

Abstract
The aim of this study was to investigate the role of osteopontin (OPN) in hematopoietic stem cell (HPSC) mobilization to the liver and its contribution to alcoholic liver disease (ALD). We analyzed young (14‐16 weeks) and old (>1.5 years) wild‐type (WT) littermates and global Opn knockout (Opn−/−) mice for HPSC mobilization to the liver. In addition, WT and Opn−/− mice were chronically fed the Lieber–DeCarli diet for 7 weeks. Bone marrow (BM), blood, spleen, and liver were analyzed by flow cytometry for HPSC progenitors and polymorphonuclear neutrophils (PMNs). Chemokines, growth factors, and cytokines were measured in serum and liver. Prussian blue staining for iron deposits and naphthol AS‐D chloroacetate esterase staining for PMNs were performed on liver sections. Hematopoietic progenitors were lower in liver and BM of young compared to old Opn−/− mice. Granulocyte colony‐stimulating factor and macrophage colony‐stimulating factor were increased in Opn−/− mice, suggesting potential migration of HPSCs from the BM to the liver. Furthermore, ethanol‐fed Opn−/− mice showed significant hepatic PMN infiltration and hemosiderin compared to WT mice. As a result, ethanol feeding caused greater liver injury in Opn−/− compared to WT mice. Conclusion: Opn deletion promotes HPSC mobilization, PMN infiltration, and iron deposits in the liver and thereby enhances the severity of ALD. The age‐associated contribution of OPN to HPSC mobilization to the liver, the prevalence of PMNs, and accumulation of hepatic iron, which potentiates oxidant stress, reveal novel signaling mechanisms that could be targeted for therapeutic benefit in patients with ALD. (Hepatology Communications 2018;2:84–98)
Abbreviations
- ALD
alcoholic liver disease
- BM
bone marrow
- CD
cluster of differentiation
- CLP
common lymphoid progenitor
- CXCL
chemokines (C‐X‐C motif) ligand
- CXCR
CXC chemokine receptor
- EPO
erythropoietin
- GCSF
granulocyte‐colony stimulating factor
- HIF
hypoxia inducible factor
- HPSC
hematopoietic stem cell
- IL
interleukin
- Kitl
kit‐ligand
- MCSF
macrophage‐colony stimulating factor
- MDA
malondialdehyde
- MF
macrophage
- MIP1α
macrophage inflammatory protein 1α
- Mo
monocytes
- mRNA
messenger RNA
- OPN
osteopontin
- Opn−/−
global osteopontin knockout
- PMN
polymorphonuclear neutrophil
- RANTES
regulated on activation normal T cell expressed and secreted
- SDF
stromal cell‐derived factor
- TIBC
total iron‐binding capacity
- TNFα
tumor necrosis factor alpha
- VEGF
vascular endothelial growth factor
- WT
wild‐type
Introduction
Many clinical studies have been conducted to investigate the safety and efficacy of bone marrow (BM)‐derived cells for treating liver disease.1 Pluripotent hematopoietic stem cells (HPSCs) can participate in the repopulation of normal tissue renewal and severe liver injury to improve its function.2 Therefore, we focused on the long‐term implication of BM‐derived HPSC mobilization to the liver due to Opn deletion and its potential contribution to alcoholic liver disease (ALD).
To mobilize BM‐derived pluripotent HPSCs, granulocyte‐colony stimulating factor (GCSF)1 has demonstrated good tolerance in patients with decompensated ALD and increases the proliferative activity of both hepatic progenitor cells and mature hepatocytes in the short term.3 However, larger studies are necessary to define the role of BM cell therapy in patients with chronic ALD and for those with the most severe forms of the disease.
Osteopontin (OPN) is a glycoprotein involved in cell adhesion, inflammation, angiogenesis, and tumor metastasis. It is a key constituent of the HPSC niche that drives HPSC localization and is a physiological negative regulator of HPSC proliferation.4 OPN binds several integrins5 and the cluster of differentiation (CD)44 receptor.6 Our laboratory has demonstrated that OPN participates in the pathogenesis of liver fibrosis through integrin αvβ3 signaling.7, 8, 9 Moreover, OPN has been implicated in the development of autoimmune10 and allergic airway diseases,11 up‐regulation of interferon‐α,12 interleukin (IL)‐18 and IL‐27 expression,13, 14 and inhibition of HPSC proliferation.15 Yet, the contribution of OPN to the mechanisms behind hematopoiesis, HPSC lodgment, and control and retention within the BM along with mobilization to the liver are currently unknown.
Because the presence of polymorphonuclear neutrophils (PMNs) correlates with ALD severity in humans and Opn deletion has been associated with increased inflammation16 and alcoholic neutrophilic hepatitis,17, 18 we hypothesized that OPN stimulates BM‐derived HPSC mobilization to the liver, creating a proinflammatory environment conducive to liver injury and ALD.
Besides the contribution of BM‐derived macrophages (MFs)19 and PMNs20 to inflammation and steatosis, iron overload also promotes alcohol‐induced inflammation.21 Hepatic iron overload, a common adverse event in patients undergoing HPSC transplantation,22 catalyzes the Fenton reaction and increases oxidative stress along with lipid peroxidation and as a result accelerates the progression of ALD. Yet, the role of OPN in regulating iron homeostasis in the liver remains undefined. Thus, the aim of this study was to investigate the contribution of OPN to HPSC mobilization to the liver and hepatic iron overload in ALD.
Materials and Methods
MICE
Global osteopontin knockout (Opn−/−) mice (C57BL/6J) and their wild‐type (WT) littermates were purchased from the Jackson Laboratories (Bar Harbor, ME). Male and female young (14‐16 weeks) and old (>1.5 years) Opn −/− mice and age‐matched WT littermates were used in this study. All animals received humane care according to the criteria outlined in the Guide for the Care and Use of Laboratory Animals prepared by the National Academy of Sciences and published by the National Institutes of Health.
GENERAL METHODOLOGY
Serum and liver growth factors, chemokines, and proinflammatory cytokines (GCSF, macrophage‐colony stimulating factor [MCSF], chemokine (C‐X‐C motif) ligand 1 [CXCL1], macrophage inflammatory protein 1 alpha [MIP1α], regulated on activation normal T cell expressed and secreted [RANTES], IL‐6, MIP2, tumor necrosis factor alpha [TNFα], IL‐1β, and vascular endothelial growth factor [VEGF]) were analyzed using the Milliplex Map Kit (EMD Millipore Corporation, Billerica, MA). Hemosiderin staining was performed with the Iron Staining Kit (Thermo Fisher Scientific, Waltham, MA). Total iron‐binding capacity (TIBC) was calculated from the quantitative determination of iron and unsaturated iron‐binding capacity with the iron/TIBC Reagent Set (Pointe Scientific, Canton, MI). Serum ferritin was measured by enzyme‐linked immunosorbent assay using a kit from Life Technologies (Carlsbad, CA). Lipid peroxidation was determined according to Yagi.23 Details on general methodology, hematoxylin and eosin, naphthol AS‐D chloroacetate esterase, and immunohistochemistry have been described in our publications.8, 9, 16, 18
STATISTICAL ANALYSIS
Data are expressed as mean ± SD. Statistical comparisons among groups were performed by a two‐factor analysis of variance. All experiments were carried out in triplicate at least 4 times, and a representative image or blot is shown in all figures.
Results
Opn −/− DISPLAY INCREASED HEPATIC CD34+ AND CD127+ HEMATOPOIETIC PROGENITORS COMPARED TO WT MICE
Hematoxylin and eosin staining revealed small aggregates of cells with intense basophilic nuclei and cell clusters of different sizes in the liver of old Opn−/− male (Fig. 1A, bottom right panel and zoomed views) and old Opn−/− female mice (Fig. 1B, bottom right panel and zoomed views), which displayed hepatosplenomegaly compared to their age‐matched WT littermates (Fig. 1C). In addition, old Opn−/− mice showed HPSC lodgment in the spleen, suggesting global HPSC mobilization (Supporting Fig. S1A). The pathology scores showed increased inflammation in the livers from old Opn −/− compared to WT mice (Fig. 1D). Quantitative polymerase chain reaction analysis of liver lysates for the stem cell factor kit‐ligand (Kitl), which plays an essential role in hematopoiesis and stem cell maintenance,24 showed messenger RNA (mRNA) up‐regulation in old Opn−/− compared to WT mice (Fig. 1E). Because CD34+ HPSCs interact with OPN through β1‐integrins,4 flow cytometry was performed to identify the hepatic HPSC population involved. Increased hepatic CD34+ and CD127+ hematopoietic progenitors were found in old (Fig. 1F) and young (Fig. 1G) Opn−/− mice, suggesting that global Opn deletion likely promotes extracellular matrix breakdown, allowing the release of HPSCs from the BM to the circulation.
Figure 1.
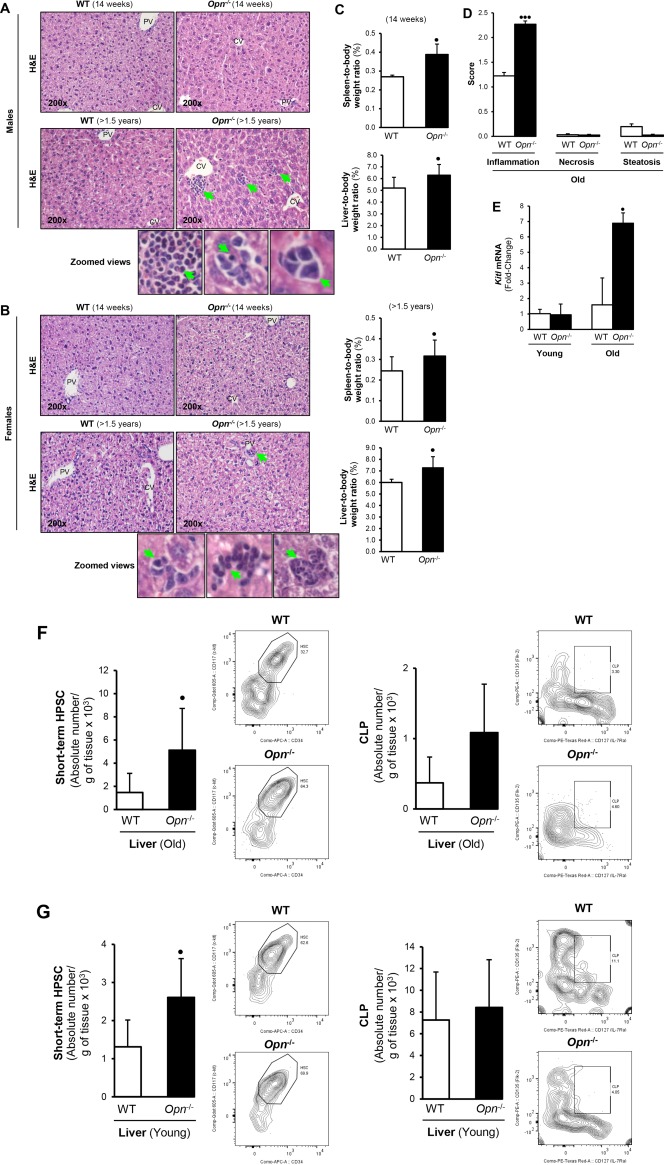
Opn −/− mice show increased hepatic hematopoietic progenitors compared to WT mice. (A,B) H&E staining reveals small aggregates of cells with intensely basophilic nuclei and clusters of cells in the liver of (top panels) young (14‐16 weeks;) and (bottom panels) old (>1.5 years; green arrows in panels and zoomed views) Opn −/− mice. (C) Spleen‐to‐body weight and liver‐to‐body weight ratios in young males and females show hepatosplenomegaly in Opn −/− compared to WT mice. (D) Pathology scores from old Opn −/− and WT mice. (E) Kitl mRNA in liver of young and old Opn −/− and WT male mice. (F) Opn deletion increases CD34+ and CD127+ cell hepatic population in old and young mice. (G) Old and young Opn −/− mice displayed increased Lin– Sca‐1+ c‐kit+ CD34+ CD135+ short‐term HPSCs and Lin– Sca‐1+ c‐kit+ CD127+ CD135+ CLPs compared to WT littermates. Bar graphs and representative flow plots are shown. Liver leukocytes were isolated and gated using SSC/FSC, viability dye (to exclude dead cells), single‐cell population (to exclude doublets), and Lin– Sca‐1+ c‐kit+ (to identify hematopoietic cells). Results are expressed as mean ± SD; n = 6/group, • P < 0.05, •• P < 0.01, and ••• P < 0.001 for Opn −/− versus WT mice (both genders). Abbreviations: APC, antigen‐presenting cell; CV, central vein; FSC, forward scatter; H&E, hematoxylin and eosin; Lin– Sca‐1+ c‐kit+, lineage‐negative, stem cell antigen‐1‐positive, and c‐kit receptor‐positive; PE, phycoerythrin; PV, portal vein; SSC, side scatter.
MYELOID PROGENITOR CELLS, CD34+ CELLS, AND CD127+ CELLS ARE LESS COMMON IN THE BM OF YOUNG COMPARED TO OLD Opn −/− MICE
Young and old Opn −/− mice and age‐matched WT littermates were sacrificed, and hematopoietic progenitors in the BM, spleen, and liver were analyzed by flow cytometry. There were fewer common myeloid progenitors, such as granulocyte macrophage progenitors, macrophage dendritic cell progenitors, and common dendritic cell progenitors, in the BM of young compared to old Opn−/− mice (Fig. 2A,B). Short‐term CD34+ CD135+ HPSCs (Fig. 2C) and common lymphoid progenitors (CLPs) CD127+ CD135+ (Fig. 2D) were also lower in number in the BM but not in the spleen of young Opn −/− compared to WT mice (Supporting Fig. S1B‐E). These cell populations were significantly increased in old Opn −/− mice (Fig. 2E,F), suggesting a possible role for OPN in restricting HPSC mobilization. Next, we analyzed whether these cell populations were mobilized from the BM to other organs. However, no differences were observed in the lung, heart, and kidney (data not shown). Based on the characterization of these cells in the liver and the BM, next we examined the potential molecular mechanism involved in HPSC mobilization to the liver and its implications for ALD.
Figure 2.
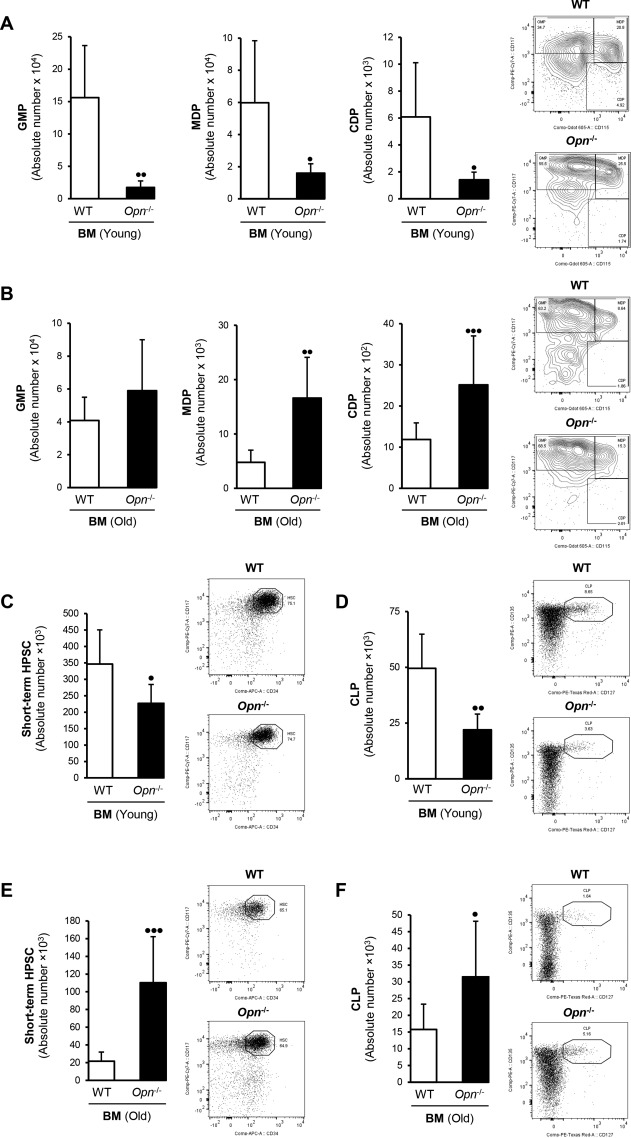
Myeloid progenitor and CD34+ and CD127+ cells were lower in the BM of young compared to old Opn −/− mice. GMP, CD117High CD115–; MDP, CD117+ CD115+; CDP, CD117–. (A) CD115+ were less in young Opn −/− but (B) more in old Opn −/−compared to aged‐matched WT mice. (C,D) Young Opn −/− mice show less short‐term CD34+ HPSCs and CLPs in the BM compared to (E,F) old Opn −/− mice. Bar graphs and representative flow plots are shown. Results are expressed as mean of absolute number of cells per femur or per gram of tissue ± SD; n = 6/group; • P < 0.05, •• P < 0.01, and ••• P < 0.001 for Opn −/− versus WT mice (both sexes). Abbreviations: CDP, common dendritic cell progenitor; GMP, granulocyte‐macrophage progenitor; MDP, macrophage‐dendritic cell progenitor; PE, phycoerythrin.
Opn −/− EXHIBIT INCREASED HEPATIC HEMATOPOIETIC GROWTH FACTORS AND CHEMOKINES
Because the ability of cells to home, proliferate, and mature in extramedullary organs involves local and systemic chemokine production, we measured growth factors, chemokines, and proinflammatory cytokines in serum and liver from WT and Opn−/− mice. Hepatic hematopoietic growth factors, such as GCSF and MCSF (Fig. 3A), CXCL1, MIP1α, RANTES, and IL‐6 (Fig. 3B) along with MIP2, TNFα, and IL‐1β (Fig. 3C), were significantly up‐regulated in Opn−/− compared to WT mice. As VEGF is required for the production of fully committed hematopoietic progenitors,25 we measured its production and found that serum (Fig. 3D) but not hepatic VEGF (not shown) was increased in young Opn−/− compared to WT mice.
Figure 3.
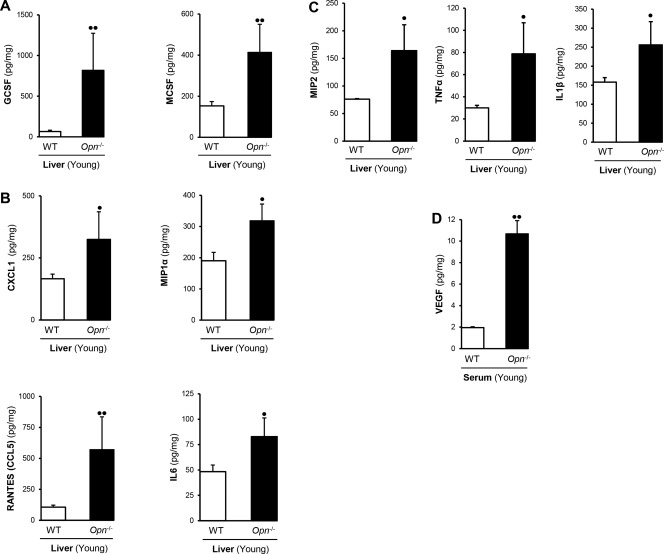
Opn −/− exhibit increased hepatic hematopoietic growth factors and chemokines. (A) Hepatic hematopoietic growth factors (GCSF and MCSF) and (B) chemokines (CXCL1, MIP1α, RANTES, and IL‐6); (C) MIP2, TNFα, and IL‐1β; (D) and serum VEGF are significantly increased in Opn −/− compared to WT mice. Results are expressed as mean ± SD; n = 6/group; • P < 0.05 and •• P < 0.01 for Opn −/− versus WT mice.
Several mechanisms and soluble factors are involved in the mobilization of HPSCs from the BM to other organs: the CXC chemokine receptor (CXCR)4/stromal cell‐derived factor 1α (SDF1α) axis,26 soluble KITL,27 VEGF,28 and induction of hypoxic factors.29 Because HPSCs express CXCR4 and are chemoattracted to and retained within the BM by SDF1α,30 the CXCR4/SDF1α signaling axis was analyzed in liver lysates of Opn −/− and WT mice; yet, there was no major difference in their mRNA levels (Supporting Fig. S2).
Taken together, these results suggest that proinflammatory and growth stimulatory factors likely regulate the differentiation of HPSCs into mature inflammatory cells,31 such as PMNs and MFs in the liver. Similarly, hepatic chemokine‐dependent signaling but not the CXCR4/SDF1α axis32 could be responsible for the trafficking of HPSCs to the liver in Opn−/− mice. Consequently, a preceding proinflammatory environment in Opn−/− mice could possibly sensitize the liver to the hepatotoxic effects of alcohol.
ALCOHOL FEEDING INCREASES LIVER PMN INFILTRATION IN Opn −/− COMPARED TO WT MICE
To directly address the contribution of HPSCs found in the liver of Opn −/− mice in the setting of ALD, blood and liver of young and old WT and Opn −/− mice were analyzed by flow cytometry for PMNs and monocytes (Mo). Under basal conditions, young Opn−/− but not old mice (not shown) showed more circulating PMNs but less Mo, lymphocyte antigen 6 complex locus C Mo, and eosinophils than WT mice (Fig. 4A‐D). Hepatic PMNs but not Mo were significantly increased in young and had a trend toward an increase in old Opn−/− compared to WT mice (Fig. 4E). Because peripheral neutrophilia and liver PMN infiltration are frequently found in patients with ALD,33 naphthol AS‐D chloroacetate esterase immunostaining, markers of neutrophil activation, such as Neutrophil cytosolic factor (NCF) 1/2/4, heme‐binding membrane glycoprotein (Gp91phox) and neutrophil cytochrome b light chain (P22Phox), together with Il‐8 (a key mediator of PMN recruitment and degranulation) and its receptor Cxr1 mRNA were analyzed in total liver. Alcohol‐fed Opn−/− showed more liver PMNs (Fig. 4F) and markers of neutrophil activation (Fig. 4G) but not increased Il‐8 or Cxr1 mRNA compared to WT mice (Supporting Fig. S3A). Moreover, no changes were observed in Cd11b mRNA in Opn−/− compared to WT mice (Supporting Fig. S3B). However, we recently demonstrated increased hepatic basal and alcohol‐induced F4/80+ cells in Opn−/− mice.18
Figure 4.

Alcohol feeding increases liver PMN infiltration in Opn −/− compared to WT mice. Bar graphs and representative flow plots of myeloid lineage‐derived cells in serum of old and young WT and Opn −/− male and female mice for (A) circulating PMNs, (B) Mo, (C) Ly‐6C+ Mo, and (D) eosinophils. (E) Hepatic PMNs and Mo of Opn −/− and WT mice. (F) NASDCA staining shows increased PMNs in alcohol‐fed Opn −/− compared to WT mice (yellow arrows; magnification ×630). (G) qPCR of markers of neutrophil activation. Results are expressed as mean ± SD; n = 6/group, • P < 0.05, •• P < 0.01, and ••• P < 0.001 for Opn −/− versus WT mice. Abbreviations: CV, central vein; Ly‐6C, lymphocyte antigen 6 complex locus C; NASDCA, naphthol AS‐D chloroacetate esterase; qPCR, quantitative polymerase chain reaction. NCF, Neutrophil cytosolic factor; Gp91phox, heme‐binding membrane glycoprotein; P22Phox, neutrophil cytochrome b light chain.
ALCOHOL FEEDING IMPAIRS ERYTHROPOIESIS AND INCREASES HEPATIC HEMOSIDERIN AND LIPID PEROXIDATION IN Opn −/− COMPARED TO WT MICE
Hypoxia‐inducible factor 1α (HIF1α) prevents hematopoietic cell damage due to overproduction of reactive oxygen species,34 suggesting that the hypoxic niche helps maintain the long lifespan of HPSCs. Because HPSCs exhibit hypoxic profiles,35, 36 we performed quantitative polymerase chain reaction analysis for Hif1α; yet, no significant changes were observed (Supporting Fig. S4). HIF1α binds to and transactivates erythropoietin (Epo), which encodes for erythropoietin, a key hormone regulator of red blood cell production and master regulator of erythroid development and iron uptake by erythrocytes.37 We analyzed the mRNA of Epo and Hamp, the latter a central player in iron homeostasis,38 in livers of young and old Opn−/− and WT mice. Opn−/− mice showed increased hepatic Epo yet decreased Hamp mRNA compared to WT mice. Hamp RNA was significantly reduced in young but not old Opn−/− mice (Fig. 5A), suggesting abnormal erythropoiesis and impaired iron homeostasis in Opn−/− compared to WT mice.
Figure 5.
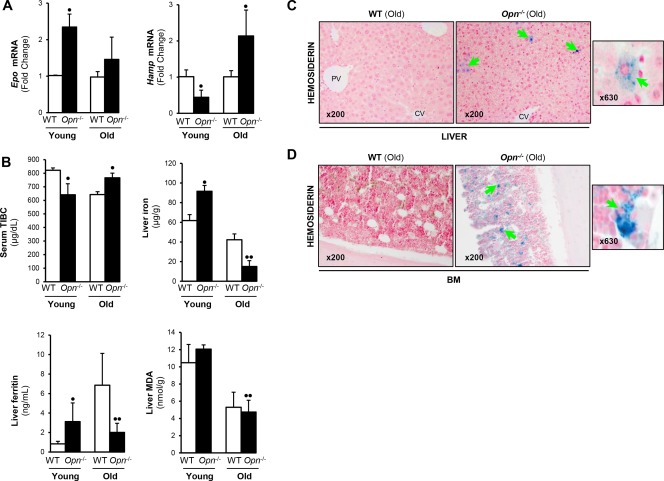
Opn −/− mice have decreased hepatic Hamp mRNA and increased iron deposits in liver and BM compared to WT mice. (A) qPCR analysis in male and female mice for hepatic Epo and Hamp mRNA in young and old Opn −/− compared to WT mice. All data were normalized to the expression of glyceraldehyde‐3‐phosphate dehydrogenase. (B) Serum TIBC decreased while liver iron, ferritin, and MDA increased in young compared to old Opn −/− mice. (C) Liver (green arrows) and (D) BM (green arrows) of young (data not shown) and old Opn −/− mice stained with Perls' Prussian blue to detect hemosiderin. Results are expressed as mean ± SD; n = 6/group, • P < 0.05 and •• P < 0.01 for Opn −/− versus WT mice. Abbreviations: qPCR, quantitative polymerase chain reaction.
Next, we looked at hepatic iron content due to its role in promoting lipid peroxidation, apoptosis, and liver injury.39 Serum TIBC decreased whereas hepatic iron, ferritin, and malondialdehyde (MDA), a lipid peroxidation product,40 increased in young compared to old Opn−/− mice (Fig. 5B). Liver and BM were stained with Perls' Prussian blue. At baseline, old Opn−/− mice had more hemosiderin deposits in the liver (Fig. 5C) compared to young Opn−/− mice (data not shown) and had far more in the BM (Fig. 5D), minimal amount in the lung, and deposits were totally absent in the heart and kidney (Supporting Fig. S5A‐C). Furthermore, ethanol‐fed Opn−/− presented more hepatic hemosiderin compared to WT mice (Fig. 6A). Low serum TIBC and increased hepatic iron, ferritin, and MDA levels were found in ethanol‐fed Opn−/− compared to WT mice (Fig. 6B). Hepatic Epo mRNA was similar, whereas Hamp mRNA decreased in alcohol‐fed Opn−/− compared to WT mice (Fig. 6C). Thus, ethanol caused greater liver injury and lipid peroxidation in Opn−/− compared to WT mice. Collectively, these results suggest that Opn deletion significantly increases HPSC mobilization to the liver, hepatic inflammation, iron deposits, and lipid peroxidation, all of which could contribute to the pathogenesis of ALD.
Figure 6.
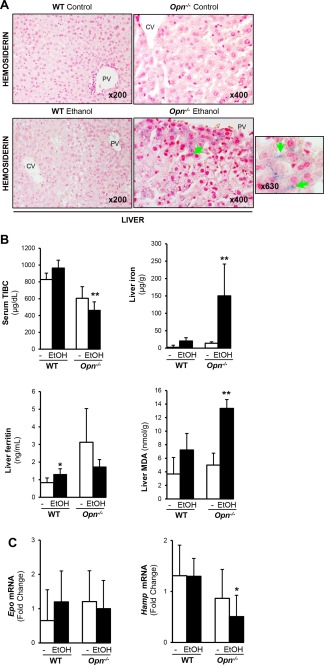
Alcohol feeding increases hepatic hemosiderin and lipid peroxidation and impairs erythropoiesis in Opn −/− compared to WT mice. (A, green arrows) Alcohol feeding in male and female mice increased hepatic hemosiderin in the liver of Opn −/− compared to WT mice. (B) Serum TIBC decreased whereas liver iron, ferritin, and MDA increased in Opn −/− ethanol‐fed compared to WT mice. (C) qPCR analysis for liver Epo and Hamp mRNA in ethanol‐fed mice compared to its age‐matched control. The expression was normalized to the expression of glyceraldehyde‐3‐phosphate dehydrogenase. Results are expressed as mean ± SD; n = 6/group, *P < 0.05 and **P < 0.01 for ethanol‐fed versus control mice. Abbreviations: CV, central vein; EtOH, ethanol; PV, portal vein; qPCR, quantitative polymerase chain reaction.
Discussion
The liver has the unique feature of inducing mobilization and engraftment of HPSCs41 in addition to supporting hematopoiesis following transplantation.42 Here, we highlight three mechanisms by which Opn deletion aggravates ALD: HPSC mobilization, hepatic PMN extravasation, and iron overload.
Among other molecules that sustain HPSC lodgment, OPN appears to have a key role. It is at present an unrecognized component of endosteally located stem cell niches with an important physiologic role in the regulation of HPSC localization and proliferation. HPSCs bind circulating OPN, which results in a decrease in their differentiation and proliferation.43, 44 We have previously shown that serum OPN levels increase in ALD.18 The increased cellularity observed in Opn−/− mice along with HPSC mobilization and hepatosplenomegaly suggest that OPN plays a prominent role in HPSC niche interactions.
Nilsson et al.4 have shown that Opn −/− mice display BM hypercellularity and increased lineage‐negative, stem cell antigen‐1‐positive, and c‐kit receptor‐positive cells compared to WT mice. They examined the ability of CD34+ HPSCs to specifically bind OPN through integrins and the Opn role on HPSC apoptosis, proliferation, and differentiation. These results, also validated by others,45 indicate a biologically relevant role for Opn deletion in the multifaceted HPSCs in the BM, promoting expansion and mobilization of BM progenitors to peripheral blood due to the fact that stromal OPN anchors HPSCs to the BM niche through β1 integrin and the CD44 receptor.46
It has been suggested that adhesion of human hematopoietic CD34+ cells to human liver is CD44 dependent and modulated by CXCR3/4.41 Hepatosplenomegaly and BM cellularity changes were observed in Opn−/−‐deficient mice, indicating that absence of extrahepatic Opn may stimulate HPSC migration from the BM to other organs as mobilization of HPSCs occurred in the liver and other anatomic sites, such as spleen, kidney, and lung, in Opn−/− mice. Yet, whether these events are mediated by CD44 or CXCR3/4 remains elusive.
The increase in GCSF, MCSF, CXCL1, MIP1α, RANTES, and IL‐6 proteins but not in Cxcr4 and Sdf1α mRNA levels, a pathway with a major role on HPSC mobilization and retention, could suggest that OPN fine‐tunes engraftment of HPSCs through hepatic production of regulatory factors involved in proliferation and differentiation of HPSCs. This may affect HPSC homeostasis in the liver and tilt the balance toward increased inflammatory cells that could enhance ethanol‐induced liver injury. In our study, Opn−/− had neutrophilia, increased neutrophil activation, elevated cytokines (i.e., TNFα, IL‐1β, and MIP2), and more hepatic PMN, albeit similar expression of Il‐8 and Cxcr1 mRNA levels compared to WT mice, suggestive of production or additional infiltration of PMNs in the liver that could support oxidative stress‐induced hepatic damage as observed by the MDA values in Opn−/− mice. The elevated TNFα and IL‐1β levels could increase E‐selectin, known to enhance PMN sequestration and transmigration into the liver.47, 48
Moreover, we show that Opn deletion increases hepatic MFs and PMNs, which are further enhanced by alcohol consumption. This, in turn, can potentiate OPN‐mediated alcohol‐induced liver injury because MFs and PMNs are critical for the progression of ALD and our data suggest neutrophil activation. Overall, these findings suggest that Opn deletion can exacerbate alcohol‐induced liver damage by inducing HPSC mobilization to the liver, PMN infiltration, iron deposition, and lipid peroxidation.
We further provide evidence that short‐term HPSCs and CLPs accumulate in the liver of young and old Opn−/− mice under steady‐state conditions. This is of singular interest as older mice are generally more susceptible to alcohol‐induced liver injury and fibrosis,49 reinforcing how aging fuels HPSC egression from the BM in Opn−/− mice. As evidenced by the controls, both cohorts of young and old Opn−/− mice had increased intrahepatic PMNs, HPSCs, and CLPs, suggesting a potential mechanism driven by OPN to enhance alcohol‐induced liver injury in Opn−/− mice. Future mechanistic studies linking OPN‐dependent PMN infiltration with ALD are needed.
Chronic iron overload worsens alcohol‐induced liver injury and vascular reactivity by increasing oxidative stress through the Fenton reaction, thereby promoting lipid peroxidation50 and accelerating the development of ALD. Hepcidin, a small polypeptide produced by hepatocytes, plays a central role in regulating iron uptake by promoting internalization and degradation of ferroportin, the only known cellular iron exporter.51 The increased hepatic expression of Epo resulting from the presence of proinflammatory cytokines could suppress Hamp expression52 and erythroid iron intake, leading to accumulation of hemichromes in the liver. This will support the likelihood of impaired iron homeostasis, uptake, and deposition in the liver of Opn−/− mice, hence increasing alcohol‐induced liver injury in young but not in old Opn−/− mice. This may also result from greater HPSC turnover and lower circulating iron in old Opn−/− mice, both of which have a tendency to increase with aging.53, 54
Because OPN‐dependent release of HPSCs from the BM promotes excessive homing to the liver, as observed by hepatic CD34+ and CD127+ HPSCs, our results suggest that Opn −/− mice develop a proinflammatory liver milieu that may predispose them to greater alcohol‐induced liver injury. Although OPN is increased in alcoholic and nonalcoholic steatohepatitis,55, 56 the ability of OPN to regulate the proinflammatory response may be indicative of its relevance when designing new therapies. Indeed, we have demonstrated that oral administration of OPN (i.e., from bovine milk) is efficient in treating early ALD in mice.16, 18
Despite some experimental and clinical work having established association among iron overload, oxidative stress, and ALD, additional studies are needed to better understand the cell‐specific mechanisms driven by OPN. One possibility is that OPN is key for maintaining gut integrity and permeability; as iron is absorbed in the gut,57 lipopolysaccharide translocation from the gut to the portal blood activates Kupffer cells to produce TNFα and OPN binds lipopolysaccharides blocking Kupffer cell activation and TNFα production,18 in that way contributing to lower alcohol‐mediated injury. These studies are under way in our laboratory to reinforce the role of cell‐specific OPN production in the pathogenesis of ALD. Overall, the present work demonstrates that global Opn deletion promotes HPSC mobilization, PMN infiltration, and iron deposits in the liver, thereby contributing to enhance the severity of ALD (Fig. 7).
Figure 7.
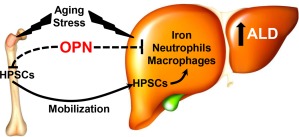
Opn deletion promotes HPSC mobilization to the liver and increases hepatic iron, neutrophil, and macrophage infiltration, thereby contributing to ALD. Besides OPN, aging, inflammatory cell infiltration, iron overload, and the accompanying lipid peroxidation are major events responsible for increased susceptibility to ALD. OPN as a negative regulator of HPSC proliferation, lodgment, and retention can regulate the response to ALD as continuous mobilization of BM‐derived HPSCs to the liver could lead to a proinflammatory environment.
Author names in bold designate shared co‐first authorship.
Supporting information
Additional Supporting Information may be found at http://onlinelibrary.wiley.com/doi/10.1002/hep4.1116/full.
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Potential conflict of interest: Nothing to report.
Supported by the Chicago Biomedical Consortium (grant PDR‐088 to F.M., H.G., N.N.) and U.S. Public Health Service Grants from the National Institute on Alcohol Abuse and Alcoholism (P20 AA017067, P20 AA017067‐01S1, P20 AA017067‐03S1, U01 AA021887) to N.N.
REFERENCES
- 1. Spahr L, Chalandon Y, Terraz S, Kindler V, Rubbia‐Brandt L, Frossard JL, et al. Autologous bone marrow mononuclear cell transplantation in patients with decompensated alcoholic liver disease: a randomized controlled trial. PLoS One 2013;8:e53719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Theise ND, Badve S, Saxena R, Henegariu O, Sell S, Crawford JM, et al. Derivation of hepatocytes from bone marrow cells in mice after radiation‐induced myeloablation. Hepatology 2000;31:235‐240. [DOI] [PubMed] [Google Scholar]
- 3. Lyra AC, Soares MB, da Silva LF, Braga EL, Oliveira SA, Fortes MF, et al. Infusion of autologous bone marrow mononuclear cells through hepatic artery results in a short‐term improvement of liver function in patients with chronic liver disease: a pilot randomized controlled study. Eur J Gastroenterol Hepatol 2010;22:33‐42. [DOI] [PubMed] [Google Scholar]
- 4. Nilsson SK, Johnston HM, Whitty GA, Williams B, Webb RJ, Denhardt DT, et al. Osteopontin, a key component of the hematopoietic stem cell niche and regulator of primitive hematopoietic progenitor cells. Blood 2005;106:1232‐1239. [DOI] [PubMed] [Google Scholar]
- 5. Lund SA, Wilson CL, Raines EW, Tang J, Giachelli CM, Scatena M. Osteopontin mediates macrophage chemotaxis via alpha4 and alpha9 integrins and survival via the alpha4 integrin. J Cell Biochem 2013;114:1194‐1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Weber GF, Ashkar S, Cantor H. Interaction between CD44 and osteopontin as a potential basis for metastasis formation. Proc Assoc Am Physicians 1997;109:1‐9. [PubMed] [Google Scholar]
- 7. Arriazu E, Ge X, Leung TM, Magdaleno F, Lopategi A, Lu Y, et al. Signalling via the osteopontin and high mobility group box‐1 axis drives the fibrogenic response to liver injury. Gut 2017;66:1123‐1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Urtasun R, Lopategi A, George J, Leung TM, Lu Y, Wang X, et al. Osteopontin, an oxidant stress sensitive cytokine, up‐regulates collagen‐I via integrin alpha(V)beta(3) engagement and PI3K/pAkt/NFkappaB signaling. Hepatology 2012;55:594‐608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wang X, Lopategi A, Ge X, Lu Y, Kitamura N, Urtasun R, et al. Osteopontin induces ductular reaction contributing to liver fibrosis. Gut 2014;63:1805‐1818. [DOI] [PubMed] [Google Scholar]
- 10. Qi Y, Li X, Ma X, Xu L, Zhang X, Jiang X, et al. The role of osteopontin in the induction of the CD40 ligand in Graves' disease. Clin Endocrinol (Oxf) 2014;80:128‐134. [DOI] [PubMed] [Google Scholar]
- 11. Samitas K, Zervas E, Vittorakis S, Semitekolou M, Alissafi T, Bossios A, et al. Osteopontin expression and relation to disease severity in human asthma. Eur Respir J 2011;37:331‐341. [DOI] [PubMed] [Google Scholar]
- 12. Shinohara ML, Lu L, Bu J, Werneck MB, Kobayashi KS, Glimcher LH, et al. Osteopontin expression is essential for interferon‐alpha production by plasmacytoid dendritic cells. Nat Immunol 2006;7:498‐506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cantor H, Shinohara ML. Regulation of T‐helper‐cell lineage development by osteopontin: the inside story. Nat Rev Immunol 2009;9:137‐141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Seita J, Asakawa M, Ooehara J, Takayanagi S, Morita Y, Watanabe N, et al. Interleukin‐27 directly induces differentiation in hematopoietic stem cells. Blood 2008;111:1903‐1912. [DOI] [PubMed] [Google Scholar]
- 15. Zhang QS, Benedetti E, Deater M, Schubert K, Major A, Pelz C, et al. Oxymetholone therapy of fanconi anemia suppresses osteopontin transcription and induces hematopoietic stem cell cycling. Stem Cell Reports 2015;4:90‐102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ge X, Lu Y, Leung TM, Sorensen ES, Nieto N. Milk osteopontin, a nutritional approach to prevent alcohol‐induced liver injury. Am J Physiol Gastrointest Liver Physiol 2013;304:G929‐G939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lazaro R, Wu R, Lee S, Zhu NL, Chen CL, French SW, et al. Osteopontin deficiency does not prevent but promotes alcoholic neutrophilic hepatitis in mice. Hepatology 2015;61:129‐140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ge X, Leung TM, Arriazu E, Lu Y, Urtasun R, Christensen B, et al. Osteopontin binding to lipopolysaccharide lowers tumor necrosis factor‐alpha and prevents early alcohol‐induced liver injury in mice. Hepatology 2014;59:1600‐1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhou H, Yu M, Zhao J, Martin BN, Roychowdhury S, McMullen MR, et al. IRAKM‐Mincle axis links cell death to inflammation: pathophysiological implications for chronic alcoholic liver disease. Hepatology 2016;64:1978‐1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Li M, He Y, Zhou Z, Ramirez T, Gao Y, Gao Y, et al. MicroRNA‐223 ameliorates alcoholic liver injury by inhibiting the IL‐6‐p47phox‐oxidative stress pathway in neutrophils. Gut 2017;66:705‐715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ioannou GN, Dominitz JA, Weiss NS, Heagerty PJ, Kowdley KV. The effect of alcohol consumption on the prevalence of iron overload, iron deficiency, and iron deficiency anemia. Gastroenterology 2004;126:1293‐1301. [DOI] [PubMed] [Google Scholar]
- 22. Pullarkat V. Iron overload in patients undergoing hematopoietic stem cell transplantation. Adv Hematol 2010;2010.pii:345756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yagi K. Simple assay for the level of total lipid peroxides in serum or plasma. Methods Mol Biol 1998;108:101‐106. [DOI] [PubMed] [Google Scholar]
- 24. Cosgun KN, Rahmig S, Mende N, Reinke S, Hauber I, Schafer C, et al. Kit regulates HSC engraftment across the human‐mouse species barrier. Cell Stem Cell 2014;15:227‐238. [DOI] [PubMed] [Google Scholar]
- 25. Pearson S, Sroczynska P, Lacaud G, Kouskoff V. The stepwise specification of embryonic stem cells to hematopoietic fate is driven by sequential exposure to Bmp4, activin A, bFGF and VEGF. Development 2008;135:1525‐1535. [DOI] [PubMed] [Google Scholar]
- 26. Petit I, Jin D, Rafii S. The SDF‐1‐CXCR4 signaling pathway: a molecular hub modulating neo‐angiogenesis. Trends Immunol 2007;28:299‐307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Heissig B, Hattori K, Dias S, Friedrich M, Ferris B, Hackett NR, et al. Recruitment of stem and progenitor cells from the bone marrow niche requires MMP‐9 mediated release of kit‐ligand. Cell 2002;109:625‐637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li B, Sharpe EE, Maupin AB, Teleron AA, Pyle AL, Carmeliet P, et al. VEGF and PlGF promote adult vasculogenesis by enhancing EPC recruitment and vessel formation at the site of tumor neovascularization. FASEB J 2006;20:1495‐1497. [DOI] [PubMed] [Google Scholar]
- 29. Mantel CR, O'Leary HA, Chitteti BR, Huang X, Cooper S, Hangoc G, et al. Enhancing hematopoietic stem cell transplantation efficacy by mitigating oxygen shock. Cell 2015;161:1553‐1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Petit I, Szyper‐Kravitz M, Nagler A, Lahav M, Peled A, Habler L, et al. G‐CSF induces stem cell mobilization by decreasing bone marrow SDF‐1 and up‐regulating CXCR4. Nat Immunol 2002;3:687‐694. [DOI] [PubMed] [Google Scholar]
- 31. Cacalano G, Lee J, Kikly K, Ryan AM, Pitts‐Meek S, Hultgren B, et al. Neutrophil and B cell expansion in mice that lack the murine IL‐8 receptor homolog. Science 1994;265:682‐684. [DOI] [PubMed] [Google Scholar]
- 32. Hoggatt J, Pelus LM. Mobilization of hematopoietic stem cells from the bone marrow niche to the blood compartment. Stem Cell Res Ther 2011;2:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Neuman MG, Schmilovitz‐Weiss H, Hilzenrat N, Bourliere M, Marcellin P, Trepo C, et al. Markers of inflammation and fibrosis in alcoholic hepatitis and viral hepatitis C. Int J Hepatol 2012;2012:231210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Takubo K, Goda N, Yamada W, Iriuchishima H, Ikeda E, Kubota Y, et al. Regulation of the HIF‐1alpha level is essential for hematopoietic stem cells. Cell Stem Cell 2010;7:391‐402. [DOI] [PubMed] [Google Scholar]
- 35. Morikawa T, Takubo K. Hypoxia regulates the hematopoietic stem cell niche. Pflugers Arch Eur J Physiol 2016;468:13‐22. [DOI] [PubMed] [Google Scholar]
- 36. Nombela‐Arrieta C, Pivarnik G, Winkel B, Canty KJ, Harley B, Mahoney JE, et al. Quantitative imaging of haematopoietic stem and progenitor cell localization and hypoxic status in the bone marrow microenvironment. Nat Cell Biol 2013;15:533‐543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Semenza GL, Wang GL. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol Cell Biol 1992;12:5447‐5454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mazur A, Feillet‐Coudray C, Romier B, Bayle D, Gueux E, Ruivard M, et al. Dietary iron regulates hepatic hepcidin 1 and 2 mRNAs in mice. Metabolism 2003;52:1229‐1231. [DOI] [PubMed] [Google Scholar]
- 39. Gutteridge JM. Iron promoters of the Fenton reaction and lipid peroxidation can be released from haemoglobin by peroxides. FEBS Lett 1986;201:291‐295. [DOI] [PubMed] [Google Scholar]
- 40. Gawel S, Wardas M, Niedworok E, Wardas P. Malondialdehyde (MDA) as a lipid peroxidation marker. [in Polish] Wiad Lek 2004;57:453‐455. [PubMed] [Google Scholar]
- 41. Crosby HA, Lalor PF, Ross E, Newsome PN, Adams DH. Adhesion of human haematopoietic (CD34+) stem cells to human liver compartments is integrin and CD44 dependent and modulated by CXCR3 and CXCR4. J Hepatol 2009;51:734‐749. [DOI] [PubMed] [Google Scholar]
- 42. Muench MO, Beyer AI, Fomin ME, Thakker R, Mulvaney US, Nakamura M, et al. The adult livers of immunodeficient mice support human hematopoiesis: evidence for a hepatic mast cell population that develops early in human ontogeny. PLoS One 2014;9:e97312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Storan MJ, Heazlewood SY, Heazlewood CK, Haylock DN, Alexander WS, Neaves RJ, et al. Brief report: factors released by megakaryocytes thrombin cleave osteopontin to negatively regulate hematopoietic stem cells. Stem Cells 2015;33:2351‐2357. [DOI] [PubMed] [Google Scholar]
- 44. Grassinger J, Haylock DN, Storan MJ, Haines GO, Williams B, Whitty GA, et al. Thrombin‐cleaved osteopontin regulates hemopoietic stem and progenitor cell functions through interactions with alpha9beta1 and alpha4beta1 integrins. Blood 2009;114:49‐59. [DOI] [PubMed] [Google Scholar]
- 45. Stier S, Ko Y, Forkert R, Lutz C, Neuhaus T, Grunewald E, et al. Osteopontin is a hematopoietic stem cell niche component that negatively regulates stem cell pool size. J Exp Med 2005;201:1781‐1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Boyerinas B, Zafrir M, Yesilkanal AE, Price TT, Hyjek EM, Sipkins DA. Adhesion to osteopontin in the bone marrow niche regulates lymphoblastic leukemia cell dormancy. Blood 2013;121:4821‐4831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bertola A, Park O, Gao B. Chronic plus binge ethanol feeding synergistically induces neutrophil infiltration and liver injury in mice: a critical role for E‐selectin. Hepatology 2013;58:1814‐1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mathews S, Feng D, Maricic I, Ju C, Kumar V, Gao B. Invariant natural killer T cells contribute to chronic‐plus‐binge ethanol‐mediated liver injury by promoting hepatic neutrophil infiltration. Cell Mol Immunol 2016;13:206‐216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ramirez T, Li YM, Yin S, Xu MJ, Feng D, Zhou Z, et al. Aging aggravates alcoholic liver injury and fibrosis in mice by downregulating sirtuin 1 expression. J Hepatol 2017;66:601‐609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Leonard SS, Harris GK, Shi X. Metal‐induced oxidative stress and signal transduction. Free Radic Biol Med 2004;37:1921‐1942. [DOI] [PubMed] [Google Scholar]
- 51. Liu Q, Davidoff O, Niss K, Haase VH. Hypoxia‐inducible factor regulates hepcidin via erythropoietin‐induced erythropoiesis. J Clin Invest 2012;122:4635‐4644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sasaki Y, Noguchi‐Sasaki M, Yasuno H, Yorozu K, Shimonaka Y. Erythropoietin stimulation decreases hepcidin expression through hematopoietic activity on bone marrow cells in mice. Int J Hematol 2012;96:692‐700. [DOI] [PubMed] [Google Scholar]
- 53. Chambers SM, Shaw CA, Gatza C, Fisk CJ, Donehower LA, Goodell MA. Aging hematopoietic stem cells decline in function and exhibit epigenetic dysregulation. PLoS Biol 2007;5:e201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Fairweather‐Tait SJ, Wawer AA, Gillings R, Jennings A, Myint PK. Iron status in the elderly. Mech Ageing Dev 2014;136‐137:22‐28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Neuman MG, Maor Y, Nanau RM, Melzer E, Mell H, Opris M, et al. Alcoholic liver disease: role of cytokines. Biomolecules 2015;5:2023‐2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Apte UM, Banerjee A, McRee R, Wellberg E, Ramaiah SK. Role of osteopontin in hepatic neutrophil infiltration during alcoholic steatohepatitis. Toxicol Appl Pharmacol 2005;207:25‐38. [DOI] [PubMed] [Google Scholar]
- 57. Conrad ME, Umbreit JN. Pathways of iron absorption. Blood Cells Mol Dis 2002;29:336‐355. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found at http://onlinelibrary.wiley.com/doi/10.1002/hep4.1116/full.
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information