

SYNOPSIS
Primary cutaneous CD30+ lymphoproliferative disorders encompass lymphomatoid papulosis (LyP), primary cutaneous anaplastic large cell lymphoma (pcALCL) and indeterminate cases. LyP is a benign disorder characterized by recurrent crops of several to hundreds of papulonodules, red or violaceous in color and measuring up to 20mm, usually on the trunk and extremities. Patients with LyP are at increased risk of a secondary malignancy that may be diagnosed before, during or after the diagnosis of LyP and thus should receive ongoing surveillance. pcALCL is characterized by a solitary red to violaceous nodule or tumor greater than 20mm and may occur anywhere on the body. Secondary cutaneous ALCL must be excluded for any patient initially presenting with a cutaneous lesion of ALCL. LyP is benign, limited to the skin and self-resolves with a 5-year survival rate of 100%; pcALCL is usually limited to the skin and responsive to directed therapies, with a 5-year survival of over 95%. Aggressive systemic or multi-agent chemotherapeutic regimens should be avoided.
Keywords: CD30+, Cutaneous lymphoproliferative disorders, Lymphomatoid papulosis, Primary cutaneous anaplastic large cell lymphoma, Secondary cutaneous anaplastic large cell lymphoma
Introduction
Cluster of differentiation 30 (CD30), a 120 kDa type I transmembrane glycoprotein of the tumor necrosis factor receptor superfamily member 8 (TNFRSF8) gene and previously known as Ki-1 antigen, is a cell surface cytokine receptor present on activated T- and B-cells. Upon T-cell activation, CD28 and other co-stimulatory receptors, including CD30 are upregulated. CD30 expression requires CD28 or IL-4 receptor (IL-4R) signalling and, when CD30 is activated, downstream signalling augments T-cell proliferation at low levels and regulates T-cell survival.1,2
CD30 interacts with CD30 ligand (CD30L, CD153, TNFSF8), a 40-kDa type II membrane-associated glycoprotein belonging to the TNF family1,2, that is expressed on activated T-cells, primarily CD4 T-cells of both Th1 and Th2 phenotype, as well as on a subset of accessory cells1,3,4 and B cells5,6. CD30 signalling ultimately leads to nuclear factor (NF)-kB activation through both TNFR-associated factor (TRAF) 2-dependent and TRAF2-independent pathways that can inhibit effector cell activity, promote apoptosis or promote survival depending on the cell type and different intracellular signaling pathways activated.7,8,9 For example, ligation of CD30 signals can downregulate the cytotoxic effector molecules Fas ligand, perforin, granzyme B and inhibit cytotoxicity.4 CD30 signalling also promotes apoptosis by strongly inhibiting the expression of the oncogene c-myc and upregulating Fas (TNFRSF6), death receptor 3 (TNFRSF25) and TNF-related apoptosis-inducing ligand (TNFSF10, TRAIL).10 In addition to increasing the cell’s susceptibility to apoptosis, CD30 signalling strongly upregulates chemokine receptor 7 (CCR7), a homing molecule that enhances the cell’s ability to home to lymphoid organs. A variety of causes have been implicated in the induction of CD30 expression in human cutaneous reactive and neoplastic lymphocytic processes including infectious, exogenous, inflammatory and lymphoproliferative disorders (Table 1).
TABLE 1.
CD30+ Lymphocytic Disorders
| Neoplastic | Lymphomatoid papulosis (LyP) Primary cutaneous anaplastic large cell lymphoma (pcALCL) br1>Systemic cutaneous anaplastic large cell lymphoma (scALCL) Mycosis fungoides or Sezary syndrome with CD30+ large cell transformation Cutaneous Hodgkin’s disease CD30+ large B-cell lymphoma Epstein Barr virus (EBV)+ hydroa vacciniforme-like T-cell lymphoma Human T-cell Lymphotropic virus type 1 (HTLV-1) associated adult T-cell lymphoma/leukemia Eruptive keratoacantomas |
| Exogeneous | Drug-induced reactive lymphoid hyperplasia44,45,46,47,48,49, Insect bite reaction50 Scabies infestation51 |
| Infection associated | EBV52 HTLV-1/2 Mycobacteria53 Herpes simplex virus (HSV)50 Human immunodeficiency virus54 Other infections: leishmaniasis, syphilis, varicella zoster virus, molluscum contagiosum virus and parapox virusError! Bookmark not defined. |
| Inflammatory conditions | Pityriasis lichenoides
Atopic dermatitis56 |
Adapted from: LeBoeuf NR, McDermott S, Harris NL. Case records of the Massachusetts General Hospital. Case 5–2015. A 69-year-old woman with recurrent skin lesions after treatment for lymphoma. N Engl J Med. 2015 Feb 12;372(7):650–9, with permission.
CD30 is expressed in a subset of B-cell lymphoid malignancies (approximately 20%) and T-cell lymphoid malignancies (30%), including the most common CD30+ lymphoid malignancies, Hodgkin’s lymphoma and systemic anaplastic large cell lymphoma.11,12,13 However, there is a subset of non-Hodgkin’s lymphoma where the disease process is contained within the skin with no other evidence of blood and lymph node involvement. This article will focus on the diagnosis, clinical presentation and management of these primary cutaneous CD30 lymphoproliferative disorders.
Primary cutaneous CD30+ lymphoproliferative disorders (LPD) comprise a spectrum of conditions with similar histologic and molecular features, but different clinical presentations. According to the World Health Organization (WHO) and European Organization for Research and Treatment (EORTC) classification, this group accounts for 20% of all cutaneous lymphomas, second most common cutaneous T-cell lymphoma (CTCL) behind mycosis fungoides.14 The primary cutaneous CD30+ LPD include lymphomatoid papulosis (LyP), primary cutaneous anaplastic large cell lymphoma (pcALCL) and borderline or indeterminate cases.
pcALCL and LyP can be thought of as a clinical spectrum where indeterminate cases may have clinical and histological features of either (Figure 1). As will be discussed, patients with LyP may have a co-existing secondary lymphoma, including pcALCL and therefore both conditions may exist simultaneously in an individual patient. Indeterminate cases tend to eventually develop clinical features of either LyP or pcALCL over time. Historical diagnoses that may capture the full spectrum of CD30 LPD include regressing atypical histiocytosis or indolent primary cutaneous Hodgkin lymphoma.15
Figure 1.

Primary cutaneous CD30+ LPD clinical spectrum
Lymphomatoid papulosis (LyP)
Epidemiology
Macaulay first described lymphomatoid papulosis in 1968, aptly referring to the recurring self-healing eruption as clinically benign but histologically malignant.16 LyP is the most common CD30+ LPD, more common than pcALCL. Best estimates suggest that there are approximately 1.2 to 1.9 cases per million persons of LyP in the US.17 It presents in men more often than women with an average age of onset of 35 to 45 years18. (Table 2). The etiology of LyP is not known although hypotheses implicating reactive phenomena inducing over expression of CD30 have been proposed.
TABLE 2.
Epidemiology of LyP and pcALCL
| LyP | pcALCL | |
|---|---|---|
| % of primary cutaneous lymphomas | 12% | 8% |
| 5-year survival | 100% | >95% |
| Incidence | 1.2–1.9 cases per 1,000,000 | Unknown |
| M:F Ratio | 1.4:1 | 3:1 |
| Median Age of Diagnosis (years) | 45.5 | 60 |
| Age range (years) | 4–88 | 16–89 |
Prognosis
The 5-year survival of patients with LyP is close to 100%, despite the increased risk of secondary malignancy that may be diagnosed before, during or after the diagnosis of LyP.14 This secondary malignancy risk can affect 5–30% of LyP patients19,20,21,22 although, several larger retrospective series indicated that the incidence of a secondary malignancy may actually be closer to 40–60%.23,24 Mycosis fungoides (MF) and ALCL are the first and second most commonly associated malignancies respectively, accounting for over 90% of secondary malignancies in one series.24 The majority of the secondary cases of MF are early stage, IA or IB. Other secondary hematologic malignancies that have been reported to occur with LyP are listed in Table 3. Male sex and advanced age are risk factors associated with a higher risk of a secondary LPD; additional factors are noted in Table 4.
TABLE 3.
Secondary malignancies associated with LyP
| Most common (>90% of cases) | Mycosis fungoides, stage IA or IB > later stage Anaplastic large cell lymphoma, primary cutaneous > nodal Hodgkin’s disease |
| Rare reports (<10% of cases) | Chronic lymphocytic leukemia Acute myeloid leukemia B-cell lymphoma T-cell large granular lymphocytic leukemia Multiple myeloma Myelodysplastic syndrome |
Data from de Souza A, el-Azhary RA, Camilleri MJ, Wada DA, Appert DL, Gibson LE. In search of prognostic indicators for lymphomatoid papulosis: a retrospective study of 123 patients. J Am Acad Dermatol. 2012 Jun;66(6):928–37 and Wieser I, Tetzlaff MT, Torres Cabala CA, Duvic M. Primary cutaneous CD30(+) lymphoproliferative disorders. J Dtsch Dermatol Ges. 2016 Aug;14(8):767–82.
TABLE 4.
Risk factors for developing a secondary malignancy with LyP
Presentation
Clinically, LyP is characterized by recurrent crops of red to violaceous papules and nodules measuring up to 20mm but typically 3–10mm in diameter (Table 5). Patients often present with lesions in various stages due to the recurrent and successive crops of papules and nodules, with hyperpigmented macules and varioliform scars in the background (Figure 2). The number of lesions can range from a few to hundreds at a time. Lesions typically are generalized with the majority on the trunk and extremities. Localized presentations of crops within regional areas or in agminated plaques have also been described.25,26 Approximately half of all patients are asymptomatic while others experience pruritus and/or pain secondary to ulceration, crusting and central necrosis.27 LyP is not associated with systemic symptoms. The resolving lesions often display post-inflammatory hypo- or hyperpigmented macules. Necrotic lesions may leave varioliform scars, usually smaller than the original papules. Typically, lesions spontaneously resolve within 1–4 months28, most often between 2 and 8 weeks. The self-healing nature of LyP is the critical clinical pearl required for making the diagnosis, particularly in the face of a concerning pathology report. It is hypothesized that the feature of resolution may be associated with CD30L expression on the neoplastic cells, causing the CD30 expressing cells to undergo apoptosis either through CD30-CD30L inhibition of neoplastic cell growth and/or increased sensitivity of the neoplastic cells to Fas-FasL mediated apoptosis.8 Patients with LyP may develop recurrent crops over several months or for decades, with cases reported to last over 40 years.18
TABLE 5.
Clinical features of LyP and pcALCL
| LyP | pcALCL | |
|---|---|---|
| Size | 5–10mm | >20mm |
| Number of lesions | Several to hundreds | Solitary or localized |
| Distribution | Trunk and limbs | Anywhere |
| Duration of lesions | 3–8 weeks | >12 weeks |
| Self-resolving | 100% | 28% (0–44%) |
| Extracutaneous disease | 0% | 13% (0–24%) |
Figure 2.
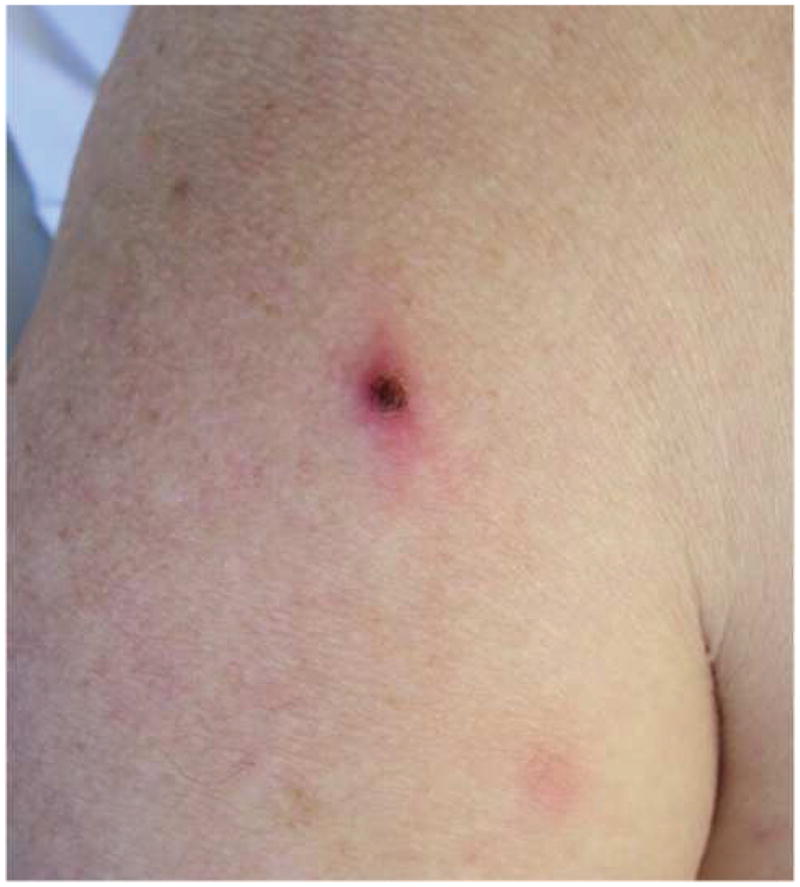

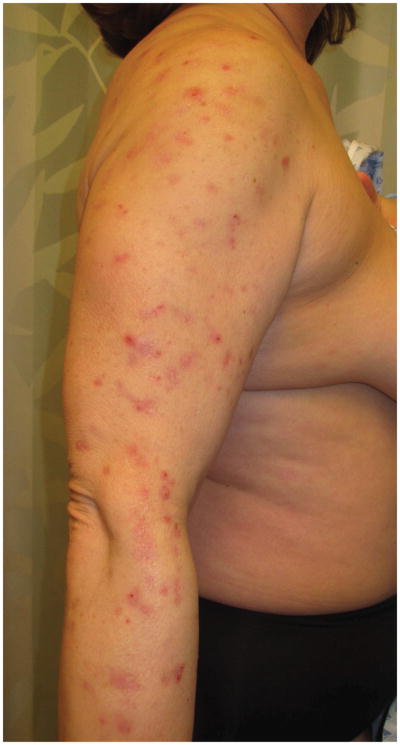

- 2A – Typical lesion of LyP: 6mm violaceous papule with necrotic center
- 2B, C – Crops of LyP in various stages of evolution
- 2D – Inflamed lesion of LyP, with a surrounding crop of more typical lesions
Differential diagnosis
With histologic evidence of a CD30+ infiltrate, all entities in Table 1 should be considered. Coupled with a clinical presentation of recurrent self-resolving crops of papulonodules, the main differential diagnoses include:
Pityriasis lichenoides
Borderline CD30 LPD
Cutaneous anaplastic large cell lymphoma
-
Reactive lymphoid hyperplasia (lymphocytoma cutis) secondary to:
Arthropod assault
Scabies
Medications
Herpes simplex virus
Varicella zoster virus
Pathology
When considering LyP, skin biopsy is recommended for histologic and immunohistochemical (IHC) evaluation to classify the cellular subtypes and rule out infectious entities. DNA should be sent for T-cell gene rearrangement polymerase chain reaction (PCR) to assess for clonality.
There are currently 5 generally accepted histologic subtypes of LyP (A-E), as well as a recently proposed 6th subtype (F) (Table 6). CD30+ T-cell lymphocytes are the hallmark of all histologic types of LyP, although type B has variable positivity, reported to range from 0 to 77% of the infiltrate.29 Subtypes may occur concurrently within the same biopsy or within different specimens taken from the same patient. Other rare pathological variants include γ/δ-type30 and LyP with 6p25.3 re-arrangement-type31. The significance of the γ/δ-variant is unknown. The 6p25.3-type is described as biphasic with small-medium lymphocytes in the epidermis and larger pleomorphic lymphocytes in the dermis. Clinically, this variant presents in older individuals (mean 75 years) and has a higher male predominance (3:1 M:F ratio). Otherwise, the course follows the same natural history of all other variants of LyP.
Table 6.
Histological morphologies of LyP
| Type | % of cases | Histologically Mimicks | Description |
|---|---|---|---|
| A | 47–82% | Hodgkin lymphoma Transformed MF |
• Large Reed-Sternberg-like atypical lymphocytes • Wedge-shaped heterogeneous infiltrate with lymphocytes, neutrophils, eosinophils and histiocytes |
| B | 4–17% | MF | • Epidermotropic band-like infiltrate • Small irregular lymphocytes • Cerebriform nuclei |
| C | 7–22% | ALCL | • Sheets or clustered infiltrate • Large atypical lymphocytes • Few inflammatory cells |
| D | ~8% | Primary cutaneous aggressive CD8+ cytotoxic T-cell lymphoma (TCL), PLC/PLEVA Pagetoid reticulosis Cutaneous gamma/delta TCL |
• Epidermotrophic infiltrate • CD8+ • Small to medium atypical lymphocytes |
| E | ~0.6% | Angiocentric: Extranodal NK/T-cell lymphoma, nasal type Cutaneous gamma/delta TCL ALCL variant with angiocentric and/or angiodestructive growth |
• Small- to medium-sized lymphocytes • Angiocentric: CD8+ infiltrating walls of small to medium-sized vessels • Vasculitis: fibrin, thromboses and extravasation of red blood cells |
| F58 | 5–10% | Folliculotropic: Folliculotropic MF Pseudolymphoma Connective tissue diseases |
• Perifollicular infiltrate • Medium to large lymphoid cells • Follicular mucinosis • Neutrophils within infundibula |
| Mixed | 4–9% | • More than 1 histological type in the same patient or lesion |
The clinical significance of the histological subtypes remains unclear, with rare exceptions. As noted, type B can be CD30 negative and histologically resemble MF. In this setting, the clinical morphology and behaviour is then required to distinguish the two and render a diagnosis of LyP. Additionally, subtypes B and C have been shown to be associated with a higher risk of secondary malignancy.24
Immunohistochemistry is required to characterize the infiltrate (Table 7). The majority of LyP cases are CD4+ and CD45RO+; however, type D, type E and LyP in children are CD4− CD8+.29 CD45RO helps to differentiate LyP type D from aggressive epidermotropic CD8+ CTCL with the former being CD45RO+ and the latter being CD45RO-.14
Table 7.
Immunohistochemical profile of LyP and ALCL
| LyP | pcALCL | scALCL | |
|---|---|---|---|
| Clonality | 40–100% | >90% | ~90% |
| CD30 | + (type B, variably) | >75% + (required for diagnosis) | + |
| CD56 | ~10% | 12–75% | + (worst prognosis) |
| Bcl-2 | − | 30% | + |
| Cytotoxic molecules: TIA-1, Granzyme B or Perforin | + | ~50% | + |
| ALK | − | Rare | 50% |
| CLA | + | Variable | − |
| EMA | − | − | + |
| t(2;5)(pq23;q35) translocation | − | <10% | 70–75% |
Data from references 30, 59, and 60
T-cell receptor (TCR) gene rearrangement demonstrates clonality in 40–100% of cases of LyP, despite its benignity.29 The significance of the clonality in the risk of developing a secondary malignancy remains to be determined. The majority of cases have α/β T-cell receptor (TCR) clones with few reports of γ/δ TCR clonality, particularly in type D LyP.32
Work-up
The evaluation of a patient with a suspected or biopsy confirmed case of CD30 LPD is outlined in Figure 3 and Table 8. Despite being a disorder visible on exam, definitive diagnosis is often delayed by 1 to 3 years.33 In most cases, a diagnosis of LyP can be rendered based on history and thorough dermatologic physical exam alone, with biopsy providing diagnostic confirmation. For accuracy of diagnosis and to exclude those entities described in the differential diagnosis, the following investigations are recommended:
Figure 3.

Diagnosis algorithm for primary cutaneous CD30+ LPD
Table 8.
CD30+ Work-up
| LyP | pcALCL | scALCL | |
|---|---|---|---|
| History | |||
| Spontaneous regression | ✓ | ✓ | x |
| Previous Lymphoid Neoplasms (MF, nodal ALCL or Hodgkin lymphoma) | ✓ | x | ✓ |
| Immunosuppression | x | x | x |
| B symptoms | x | x | ✓ |
| Physical exam | |||
| Solitary lesion | x | ✓ | ✓ |
| Many lesions | ✓ | x | ✓ |
| Patches/plaques of MF | ✓ | x | x |
| Enlarged Lymph Nodes | x | x | ✓ |
| Hepatosplenomegaly | x | x | ✓ |
| Laboratory investigations | |||
| Abnormal CBC with differential | x | x | ✓ |
| Abnormal LDH | x | x | ✓ |
| Serology for HTLV-1/2 | x | x | x |
| Other investigations | |||
| Contrast enhanced CT ± PET of chest, abdomen and pelvis or whole body integrated PET-CT | x | ✓ | ✓ |
| Bone marrow aspirate or biopsy | x | Only if radiologic evidence of extracutaneous disease | ✓ |
| Lymph node biopsy if > 1.5cm | x | If >1.5cm palpable or evidence on imaging | ✓ |
Adapted from: Kempf W, Pfaltz K, Vermeer MH, et al. EORTC, ISCL, and USCLC consensus recommendations for the treatment of primary cutaneous CD30-positive lymphoproliferative disorders: lymphomatoid papulosis and primary cutaneous anaplastic large-cell lymphoma. Blood 2011; 118:4024, with permission.
Skin biopsy, including IHC and TCR gene rearrangement
Complete blood count (CBC) with differential
Lactate dehydrogenase (LDH)
Serology for HTLV 1 and 2 for patients in endemic areas
Imaging, if indicated based on history or exam
Lymph node biopsy, if enlarged
It is suggested to biopsy 2 or more papules that are inflammatory but, have not yet undergone necrosis.
Treatment
Management of LyP depends on clinical severity and symptoms. Indications to treat include cases that are diffuse or progressive, are physically symptomatic or lead to disfigurement from significant scarring or pigmentary change.
With limited disease burden, active non-treatment may be appropriate and considered first-line. Patients can be reassured that treatment has not been reported to alter the natural course of LyP or the risk of developing a secondary malignancy.33
If active treatment is required, a therapeutic ladder for treating LyP is outlined in Table 9. As with any therapy, and of particular importance in the setting of a recurrent, self-resolving disorder, the benefits of the treatment must outweigh the associated risks. The goal of treatment is to prevent new outbreaks, accelerate resolution of lesions and prevent secondary scarring and pigmentary change. With all treatments, recurrence occurs in over 40% of patients, typically within weeks of discontinuing or decreasing treatment.34 While Table 9 lists many therapeutic options, the majority of patients with few lesions are managed with potent topical steroids at the first sign of a new papule; those with more diffuse disease respond to low dose methotrexate or phototherapy. Additional agents are rarely required and multi-agent chemotherapy regimens are not indicated or effective at inducing a prolonged remission in this benign disorder. The use of systemic chemotherapy has been associated with rapid recurrence of LyP either during or after treatment.34
TABLE 9.
Treatment of LyP
| 1st Line | Active non-treatment Topical corticosteroids Class I-III for trunk and extremities; Class IV, V for face, genitals and axillae Phototherapy Methotrexate, 5–25mg/week* |
| 2nd Line | Topical tacrolimus Topical nitrogen mustard Topical retinoids (bexarotene) Topical carmustine |
| 3rd Line | Radiotherapy* Imiquimod 5% cream Interferon-a, Interferon-g Brentuximab vendotin (anti-CD30 monoclonal antibody)** Antiobiotics: tetracyclines, penicillin, erythromycin Sulfones Surgical excision* |
Generally accepted regimens include starting doses 7.5–12.5mg per week, increasing as tolerated every 8–12 weeks until clear up to 25mg per week. Once control has been maintained for 8–12 weeks with no new lesions, the dose is titrated down in a similar fashion to the lowest dose attainable without flares.
For larger, refractory and persistent lesions
For multifocal disease
Adapted from: Klein, RS, Singer E, Junkins-Hopkins JM, Vittorio CC, Rook AH, Kim EJ. “141: Lymphomatoid Papulosis.” Treatment of skin disease: Comprehensive therapeutic strategies. By Lebwohl, M. G., Heymann, W., Berth-Jones, J., & Coulson, I. 4th ed. Edinburgh: Saunders. 2014. 430–434, with permission.
Regardless of treatment plan, patients living with LyP should have life-long follow-up to monitor for the development of a secondary hematologic malignancy. Additionally, any lesion that is persistent and/or greater than 2cm should be biopsied to rule out concomitant ALCL or other secondary neoplasms.
Cutaneous anaplastic large cell lymphoma (cALCL)
cALCL can be divided into primary cutaneous ALCL (pcALCL) and secondary cutaneous ALCL (scALCL). scALCL is systemic ALCL with skin involvement, where the skin is the most common extra-nodal site.35 Please see Dai Chihara and Michelle A. Fanale’s “Management of Anaplastic Large Cell Lymphoma,” in this issue for a more in depth discussion of Systemic ALCL.
Epidemiology
Patients with pcALCL have an older median age of onset (60 years) than their LyP counterparts, and it affects males more than females at a ratio of 3:1 (Table 2).20 scALCL has a bimodal age distribution that varies with anaplastic lymphoma kinase (ALK) expressivity; patients with ALK positive scALCL present at a median age of 34 years; whereas, those that are ALK negative present at a median age of 58 years.36
Prognosis
Similar to LyP, pcALCL has a favourable prognosis with greater than 95% survival at 5 and 10 years. With draining lymph node involvement in greater than 1 nodal basin, survival decreases to 76–96% at 5 years; however, involvement of nodes in a single draining basin has a prognosis similar to patients with disease isolated to the skin.20 Conversely, patients with systemic ALCL have a less favourable prognosis; those with ALK positive disease tend to be younger and have a 5-year survival of 70% while ALK negative disease tends to occur in older patients and has a 49% 5-year survival. Extra-nodal involvement of sALCL, such as cutaneous involvement, is a poor prognostic sign.36
Clinical
In contrast to the successive crops of small self-healing papulonodules of LyP, pcALCL most often presents with a solitary or local group of nodules or tumors, larger than 2cm. Patients describe a rapidly growing, red to violaceous nodule or tumor that may ulcerate (Figure 4).15 While alarming, these lesions are generally asymptomatic and patients are systemically well, without fevers, chills, fatigue, night sweats or weight loss. Such B symptoms should raise suspicion of a systemic lymphoma.
Figure 4.




- 4A – Typical pcALCL tumour: Red, friable 2.5cm tumor, well-defined with central crusting
- 4B -- pcALCL tumor measuring 5.5 x 7.2cm with central clearing and hemorrhagic crust
- 4C – Early pink plaque of pcALCL
- 4D – Multifocal, localized and ulcerative pcALCL
While the majority of patients present with a solitary lesion, approximately 25% of cases of pcALCL present with a localized group of nodulo-tumors and up to 22% of cases may have multifocal (usually two) lesions at different anatomic sites.20 Regression, either partial or total, is variable and occurs in approximately 28% of cases with a range of 0—44%. This feature may highlight indeterminate cases or those that are confused with LyP. Spread of pcALCL to extracutaneous sites is uncommon, but has been reported in approximately 13% of cases with a range of 0–24% depending on the series.37
Importantly, systemic ALCL commonly presents with B symptoms and approximately 20% of cases of sALCL will develop skin lesions (Figure 5).38 The lesions tend to be multifocal or generalized in contrast to pcALCL.
Figure 5.

Cutaneous involvement of ALK- systemic ALCL, with annular plaques and tumors. Note evidence of scarring at prior sites on the arm and trunk.
Pathology
In the majority of cases, routine histopathology of pcALCL demonstrates a dense dermal nodular infiltrate with sheets of atypical large anaplastic lymphocytes. The epidermis is generally uninvolved unless there is ulceration present. Anaplastic cells refer to cells with irregular nuclei that are often horseshoe-shaped, have eosinophilic nucleoli and abundant cytoplasm.14 Importantly, the sheets of anaplastic lymphocytes cannot be distinguished from LyP type C histologically and differentiation of the two entities is made based on the clinical presentation. In 20–25% of cases, pcALCL presents with a non-anaplastic pleomorphic or immunoblastic histopathology. In these cases, which show a heterogeneous inflammatory infiltrate including neutrophils and eosinophils, differentiation from LyP type A is made based on the clinical presentation. Interestingly and sometimes complicating the clinicopathologic correlation, cases of pcALCL with LyP-like histopathology are more likely to completely regress.20
By definition, at least 75% of the tumor cells must express CD30.39 In pcALCL, anaplastic lymphoma kinase (ALK) is almost always negative. Importantly, in scALCL, ALK is positive in only 50% of cases40 and therefore, ALK negativity does not rule out scALCL. Additionally, cutaneous lymphocyte antigen (CLA) is generally positive in pcALCL whereas epithelial membrane antigen (EMA) is typically negative.20 In contrast, expression of CLA is usually negative and EMA is positive in scALCL. (Table 7) Please see Dai Chihara and Michelle A. Fanale’s “Management of Anaplastic Large Cell Lymphoma,” in this issue for additional details on the pathology of scALCL.
Differential diagnosis
Again, with histologic evidence of a CD30+ infiltrate, all entities in Table 1 should be considered. However, the main differential in nodulo-tumors > 2cm that variably self-resolve are:
scALCL
LyP
Transformed mycosis fungoides
Other systemic lymphomas including Adult T-cell leukemia-lymphoma (ATLL) or Hodgkin disease with cutaneous involvement
Nodular reactive lymphoid hyperplasia due to arthropod bite, medication or infection
Work-up
The work-up for cALCL is more extensive than LyP owing to the greater possibility of extracutaneous involvement. A comprehensive history and physical exam along with a biopsy of suspicious lesions remain the first steps in diagnosis. Similar to LyP, the skin biopsy should be performed and examined for histopathologic appearance, classification of the infiltrate using IHC, and T-cell gene rearrangement to assess for clonality.
Following the establishment of a diagnosis of cALCL based on clinical and pathologic features, systemic involvement must be ruled out. Lack of B-symptoms is supportive of a diagnosis of pcALCL; however, complete evaluation is recommended to evaluate for extracutaneous disease of all subsets. The following are recommended:
CBC with differential
LDH
Contrast enhanced computed tomography (CT) with positron emission tomography (PET/CT) is preferred over CT of the chest, abdomen and pelvis
Biopsy of any avid lymph nodes and those larger than 1.5cm
Bone marrow biopsy is considered in the setting of diffuse or multifocal tumors, abnormal hematologic exam or documented extracutaneous disease.
Treatment
The approach to pcALCL therapy is determined by clinical presentation (Table 10: Treatment of pcALCL). The mainstay of treatment for solitary to few lesions of pcALCL is radiotherapy or surgical excision. Given the inherent difficulty in determining margins for cutaneous LPD, radiotherapy is preferred. There are no recommended surgical margins for pcALCL. The optimal dose for radiotherapy also has not been identified but generally 36–40 Gy in 2–3 fractions are used with a margin of 2–3cm, with complete responses ranging from 86% to 100%.41
TABLE 10.
Treatment of pcALCL
| Solitary or grouped lesions | Local radiotherapy, first line Excision |
|---|---|
| Multifocal | Low dose methotrexate (5–25mg/week) Systemic retinoids Pralatrexate Brentuximab vedotin Monitor for spontaneous resolution |
| Extracutaneous spread | Nodal radiation, if single basin Brentuximab vedotin Low dose methotrexate (5–25mg/week) Pralatrexate Multi-agent doxorubicin based chemotherapy |
Data from National Comprehensive Cancer Network. Non-Hodgkin lymphomas version 3.2016 (https://www.nccn.org/professionals/physician_gls/PDF/nhl.pdf).
Historically treatment for multiple lesions involved multi-agent chemotherapy. Based on the overall prognosis, natural history of pcALCL and high rates of relapse after systemic treatment (40–70%)34,37, multi-agent chemotherapy is not considered first line; there does not appear to be added benefit beyond less toxic alternatives. Low-dose (less than 25mg/week) methotrexate is considered first-line for multifocal pcALCL where radiotherapy is not feasible.28 Brentuximab vedotin has been used off-label for multifocal, refractory, extracutaneous or relapsed pcALCL42 and is increasingly being used early in the treatment course. It is currently FDA approved for treatment of patients with systemic ALCL after failure of at least one prior multi-agent chemotherapy regimen.43 In the case of pcALCL with nodal involvement to a single region, radiotherapy to the primary site and nodal basin may be employed.
Conclusion
CD30 lymphoproliferative disorders of the skin are comprised of a spectrum of benign and malignant diseases encompassing LyP, pcALCL and borderline cases. Accurate diagnosis of these conditions requires a thorough history and complete dermatologic and nodal exam, noting the natural course of the lesions and the status of lymph nodes and systemic symptoms. The accurate description of the morphology, distribution and behaviour of lesions is crucial for reaching the correct diagnosis. While all cases of CD30 LPD may look histologically malignant, their behaviour and knowledge of the natural course of LyP and pcALCL (5-year survival of 100% and greater than 95% respectively) allows clinicians to avoid aggressive treatment with high recurrence rates. Importantly, ongoing surveillance of these patients is still required to monitor for secondary malignancy with LyP and recurrence or extracutaneous spread of ALCL.
KEY POINTS.
Primary cutaneous CD30+ lymphoproliferative disorders encompass a spectrum of benign to malignant phenotypes including lymphomatoid papulosis (LyP), primary cutaneous anaplastic large cell lymphoma (pcALCL) and borderline cases.
LyP is characterized by recurrent crops of several to hundreds of red to violaceous papulonodules measuring up to 20mm, usually on the trunk and extremities.
pcALCL is characterized by a solitary or localized red to violaceous nodulotumor greater than 20mm that may occur anywhere on the body.
Patients with LyP are at increased risk of secondary malignancy, most often mycosis fungoides or ALCL, may be diagnosed before, during or after the diagnosis of LyP and should undergo ongoing surveillance.
Patients presenting with cutaneous ALCL should be worked-up to ensure it is primary cutaneous and not secondary cutaneous involvement of systemic ALCL.
Footnotes
DISCLOSURE STATEMENT
The authors have nothing to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gilfillan MC, Noel PJ, Podack ER, Reiner SL, Thompson CB. Expression of the costimulatory receptor CD30 is regulated by both CD28 and cytokines. J Immunol. 1998;160(5):2180–7. [PubMed] [Google Scholar]
- 2.Smith CA, Gruss HJ, Davis T, Anderson D, Farrah T, Baker E, Sutherland GR, Brannan CI, Copeland NG, Jenkins NA, et al. CD30 antigen, a marker for Hodgkin's lymphoma, is a receptor whose ligand defines an emerging family of cytokines with homology to TNF. Cell. 1993 Jul 2;73(7):1349–60. doi: 10.1016/0092-8674(93)90361-s. [DOI] [PubMed] [Google Scholar]
- 3.Bowen MA, Lee RK, Miragliotta G, Nam SY, Podack ER. Structure and expression of murine CD30 and its role in cytokine production. J Immunol. 1996 Jan 15;156(2):442–9. [PubMed] [Google Scholar]
- 4.Muta H, Podack ER. CD30: from basic research to cancer therapy. Immunol Res. 2013 Dec;57(1–3):151–8. doi: 10.1007/s12026-013-8464-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Younes A, Consoli U, Snell V, Clodi K, Kliche KO, Palmer JL, Gruss HJ, Armitage R, Thomas EK, Cabanillas F, Andreeff M. CD30 ligand in lymphoma patients with CD30+ tumors. J Clin Oncol. 1997 Nov;15(11):3355–62. doi: 10.1200/JCO.1997.15.11.3355. [DOI] [PubMed] [Google Scholar]
- 6.Cerutti A, Schaffer A, Goodwin RG, Shah S, Zan H, Ely S, Casali P. Engagement of CD153 (CD30 ligand) by CD30+ T cells inhibits class switch DNA recombination and antibody production in human IgD+ IgM+ B cells. J Immunol. 2000 Jul 15;165(2):786–94. doi: 10.4049/jimmunol.165.2.786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Song HY, Regnier CH, Kirschning CJ, Goeddel DV, Rothe M. Tumor necrosis factor (TNF)-mediated kinase cascades: bifurcation of nuclear factor-kappaB and c-jun N-terminal kinase (JNK/SAPK) pathways at TNF receptor-associated factor 2. Proc Natl Acad Sci U S A. 1997 Sep 2;94(18):9792–6. doi: 10.1073/pnas.94.18.9792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mori M, Manuelli C, Pimpinelli N, Mavilia C, Maggi E, Santucci M, Bianchi B, Cappugi P, Giannotti B, Kadin ME. CD30-CD30 ligand interaction in primary cutaneous CD30(+) T-cell lymphomas: A clue to the pathophysiology of clinical regression. Blood. 1999 Nov 1;94(9):3077–83. [PubMed] [Google Scholar]
- 9.Bargou RC, Emmerich F, Krappmann D, Bommert K, Mapara MY, Arnold W, Royer HD, Grinstein E, Greiner A, Scheidereit C, Dorken B. Constitutive nuclear factor-kappaB-RelA activation is required for proliferation and survival of Hodgkin's disease tumor cells. J Clin Invest. 1997 Dec 15;100(12):2961–9. doi: 10.1172/JCI119849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Muta H, Boise LH, Fang L, Podack ER. CD30 signals integrate expression of cytotoxic effector molecules, lymphocyte trafficking signals, and signals for proliferation and apoptosis. J Immunol. 2000 Nov 1;165(9):5105–11. doi: 10.4049/jimmunol.165.9.5105. [DOI] [PubMed] [Google Scholar]
- 11.Falini B, Pileri S, Pizzolo G, Durkop H, Flenghi L, Stirpe F, Martelli MF, Stein H. CD30 (Ki-1) molecule: a new cytokine receptor of the tumor necrosis factor receptor superfamily as a tool for diagnosis and immunotherapy. Blood. 1995 Jan 1;85(1):1–14. [PubMed] [Google Scholar]
- 12.Bhatt G, Maddocks K, Christian B. CD30 and CD30-Targeted Therapies in Hodgkin Lymphoma and Other B cell Lymphomas. Curr Hematol Malig Rep. 2016 Sep 9; doi: 10.1007/s11899-016-0345-y. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 13.Stein H, Mason DY, Gerdes J, O'Connor N, Wainscoat J, Pallesen G, Gatter K, Falini B, Delsol G, Lemke H, et al. The expression of the Hodgkin's disease associated antigen Ki-1 in reactive and neoplastic lymphoid tissue: evidence that Reed-Sternberg cells and histiocytic malignancies are derived from activated lymphoid cells. Blood. 1985 Oct;66(4):848–58. [PubMed] [Google Scholar]
- 14.Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005 May 15;105(10):3768–85. doi: 10.1182/blood-2004-09-3502. [DOI] [PubMed] [Google Scholar]
- 15.Stein H, Foss HD, Durkop H, Marafioti T, Delsol G, Pulford K, Pileri S, Falini B. CD30(+) anaplastic large cell lymphoma: a review of its histopathologic, genetic, and clinical features. Blood. 2000 Dec 1;96(12):3681–95. [PubMed] [Google Scholar]
- 16.Macaulay WL. Lymphomatoid papulosis: A continuing self-healing eruption, clinically benign-histologically malignant. Arch Dermatol. 1968 Jan;97(1):23–30. doi: 10.1001/archderm.97.1.23. [DOI] [PubMed] [Google Scholar]
- 17.Wang HH, Lach L, Kadin ME. Epidemiology of lymphomatoid papulosis. Cancer. 1992;70(12):2951–2957. doi: 10.1002/1097-0142(19921215)70:12<2951::aid-cncr2820701236>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 18.Willemze Rein. 120: Cutaneous T-Cell Lymphoma. In: Bolognia Jean, Jorizzo Joseph L, Schaffer Julie V., editors. Dermatology. 3. Vol. 2. Philadelphia: Elsevier Saunders; 2012. pp. 2029–032. Print. [Google Scholar]
- 19.Wang HH, Myers T, Lach LJ, et al. Increased risk of lymphoid and nonlymphoid malignancies in patients with lymphomatoid papulosis. Cancer. 1999;86:1240. [PubMed] [Google Scholar]
- 20.Bekkenk MW, Geelen FA, van Voorst Vader PC, et al. Primary and secondary cutaneous CD30(+) lymphoproliferative disorders: a report from the Dutch Cutaneous Lymphoma Group on the long-term follow-up data of 219 patients and guidelines for diagnosis and treatment. Blood. 2000;95:3653. [PubMed] [Google Scholar]
- 21.Gruber R, Sepp NT, Fritsch PO, Schmuth M. Prognosis of lymphomatoid papulosis. Oncologist. 2006 Sep;11(8):955–7. doi: 10.1634/theoncologist.11-8-955. author reply 957. [DOI] [PubMed] [Google Scholar]
- 22.de Souza A, el-Azhary RA, Camilleri MJ, Wada DA, Appert DL, Gibson LE. In search of prognostic indicators for lymphomatoid papulosis: a retrospective study of 123 patients. J Am Acad Dermatol. 2012 Jun;66(6):928–37. doi: 10.1016/j.jaad.2011.07.012. [DOI] [PubMed] [Google Scholar]
- 23.Kunishige JH, McDonald H, Alvarez G, et al. Lymphomatoid papulosis and associated lymphomas: a retrospective case series of 84 patients. Clin Exp Dermatol. 2009;34:576. doi: 10.1111/j.1365-2230.2008.03024.x. [DOI] [PubMed] [Google Scholar]
- 24.Wieser I, Oh CW, Talpur R, Duvic M. Lymphomatoid papulosis: Treatment response and associated lymphomas in a study of 180 patients. J Am Acad Dermatol. 2016 Jan;74(1):59–67. doi: 10.1016/j.jaad.2015.09.013. [DOI] [PubMed] [Google Scholar]
- 25.Scarisbrick JJ, Evans AV, Woolford AJ, Black MM, Russell-Jones R. Regional lymphomatoid papulosis: a report of four cases. Br J Dermatol. 1999 Dec;141(6):1125–8. doi: 10.1046/j.1365-2133.1999.03218.x. [DOI] [PubMed] [Google Scholar]
- 26.Chan DV, Staidle J, Tamburro J, Mostow E. Rapid cutaneous dissemination of persistently agminated lymphomatoid papulosis in a 9-year-old boy. Arch Dermatol. 2011 Nov;147(11):1340–2. doi: 10.1001/archdermatol.2011.348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wieser I, Tetzlaff MT, Torres Cabala CA, Duvic M. Primary cutaneous CD30(+) lymphoproliferative disorders. J Dtsch Dermatol Ges. 2016 Aug;14(8):767–82. doi: 10.1111/ddg.13117. [DOI] [PubMed] [Google Scholar]
- 28.LeBoeuf NR, McDermott S, Harris NL. Case records of the Massachusetts General Hospital. Case 5–2015. A 69-year-old woman with recurrent skin lesions after treatment for lymphoma. N Engl J Med. 2015 Feb 12;372(7):650–9. doi: 10.1056/NEJMcpc1314241. [DOI] [PubMed] [Google Scholar]
- 29.Kempf W. Cutaneous CD30-positive lymphoproliferative disorders. Surg Pathol Clin. 2014;7(2):203–28. doi: 10.1016/j.path.2014.02.001. [DOI] [PubMed] [Google Scholar]
- 30.Morimura S, Sugaya M, Tamaki Z, et al. Lymphomatoid papulosis showing γδ T-cell phenotype. Acta Derm Venereol. 2011;91:712–3. doi: 10.2340/00015555-1145. [DOI] [PubMed] [Google Scholar]
- 31.Karai LJ, Kadin ME, Hsi ED, et al. Chromosomal rearrangements of 6p25.3 define a new subtype of lymphomatoid papulosis. Am J Surg Pathol. 2013;37:1173. doi: 10.1097/PAS.0b013e318282d01e. [DOI] [PubMed] [Google Scholar]
- 32.Rodriguez-Pinilla SM1, Ortiz-Romero PL, Monsalvez V, Tomas IE, Almagro M, Sevilla A, Camacho G, Longo MI, Pulpillo A, Diaz-Perez JA, Montes-Moreno S, Castro Y, Echevarria B, Trebol I, Gonzalez C, Sanchez L, Otin AP, Requena L, Rodriguez-Peralto JL, Cerroni L, Piris MA. TCR-γ expression in primary cutaneous T-cell lymphomas. Am J Surg Pathol. 2013 Mar;37(3):375–84. doi: 10.1097/PAS.0b013e318275d1a2. [DOI] [PubMed] [Google Scholar]
- 33.Kadin ME. Current management of primary cutaneous CD30+ T-cell lymphoproliferative disorders. Oncology (Williston Park) 2009;23:1158. [PubMed] [Google Scholar]
- 34.Kempf W, Pfaltz K, Vermeer MH, et al. EORTC, ISCL, and USCLC consensus recommendations for the treatment of primary cutaneous CD30-positive lymphoproliferative disorders: lymphomatoid papulosis and primary cutaneous anaplastic large-cell lymphoma. Blood. 2011;118:4024. doi: 10.1182/blood-2011-05-351346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kadin ME, Carpenter C. Systemic and primary cutaneous anaplastic large cell lymphomas. Semin Hematol. 2003 Jul;40(3):244–56. doi: 10.1016/s0037-1963(03)00138-0. [DOI] [PubMed] [Google Scholar]
- 36.Savage KJ, Harris NL, Vose JM, Ullrich F, Jaffe ES, Connors JM, Rimsza L, Pileri SA, Chhanabhai M, Gascoyne RD, Armitage JO, Weisenburger DD International Peripheral T-Cell Lymphoma Project. ALK-anaplastic large-cell lymphoma is clinically and immunophenotypically different from both ALK+ ALCL and peripheral T-cell lymphoma, not otherwise specified: report from the International Peripheral T-Cell Lymphoma Project. Blood. 2008;111(12):5496. doi: 10.1182/blood-2008-01-134270. [DOI] [PubMed] [Google Scholar]
- 37.Liu HL, Hoppe RT, Kohler S, et al. CD30+ cutaneous lymphoproliferative disorders: the Stanford experience in lymphomatoid papulosis and primary cutaneous anaplastic large cell lymphoma. J Am Acad Dermatol. 2003;49:1049. doi: 10.1016/s0190-9622(03)02484-8. [DOI] [PubMed] [Google Scholar]
- 38.Falini B, Pileri S, Zinzani PL, Carbone A, Zagonel V, Wolf-Peeters C, Verhoef G, Menestrina F, Todeschini G, Paulli M, Lazzarino M, Giardini R, Aiello A, Foss HD, Araujo I, Fizzotti M, Pelicci PG, Flenghi L, Martelli MF, Santucci A. ALK+ lymphoma: clinico-pathological findings and outcome. Blood. 1999 Apr 15;93(8):2697–706. [PubMed] [Google Scholar]
- 39.Willemze R, Beljaards RC. Spectrum of primary cutaneous CD30 lymphoproliferative disorders: a proposal for classification and guidelines for management and treatment. J Am Acad Dermatol. 1993;28:973–980. doi: 10.1016/0190-9622(93)70140-o. [DOI] [PubMed] [Google Scholar]
- 40.Gascoyne RD, Aoun P, Wu D, Chhanabhai M, Skinnider BF, Greiner TC, Morris SW, Connors JM, Vose JM, Viswanatha DS, Coldman A, Weisenburger DD. Prognostic significance of anaplastic lymphoma kinase (ALK) protein expression in adults with anaplastic large cell lymphoma. Blood. 1999 Jun 1;93(11):3913–21. [PubMed] [Google Scholar]
- 41.Yu JB, McNiff JM, Lund MW, Wilson LD. Treatment of primary cutaneous CD30 anaplastic large-cell lymphoma with radiation therapy. Int J Radiat Oncol Biol Phys. 2008;70(5):1542–1545. doi: 10.1016/j.ijrobp.2007.08.077. [DOI] [PubMed] [Google Scholar]
- 42.Duvic M, Tetzla MT, Gangar P, et al. Results of a phase II trial of brentuximab vedotin for CD30+ cutaneous T-cell lymphoma and lymphomatoid papulosis. J Clin Oncol. 2015;33:3759–65. doi: 10.1200/JCO.2014.60.3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Seattle Genetics Inc. ADCETRISR full prescribing information including boxed warning – U.S. Bothell, Washington: Sep, 2016. [Google Scholar]
- 44.Yeo W, Chow J, Wong N, Chan AT, Johnson PJ. Carbamazepine-induced lymphadenopathy mimicking Ki-1 (CD301) T-cell lymphoma. Pathology. 1997;29:64. doi: 10.1080/00313029700169564. [DOI] [PubMed] [Google Scholar]
- 45.Nathan DL, Belsito DV. Carbamazepine-induced pseudolymphoma with CD-30 positive cells. J Am Acad Dermatol. 1998;38:806. doi: 10.1016/s0190-9622(98)70463-3. [DOI] [PubMed] [Google Scholar]
- 46.Saeed SA, Bazza M, Zaman M, Ryatt KS. Cefuroxime induced lymphomatoid hypersensitivity reaction. Postgrad Med J. 2000;76:577. doi: 10.1136/pmj.76.899.577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Marucci G, Sgarbanti E, Maestri A, Calandri C, Collina G. Gemcitabine-associated CD81 CD301 pseudolymphoma. Br J Dermatol. 2001;145:650. doi: 10.1046/j.1365-2133.2001.04461.x. [DOI] [PubMed] [Google Scholar]
- 48.Magro CM, Crowson AN, Kovatich AJ, Burns F. Drug-induced reversible lymphoid dyscrasia: a clonal lymphomatoid dermatitis of memory and activated T cells. Hum Pathol. 2003 Feb;34(2):119–29. doi: 10.1053/hupa.2003.4. [DOI] [PubMed] [Google Scholar]
- 49.Kim KJ, Lee MW, Choi JH, Sung KJ, Moon KC, Koh JK. CD30-positive T-cell-rich pseudolymphoma induced by gold acupuncture. Br J Dermatol. 2002;146:882. doi: 10.1046/j.1365-2133.2002.04649.x. [DOI] [PubMed] [Google Scholar]
- 50.Cepeda LT, Pieretti M, Chapman SF, Horenstein MG. CD30-positive atypical lymphoid cells in common nonneoplastic cutaneous infiltrates rich in neutrophils and eosinophils. Am J Surg Pathol. 2003;27:912. doi: 10.1097/00000478-200307000-00006. [DOI] [PubMed] [Google Scholar]
- 51.Gallardo F, Barranco C, Toll A, Pujol RM. CD30 antigen expression in cutaneous inflammatory infiltrates of scabies: a dynamic immunophenotypic pattern that should be distinguished from lymphomatoid papulosis. J Cutan Pathol. 2002 Jul;29(6):368–73. doi: 10.1034/j.1600-0560.2002.290608.x. [DOI] [PubMed] [Google Scholar]
- 52.Chai C, White WL, Shea CR, Prieto VG. Epstein Barr virus-associated lymphoproliferative-disorders primarily involving the skin. J Cutan Pathol. 1999;26:242. doi: 10.1111/j.1600-0560.1999.tb01837.x. [DOI] [PubMed] [Google Scholar]
- 53.Massi D, Trotta M, Franchi A, Pimpinelli N, Santucci M. Atypical CD301 cutaneous lymphoid proliferation in a patient with tubercolosis infection. Am J Dermatopathol. 2004;26:234. doi: 10.1097/00000372-200406000-00013. [DOI] [PubMed] [Google Scholar]
- 54.Smith KJ, Barrett TL, Neafie R, et al. Is CD30 (Ki-1) immunostaining in cutaneous eruptions useful as a marker of Th1 to Th2 cytokine switching and/or as a marker of advanced HIV-1 disease? Br J Dermatol. 1998;138:774. doi: 10.1046/j.1365-2133.1998.02212.x. [DOI] [PubMed] [Google Scholar]
- 55.Horn T, Lehmkuhle MA, Gore S, Hood A, Burke P. Systemic cytokine administration alters the histology of the eruption of lymphocyte recovery. J Cutan Pathol. 1996;23:242. doi: 10.1111/j.1600-0560.1996.tb01473.x. [DOI] [PubMed] [Google Scholar]
- 56.Dummer W, Rose C, Brocker EB. Expression of CD30 on T helper cells in the inflammatory infiltrate of acute atopic dermatitis but not of allergic contact dermatitis. Arch Dermatol Res. 1998;290:598. doi: 10.1007/s004030050358. [DOI] [PubMed] [Google Scholar]
- 57.Cordel N, Tressieres B, D'Incan M, et al. Frequency and Risk Factors for Associated Lymphomas in Patients With Lymphomatoid Papulosis. Oncologist. 2016;21:76. doi: 10.1634/theoncologist.2015-0242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kempf W, Kazakov DV, Baumgartner H-P, Kutzner H. Follicular lymphomatoid papulosis revisited: a study of 11 cases, with new histopathological findings. J Am Acad Dermatol. 2013;68(5):809–816. doi: 10.1016/j.jaad.2012.12.952. [DOI] [PubMed] [Google Scholar]