

Significance
Unimolecular hydrogen shift reactions to peroxy radicals have been shown to be important in the atmospheric photooxidation of isoprene and α-pinene. These studies also report the efficient generation of highly oxidized organic molecules known to contribute to particle formation and growth. Here, we quantify the rate of this oxidation pathway for peroxy radicals produced in the oxidation of n-hexane under conditions relevant to the atmosphere. The results suggest that autoxidation pathways are competitive against bimolecular reactions for a broad range of substrates, including many that result from urban emissions. The formation of organic hydroperoxides from atmospheric autoxidation has unknown implications for air quality.
Keywords: atmospheric chemistry, air pollution, autoxidation
Abstract
Gas-phase autoxidation—regenerative peroxy radical formation following intramolecular hydrogen shifts—is known to be important in the combustion of organic materials. The relevance of this chemistry in the oxidation of organics in the atmosphere has received less attention due, in part, to the lack of kinetic data at relevant temperatures. Here, we combine computational and experimental approaches to investigate the rate of autoxidation for organic peroxy radicals (RO2) produced in the oxidation of a prototypical atmospheric pollutant, n-hexane. We find that the reaction rate depends critically on the molecular configuration of the RO2 radical undergoing hydrogen transfer (H-shift). RO2 H-shift rate coefficients via transition states involving six- and seven-membered rings (1,5 and 1,6 H-shifts, respectively) of α-OH hydrogens (HOC-H) formed in this system are of order 0.1 s−1 at 296 K, while the 1,4 H-shift is calculated to be orders of magnitude slower. Consistent with H-shift reactions over a substantial energetic barrier, we find that the rate coefficients of these reactions increase rapidly with temperature and exhibit a large, primary, kinetic isotope effect. The observed H-shift rate coefficients are sufficiently fast that, as a result of ongoing NOx emission reductions, autoxidation is now competing with bimolecular chemistry even in the most polluted North American cities, particularly during summer afternoons when NO levels are low and temperatures are elevated.
The gas-phase oxidation of organic compounds in the atmosphere proceeds through a number of reactive pathways. It is well established that reactions are initiated by oxidants including OH, NO3, and O3, and that, in the presence of oxygen, organic peroxy radicals (RO2) are usually formed (1). The subsequent chemistry of RO2 is diverse and depends on the chemical state of the atmosphere. Traditionally, bimolecular reaction with NO, HO2, or other RO2 has been assumed to dominate the fate of these radicals. Gas-phase autoxidation previously received significant attention only in combustion chemistry where high temperatures promote the process by permitting the reactants to overcome sizeable energetic barriers. The critical reaction in autoxidation, which generally governs the overall kinetics, is an intramolecular hydrogen shift to the RO2. This reaction produces hydroperoxyalkyl radicals (often denoted QOOH), which are known intermediates in autoignition (2, 3). QOOH have only recently been observed (4). While several studies conducted at elevated temperatures have suggested that autoxidation is important in tropospheric chemistry (5–9), experimental kinetic measurements at atmospherically relevant temperatures have been lacking.
Several studies now report the atmospheric significance of RO2 H-shift chemistry (10–24). Crounse et al. (15) suggested that this mechanism may explain the rapid oxygenation of hydrocarbons that contribute to particle growth. Subsequently, autoxidation was implicated in the generation of low-volatility molecules resulting from a single addition of OH or O3 to monoterpenes (25–30). In these systems, autoxidation reactions proceed through successive isomerizations and O2 additions, resulting in the formation of molecules with high O/C ratios and, often, multiple hydroperoxide groups (31). Such compounds have recently been observed to undergo gas–particle transfer (32) and have been shown to be important in particle nucleation (33–50).
While appreciation for the importance of autoxidation is increasing, significant shortcomings exist that preclude adequate characterization of its impact. Research to date has failed to fully describe the autoxidation mechanism of monoterpenes. Attempts have been made to explore autoxidation using cyclohexene as a model system (51, 52). Even in such simplified systems, however, multiple QOOH are formed, each of which can proceed through a large number of possible pathways to produce closed-shell products. Thus, elucidation of the mechanisms has proved challenging.
Here, we use both experimental and computational methods to determine the room temperature rate constants for autoxidation in a simple system—peroxy radicals produced via photooxidation of n-hexane in the presence of NO. The existence of an autoxidation pathway in this system has previously been demonstrated at elevated temperature (5). In the atmosphere, oxidation of hexane by the hydroxyl radical (OH) in the presence of NO produces alkoxy radicals, an example of which is shown in Scheme 1. Some of these alkoxy radicals can isomerize and react with O2 to yield hydroxyperoxy radicals. For simplicity, our experiments use 2-hexanol as the precursor to produce a suite of these hydroxyperoxy radicals. In urban regions, the expectation is that these RO2 react further with NO, ultimately producing hydroxy carbonyls, HO2, and NO2. Autoxidation, on the other hand, competes with the second NO reaction as shown in Scheme 1. In contrast to the mechanism proposed to explain the highly oxidized products observed in the OH and O3 initiated oxidation of monoterpenes (25–30), the RO2 in the hexane system primarily proceed through only a single H-shift yielding QOOH, which react with O2 to produce ketohydroperoxides and HO2. This simplification enables unambiguous experimental constraints for the specific RO2 H-shift rate coefficients.
Scheme 1.

The mechanism to produce the 2,5 RO2 (orange box) from n-hexane in the atmosphere is shown. This RO2 reacts with NO to produce a hydroxy ketone (RONO2, as shown in Scheme 3, are also produced in a minor channel). Competing with this chemistry is a unimolecular 1,6 RO2 H-shift (autoxidation), which produces a ketohydroperoxide and HO2, after further reaction with O2. The first-order rate constants are provided at 300 K and 1 atm of pressure.
Results and Discussion
Computational Approach.
We calculate the rate constants of the H-shift reactions using multiconformer transition state theory (MC-TST) (53–56). The MC-TST expression for a rate constant is given by the following:
where kB is the Boltzmann constant, h is Planck’s constant, T is the temperature, QTS,i is the partition function for the ith transition state (TS) conformer, and ΔEi is the difference in zero-point corrected energy between the ith TS conformer and the lowest energy TS conformer. ETS,0 is the zero-point corrected energy of the lowest energy TS conformer. The analogous symbols apply for the reactant conformers. is the tunneling correction factor. Here, we use the 1D Eckart tunneling approximation, which takes the forward and reverse barrier height and the imaginary frequency of the TS as input (57). The partition functions, energies, barrier heights, and imaginary frequencies needed to calculate kMC-TST are obtained following the approach described by Møller et al. (56). Briefly, ωB97X-D/aug-cc-pVTZ was used for the geometries, frequencies, partition functions, zero-point energy corrections, and relative energies between unique conformers. The conformers were located by a systematic conformer search using molecular mechanics methods (58–60). CCSD(T)-F12a/VDZ-F12 single-point energy calculations were performed for more accurate electronic energies in the barrier heights (61–65). See SI Appendix for a detailed description.
Experimental Approach.
The RO2 studied in this work were prepared via oxidation of 2-hexanol by OH (Scheme 2) in a ∼1-m3 environmental chamber made of Teflon. To determine the rate constants of the H-shifts, we studied the competition between bimolecular and unimolecular chemistry in a suite of experiments with differing concentrations of NO and HO2, thereby producing a range of RO2 bimolecular lifetimes
Determination of the concentrations of NO and HO2 in our experiments is described in SI Appendix. The rate constants ( and ) are taken from the literature (66) and described further in SI Appendix. We assume the ratio of the rate constants is isomer independent.
Scheme 2.

We oxidize 2-hexanol with OH in air to produce a suite of RO2 radicals including the 2,5 RO2 (orange box) shown in Scheme 1.
We use measurements of the organonitrates (RONO2), produced as minor products in the RO2 + NO channel, to probe the bimolecular chemistry. Following oxidation, the RONO2 isomers were separated by gas chromatography and detected with chemical ionization mass spectrometry (GC-CIMS) using CF3O− (m/z = 85) as a reagent ion (Fig. 1). GC-CIMS has been documented extensively (14, 15, 24, 67–70). A deconvolution algorithm described in SI Appendix was required to analyze the chromatograms as it was not possible to increase the column length to fully separate the RONO2 isomers without suffering significant isomer-specific transmission losses. Quantification became more difficult (with resulting higher uncertainty) for experiments at elevated temperature and/or long bimolecular lifetimes due to the higher water vapor concentration (∼300 ppmv) that results from diffusion of water through the Teflon chamber walls.
Fig. 1.

A chromatogram of five first-generation organonitrate (RONO2) isomers formed in the OH oxidation of 2-hexanol. We measure the isomer distribution relative to the 2,3 isomer at different bimolecular lifetimes (τbimolecular). Loss of the 2,4 and 2,5 RONO2 is evident in the experiment at τbimolecular ∼ 4 s (black) compared with the experiment at τbimolecular < 0.03 s (red). Details on structural assignment can be found in SI Appendix.
The H-shifts from the 2,3 RO2 are calculated to be orders of magnitude slower than bimolecular chemistry for all experiments reported here (Table 1 and SI Appendix, Table S5). We therefore assume that . For the 2,4 and 2,5 RO2 isomers, however, we find that the amount of time available to react with NO is shortened by unimolecular chemistry:
As a result, the yields of the 2,4 and 2,5 RONO2 relative to that of the 2,3 RONO2 serve as a sensitive probe of unimolecular chemistry. A depiction of the method is shown in Scheme 3.
Table 1.
H-shift rate coefficients (seconds−1) and factors derived from theory and experiment
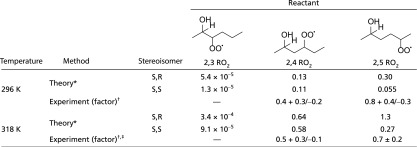 |
For the temperature-dependent rate expressions, refer to SI Appendix. Uncertainty in the calculated rate coefficients is estimated to be less than a factor of 10.
Reported values are scaling factors that afford the best fit to the experimental data, assuming a fixed ratio between the calculated diastereomer rate constants and an initial racemic mixture of isomers. Uncertainty is estimated as described in SI Appendix. The experiments are unable to differentiate the S,R and S,S isomers.
Data for 318 K are available in SI Appendix.
Scheme 3.

Peroxy radicals produced in the reaction of OH with 2-hexanol produce organonitrates (RONO2, blue boxes) after reaction with NO, in a branching ratio measured here to be ∼25% at 296 K. Reaction with HO2 produces hydroperoxides (ROOH). In competition with bimolecular chemistry, the 2,4 RO2 undergoes a unimolecular H-shift to produce QOOH. By measuring the changing yield of the 2,4 and 2,5 RONO2 isomers relative to that of the 2,3 RONO2, an isomer that isomerizes negligibly under the conditions of these experiments, we experimentally constrain the H-shift rate constant. The chemistry of the 2,5 RO2 radical (not shown) parallels that of the 2,4 isomer.
H-Shift Rate Coefficients.
As shown in Fig. 2, the observed ratio of the 2,4 and 2,5 RONO2 to the 2,3 RONO2 decreases as bimolecular lifetimes extend beyond 1 s. At 296 K, the equivalence point in the ratio of the 2,4 and 2,5 RONO2 relative to the 2,3 RONO2 observed at is consistent with unimolecular chemistry occurring at a rate of ∼0.1 s−1.
Fig. 2.

Comparison of experimental (black dots) and computational (red lines) results at 296 K for the 1,5 H-shift (Top) and 1,6 H-shift (Bottom). We demonstrate the difference in calculated 2,5 RO2 H-shift rate coefficients between S,R and S,S diastereomers with the two dashed red lines that, when combined assuming an initial racemic mixture, produce the solid red line. The gray shaded region represents the range of uncertainty in the calculated rate coefficients.
Consistent with a large energetic barrier encountered along the H-shift reaction coordinate, the falloff occurs at a shorter bimolecular lifetime at elevated temperature (71) (Table 1 and SI Appendix). Providing further evidence for our assignment of the mechanism, the falloff occurs at a bimolecular lifetime more than 20 times greater when deuterium is substituted α to the OH group (HOC-D; SI Appendix). This is consistent with the expected primary kinetic isotope effect for RO2 H-shifts (25, 52, 72).
In Fig. 2, comparisons are shown between the measurements and a model using the calculated rate coefficients for the H-shift reactions (Table 1). The 1,4 H-shift from the 2,3 isomer is calculated to be very slow (<10−4 s−1 at T = 296 K) and, as discussed in SI Appendix, a 1,5 H-shift from this isomer was considered but is also expected to be slow. In contrast, the calculated rate coefficients of the 1,5 and 1,6 H-shifts from the 2,4 and 2,5 RO2 are orders of magnitude faster. Surprisingly, the calculated H-shift rate coefficients of the S,S and S,R isomers of the 2,5 RO2 differ significantly. As seen in Fig. 3, the origin of this difference is a change in the hydrogen bond-like interaction between the hydroxy group and the carbon-bonded oxygen atom of the peroxy radical moiety between the reactant and the TS in the S,R diastereomer. This produces a ∼1 kcal/mol difference in barrier height and enhances the rate constant of the S,R diastereomer by a factor of ∼5. However, illustrating the difficulty in generalizing such behavior, opposing effects related to the barrier height, partition functions, and tunneling result in similar calculated rate coefficients for the 1,5 H-shift from S,R and S,S isomers of the 2,4 RO2 (SI Appendix).
Fig. 3.

ωB97X-D/aug-cc-pVTZ optimized structures of the lowest-energy conformers of the reactant and TS. The S,R diastereomer of the 2,5 RO2 exhibits a hydrogen bond-like interaction which stabilizes the TS. Consequently, the H-shift rate of this isomer is enhanced by a factor of ∼5. Green halos highlight atoms involved in the hydrogen bond-like interaction. Blue halos are used when no such interaction exists.
In the model used in Fig. 2 (solid red line), we assume that a racemic mixture is produced when O2 adds to the alkyl radical and that the rate coefficients of RO2 H-shifts for enantiomeric pairs are identical (Table 1). Simulations where the peroxy radicals are either entirely (R,R; S,S) or (R,S; S,R) are shown as dashed lines.
The experimental factors in Table 1 are the multiplicative scaling of the calculated rate constants required to best fit the experimental data (SI Appendix, Figs. S8–S10). For instance, a factor of 1.0 would indicate that a best fit was achieved without scaling the calculated rate constants. The best fit to the experimental data at 296 K require scaling of the calculated values by less than a factor of 3 for both the 1,5 and 1,6 H-shifts, well within the combined uncertainties. The reasonable agreement between the experimental and computational results for these H-shifts suggests that the computational approach used here, and described fully by Møller et al. (56), provides an efficient method applicable to a much broader range of substrates than can plausibly be investigated in the laboratory.
Autoxidation Products.
We observe a CIMS signal at m/z 217 (a cluster of CF3O− with a compound of molecular weight 132 amu), corresponding to the mass of expected autoxidation products, ketohydroperoxides. We assign this signal to the two RO2 α-OH H-shift reactions shown in Scheme 4. Consistent with an autoxidation mechanism, the absolute yield of m/z 217 at similar bimolecular lifetimes increases with temperature.
Scheme 4.

Formation mechanism of ketohydroperoxides (blue highlighted). Hydrogen atoms involved in the H-shifts are orange highlighted. Also shown is the route to hydroxy ketohydroperoxides, suspected products of a 1,5 H-shift from the 2,5 RO2. Approximate, calculated rate coefficients are provided where available at 296 K.
An additional signal at m/z 233 (m/z 234 with D substitution) was observed, consistent with formation of a hydroxy ketohydroperoxide that arises from the 2,5 RO2 through successive isomerizations as indicated in Scheme 4. The formation of this compound was enhanced by deuterium substitution at the α-OH center, which slows the 1,6 H-shift channel. The signal is much smaller in the nondeuterated experiments as the 1,6 H-shift outruns this chemistry. See SI Appendix for further details.
Similar ketohydroperoxides were previously detected in the low NO oxidation of C12 alkanes (73–75). Although autoxidation was not discussed, second-generation alkoxy radicals form hydroxyperoxy radicals that almost certainly undergo H-shifts at rates comparable to those reported here. The ketohydroperoxides were observed to partition to the particle phase with simultaneous conversion to peroxyhemiacetals. Additionally, a cyclization pathway from γ-ketohydroperoxides to form endoperoxides was suggested, and is similar to a pathway discussed in SI Appendix and observed elsewhere (9, 76).
Atmospheric Implications.
In the atmosphere, the bimolecular lifetimes of peroxy radicals typically range from 1 s to more than 100 s. The lowest radical abundances (and therefore long bimolecular RO2 lifetimes) are characteristic of attenuated UV environments (e.g., at night or in shaded regions below thick cloud or tree canopies) or regions remote from anthropogenic NOx emissions. Due to emission reductions from power generation and transportation, however, NOx levels are declining rapidly across North America, reaching levels unprecedented in the past several decades (77, 78). The 2013 SENEX and SEAC4RS aircraft campaigns, for example, sampled large swaths of the southeastern United States including areas significantly influenced by urban emissions. NO mixing ratios were often <100 pptv and nearly always <1 ppbv (79, 80). Even more impressive, NOx levels in Pasadena, California, declined by more than a factor of 2 between 2010 and 2017 (81). In August 2017, we measured [NO] below 500 pptv on several weekend afternoons, corresponding to RO2 lifetimes longer than 10 s. With afternoon temperatures typically exceeding 305 K, the autoxidation chemistry described here is now competing with reactions between peroxy radicals and NO in the middle of one of North America’s most polluted cities.
While the rate of autoxidation is highly dependent on the substrate, this chemistry is undoubtedly important for many of the organic compounds emitted into the urban atmosphere. A recent review of vehicle emissions and urban aerosol formation speculated that autoxidation might play a role in the degradation of certain unsaturated compounds (82), but its role in alkane oxidation was not appreciated. Alkanes constitute a substantial fraction of urban nonmethane hydrocarbon emissions (83–87), and n-alkanes with greater than five carbons are known emissions from gasoline- and diesel-powered vehicles arising from both incomplete combustion and fuel evaporation (88, 89). These compounds will undergo oxidation pathways that are nearly identical to those reported here for n-hexane.
The importance of organic hydroperoxide formation via autoxidation will depend on the fate and toxicity of these compounds. Peroxides are reactive oxygen species that are known to produce oxidative stress in plants and animals (90–92). Additionally, the rate of oxidant (e.g., ozone) and aerosol formation is almost certainly sensitive to autoxidation. Because autoxidation leads to the degradation of volatile organic compounds without converting NO to NO2, ozone formation may be slower when this chemistry is important. While the impact on aerosol formation is less clear, it is likely to be enhanced because autoxidation adds oxygen with minimal fragmentation. For example, Zhao et al. (93) recently showed that while NOx emissions are lower with the latest vehicle emissions control technology, the organic aerosol yield is greater than from the emissions using older-generation technology. Although the authors do not provide a mechanistic explanation, the higher yields are likely attributable, in part, to hydroperoxide formation via an autoxidation mechanism.
As a result of highly successful policies to reduce emissions of NOx, our results suggest that autoxidation is now becoming an important pathway for urban photochemistry across North America. However, the photochemical models that have been used to inform these policies have little if any validation in the low-NO regimes we are now experiencing. Thus, there is a risk that attainment of ozone compliance may occur at the expense of other air quality goals because of more efficient hydroperoxide and aerosol formation. It is thus imperative that our understanding of the low-NO chemistry for the suite of organic compounds typically found in the atmosphere advance quickly and that monitoring efforts to quantify low-NO processes, such as autoxidation, be undertaken with haste.
Methods
The experimental apparatus, including the GC-CIMS technique, has been previously described and is outlined in SI Appendix for these experiments (14, 15, 67–70). Experiments are performed in a ∼1-m3 Teflon environmental chamber. In nearly all experiments, the precursor used was 2-hexanol, while CH3ONO was used as a photolytic source of HO2 and NO (and thus OH). NO was added before oxidation for experiments focusing on short RO2 lifetimes and was quantified using a Teledyne 200EU chemiluminescence NOx analyzer. For experiments without additional NO added, we use established methods to estimate its abundance (and that of HO2) as detailed in SI Appendix. We attempted to replicate the method of Jorand et al. (5) near room temperature, but, for reasons described in SI Appendix, these experiments were not successful.
Experimental Uncertainty.
Considerable (>50%) experimental uncertainty arises in our estimate of for , due to imprecision in the interpretation of chromatographic peaks, and temperature fluctuations in our chamber. Details are provided in SI Appendix.
Computational Uncertainty.
We estimate the uncertainty of the calculated rate constants to be less than a factor of 10. The uncertainties arise primarily from the barrier height, tunneling correction, and the partition functions. Due to error cancelation, the ratio of the theoretical rate constants for these different H-shifts are likely more accurate than the absolute rate coefficients. This is especially true for reactions of the same or very similar molecules. See SI Appendix for details.
Supplementary Material
Acknowledgments
We thank Kristian H. Møller for helpful discussions related to the implementation of MC-TST. J.C.H. thanks the Camille and Henry Dreyfus Postdoctoral Program in Environmental Chemistry for support. We acknowledge funding from National Science Foundation Grant CHE-1508526 as well as from the University of Copenhagen.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1715540115/-/DCSupplemental.
References
- 1.Seinfeld JH, Pandis SN. Atmospheric Chemistry and Physics: From Air Pollution to Climate Change. Wiley; New York: 1998. [Google Scholar]
- 2.Zádor J, Taatjes CA, Fernandes RX. Kinetics of elementary reactions in low-temperature autoignition chemistry. Pror Energy Combust Sci. 2011;37:371–421. [Google Scholar]
- 3.Taatjes CA. Uncovering the fundamental chemistry of alkyl + O2 reactions via measurements of product formation. J Phys Chem A. 2006;110:4299–4312. doi: 10.1021/jp056997f. [DOI] [PubMed] [Google Scholar]
- 4.Savee JD, et al. Carbon radicals. Direct observation and kinetics of a hydroperoxyalkyl radical (QOOH) Science. 2015;347:643–646. doi: 10.1126/science.aaa1495. [DOI] [PubMed] [Google Scholar]
- 5.Jorand F, et al. Isomeric hexyl-ketohydroperoxides formed by reactions of hexoxy and hexylperoxy radicals in oxygen. Int J Chem Kinet. 2003;35:354–366. [Google Scholar]
- 6.Perrin O, Heiss A, Doumenc F, Sahetchian K. Homogeneous and heterogeneous reactions of the n-C5H11O, n-C5H10OH and OOC5H10OH radicals in oxygen. Analytical steady state solution by use of the Laplace transform. J Chem Soc Faraday Trans. 1998;94:2323–2335. [Google Scholar]
- 7.Blin-Simiand N, et al. Hydroperoxides with zero, one, two or more carbonyl groups formed during the oxidation of n-dodecane. Combust Flame. 2001;126:1524–1532. [Google Scholar]
- 8.Heiss A, Sahetchian K. Isomerization reactions of the n-C4H9O and n-OOC4H8OH radicals in oxygen. Int J Chem Kinet. 1996;28:531–544. [Google Scholar]
- 9.Perrin O, Heiss A, Sahetchian K, Kerhoas L, Einhorn J. Determination of the isomerization rate constant HOCH2CH2CH2CH(OO·)CH3 → HOC·HCH2CH2CH(OOH)CH3. Importance of intramolecular hydroperoxy isomerization in tropospheric chemistry. Int J Chem Kinet. 1998;30:875–887. [Google Scholar]
- 10.Peeters J, Nguyen TL, Vereecken L. HOx radical regeneration in the oxidation of isoprene. Phys Chem Chem Phys. 2009;11:5935–5939. doi: 10.1039/b908511d. [DOI] [PubMed] [Google Scholar]
- 11.Asatryan R, da Silva G, Bozzelli JW. Quantum chemical study of the acrolein (CH2CHCHO) + OH + O2 reactions. J Phys Chem A. 2010;114:8302–8311. doi: 10.1021/jp104828a. [DOI] [PubMed] [Google Scholar]
- 12.Crounse JD, Paulot F, Kjaergaard HG, Wennberg PO. Peroxy radical isomerization in the oxidation of isoprene. Phys Chem Chem Phys. 2011;13:13607–13613. doi: 10.1039/c1cp21330j. [DOI] [PubMed] [Google Scholar]
- 13.Vereecken L, Francisco JS. Theoretical studies of atmospheric reaction mechanisms in the troposphere. Chem Soc Rev. 2012;41:6259–6293. doi: 10.1039/c2cs35070j. [DOI] [PubMed] [Google Scholar]
- 14.Crounse JD, et al. Atmospheric fate of methacrolein. 1. Peroxy radical isomerization following addition of OH and O2. J Phys Chem A. 2012;116:5756–5762. doi: 10.1021/jp211560u. [DOI] [PubMed] [Google Scholar]
- 15.Crounse JD, Nielsen LB, Jørgensen S, Kjaergaard HG, Wennberg PO. Autoxidation of organic compounds in the atmosphere. J Phys Chem Lett. 2013;4:3513–3520. [Google Scholar]
- 16.Knap HC, Jørgensen S, Kjaergaard HG. Theoretical investigation of the hydrogen shift reactions in peroxy radicals derived from the atmospheric decomposition of 3-methyl-3-buten-1-ol (MBO331) Chem Phys Lett. 2015;619:236–240. [Google Scholar]
- 17.Jokinen T, et al. Rapid autoxidation forms highly oxidized RO2 radicals in the atmosphere. Angew Chem Int Ed Engl. 2014;53:14596–14600. doi: 10.1002/anie.201408566. [DOI] [PubMed] [Google Scholar]
- 18.Kurtén T, et al. Computational study of hydrogen shifts and ring-opening mechanisms in α-pinene ozonolysis products. J Phys Chem A. 2015;119:11366–11375. doi: 10.1021/acs.jpca.5b08948. [DOI] [PubMed] [Google Scholar]
- 19.Hyttinen N, et al. Unimolecular HO2 loss from peroxy radicals formed in autoxidation is unlikely under atmospheric conditions. J Phys Chem A. 2016;120:3588–3595. doi: 10.1021/acs.jpca.6b02281. [DOI] [PubMed] [Google Scholar]
- 20.Jørgensen S, et al. Rapid hydrogen shift scrambling in hydroperoxy-substituted organic peroxy radicals. J Phys Chem A. 2016;120:266–275. doi: 10.1021/acs.jpca.5b06768. [DOI] [PubMed] [Google Scholar]
- 21.Richters S, Herrmann H, Berndt T. Highly oxidized RO2 radicals and consecutive products from the ozonolysis of three sesquiterpenes. Environ Sci Technol. 2016;50:2354–2362. doi: 10.1021/acs.est.5b05321. [DOI] [PubMed] [Google Scholar]
- 22.Knap HC, Jørgensen S. Rapid hydrogen shift reactions in acyl peroxy radicals. J Phys Chem A. 2017;121:1470–1479. doi: 10.1021/acs.jpca.6b12787. [DOI] [PubMed] [Google Scholar]
- 23.Wang S, Wu R, Berndt T, Ehn M, Wang L. Formation of highly oxidized radicals and multifunctional products from the atmospheric oxidation of alkylbenzenes. Environ Sci Technol. 2017;51:8442–8449. doi: 10.1021/acs.est.7b02374. [DOI] [PubMed] [Google Scholar]
- 24.Teng AP, Crounse JD, Wennberg PO. Isoprene peroxy radical dynamics. J Am Chem Soc. 2017;139:5367–5377. doi: 10.1021/jacs.6b12838. [DOI] [PubMed] [Google Scholar]
- 25.Ehn M, et al. A large source of low-volatility secondary organic aerosol. Nature. 2014;506:476–479. doi: 10.1038/nature13032. [DOI] [PubMed] [Google Scholar]
- 26.Zhang X, et al. Formation and evolution of molecular products in α-pinene secondary organic aerosol. Proc Natl Acad Sci USA. 2015;112:14168–14173. doi: 10.1073/pnas.1517742112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Berndt T, et al. Hydroxyl radical-induced formation of highly oxidized organic compounds. Nat Commun. 2016;7:13677. doi: 10.1038/ncomms13677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jokinen T, et al. Production of extremely low volatile organic compounds from biogenic emissions: Measured yields and atmospheric implications. Proc Natl Acad Sci USA. 2015;112:7123–7128. doi: 10.1073/pnas.1423977112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mentel TF, et al. Formation of highly oxidized multifunctional compounds: Autoxidation of peroxy radicals formed in the ozonolysis of alkenes—deduced from structure–product relationships. Atmos Chem Phys. 2015;15:6745–6765. [Google Scholar]
- 30.Zhang X, et al. Highly oxygenated multifunctional compounds in α-pinene secondary organic aerosol. Environ Sci Technol. 2017;51:5932–5940. doi: 10.1021/acs.est.6b06588. [DOI] [PubMed] [Google Scholar]
- 31.Wang Z, et al. Unraveling the structure and chemical mechanisms of highly oxygenated intermediates in oxidation of organic compounds. Proc Natl Acad Sci USA. November 6, 2017;114(50):13102–13107. doi: 10.1073/pnas.1707564114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mutzel A, et al. Highly oxidized multifunctional organic compounds observed in tropospheric particles: A field and laboratory study. Environ Sci Technol. 2015;49:7754–7761. doi: 10.1021/acs.est.5b00885. [DOI] [PubMed] [Google Scholar]
- 33.Kirkby J, et al. Ion-induced nucleation of pure biogenic particles. Nature. 2016;533:521–526. doi: 10.1038/nature17953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schobesberger S, et al. Molecular understanding of atmospheric particle formation from sulfuric acid and large oxidized organic molecules. Proc Natl Acad Sci USA. 2013;110:17223–17228. doi: 10.1073/pnas.1306973110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Riccobono F, et al. Oxidation products of biogenic emissions contribute to nucleation of atmospheric particles. Science. 2014;344:717–721. doi: 10.1126/science.1243527. [DOI] [PubMed] [Google Scholar]
- 36.Zhao J, Ortega J, Chen M, McMurry PH, Smith JN. Dependence of particle nucleation and growth on high-molecular-weight gas-phase products during ozonolysis of α-pinene. Atmos Chem Phys. 2013;13:7631–7644. [Google Scholar]
- 37.Bianchi F, et al. New particle formation in the free troposphere: A question of chemistry and timing. Science. 2016;352:1109–1112. doi: 10.1126/science.aad5456. [DOI] [PubMed] [Google Scholar]
- 38.Kulmala M, et al. Direct observations of atmospheric aerosol nucleation. Science. 2013;339:943–946. doi: 10.1126/science.1227385. [DOI] [PubMed] [Google Scholar]
- 39.Jimenez JL, et al. Evolution of organic aerosols in the atmosphere. Science. 2009;326:1525–1529. doi: 10.1126/science.1180353. [DOI] [PubMed] [Google Scholar]
- 40.Shrivastava M, et al. Recent advances in understanding secondary organic aerosol: Implications for global climate forcing. Rev Geophys. 2017;55:509–559. [Google Scholar]
- 41.Kroll JH, Seinfeld JH. Chemistry of secondary organic aerosol: Formation and evolution of low-volatility organics in the atmosphere. Atmos Environ. 2008;42:3593–3624. [Google Scholar]
- 42.Chacon-Madrid HJ, Donahue NM. Fragmentation vs. functionalization: Chemical aging and organic aerosol formation. Atmos Chem Phys. 2011;11:10553–10563. [Google Scholar]
- 43.Donahue NM, et al. How do organic vapors contribute to new-particle formation? Faraday Discuss. 2013;165:91–104. doi: 10.1039/c3fd00046j. [DOI] [PubMed] [Google Scholar]
- 44.Huang R-J, et al. High secondary aerosol contribution to particulate pollution during haze events in China. Nature. 2014;514:218–222. doi: 10.1038/nature13774. [DOI] [PubMed] [Google Scholar]
- 45.Ng NL, et al. Organic aerosol components observed in northern hemispheric datasets from aerosol mass spectrometry. Atmos Chem Phys. 2010;10:4625–4641. [Google Scholar]
- 46.Donahue NM, et al. Aging of biogenic secondary organic aerosol via gas-phase OH radical reactions. Proc Natl Acad Sci USA. 2012;109:13503–13508. doi: 10.1073/pnas.1115186109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Monge ME, et al. Alternative pathway for atmospheric particles growth. Proc Natl Acad Sci USA. 2012;109:6840–6844. doi: 10.1073/pnas.1120593109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Barsanti KC, Kroll JH, Thornton JA. Formation of low-volatility organic compounds in the atmosphere: Recent advancements and insights. J Phys Chem Lett. 2017;8:1503–1511. doi: 10.1021/acs.jpclett.6b02969. [DOI] [PubMed] [Google Scholar]
- 49.Daumit KE, Kessler SH, Kroll JH. Average chemical properties and potential formation pathways of highly oxidized organic aerosol. Faraday Discuss. 2013;165:181–202. doi: 10.1039/c3fd00045a. [DOI] [PubMed] [Google Scholar]
- 50.Elm J, Myllys N, Kurtén T. What is required for highly oxidized molecules to form clusters with sulfuric acid? J Phys Chem A. 2017;121:4578–4587. doi: 10.1021/acs.jpca.7b03759. [DOI] [PubMed] [Google Scholar]
- 51.Rissanen MP, et al. Effects of chemical complexity on the autoxidation mechanisms of endocyclic alkene ozonolysis products: From methylcyclohexenes toward understanding α-pinene. J Phys Chem A. 2015;119:4633–4650. doi: 10.1021/jp510966g. [DOI] [PubMed] [Google Scholar]
- 52.Rissanen MP, et al. The formation of highly oxidized multifunctional products in the ozonolysis of cyclohexene. J Am Chem Soc. 2014;136:15596–15606. doi: 10.1021/ja507146s. [DOI] [PubMed] [Google Scholar]
- 53.Eyring H. The activated complex and the absolute rate of chemical reactions. Chem Rev. 1935;17:65–77. [Google Scholar]
- 54.Evans MG, Polanyi M. Some applications of the transition state method to the calculation of reaction velocities, especially in solution. J Chem Soc, Faraday Trans. 1935;31:875–894. [Google Scholar]
- 55.Vereecken L, Peeters J. The 1,5-H-shift in 1-butoxy: A case study in the rigorous implementation of transition state theory for a multirotamer system. J Chem Phys. 2003;119:5159–5170. [Google Scholar]
- 56.Møller KH, Otkjær RV, Hyttinen N, Kurtén T, Kjaergaard HG. Cost-effective implementation of multiconformer transition state theory for peroxy radical hydrogen shift reactions. J Phys Chem A. 2016;120:10072–10087. doi: 10.1021/acs.jpca.6b09370. [DOI] [PubMed] [Google Scholar]
- 57.Eckart C. The penetration of a potential barrier by electrons. Phys Rev. 1930;35:1303–1309. [Google Scholar]
- 58.Chai J-D, Head-Gordon M. Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections. Phys Chem Chem Phys. 2008;10:6615–6620. doi: 10.1039/b810189b. [DOI] [PubMed] [Google Scholar]
- 59.Dunning TH. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J Chem Phys. 1989;90:1007–1023. [Google Scholar]
- 60.Kendall RA, Dunning TH, Harrison RJ. Electron affinities of the first‐row atoms revisited. Systematic basis sets and wave functions. J Chem Phys. 1992;96:6796–6806. [Google Scholar]
- 61.Watts JD, Gauss J, Bartlett RJ. Coupled‐cluster methods with noniterative triple excitations for restricted open‐shell Hartree–Fock and other general single determinant reference functions. Energies and analytical gradients. J Chem Phys. 1993;98:8718–8733. [Google Scholar]
- 62.Adler TB, Knizia G, Werner H-J. A simple and efficient CCSD(T)-F12 approximation. J Chem Phys. 2007;127:221106. doi: 10.1063/1.2817618. [DOI] [PubMed] [Google Scholar]
- 63.Knizia G, Adler TB, Werner H-J. Simplified CCSD(T)-F12 methods: Theory and benchmarks. J Chem Phys. 2009;130:054104. doi: 10.1063/1.3054300. [DOI] [PubMed] [Google Scholar]
- 64.Werner H-J, Knizia G, Manby FR. Explicitly correlated coupled cluster methods with pair-specific geminals. Mol Phys. 2011;109:407–417. [Google Scholar]
- 65.Peterson KA, Adler TB, Werner H-J. Systematically convergent basis sets for explicitly correlated wavefunctions: The atoms H, He, B-Ne, and Al-Ar. J Chem Phys. 2008;128:084102. doi: 10.1063/1.2831537. [DOI] [PubMed] [Google Scholar]
- 66.Saunders SM, Jenkin ME, Derwent RG, Pilling MJ. Protocol for the development of the Master Chemical Mechanism, MCM v3 (Part A): Tropospheric degradation of non-aromatic volatile organic compounds. Atmos Chem Phys. 2003;3:161–180. [Google Scholar]
- 67.St Clair JM, et al. Kinetics and products of the reaction of the first-generation isoprene hydroxy hydroperoxide (ISOPOOH) with OH. J Phys Chem A. 2016;120:1441–1451. doi: 10.1021/acs.jpca.5b06532. [DOI] [PubMed] [Google Scholar]
- 68.Bates KH, et al. Gas phase production and loss of isoprene epoxydiols. J Phys Chem A. 2014;118:1237–1246. doi: 10.1021/jp4107958. [DOI] [PubMed] [Google Scholar]
- 69.Praske E, et al. Atmospheric fate of methyl vinyl ketone: Peroxy radical reactions with NO and HO2. J Phys Chem A. 2015;119:4562–4572. doi: 10.1021/jp5107058. [DOI] [PubMed] [Google Scholar]
- 70.Crounse JD, McKinney KA, Kwan AJ, Wennberg PO. Measurement of gas-phase hydroperoxides by chemical ionization mass spectrometry. Anal Chem. 2006;78:6726–6732. doi: 10.1021/ac0604235. [DOI] [PubMed] [Google Scholar]
- 71.Orlando JJ, Tyndall GS. Laboratory studies of organic peroxy radical chemistry: An overview with emphasis on recent issues of atmospheric significance. Chem Soc Rev. 2012;41:6294–6317. doi: 10.1039/c2cs35166h. [DOI] [PubMed] [Google Scholar]
- 72.Muchalski H, Levonyak AJ, Xu L, Ingold KU, Porter NA. Competition H(D) kinetic isotope effects in the autoxidation of hydrocarbons. J Am Chem Soc. 2015;137:94–97. doi: 10.1021/ja511434j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yee LD, et al. Effect of chemical structure on secondary organic aerosol formation from C12 alkanes. Atmos Chem Phys. 2013;13:11121–11140. [Google Scholar]
- 74.Yee LD, et al. Secondary organic aerosol formation from low-NOx photooxidation of dodecane: Evolution of multigeneration gas-phase chemistry and aerosol composition. J Phys Chem A. 2012;116:6211–6230. doi: 10.1021/jp211531h. [DOI] [PubMed] [Google Scholar]
- 75.Shiraiwa M, et al. Size distribution dynamics reveal particle-phase chemistry in organic aerosol formation. Proc Natl Acad Sci USA. 2013;110:11746–11750. doi: 10.1073/pnas.1307501110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jalan A, et al. New pathways for formation of acids and carbonyl products in low-temperature oxidation: The Korcek decomposition of γ-ketohydroperoxides. J Am Chem Soc. 2013;135:11100–11114. doi: 10.1021/ja4034439. [DOI] [PubMed] [Google Scholar]
- 77.US Environmental Protection Agency 2011 Benefits and Costs of the Clean Air Act 1990–2020, the Second Prospective Study. Available at www.epa.gov/clean-air-act-overview/benefits-and-costs-clean-air-act-1990-2020-second-prospective-study. Accessed August 8, 2017. [PubMed]
- 78.Russell AR, Valin LC, Cohen RC. Trends in OMI NO2 observations over the United States: Effects of emission control technology and the economic recession. Atmos Chem Phys. 2012;12:12197–12209. [Google Scholar]
- 79.Wolfe GM, et al. Formaldehyde production from isoprene oxidation across NOx regimes. Atmos Chem Phys. 2016;16:2597–2610. doi: 10.5194/acp-16-2597-2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yu K, et al. Sensitivity to grid resolution in the ability of a chemical transport model to simulate observed oxidant chemistry under high-isoprene conditions. Atmos Chem Phys. 2016;16:4369–4378. [Google Scholar]
- 81.California Air Resources Board 2017 Air Quality and Meteorological Information System. Available at https://www.arb.ca.gov/aqmis2/aqdselect.php. Accessed August 8, 2017.
- 82.Gentner DR, et al. Review of urban secondary organic aerosol formation from gasoline and diesel motor vehicle emissions. Environ Sci Technol. 2017;51:1074–1093. doi: 10.1021/acs.est.6b04509. [DOI] [PubMed] [Google Scholar]
- 83.Baker AK, et al. Measurements of nonmethane hydrocarbons in 28 United States cities. Atmos Environ. 2008;42:170–182. [Google Scholar]
- 84.Warneke C, et al. Determination of urban volatile organic compound emission ratios and comparison with an emissions database. J Geophys Res Atmos. 2007;112:D10S47. [Google Scholar]
- 85.Warneke C, et al. Multiyear trends in volatile organic compounds in Los Angeles, California: Five decades of decreasing emissions. J Geophys Res Atmos. 2012;117:D00V17. [Google Scholar]
- 86.von Schneidemesser E, Monks PS, Plass-Duelmer C. Global comparison of VOC and CO observations in urban areas. Atmos Environ. 2010;44:5053–5064. [Google Scholar]
- 87.Boynard A, et al. Spatial and seasonal variability of measured anthropogenic non-methane hydrocarbons in urban atmospheres: Implication on emission ratios. Atmos Environ. 2014;82:258–267. [Google Scholar]
- 88.Watson JG, Chow JC, Fujita EM. Review of volatile organic compound source apportionment by chemical mass balance. Atmos Environ. 2001;35:1567–1584. [Google Scholar]
- 89.Schauer JJ, Kleeman MJ, Cass GR, Simoneit BRT. Measurement of emissions from air pollution sources. 2. C1 through C30 organic compounds from medium duty diesel trucks. Environ Sci Technol. 1999;33:1578–1587. [Google Scholar]
- 90.Gebicki JM. Protein hydroperoxides as new reactive oxygen species. Redox Rep. 1997;3:99–110. doi: 10.1080/13510002.1997.11747096. [DOI] [PubMed] [Google Scholar]
- 91.Yin H, Xu L, Porter NA. Free radical lipid peroxidation: Mechanisms and analysis. Chem Rev. 2011;111:5944–5972. doi: 10.1021/cr200084z. [DOI] [PubMed] [Google Scholar]
- 92.Zielinski ZAM, Pratt DA. Lipid peroxidation: Kinetics, mechanisms, and products. J Org Chem. 2017;82:2817–2825. doi: 10.1021/acs.joc.7b00152. [DOI] [PubMed] [Google Scholar]
- 93.Zhao Y, et al. Reducing secondary organic aerosol formation from gasoline vehicle exhaust. Proc Natl Acad Sci USA. 2017;114:6984–6989. doi: 10.1073/pnas.1620911114. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.