI activation by thrombin. Blood Coagul Fibrinolysis. 1993;4:15–20. [PubMed] [Google Scholar]
- 107.von dem Borne PA, Meijers JC, Bouma BN. Effect of heparin on the activation of factor XI by fibrin-bound thrombin. Thromb Haemost. 1996;76:347–353. [PubMed] [Google Scholar]
- 108.Baglia FA, Walsh PN. A binding site for thrombin in the apple 1 domain of factor XI. J Biol Chem. 1996;271:3652–3658. doi: 10.1074/jbc.271.7.3652. [DOI] [PubMed] [Google Scholar]
- 109.Kravtsov DV, Matafonov A, Tucker EI, et al. Factor XI contributes to thrombin generation in the absence of factor XII. Blood. 2009;114:452–458. doi: 10.1182/blood-2009-02-203604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Lane DA, Philippou H, Huntington JA. Directing thrombin. Blood. 2005 Oct 15;106(8):2605–12. doi: 10.1182/blood-2005-04-1710. [DOI] [PubMed] [Google Scholar]
- 111.Pathak M, Wilmann P, Awford J, Li C, Hamad BK, Fischer PM, Dreveny I, Dekker LV, Emsley J. Coagulation factor XII protease domain crystal structure. J Thromb Haemost. 2015;13:580–591. doi: 10.1111/jth.12849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Hamad BK, Pathak M, Manna R, Fischer PM, Emsley J, Dekker LV. Assessment of the protein interaction between coagulation factor XII and corn trypsin inhibitor by molecular docking and biochemical validation. J Thromb Haemost. 2017;15:1818–1828. doi: 10.1111/jth.13773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Vadivel K, Schmidt AE, Marder VJ, Krishnaswamy S, Bajaj SP. Hemostasis and Thrombosis: basic principles and clinical practice. 6th. Philadelphia: Lippincott, Williams & Wilkins; 2012. Structure and function of vitamin K-dependent coagulation and anticoagulation proteins; pp. 208–232. [Google Scholar]
- 114.Vadivel K, Bajaj SP. Structural biology of factor VIIa/tissue factor initiated coagulation. Front Biosci (Landmark Ed) 2012;17:2476–2494. doi: 10.2741/4066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Wolberg AS, Morris DP, Stafford DW. Factor IX activation by factor XIa proceeds without release of a free intermediate. Biochemistry. 1997;36:4074–4079. doi: 10.1021/bi962274y. [DOI] [PubMed] [Google Scholar]
- 116.Geng Y, Verhamme IM, Messer A, Sun MF, Smith SB, Bajaj SP, et al. A sequential mechanism for exosite-mediated factor IX activation by factor XIa. J Biol Chem. 2012;287:38200–38209. doi: 10.1074/jbc.M112.376343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Geng Y, Verhamme IM, Sun MF, Bajaj SP, Emsley J, Gailani D. Analysis of the factor XI variant Arg184Gly suggests a structural basis for factor IX binding to factor XIa. J Thromb Haemost. 2013;11:1374–1384. doi: 10.1111/jth.12275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Sun Y, Gailani D. Identification of a factor IX binding site on the third apple domain of activated factor XI. J Biol Chem. 1996;271:29023–29028. doi: 10.1074/jbc.271.46.29023. [DOI] [PubMed] [Google Scholar]
- 119.Sun MF, Zhao M, Gailani D. Identification of amino acids in the factor XI apple 3 domain required for activation of factor IX. J Biol Chem. 1999;274:36373–36378. doi: 10.1074/jbc.274.51.36373. [DOI] [PubMed] [Google Scholar]
- 120.Ogawa T, Verhamme IM, Sun MF, Bock PE, Gailani D. Exosite-mediated substrate recognition of factor IX by factor XIa. The factor XIa heavy chain is required for initial recognition of factor IX. J Biol Chem. 2005;280:23523–23530. doi: 10.1074/jbc.M500894200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Aktimur A, Gabriel MA, Gailani D, Toomey JR. The factor IX gamma-carboxyglutamic acid (Gla) domain is involved in interactions between factor IX and factor XIa. J Biol Chem. 2003;278:7981–7987. doi: 10.1074/jbc.M212748200. [DOI] [PubMed] [Google Scholar]
- 122.Matafonov A, Cheng Q, Geng Y, Verhamme IM, Umunakwe O, Tucker EI, Sun MF, Serebrov V, Gruber A, Gailani D. Evidence for factor IX-independent roles for factor XIa in blood coagulation. J Thromb Haemost. 2013;11:2118–2127. doi: 10.1111/jth.12435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Whelihan MF, Orfeo T, Gissel MT, Mann KG. Coagulation procofactor activation by factor XIa. J Thromb Haemost. 2010;8:1532–1539. doi: 10.1111/j.1538-7836.2010.03899.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Choi SH, Smith SA, Morrissey JH. Polyphosphate accelerates factor V activation by factor XIa. Thromb Haemost. 2015;113:599–604. doi: 10.1160/TH14-06-0515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Griffin JH. Role of surface in surface-dependent activation of Hageman factor (blood coagulation factor XII) Proc Natl Acad Sci USA. 1978;75:1998–2002. doi: 10.1073/pnas.75.4.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Scott CF, Silver LD, Purdon AD, Colman RW. Cleavage of human high molecular weight kininogen by factor XIa in vitro. Effect on structure and function. J Biol Chem. 1985;260:10856–10863. [PubMed] [Google Scholar]
- 127.Puy C, Tucker EI, Matafonov A, Cheng Q, Zientek KD, Gailani D, Gruber A, McCarty OJ. Activated factor XI increases the procoagulant activity of the extrinsic pathway by inactivating tissue factor pathway inhibitor. Blood. 2015;125:1488–1496. doi: 10.1182/blood-2014-10-604587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Puy C, Tucker EI, Ivanov IS, Gailani D, Smith SA, Morrissey JH, Gruber A, McCarty OJ. Platelet-Derived Short-Chain Polyphosphates Enhance the Inactivation of Tissue Factor Pathway Inhibitor by Activated Coagulation Factor XI. PLoS One. 2016;11:e0165172. doi: 10.1371/journal.pone.0165172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Puy C, Garland KS, Shirai T, Reitsma SE, Zilberman-Rudenko J, Gruber A, McCarty OJT. Activated FXI regulates the catalytic activity of ADAMTS13 by removing the CUB domains. RPTH. 2017;1(Suppl. 1):98. [Google Scholar]
- 130.Tucker EI, Marzec UM, White TC, Hurst S, Rugonyi S, McCarty OJ, Gailani D, Gruber A, Hanson SR. Prevention of vascular graft occlusion and thrombus-associated thrombin generation by inhibition of factor XI. Blood. 2009;113:936–944. doi: 10.1182/blood-2008-06-163675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Zilberman-Rudenko J, Itakura A, Wiesenekker CP, Vetter R, Maas C, Gailani D, Tucker EI, Gruber A, Gerdes C, McCarty OJ. Coagulation Factor XI Promotes Distal Platelet Activation and Single Platelet Consumption in the Bloodstream Under Shear Flow. Arterioscler Thromb Vasc Biol. 2016;36:510–517. doi: 10.1161/ATVBAHA.115.307034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Zhu S, Travers RJ, Morrissey JH, Diamond SL. FXIa and platelet polyphosphate as therapeutic targets during human blood clotting on collagen/tissue factor surfaces under flow. Blood. 2015;126:1494–1502. doi: 10.1182/blood-2015-04-641472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Gruber A, Hanson SR. Factor XI-dependence of surface- and tissue factor-initiated thrombus propagation in primates. Blood. 2003;102:953–955. doi: 10.1182/blood-2003-01-0324. [DOI] [PubMed] [Google Scholar]
- 134.Greengard JS, Heeb MJ, Ersdal E, Walsh PN, Griffin JH. Binding of coagulation factor XI to washed human platelets. Biochemistry. 1986;25:3884–3890. doi: 10.1021/bi00361a022. [DOI] [PubMed] [Google Scholar]
- 135.Ho DH, Baglia FA, Walsh PN. Factor XI binding to activated platelets is mediated by residues R(250), K(255), F(260), and Q(263) within the apple 3 domain. Biochemistry. 2000;39:316–323. doi: 10.1021/bi991851q. [DOI] [PubMed] [Google Scholar]
- 136.Kossmann S, Lagrange J, Jäckel S, Jurk K, Ehlken M, Schönfelder T, Weihert Y, Knorr M, Brandt M, Xia N, Li H, Daiber A, Oelze M, Reinhardt C, Lackner K, Gruber A, Monia B, Karbach SH, Walter U, Ruggeri ZM, Renné T, Ruf W, Münzel T, Wenzel P. Platelet-localized FXI promotes a vascular coagulation-inflammatory circuit in arterial hypertension. Sci Transl Med. 2017;9 doi: 10.1126/scitranslmed.aah4923. pii:eaah4923. [DOI] [PubMed] [Google Scholar]
- 137.Baglia FA, Gailani D, López JA, Walsh PN. Identification of a binding site for glycoprotein Ibalpha in the Apple 3 domain of factor XI. J Biol Chem. 2004;279:45470–45476. doi: 10.1074/jbc.M406727200. [DOI] [PubMed] [Google Scholar]
- 138.Baglia FA, Shrimpton CN, Emsley J, Kitagawa K, Ruggeri ZM, López JA, Walsh PN. Factor XI interacts with the leucine-rich repeats of glycoprotein Ibalpha on the activated platelet. J Biol Chem. 2004;279:49323–49329. doi: 10.1074/jbc.M407889200. [DOI] [PubMed] [Google Scholar]
- 139.White-Adams TC, Berny MA, Tucker EI, Gertz JM, Gailani D, Urbanus RT, de Groot PG, Gruber A, McCarty OJ. Identification of coagulation factor XI as a ligand for platelet apolipoprotein E receptor 2 (ApoER2) Arterioscler Thromb Vasc Biol. 2009;29:1602–1607. doi: 10.1161/ATVBAHA.109.187393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Walsh PN. Prothrombin is a cofactor for the binding of factor XI to the platelet surface and for platelet-mediated factor-XI activation by thrombin. Biochemistry. 2007;46:12886–12887. doi: 10.1021/bi701501k. [DOI] [PubMed] [Google Scholar]
- 141.Verhoef JJ, Barendrecht AD, Nickel KF, Dijkxhoorn K, Kenne E, Labberton L, McCarty OJ, Schiffelers R, Heijnen HF, Hendrickx AP, Schellekens H, Fens MH, de Maat S, Renné T, Maas C. Polyphosphate nanoparticles on the platelet surface trigger contact system activation. Blood. 2017;129:1707–1717. doi: 10.1182/blood-2016-08-734988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Huang M, Furie BC, Furie B. Crystal structure for the calcium-stabilized human factor IX Gla-domain bound to a conformation-specific anti-factor IX antibody. J Biol Chem. 2004;279:14338–14346. doi: 10.1074/jbc.M314011200. [DOI] [PubMed] [Google Scholar]