

Abstract
Nonalcoholic fatty liver disease (NAFLD) contributes to the pathogenesis of type 2 diabetes and cardiovascular disease, and patients with nonalcoholic steatohepatitis (NASH) are also at risk of developing cirrhosis, liver failure, and hepatocellular carcinoma. To date, no specific therapy exists for NAFLD/NASH, which has been recognized as one of the major unmet medical needs of the twenty‐first century. We recently identified serine/threonine protein kinase (STK)25 as a critical regulator of energy homeostasis and NAFLD progression. Here, we investigated the effect of antisense oligonucleotides (ASOs) targeting Stk25 on the metabolic and molecular phenotype of mice after chronic exposure to dietary lipids. We found that Stk25 ASOs efficiently reversed high‐fat diet‐induced systemic hyperglycemia and hyperinsulinemia, improved whole‐body glucose tolerance and insulin sensitivity, and ameliorated liver steatosis, inflammatory infiltration, apoptosis, hepatic stellate cell activation, and nutritional fibrosis in obese mice. Moreover, Stk25 ASOs suppressed the abundance of liver acetyl‐coenzyme A carboxylase (ACC) protein, a key regulator of both lipid oxidation and synthesis, revealing the likely mechanism underlying repression of hepatic fat accumulation by ASO treatment. We also found that STK25 protein levels correlate significantly and positively with NASH development in human liver biopsies, and several common nonlinked single‐nucleotide polymorphisms in the human STK25 gene are associated with altered liver fat, supporting a critical role of STK25 in the pathogenesis of NAFLD in humans. Conclusion: Preclinical validation for the metabolic benefit of pharmacologically inhibiting STK25 in the context of obesity is provided. Therapeutic intervention aimed at reducing STK25 function may provide a new strategy for the treatment of patients with NAFLD, type 2 diabetes, and related complex metabolic diseases. (Hepatology Communications 2018;2:69–83)
Abbreviations
- ACC
acetyl‐coenzyme A carboxylase
- ALT
alanine aminotransferase
- ASO
antisense oligonucleotide
- AMPK
adenosine monophosphate‐activated protein kinase
- bEND.3
brain endothelial
- CoA
coenzyme A
- GTT
glucose tolerance test
- HOMA‐IR
model assessment score of insulin resistance
- mRNA
messenger RNA
- NAFL
nonalcoholic fatty liver
- NAFLD
nonalcoholic fatty liver disease
- NAS
nonalcoholic fatty liver disease activity score
- NASH
nonalcoholic steatohepatitis
- PBS
phosphate‐buffered saline
- PCR
polymerase chain reaction
- SNP
single nucleotide polymorphism
- STK25
serine/threonine protein kinase 25
- TAG
triacylglycerol
- TUNEL
terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick‐end labeling
- VLDL
very low‐density lipoprotein
- vol.
volume
Introduction
Nonalcoholic fatty liver disease (NAFLD), comprising nonalcoholic fatty liver (NAFL) and nonalcoholic steatohepatitis (NASH), is emerging as a leading cause of advanced liver disease worldwide. NAFLD has an estimated prevalence ranging from 20% to 30% in Europe and the Middle East and affects one third of the adult population in the United States.1 In addition to benign lipid accumulation in the liver (steatosis) characterizing NAFL, NASH patients display hepatic inflammation, fibrosis, and hepatocellular damage in the form of ballooning and apoptosis. NAFLD actively contributes to the pathogenesis of type 2 diabetes and cardiovascular disease, and NASH is emerging as a dominant cause of hepatocellular carcinoma, the third leading cause of cancer mortality.2, 3 To date, no pharmacological treatment exists for NAFLD, which is projected to become the major underlying etiology for liver transplantation by 2020.2 With this growing burden of NAFLD, it is critical to understand the molecular pathogenesis of the disease and to develop safe and effective pharmacologic therapies for its prevention and treatment.
We recently identified serine/threonine protein kinase (STK)25, a member of the sterile 20 kinase superfamily,4 as a critical regulator of whole‐body energy homeostasis.5, 6, 7, 8, 9, 10, 11, 12 We found that genetic disruption of STK25 in mice challenged with a high‐fat diet prevents liver steatosis and is accompanied by better preserved systemic glucose tolerance and insulin sensitivity compared to wild‐type littermates.5 In contrast, high‐fat‐fed transgenic mice overexpressing STK25 develop aggravated liver steatosis combined with more severe whole‐body glucose intolerance and insulin resistance compared to wild‐type mice.6, 9 Furthermore, we found that Stk25 –/– mice are protected against NASH induced by a methionine and choline‐deficient diet, as evidenced by dramatic suppression of hepatic lipid storage, oxidative stress, hepatocellular damage, inflammation, and nutritional fibrosis in the liver.7 Conversely, methionine and choline‐deficient diet‐induced NASH is markedly exacerbated in Stk25 transgenic mice compared with their wild‐type littermates.7 Consistent with these results from mouse models, we observed that knockdown of STK25 by small interfering RNA in human cultured hepatocytes attenuates lipid accumulation and improves insulin action, whereas the reciprocal effect was seen with STK25 overexpression.8 We also found a statistically significant positive correlation between STK25 messenger RNA (mRNA) expression and three individual histologic components of the NAFLD activity score (NAS; steatosis, lobular inflammation, and hepatocellular ballooning) in human liver biopsies.7 Interestingly, both in human and mouse liver cells, STK25 protein coats the surface of intrahepatocellular lipid droplets.6, 8
Our previous studies identified STK25 as a potential new‐in‐class drug target for the treatment of NAFLD, type 2 diabetes, and related metabolic diseases. The objective of the current study was to demonstrate in vivo proof‐of‐principle for the metabolic benefit of pharmacologically inhibiting STK25 function in mice by using generation 2.5 antisense oligonucleotides (ASOs).13 Antisense technology exploits a cellular ribonuclease H mechanism to specifically degrade the target mRNA in an mRNA–ASO duplex.14 Second‐generation ASOs are potent and specific inhibitors of gene expression,14 and mipomersen (Kynamro), a second‐generation ASO targeting liver‐derived apolipoprotein B‐100 for the treatment of homozygous familial hypercholesterolemia, was the first systemically administered ASO to achieve U.S. Federal Drug Administration approval.15 Recently, generation 2.5 ASO chemistry used in this study was developed; this enhances ASO potency 3‐fold to 5‐fold.13, 16, 17 Previous pharmacokinetic studies have shown that using generation 2.5 ASOs results in significant target reduction in peripheral organs, including liver, muscle, and adipose tissue, without penetrating the blood–brain barrier.17 This chemical class of ASOs has been widely used in preclinical experimental models18, 19, 20 and is also currently being evaluated in various clinical trials20 (ClinicalTrials.gov numbers NCT02983578, NCT02499328, NCT02549651, NCT02144051).
The results of the present study demonstrate that repressing STK25 abundance in mice by ASOs effectively reverses the diet‐induced impairment in glucose and insulin homeostasis and ameliorates liver steatosis, inflammation, and fibrosis in the context of obesity. Further investigations of the potential therapeutic benefit of pharmacological STK25 inhibitors as new‐in‐class drug candidates for NAFLD, type 2 diabetes, and related metabolic diseases in humans are warranted.
Materials and Methods
GENERATION OF ASOs
The oligonucleotides used in this study were 16 nucleotides in length and chemically modified with phosphorothioates in the backbone, three 2′‐4′ constrained ethyl residues at each terminus, and a central deoxynucleotide region of 10 residues (3‐10‐3 gapmer design). Oligonucleotides were synthesized as previously described.13 Stk25 ASO sequences used in this study were ASO#1 5′‐GGACGATTCGAGTACT‐3′ and ASO#2 5′‐GCATAATCCCCTAGGC‐3′. ASOs were dissolved in phosphate‐buffered saline (PBS) (without Ca2+ or Mg2+; Invitrogen, Carlsbad, CA) for in vivo experiments.
CELL CULTURE EXPERIMENTS
Mouse brain endothelial (bEND.3) cells (American Type Culture Collection, Manassas, VA) were cultured in Dulbecco's modified Eagle's medium with high glucose (Lonza, Basel, Switzerland) supplemented with 10% (volume [vol.]/vol.) fetal bovine serum (Invitrogen) at 37°C and 5% CO2. ASOs were administered to cells at the indicated concentrations by electroporation using the HT‐200 BTX Electroporator with the ElectroSquare Porator (ECM 830) voltage source in 96‐well BTX electroporation plates (2 mm; Harvard Apparatus, Holliston, MA), and cells were plated. Cells were harvested 16 hours after electroporation, and total RNA was isolated using glass fiber filter plates (Pall Corporation, Port Washington, NY). The amount of Stk25 mRNA was quantified using a quantitative real‐time polymerase chain reaction (PCR) assay on the QuantStudio 7 instrument (Applied Biosystems, Foster City, CA). The sequences of primers and probes used to detect Stk25 mRNA were as follows: forward: 5′‐GTCCTTATATCACCCGCTACTTC‐3′, reverse: 5′‐AGGTTTCAGCAAGTCCAGTG‐3′; probe: 5′‐Fam‐ACAGCTTGGTGCTCTTCAGGTAGG‐Iowa Black‐3′ (IDT, San Diego, CA). PCR results were normalized to total RNA measured with Quant‐iT RiboGreen RNA reagent (Molecular Probes, Eugene, OR).
ANIMAL EXPERIMENTS
Male mice of C57BL/6J strain (Charles River, Sulzfeld, Germany; Jackson, Sacramento, CA) were housed four to eight animals per cage in a temperature‐controlled (21°C) facility with a 12‐hour light–dark cycle with free access to water and food. To assess the efficacy of Stk25 ASOs in reducing the target gene expression, 6‐week‐old mice fed a regular chow diet were treated with Stk25 ASO#1 or ASO#2 in PBS at the indicated concentrations or placebo (PBS), twice weekly for 4 weeks by intraperitoneal injections. At the age of 10 weeks, the mice were killed. Blood was collected by heart puncture for analysis of plasma chemistry, and body and organ weights were recorded. Liver samples were snap frozen in liquid nitrogen and stored at –80°C for analysis of Stk25 mRNA expression.
To assess the metabolic effect of Stk25 ASOs, 6‐week‐old mice were fed a high‐fat diet (45 kcal% fat; D12451; Research Diets, New Brunswick, NJ) for 21 weeks and were treated with Stk25 ASO#1 or ASO#2 in PBS (50 mg/kg/week) or placebo (PBS), by intraperitoneal injections twice weekly for the last 6 weeks (Supporting Fig. S1). Body weight was recorded weekly during the treatment period. Baseline glucose and insulin levels were measured at different time points during the study using blood samples collected by tail bleed after 4 hours of food withdrawal. At the age of 27 weeks, the mice were killed after 4 hours of food withdrawal. Blood was collected by heart puncture for analysis of triacylglycerol (TAG) and cholesterol. Liver samples were collected for histologic analysis (see below). Liver, gastrocnemius skeletal muscle, and subcutaneous white adipose tissue were also snap frozen in liquid nitrogen and stored at –80°C for biochemical assays and analysis of gene and/or protein expression.
All animal experiments were performed after prior approval from the local Ethics Committee for Animal Studies at the Administrative Court of Appeals in Gothenburg, Sweden, or by the Ionis Pharmaceuticals Institutional Animal Care and Use Committee, Carlsbad, CA, following appropriate guidelines.
BIOCHEMICAL ASSAYS
Plasma albumin, bilirubin, blood urea nitrogen, creatinine, alanine aminotransferase (ALT), and aspartate aminotransferase (AST) were measured using an AU480 Clinical Chemistry Analyzer (Beckman Coulter, Providence, RI). Basal blood glucose and plasma insulin were assessed using an Accu‐Chek glucometer (Roche Diagnostics, Basel, Switzerland) and the Ultrasensitive Mouse Insulin Enzyme‐Linked Immunosorbent Assay kit (Crystal Chem, Downers Grove, IL), respectively. Plasma levels of TAG and cholesterol were measured using the Infinity TAG and the Infinity Cholesterol kits, respectively (Thermo Fisher Scientific, Waltham, MA), with Multiconstituent Calibrator 1E65‐04 (Abbott, North Chicago, IL) used as reference. For hepatic TAG measurements, lipids were extracted from liver samples and analyzed using straight‐phase high‐performance liquid chromatography as described.21 Hepatic collagen deposition was assessed in liver homogenates using the Hydroxyproline Colorimetric Assay kit (Sigma‐Aldrich, St. Louis, MO).
GLUCOSE AND INSULIN TOLERANCE TESTS
Following 4 hours of food withdrawal, mice were injected with glucose (1 g/kg; Fresenius Kabi, Bad Homburg, Germany) or human recombinant insulin (1 U/kg; Actrapid Penfill; Novo Nordisk, Bagsværd, Denmark) intraperitoneally at time 0 for the glucose tolerance test (GTT) and insulin tolerance test (ITT), respectively. Blood was taken from the tail tip to determine blood glucose concentrations at 0, 15, 30, 60, 90, and 120 minutes postinjection, using an Accu‐Chek glucometer.
HISTOLOGY
Liver samples were fixed with 4% (vol./vol.) phosphate‐buffered formaldehyde (Histolab Products, Gothenburg, Sweden), embedded in paraffin, sectioned, and stained with hematoxylin and eosin. Total hepatic lipid area was assessed in five randomly selected 200× microscopic fields per mouse, using ImageJ version 1.47 (Wayne Rasband, National Institutes of Health, Bethesda, MD). Ten randomly selected 200× microscopic fields per mouse were assessed for the NAS according to Kleiner/Brunt criteria.22, 23, 24, 25 To estimate the degree of fibrosis, liver sections were stained with picrosirius red (Histolab Products), counterstained with Fast Green (Sigma‐Aldrich), and the stained area was measured in five randomly selected 200× microscopic fields per mouse. Ten randomly selected 200× microscopic fields per mouse were also assessed for fibrosis score according to Kleiner/Brunt criteria.22, 23, 26 In parallel, liver samples were embedded in optimal cutting temperature mounting medium (Histolab Products) and frozen in liquid nitrogen, followed by cryosectioning and staining with Oil Red O (Sigma‐Aldrich).
IMMUNOHISTOCHEMISTRY AND IMMUNOFLUORESCENCE
Liver sections were incubated with primary antibodies, followed by incubation with biotinylated secondary antibodies. See Supporting Material for antibody information.
QUANTITATIVE REAL‐TIME PCR AND WESTERN BLOT ANALYSIS IN MOUSE TISSUE SAMPLES
See Supporting Material for details.
WESTERN BLOT ANALYSIS IN LIVER BIOPSIES OF HUMAN PARTICIPANTS
The protein abundance of STK25 was measured in liver tissue samples obtained from 10 Caucasian individuals who fulfilled the following inclusion criteria: NAFL verified by computed tomography and/or ultrasound, laboratory signs (elevated aminotransferases), and/or liver elastography findings (fibrosis 2‐3) suggestive of NASH. A typical ultrasound‐guided Menghini liver biopsy was performed, of which one part was transferred to ice‐cold 4‐(2‐hydroxyethyl)‐1‐piperazine ethanesulfonic acid buffer, and stored at −80°C until further preparations. All liver biopsies were collected between 8 am and 10 am after an overnight fast. NAS was assessed on liver sections by a certified pathologist, according to the Brunt scoring system.27 Western blot analysis was carried out as described above. All participants gave their written informed consent before taking part in the study. All investigations were approved by the Ethics Committee of the University of Gothenburg, Sweden (Dnr. 1062‐11) and were carried out in accordance with the Declaration of Helsinki.
GENETIC ASSOCIATION ANALYSIS IN HUMAN PARTICIPANTS
From the ongoing Tübingen Family study for type 2 diabetes,28 we selected 430 healthy subjects from the southern part of Germany who were at risk for type 2 diabetes (i.e., with family history of diabetes, body mass index ≥27, impaired fasting glycemia, and/or previous gestational diabetes) and who had liver fat measured by 1H‐magnetic resonance spectroscopy (localized stimulated echo acquisition mode). The subjects had a mean age of 43 years (range, 18‐69 years), a mean body mass index of 30 kg/m2 (range, 19‐47 kg/m2), a mean liver fat content of 5.9% (range, 0.2%‐30.1%), and 65% were female. This study population was genotyped for 15 intronic tagging single nucleotide polymorphisms (SNPs) selected based on linkage disequilibrium information from the 1000 Genomes project covering 97% of the common genetic variation (minor allele frequency ≥0.1) in the STK25 gene. Genotyping was performed by mass spectrometry using the massARRAY system from Sequenom and the manufacturer's iPLEX software (Sequenom, Hamburg, Germany). Assay design for a sixteenth SNP necessary to cover 100% of the common variation failed. Four of the 15 genotyped SNPs were not in Hardy–Weinberg equilibrium and were excluded from analyses.
STATISTICAL ANALYSIS
Statistical significance between the groups was evaluated using the unpaired two‐tailed Student t test and among more than two groups by analysis of one‐way analysis of variance with the t test for post‐hoc analysis. Differences were considered statistically significant at P < 0.05. Statistical analyses were performed using SPSS Statistics version 22 (IBM Corp., Armonk, NY). For details on SNP analyses, see Supporting Fig. S5.
Results
Stk25 ASO TREATMENT EFFECTIVELY REDUCES Stk25 mRNA LEVELS IN VITRO AND IN VIVO
We synthesized over 400 generation 2.5 constrained ethyl ASOs,13 each 16 base pairs in length and designed to target the mouse Stk25 gene. We then evaluated their ability to reduce Stk25 mRNA expression in mouse bEND.3 cells. We found that numerous ASOs were active in repressing Stk25 mRNA levels (data not shown); however, two ASOs (Stk25 ASO#1 and #2), complementary to the 3′ untranslated region and intronic region of the Stk25 gene, respectively, demonstrated the highest potency, exhibiting more than 90% target reduction with doses higher than 2.2 μM (Fig. 1A).
Figure 1.

Stk25 ASOs effectively repressed STK25 levels in vitro and in vivo. (A) Dose‐dependent reduction of Stk25 mRNA in mouse bEND.3 cells. Data are mean ± SD (n = 4) compared with sham‐transfected control. (B) Dose‐dependent reduction of Stk25 mRNA in the liver of chow‐fed lean mice treated with Stk25 ASOs for 4 weeks. Data are mean ± SD (n = 4) compared with control group of mice treated with PBS. (C,D) Reduction of (C) Stk25 mRNA and (D) protein abundance in the liver of high‐fat‐fed mice treated with Stk25 ASOs (50 mg/kg/week) for 6 weeks. Data in (C) are mean ± SEM (n = 8) compared with control group of mice treated with PBS. In (D) representative western blots are shown with β‐actin used as loading control. **P < 0.01. Abbreviations: TG, Stk25 transgenic mice; w, week.
To evaluate the in vivo efficacy of Stk25 ASOs, we treated mice fed a regular chow diet twice weekly for 4 weeks with 12.5, 25.0, or 50.0 mg/kg/week of Stk25 ASO#1 or ASO#2. At study termination, we observed a dose‐dependent reduction of hepatic Stk25 mRNA with both ASOs (Fig. 1B), with over 90% repression in mRNA expression at the high‐dose level. Body weights, organ (liver, kidney, and spleen) weights, and plasma chemistry parameters (albumin, bilirubin, blood urea nitrogen, creatinine, ALT, and aspartate aminotransferase) remained within normal reference values in both treatment groups (data not shown).
TREATMENT WITH Stk25 ASOs REVERSES METABOLIC DEFECTS IN GLUCOSE AND INSULIN HOMEOSTASIS IN A DIET‐INDUCED TYPE 2 DIABETES MODEL IN MICE
To evaluate the metabolic effect of Stk25 ASOs in vivo, we treated mice on a high‐fat diet (for 21 weeks) with Stk25 ASO#1 or ASO#2 in PBS (50 mg/kg/week) or placebo (PBS), twice weekly for the last 6 weeks of the diet (Supporting Fig. S1). Hepatic Stk25 mRNA expression was markedly lower in groups treated with ASO#1 or ASO#2 compared with the placebo group of mice (Fig. 1C), whereas the protein abundance of STK25 was below the detection limit of western blot in the livers of ASO‐treated mice (Fig. 1D). Notably, STK25 protein was readily detected by western blot in skeletal muscle samples of control as well as ASO‐treated mice, without any difference between the groups (Supporting Fig. S2A). We observed considerable variation in STK25 protein levels in white adipose tissue in the control group of mice, and STK25 levels were below the detection limit of western blot in a number of mice both in PBS‐ and ASO‐treated groups (Supporting Fig. S2B).
We measured fasting circulating levels of glucose and insulin at different time points during the study. As expected, significant hyperglycemia and hyperinsulinemia were established in mice during 12‐14 weeks of dietary stress challenge (i.e., before ASO treatment was initiated; Fig. 2A,B). Notably, plasma insulin levels were dramatically reduced in mice by treatment with ASO#1 or ASO#2 compared with the placebo group, while blood glucose was significantly lowered only by Stk25 ASO#2 (Fig. 2A,B). In fact, fasting levels of glucose and insulin at the end of the treatment period with ASO#2 were significantly decreased compared to the values measured prior to administration and were similar to the values before high‐fat diet feeding was initiated, suggesting that the diet‐induced hyperglycemia and hyperinsulinemia were fully reversed by ASO#2 (Fig. 2A,B). Consistently, the homeostasis model assessment score of insulin resistance (HOMA‐IR) was also lower throughout the treatment period with both ASOs compared with the placebo group of mice (Fig. 2C). At the end of the treatment period, the homeostasis model assessment score of insulin resistance (HOMA‐IR) in mice treated with ASO#2 was reduced to levels previously measured in age‐matched, lean, chow‐fed mice of the same genetic background.5
Figure 2.

Stk25 ASO treatment normalized glucose and insulin homeostasis in mice fed a high‐fat diet. (A‐C) Fasting circulating levels of (A) glucose and (B) insulin; (C) HOMA‐IR was calculated using the equation (fasting glucose [mg/dL] × fasting insulin [ng/mL])/405. The number of weeks on a high‐fat diet is show for each measurement; Stk25 ASO treatment was initiated after 14 weeks of high‐fat diet feeding. (D‐F) Intraperitoneal (D,E) GTT and (F) ITT in mice treated with ASO#1, ASO#2, or placebo for 5 weeks. The AUC for each test is shown. Data are mean ± SEM from six to eight mice per group. In bar diagrams, *P < 0.05, **P < 0.01, † P = 0.08. In line diagrams, ‡ P < 0.05 and ‡ ‡ P < 0.01 for ASO#1‐treated versus placebo group; # P < 0.05 and ## P < 0.01 for ASO#2‐treated versus placebo group. Abbreviations: AUC, area under the curve; HOMA‐IR, homeostasis model assessment score of insulin resistance; IPGTT, intraperitoneal glucose tolerance test; IPITT, intraperitoneal insulin tolerance test; w, week.
In the GTT experiment, blood glucose levels returned to normal more rapidly in Stk25 ASO‐treated mice than in placebo controls (Fig. 2D), revealing a better‐preserved glucose tolerance. Interestingly, in contrast to results shown in Fig. 2A, a significant reduction of basal blood glucose was recorded in the GTT experiment in both ASO‐treated groups, suggesting that ASO#1, similar to ASO#2, lowers fasting glucose levels, although this is not evident until later time points. Of note, glucose‐stimulated insulin secretion was significantly increased in both ASO‐treated groups during the GTT, despite lower net change in blood glucose concentration compared with the placebo group, suggesting improved β‐cell function (Fig. 2E). The insulin tolerance test (ITT) revealed marked improvement in insulin sensitivity in groups treated with Stk25 ASO#1 or #2 compared with the placebo group (Fig. 2F). Stk25 ASO treatment did not affect body weight (Supporting Fig. S3), and we observed no signs of local irritation or inflammation with macroscopic examination of the injection sites.
Stk25 ASO TREATMENT AMELIORATES LIVER STEATOSIS INDUCED BY FEEDING MICE A HIGH‐FAT DIET
Chronic exposure to dietary lipids is known to promote ectopic lipid accumulation in the liver.2 Consistently, upon dissections the livers of the placebo group of mice fed a high‐fat diet were visibly lipid laden, displaying a milky pale appearance (Fig. 3A). In contrast, the livers of Stk25 ASO‐treated mice challenged with a high‐fat diet remained similar in appearance to the lean chow‐fed mice (Fig. 3A). In line with this finding, microscopic examination of hematoxylin and eosin and Oil Red O‐stained liver sections revealed pronounced steatosis in the placebo group of mice, while the density and size of intrahepatocellular lipid droplets were dramatically reduced in Stk25 ASO‐treated mice (Fig. 3A). Of note, the histologic appearance of the livers of mice treated with ASO#1 or ASO#2 closely resembled that of age‐matched lean mice fed a regular chow diet as well as the livers of high‐fat‐fed Stk25 –/– mice of the same genetic background (Fig. 3A,B).
Figure 3.

Stk25 ASO treatment ameliorated diet‐induced liver steatosis. (A) Representative images of whole liver; representative liver sections stained with H&E or Oil Red O for lipids. Scale bars, 100 μm. (B) Representative liver sections stained with H&E from age‐matched, lean, chow‐fed mice as well as Stk25 knockout mice and their wild‐type littermates fed a high‐fat diet. Stk25 knockout mice were a gift from B. Howell, State University of New York Upstate Medical University, Syracuse, NY. Scale bars, 100 μm. (C) Total lipid area in liver sections. (D) TAG content in liver extract. Age‐matched, lean, chow‐fed mice of the same genetic background are included as a reference group. Data in (C,D) are mean ± SEM from six to eight mice per group. *P < 0.05, **P < 0.01, † P = 0.08. Abbreviations: CD, chow diet; H&E, hematoxylin and eosin; HFD, high‐fat diet; KO, knockout; WT, wild‐type.
Quantification of the total lipid area confirmed a 2.2 ± 0.1‐fold and 5.0 ± 0.1‐fold reduction in the liver sections of mice treated with Stk25 ASO#1 and ASO#2, respectively, compared with the placebo group (Fig. 3C). Notably, the hepatic lipid area in liver sections of mice treated with ASO#2 was similar to that previously measured in age‐matched, lean, chow‐fed mice (chow‐fed mice, 5.5% ± 0.4%7; high‐fat‐fed ASO#2‐treated mice, 5.0% ± 0.5%). Consistent with these histologic findings, biochemical analysis showed that the TAG content in liver extracts was significantly decreased in the ASO‐treated versus placebo group, although the TAG level in ASO‐treated mice remained higher compared to age‐matched, lean, chow‐fed mice (Fig. 3D).
The liver exports TAG to extrahepatic tissues through very low‐density lipoprotein (VLDL) secretion, and the formation of VLDL‐TAG particles is highly dependent on the availability of cytosolic TAG.29, 30 Consistent with this strong correlation between VLDL secretion and the levels of intrahepatic lipids, we found that fasting plasma concentrations of TAG were slightly lower in the ASO‐treated groups compared with the placebo group of mice (Supporting Fig. S4).
Stk25 ASO TREATMENT IN MICE FED A HIGH‐FAT DIET PROTECTS AGAINST LIVER FIBROSIS, META‐INFLAMMATION, AND HEPATOCELLULAR INJURY
We next evaluated the impact of changes in liver lipid deposition by Stk25 ASO administration on hepatic fibrosis, inflammation, and cell damage. We found that the Stk25 ASO treatment protected from diet‐induced perivenular/pericellular fibrosis, as studied by picrosirius red staining for collagen fibers (Fig. 4A,B). Moreover, immunostaining for α‐smooth muscle actin, a marker for activated hepatic stellate cells responsible for liver collagen deposition, was similarly reduced in ASO‐treated livers (Fig. 4A). Consistent with these histologic findings, the hydroxyproline content, a marker of collagen deposition, was 1.5 ± 0.1‐fold and 1.3 ± 0.04‐fold lower in the liver lysates from mice treated with ASO#1 and ASO#2, respectively, compared with placebo (Fig. 4C). Notably, the hepatic hydroxyproline levels in ASO‐treated mice were similar to those measured in age‐matched, lean, chow‐fed mice of the same genetic background (Fig. 4C).
Figure 4.

Stk25 ASO treatment protected against diet‐induced fibrosis, inflammation, and cellular damage in the liver. (A) Representative liver sections stained with picrosirius red or TUNEL. Sections were processed for immunohistochemistry with anti‐α‐SMA antibodies or for immunofluorescence with anti‐Gr1 (Ly6C) antibodies (pink) with nuclei stained with DAPI (blue). Scale bars, 100 μm. (B) Quantification of picrosirius red staining. (C) Hydroxyproline content in liver extract. Age‐matched, lean, chow‐fed mice of the same genetic background are included as a reference group. (D) Quantification of Gr1 (Ly6C) staining. (E) H&E‐stained liver sections of the placebo group of mice showing the presence of micro‐ and lipogranuloma, hepatocellular ballooning, and enlarged nuclei containing intranuclear vacuoles. Scale bars, 100 μm. Data in (B‐D) are mean ± SEM from six to eight mice per group. *P < 0.05, **P < 0.01. Abbreviations: CD, chow diet; DAPI, 4',6‐diamidino‐2‐phenylindole; H&E, hematoxylin and eosin; SMA, smooth muscle actin; TUNEL, terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick‐end labeling.
Several reports have suggested that freshly infiltrating monocyte‐derived macrophages characterized by high expression of Gr1 (Ly6C) have a vital role in the development and progression of NASH.31, 32, 33 We found that Gr1 (Ly6C)‐positive areas were approximately 2‐fold reduced in Stk25 ASO‐treated livers compared with the livers of the placebo group of mice (Fig. 4A,D). As a reference, our previous studies have shown only a very low level of Gr1 (Ly6C)‐positive inflammatory cells in the livers of chow‐fed lean mice (Gr1 [Ly6C]‐positive area, 0.1% ± 0.04%7). Furthermore, we observed frequent microgranulomatous lesions and lipogranulomas in the livers of the placebo group (Fig. 4E) but not in ASO‐treated mice, suggesting attenuation of lobular inflammation by Stk25 ASO administration.
Evidence of severe hepatocellular damage was readily observed in the placebo group of mice, as demonstrated by the presence of apoptotic hepatocytes detected by terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick‐end labeling (TUNEL) assay (Fig. 4A), ballooning degradation of hepatocytes, and enlarged nuclei containing intranuclear vacuoles (Fig. 4E); we did not detect these features in mice treated with Stk25 ASOs.
TREATMENT WITH Stk25 ASOs AMELIORATES DIET‐INDUCED NAFLD IN MICE
We further compared the liver pathology in ASO‐treated versus placebo‐treated mice by using NAS as well as fibrosis score based on the Kleiner/Brunt criteria adapted to rodents.22, 23, 24, 25, 26 We found that mice treated with Stk25 ASOs scored markedly lower both for NAS and fibrosis compared with the placebo group, while the differences were most pronounced in mice treated with ASO#2 (Fig. 5).
Figure 5.
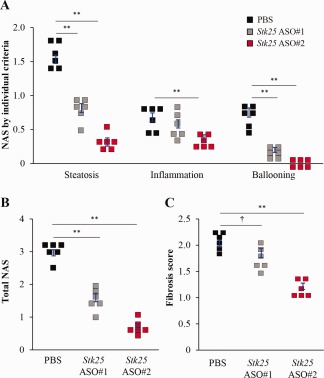
Stk25 ASO treatment in mice fed a high‐fat diet ameliorated the progression of NAFLD. (A,B) Quantification of NAS based on three histologic features (steatosis 0‐3, inflammation 0‐3, hepatocellular ballooning 0‐2) in H&E‐stained liver sections. (C) Quantification of fibrosis score based on Kleiner/Brunt criteria adapted to rodents (0, no fibrosis; 1, focal pericellular fibrosis in zone 3; 2, perivenular and pericellular fibrosis confined to zones 2 and 3; 3, bridging fibrosis; 4, cirrhosis) in liver sections stained with picrosirius red. Data are mean ± SEM from six mice per group. *P < 0.05, **P < 0.01, † P = 0.07. Abbreviation: H&E, hematoxylin and eosin.
Stk25 ASO#2 REPRESSES THE PROTEIN ABUNDANCE OF ACETYL‐COENZYME A CARBOXYLASE
Our previous results demonstrate decreased hepatic protein levels of acetyl‐coenzyme A (CoA) carboxylase (ACC), a key controller of the balance of lipid storage versus lipid oxidation, in high‐fat‐fed Stk25 –/– mice.5 Consistently, we found that the total protein abundance of ACC was 2.5 ± 0.5‐fold lower in Stk25 ASO#2‐treated livers compared with livers from the placebo group of mice (Fig. 6). ACC activity is regulated by phosphorylation, where kinases, such as adenosine monophosphate‐activated protein kinase (AMPK), directly phosphorylate and inactivate ACC.34 Similar to total protein, the levels of phosphorylated ACC were also reduced in Stk25 ASO#2‐treated livers, and there was no change in the phosphorylated ACC/ACC ratio (Fig. 6).
Figure 6.

Stk25 ASO treatment reduced ACC protein levels in high‐fat‐fed mice. Liver protein lysates were analyzed by western blot using antibodies specific for total ACC or phospho‐ACC (Ser79). Protein levels were analyzed by densitometry, and data are shown as the total and phospho‐protein abundance as well as the ratio of phospho‐protein to total protein, with values in the placebo group of mice set to 1. Representative western blots are shown with β‐actin used as a loading control. Data are mean ± SEM from seven to eight mice per group. *P < 0.05, **P < 0.01. Abbreviations: p‐ACC, phosphorylated ACC.
STK25 PROTEIN ABUNDANCE CORRELATES SIGNIFICANTLY AND POSITIVELY WITH NASH IN HUMAN LIVER
We previously reported a significant positive correlation in human liver biopsies between STK25 mRNA expression and NASH development.7 Here, we examined the levels of STK25 protein in relation to the development of NASH in liver biopsy material, where NAFLD was characterized by using the widely applied semiquantitative histologic scoring NAS.27 Consistent with the mRNA data, comparison of the patients with NAFL (i.e., those with a NAS of 1‐3/8) versus the patients with NASH (i.e., those with a NAS of 4‐8/8) showed that NASH patients displayed 2.6 ± 0.3‐fold higher STK25 protein levels (Fig. 7).
Figure 7.

Protein abundance of STK25 in human liver was found to be significantly increased in subjects with NASH. (A) Characteristics of NAFL and NASH subjects. Data are mean ± SEM. (B) Protein lysates from liver biopsies of individuals with NAFL (NAS of 1‐3/8; n = 3) versus NASH (NAS of 4‐8/8; n = 7) were analyzed by western blot using antibodies specific for STK25. Protein levels were analyzed by densitometry, and data are mean ± SEM with values in the NAFL group of subjects set to 1; *P < 0.05. Abbreviations: BMI, body mass index; T2DM, type 2 diabetes mellitus.
COMMON GENETIC VARIANTS IN THE HUMAN STK25 GENE ASSOCIATE WITH LIVER FAT CONTENT
Finally, we genotyped 430 participants of the Tübingen Family study for 11 common tagging SNPs covering major parts of the human STK25 gene and assessed their associations with liver fat content measured by magnetic resonance spectroscopy. After adjustment for sex and age, three SNPs (rs34506685, rs34751932, rs4675810) showed nominal and one SNP (rs6757649) showed significant associations. Homozygous minor allele carriers of rs34751932 and rs4675810 revealed 1.4‐fold and 1.7‐fold increased liver fat content, respectively, compared to homozygous major allele carriers (Supporting Fig. S5A,B), and homozygous minor allele carriers of rs6757649 and rs34506685 revealed 35% and 31% decreased liver fat content, respectively, compared to homozygous major allele carriers (Supporting Fig. S5C,D).
Discussion
This study found that systemic administration of Stk25 ASOs in mice significantly suppresses hepatic STK25 mRNA and protein abundance compared with placebo‐treated mice, reverses high‐fat diet‐induced systemic hyperglycemia and hyperinsulinemia, improves whole‐body glucose tolerance and insulin sensitivity, and ameliorates steatosis, inflammatory infiltration, nutritional fibrosis, and hepatocellular damage in the liver (Fig. 8). We observed no differences in body mass between the Stk25 ASO‐treated versus placebo groups, suggesting that repression of STK25 levels protects against the metabolic consequences of chronic exposure to dietary lipids independent of changes in obesity. The Stk25 ASOs were well tolerated and no systemic toxicity or local tolerability concerns were observed.
Figure 8.
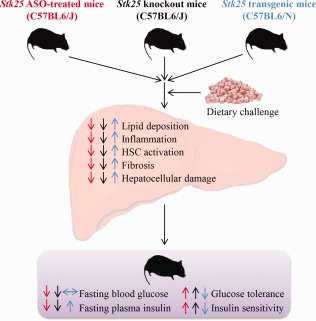
Schematic illustration of reciprocal metabolic responses in the liver as well as at the whole‐body level in mice with repressed STK25 function by either Stk25 ASO treatment or genetic disruption (indicated by red and black arrows, respectively) and transgenic mice overexpressing STK25 (indicated by blue arrows) compared with the corresponding control groups of mice. The phenotypes of Stk25 –/– and transgenic mice have been described.5, 6, 7, 9 Abbreviation: HSC, hepatic stellate cell.
STK25 is broadly expressed during embryonic development.35 Therefore, discrepancies between the phenotypic consequences of pharmacologically inhibiting STK25 by ASO treatment in adult mice described in this study versus the previously reported conventional Stk25 knockout phenotype5, 7 could be expected, attributed to possible developmental effects of this target. However, we found that the metabolic phenotype of Stk25 ASO‐treated mice was fully consistent with that of Stk25 –/– mice,5, 7 including observations of improved glucose and insulin homeostasis as well as attenuated liver lipid storage, inflammation, hepatic stellate cell activation, fibrosis, and hepatocellular damage (Fig. 8). Thus, STK25 knockdown early in development is not critical for its metabolic effects. The similarity in phenotype between treatment with Stk25 ASOs, which achieve the highest target inhibition in the liver, and whole‐body depletion of STK25 in knockout mice5, 7 also suggests that hepatic inhibition of STK25 is sufficient for its metabolic effects. Importantly, our current study extends our previous observations in Stk25 –/– mice, which showed prevention of high‐fat diet‐induced metabolic complications,5, 7 by demonstrating that STK25 inhibition can also reverse NAFLD and defects in systemic glucose and insulin homeostasis in adult mice after chronic exposure to dietary lipids. Of note, a striking pattern of opposing metabolic alterations is revealed when comparing the phenotype of Stk25 ASO‐treated mice described in this study to the previously reported phenotype of STK25‐overexpressing transgenic mice,6, 7, 9 further supporting the validity of the results (Fig. 8).
We previously reported that hepatic ACC protein abundance is significantly reduced in high‐fat‐fed Stk25 –/– versus wild‐type mice.5 This provides a likely mechanism underlying the reduced lipid accumulation in Stk25 –/– livers because ACC regulates both lipogenesis and β‐oxidation through its enzymatic product malonyl‐CoA. Malonyl‐CoA is a precursor of fatty acid synthesis and also represses long‐chain fatty acyl‐CoA uptake and oxidation in mitochondria through allosteric inhibition of carnitine palmitoyltransferase 1.36 Consistent with our observations in Stk25 –/– livers,5 hepatic ACC protein levels were significantly suppressed by Stk25 ASO treatment, suggesting that not only the phenotypic consequences but also their underlying molecular mechanisms are identical in Stk25 ASO‐treated and knockout mice.
Several lines of recent evidence support that STK25 has a similar role in the pathogenesis of NAFLD in human liver as described in mouse models. We found a significant positive correlation in human liver biopsies between NASH development and STK25 mRNA7 and protein abundance (in this study). In addition, we previously reported that STK25 knockdown in human hepatocytes suppresses lipid accumulation and increases insulin sensitivity8 and also represses NASH features, such as ALT secretion and oxidative damage.7 Furthermore, in this study we identified four common nonlinked intronic SNPs in the human STK25 gene that were associated with altered liver fat; two were associated with increased and two with decreased hepatic fat content. The divergent directionality of SNP effects may be explained by strengthening versus weakening effects of the independent noncoding base exchanges on transcription factor binding. Even though these human genetic data need replication and deeper functional exploration, they are in line with our findings in mice.
The results of this study warrant further investigation of the potential therapeutic benefit of pharmacologic STK25 inhibitors for the treatment of NAFLD. To date, no specific pharmacologic therapy is approved for NAFLD/NASH by the European Medicines Agency or the U.S. Food and Drug Administration. The results of phase II trials for the most advanced products in clinical development for NASH have recently been reported.37 Obeticholic acid, an agonist of farnesoid X receptor, achieved statistically significant improvement in the primary study endpoint (i.e., NAS without worsening of fibrosis) in a phase II trial, although a principal concern was the observation of pruritus38 (ClinicalTrials.gov number NCT01265498). Elafibranor, a dual peroxisome proliferator‐activated receptor‐α/δ agonist, failed to meet the predefined endpoint (i.e., resolution of NASH without fibrosis worsening) in the intention‐to‐treat population in a phase II trial; however, a post‐hoc analysis accounting for baseline patient heterogeneity and severity of disease demonstrated a dose‐dependent improvement in liver histology39 (ClinicalTrials.gov number NCT01694849). Interestingly, in a recently reported initial proof‐of‐concept trial in NASH patients, oral treatment with liver‐targeted, selective inhibitor of ACC GS‐0976 was shown to significantly improve liver steatosis as well as circulating biomarkers of liver fibrosis and cell death.40 These data suggest that the repression of hepatic ACC levels by Stk25 ASOs, which we observed in this study, may constitute a key mechanism for the effect of Stk25 ASOs not only on reducing liver steatosis but also for suppressing fibrosis and hepatocellular damage.
We found that the metabolic effect of ASO#2 was slightly more pronounced compared with ASO#1 for most of the parameters measured. We observed no evidence that ASO#2 was more efficient in suppressing the hepatic STK25 mRNA or protein levels at the time of study termination compared with ASO#1; however, it is possible that ASO#2 is slightly superior in pharmacokinetic properties, such as improved distribution to the disease‐relevant cell type in liver.
Taken together, the results of metabolic and molecular characterization of mice treated with Stk25 ASOs provide preclinical in vivo proof‐of‐principle for the metabolic benefit of pharmacologic STK25 inhibitors in conditions of excess dietary fuels in a mouse model and suggest that therapeutic intervention aimed at reducing STK25 function may provide a new strategy for the treatment of patients with NAFLD, type 2 diabetes, and related metabolic diseases.
Author names in bold designate shared co‐first authorship.
Supporting information
Additional Supporting Information may be found at onlinelibrary.wiley.com/doi/10.1002/hep4.1128/full.
Supporting Information 1
Supporting Information Figure 1
Supporting Information Figure 2
Supporting Information Figure 3
Supporting Information Figure 4
Supporting Information Figure 5
Potential conflict of interest: Nothing to report.
Supported by grants from the Swedish Research Council, the Novo Nordisk Foundation, the Swedish Heart and Lung Foundation, the Diabetes Wellness Network Sweden, the Estonian Research Council, the Swedish Diabetes Foundation, the Royal Society of Arts and Sciences in Gothenburg, the Wiberg Foundation, the Adlerbert Research Foundation, the I. Hultman Foundation, the S. and E. Goljes Foundation, the West Sweden ALF Program, the F. Neubergh Foundation, the I.‐B. and A. Lundbergs Research Foundation, the Swedish Innovation Agency Vinnova, the L. and J. Grönbergs Foundation, and the European Foundation for the Study of Diabetes and Novo Nordisk Partnership for Diabetes Research in Europe.
REFERENCES
- 1. Loomba R, Sanyal AJ. The global NAFLD epidemic. Nat Rev Gastroenterol Hepatol 2013;10:686‐690. [DOI] [PubMed] [Google Scholar]
- 2. Anstee QM, Targher G, Day CP. Progression of NAFLD to diabetes mellitus, cardiovascular disease or cirrhosis. Nat Rev Gastroenterol Hepatol 2013;10:330‐344. [DOI] [PubMed] [Google Scholar]
- 3. Rinella ME. Nonalcoholic fatty liver disease: a systematic review. JAMA 2015;313:2263‐2273. [DOI] [PubMed] [Google Scholar]
- 4. Thompson BJ, Sahai E. MST kinases in development and disease. J Cell Biol 2015;210:871‐882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Amrutkar M, Cansby E, Chursa U, Nunez‐Duran E, Chanclon B, Stahlman M, et al. Genetic disruption of protein kinase STK25 ameliorates metabolic defects in a diet‐induced type 2 diabetes model. Diabetes 2015;64:2791‐2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Amrutkar M, Cansby E, Nunez‐Duran E, Pirazzi C, Stahlman M, Stenfeldt E, et al. Protein kinase STK25 regulates hepatic lipid partitioning and progression of liver steatosis and NASH. FASEB J 2015;29:1564‐1576. [DOI] [PubMed] [Google Scholar]
- 7. Amrutkar M, Chursa U, Kern M, Nunez‐Duran E, Stahlman M, Sutt S, et al. STK25 is a critical determinant in nonalcoholic steatohepatitis. FASEB J 2016;30:3628‐3643. [DOI] [PubMed] [Google Scholar]
- 8. Amrutkar M, Kern M, Nunez‐Duran E, Stahlman M, Cansby E, Chursa U, et al. Protein kinase STK25 controls lipid partitioning in hepatocytes and correlates with liver fat content in humans. Diabetologia 2016;59:341‐353. [DOI] [PubMed] [Google Scholar]
- 9. Cansby E, Amrutkar M, Manneras Holm L, Nerstedt A, Reyahi A, Stenfeldt E, et al. Increased expression of STK25 leads to impaired glucose utilization and insulin sensitivity in mice challenged with a high‐fat diet. FASEB J 2013;27:3660‐3671. [DOI] [PubMed] [Google Scholar]
- 10. Nerstedt A, Cansby E, Andersson CX, Laakso M, Stancakova A, Bluher M, et al. Serine/threonine protein kinase 25 (STK25): a novel negative regulator of lipid and glucose metabolism in rodent and human skeletal muscle. Diabetologia 2012;55:1797‐1807. [DOI] [PubMed] [Google Scholar]
- 11. Chursa U, Nunez‐Duran E, Cansby E, Amrutkar M, Sutt S, Stahlman M, et al. Overexpression of protein kinase STK25 in mice exacerbates ectopic lipid accumulation, mitochondrial dysfunction and insulin resistance in skeletal muscle. Diabetologia 2017;60:553‐567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nunez‐Duran E, Chanclon B, Sutt S, Real J, Marschall HU, Wernstedt Asterholm I, et al. Protein kinase STK25 aggravates the severity of non‐alcoholic fatty pancreas disease in mice. J Endocrinol 2017;234:15‐27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Seth PP, Siwkowski A, Allerson CR, Vasquez G, Lee S, Prakash TP, et al. Short antisense oligonucleotides with novel 2′‐4′ conformationaly restricted nucleoside analogues show improved potency without increased toxicity in animals. J Med Chem 2009;52:10‐13. [DOI] [PubMed] [Google Scholar]
- 14. Bennett CF, Swayze EE. RNA targeting therapeutics: molecular mechanisms of antisense oligonucleotides as a therapeutic platform. Annu Rev Pharmacol Toxicol 2010;50:259‐293. [DOI] [PubMed] [Google Scholar]
- 15. Raal FJ, Santos RD, Blom DJ, Marais AD, Charng MJ, Cromwell WC, et al. Mipomersen, an apolipoprotein B synthesis inhibitor, for lowering of LDL cholesterol concentrations in patients with homozygous familial hypercholesterolaemia: a randomised, double‐blind, placebo‐controlled trial. Lancet 2010;375:998‐1006. [DOI] [PubMed] [Google Scholar]
- 16. Murray S, Ittig D, Koller E, Berdeja A, Chappell A, Prakash TP, et al. TricycloDNA‐modified oligo‐2′‐deoxyribonucleotides reduce scavenger receptor B1 mRNA in hepatic and extra‐hepatic tissues‐‐a comparative study of oligonucleotide length, design and chemistry. Nucleic Acids Res 2012;40:6135‐6143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hung G, Xiao X, Peralta R, Bhattacharjee G, Murray S, Norris D, et al. Characterization of target mRNA reduction through in situ RNA hybridization in multiple organ systems following systemic antisense treatment in animals. Nucleic Acid Ther 2013;23:369‐378. [DOI] [PubMed] [Google Scholar]
- 18. Donner AJ, Yeh ST, Hung G, Graham MJ, Crooke RM, Mullick AE. CD40 generation 2.5 antisense oligonucleotide treatment attenuates doxorubicin‐induced nephropathy and kidney inflammation. Mol Ther Nucleic Acids 2015;4:e265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yamamoto Y, Loriot Y, Beraldi E, Zhang F, Wyatt AW, Al Nakouzi N, et al. Generation 2.5 antisense oligonucleotides targeting the androgen receptor and its splice variants suppress enzalutamide‐resistant prostate cancer cell growth. Clin Cancer Res 2015;21:1675‐1687. [DOI] [PubMed] [Google Scholar]
- 20. Hong D, Kurzrock R, Kim Y, Woessner R, Younes A, Nemunaitis J, et al. AZD9150, a next‐generation antisense oligonucleotide inhibitor of STAT3 with early evidence of clinical activity in lymphoma and lung cancer. Sci Transl Med 2015;7:314ra185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Homan R, Anderson MK. Rapid separation and quantitation of combined neutral and polar lipid classes by high‐performance liquid chromatography and evaporative light‐scattering mass detection. J Chromatogr B Biomed Sci Appl 1998;708:21‐26. [DOI] [PubMed] [Google Scholar]
- 22. Takahashi Y, Soejima Y, Fukusato T. Animal models of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. World J Gastroenterol 2012;18:2300‐2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, et al.; Nonalcoholic Steatohepatitis Clinical Research Network . Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005;41:1313‐1321. [DOI] [PubMed] [Google Scholar]
- 24. Xie L, Yui J, Hatori A, Yamasaki T, Kumata K, Wakizaka H, et al. Translocator protein (18 kDa), a potential molecular imaging biomarker for non‐invasively distinguishing non‐alcoholic fatty liver disease. J Hepatol 2012;57:1076‐1082. [DOI] [PubMed] [Google Scholar]
- 25. Chen L, Shu Y, Liang X, Chen EC, Yee SW, Zur AA, et al. OCT1 is a high‐capacity thiamine transporter that regulates hepatic steatosis and is a target of metformin. Proc Natl Acad Sci U S A 2014;111:9983‐9988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Behari J, Yeh TH, Krauland L, Otruba W, Cieply B, Hauth B, et al. Liver‐specific beta‐catenin knockout mice exhibit defective bile acid and cholesterol homeostasis and increased susceptibility to diet‐induced steatohepatitis. Am J Pathol 2010;176:744‐753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Brunt EM, Janney CG, Di Bisceglie AM, Neuschwander‐Tetri BA, Bacon BR. Nonalcoholic steatohepatitis: a proposal for grading and staging the histological lesions. Am J Gastroenterol 1999;94:2467‐2474. [DOI] [PubMed] [Google Scholar]
- 28. Stefan N, Machicao F, Staiger H, Machann J, Schick F, Tschritter O, et al. Polymorphisms in the gene encoding adiponectin receptor 1 are associated with insulin resistance and high liver fat. Diabetologia 2005;48:2282‐2291. [DOI] [PubMed] [Google Scholar]
- 29. Adiels M, Taskinen MR, Packard C, Caslake MJ, Soro‐Paavonen A, Westerbacka J, et al. Overproduction of large VLDL particles is driven by increased liver fat content in man. Diabetologia 2006;49:755‐765. [DOI] [PubMed] [Google Scholar]
- 30. Chan DC, Watts GF, Gan S, Wong AT, Ooi EM, Barrett PH. Nonalcoholic fatty liver disease as the transducer of hepatic oversecretion of very‐low‐density lipoprotein‐apolipoprotein B‐100 in obesity. Arterioscler Thromb Vasc Biol 2010;30:1043‐1050. [DOI] [PubMed] [Google Scholar]
- 31. Heymann F, Hammerich L, Storch D, Bartneck M, Huss S, Russeler V, et al. Hepatic macrophage migration and differentiation critical for liver fibrosis is mediated by the chemokine receptor C‐C motif chemokine receptor 8 in mice. Hepatology 2012;55:898‐909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Karlmark KR, Weiskirchen R, Zimmermann HW, Gassler N, Ginhoux F, Weber C, et al. Hepatic recruitment of the inflammatory Gr1+ monocyte subset upon liver injury promotes hepatic fibrosis. Hepatology 2009;50:261‐274. [DOI] [PubMed] [Google Scholar]
- 33. Tacke F, Zimmermann HW. Macrophage heterogeneity in liver injury and fibrosis. J Hepatol 2014;60:1090‐1096. [DOI] [PubMed] [Google Scholar]
- 34. Pietrocola F, Galluzzi L, Bravo‐San Pedro JM, Madeo F, Kroemer G. Acetyl coenzyme A: a central metabolite and second messenger. Cell Metab 2015;21:805‐821. [DOI] [PubMed] [Google Scholar]
- 35. Matsuki T, Chen J, Howell BW. Acute inactivation of the serine‐threonine kinase Stk25 disrupts neuronal migration. Neural Dev 2013;8:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Fullerton MD, Galic S, Marcinko K, Sikkema S, Pulinilkunnil T, Chen ZP, et al. Single phosphorylation sites in Acc1 and Acc2 regulate lipid homeostasis and the insulin‐sensitizing effects of metformin. Nat Med 2013;19:1649‐1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rinella ME, Sanyal AJ. Management of NAFLD: a stage‐based approach. Nat Rev Gastroenterol Hepatol 2016;13:196‐205. [DOI] [PubMed] [Google Scholar]
- 38. Neuschwander‐Tetri BA, Loomba R, Sanyal AJ, Lavine JE, Van Natta ML, Abdelmalek MF, et al.; NASH Clinical Research Network . Farnesoid X nuclear receptor ligand obeticholic acid for non‐cirrhotic, non‐alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo‐controlled trial. Lancet 2015;385:956‐965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ratziu V, Harrison SA, Francque S, Bedossa P, Lehert P, Serfaty L, et al.; GOLDEN‐505 Investigator Study Group . Elafibranor, an agonist of the peroxisome proliferator‐activated receptor‐alpha and ‐delta, induces resolution of nonalcoholic steatohepatitis without fibrosis worsening. Gastroenterology 2016;150:1147‐1159.e1145. [DOI] [PubMed] [Google Scholar]
- 40. Lawitz EJ, Poordad F, Coste A, Loo N, Djedjos CS, McColgan B, et al. Acetyl‐CoA carboxylase (ACC) inhibitor GS‐0976 leads to suppression of hepatic de novo lipogenesis and significant improvements in MRI‐PDFF, MRE, and markers of fibrosis after 12 weeks of therapy in patients with NASH [Abstract]. J Hepatol 2017;66(Suppl.):S34. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found at onlinelibrary.wiley.com/doi/10.1002/hep4.1128/full.
Supporting Information 1
Supporting Information Figure 1
Supporting Information Figure 2
Supporting Information Figure 3
Supporting Information Figure 4
Supporting Information Figure 5