

Significance
The p21-activated kinase (PAK) group of serine/threonine kinases are downstream effectors of RHO GTPases and play important roles in regulation of the actin cytoskeleton, cell growth, survival, polarity, and development. Here we study the PAK PAK4 and its interaction with its small GTPase, CDC42. Using X-ray crystallography and solution scattering we discover that kinase and small GTPase bind one another in an unexpected fashion with additional unpredicted interactions. This represents an exciting precedent for kinase–GTPase interactions whereby extended interfaces, outside of the canonical GTPase binding site, can influence binding, specificity, and kinase activity.
Keywords: small GTPase, protein kinase, protein–protein interaction, signal transduction, crystal structure
Abstract
The p21-activated kinase (PAK) group of serine/threonine kinases are downstream effectors of RHO GTPases and play important roles in regulation of the actin cytoskeleton, cell growth, survival, polarity, and development. Here we probe the interaction of the type II PAK, PAK4, with RHO GTPases. Using solution scattering we find that the full-length PAK4 heterodimer with CDC42 adopts primarily a compact organization. X-ray crystallography reveals the molecular nature of the interaction between PAK4 and CDC42 and shows that in addition to the canonical PAK4 CDC42/RAC interactive binding (CRIB) domain binding to CDC42 there are unexpected contacts involving the PAK4 kinase C-lobe, CDC42, and the PAK4 polybasic region. These additional interactions modulate kinase activity and increase the binding affinity of CDC42 for full-length PAK4 compared with the CRIB domain alone. We therefore show that the interaction of CDC42 with PAK4 can influence kinase activity in a previously unappreciated manner.
The RHO family is an important subgroup of the RAS-like small GTPases and regulates functions such as cytoskeleton organization, cell morphology, cell motility, and cell cycle (1). These proteins switch between GDP-loaded inactive and GTP-loaded active conformations under the control of various factors (2). The GTP-loaded active state adopts a conformation that is able to bind downstream effector molecules. For protein kinase effectors, the interaction with GTP-loaded GTPase is often associated with increased catalytic activity and consequent activation of signal transduction. There are multiple RHO-dependent effector kinases, and of these, the p21-activated kinases (PAK), are an important family.
The six PAK family members are classified into two groups, the type I PAKs (PAK1, PAK2, and PAK3) and the type II PAKs (PAK4, PAK5, and PAK6). The PAKs play important roles in regulation of the actin cytoskeleton, cell growth, survival, polarity, and development (3–6) and have recently been found to be important for acquired drug resistance in BRAF mutant melanomas (7). All of these proteins bind to RAC1 and/or CDC42 via sequentially similar CDC42/RAC interactive binding (CRIB) domains; however, the functional result of GTPase binding differs significantly between type I and type II PAKs. For the type I PAKs, a transautoinhibited PAK homodimer is replaced by an active PAK-GTPase heterodimer (3, 8, 9). In contrast, the type II PAKs are autoregulated by the binding of a pseudosubstrate motif in cis to the kinase catalytic cleft (10–12) but GTPase binding results in modest change, if any, in catalytic activity (10, 13, 14). The small GTPase interaction with type II PAKs therefore does not seem to be sufficient for kinase activation, and further binding partners may play roles in control of kinase activation (10). Therefore, the type I PAKs are considered classical RHO GTPase effector kinases because they display increased catalytic activity upon RHO binding, but the type II PAKs represent a more complicated example of GTPase control of kinase signaling. A better understanding of how the type II PAKs are controlled by small GTPases should then facilitate new insights into RHO control of signal transduction.
Control of kinase signaling by small GTPases takes many forms. Canonical disruption of kinase autoinhibition takes place upon RAC1/CDC42 binding to type I PAKs and is found in other GTPase-kinase pairs such as RAS–RAF (15). Other mechanisms of regulation, for example how the kinase activity of the RHO kinases (ROCK1/2) is altered by interaction with RHO (16), are currently under intense investigation (17). To date, however, no structure has been determined of a full-length protein kinase in complex with its GTPase, and in light of studies in other systems where unexpected extended interfaces play roles in achieving functionally significant outcomes, such as specificity (18), probing the interactions of full-length proteins could potentially help shed light on how GTPases control kinase activity.
In this study we investigate the interactions of the type II PAKs with RHO family small GTPases. We use X-ray crystallography and small-angle X-ray scattering (SAXS) to probe the interactions of PAK4 with CDC42 and find that there is an unexpected multidentate interaction between the proteins comprised of CDC42 and the PAK4 kinase domain, CRIB domain, and N-terminal polybasic sequence. Kinase activity assays suggest that these interactions are important for modulation of catalytic activity. Our findings yield insights into the intermolecular interactions between the RHO and PAK families.
Results
The type II group of PAKs encompass distinct regions, an N-terminal polybasic motif important for membrane targeting (19), a GTPase-binding CRIB domain, an autoinhibitory pseudosubstrate motif (10–12), a flexible linker of ∼95–248 aa depending on isoform, and a C-terminal kinase domain (Fig. 1). Structural studies defining the interactions of type II PAKs with RHO-family small GTPases have previously focused on understanding the specific binding of the GTPase–CRIB interface. These interactions have been described by crystal structures of RAC3 in complex with the CRIB domain of PAK4 [Protein Data Bank (PDB) ID code 2OV2] and CDC42 with the CRIB domain of PAK6 (PDB ID code 2ODB), and in both examples a canonical GTPase is observed binding to the extended linear CRIB peptide. The mode of binding is therefore highly similar to the interactions of type I PAK1 with the RAC3 CRIB domain (PDB ID code 2QME) and PAK1 with the CDC42 CRIB domain (PDB ID code 1E0A) (20). These previous studies, therefore, provide detailed descriptions of how PAK CRIB domains bind RHO GTPases; however, no structures exist for a full-length PAK in complex with a GTPase. This is in part because GTPase-bound PAK is expected to resemble two distinct domains, a kinase domain and a CRIB–GTPase complex “domain,” connected by the flexible linker. As protein–protein interaction surfaces can sometimes extend outside of the canonical binding site and can aid specificity determination (18), we sought to test this “two distinct domains” hypothesis.
Fig. 1.

Domain structure and sequence of PAK4. (A) Domain structure of PAK4. Constructs used in this study are indicated. The polybasic, CRIB, and autoinhibitory pseudosubstrate regions are shaded and indicated. (B) N-terminal amino acid sequence of PAK4 indicating the polybasic, CRIB, and pseudosubstrate regions.
We began by conducting SAXS of full-length PAK4 (PAK4FL; PAK4 constructs indicted in Fig. 1) alone and in complex with constitutively active CDC42 (harboring Q61L mutation) (Fig. 2A and Fig. S1). We find that Guinier approximations for the radius of gyration (Rg) of the PAK4FL-CDC42 complex (∼37–39 Å) are approximately similar compared with PAK4FL alone (∼36–38 Å) (Fig. 2B) (Table S1) and that both samples display features expected for globular proteins (Fig. 2 C and D) and an asymptotic plateau in the Porod–Debye plot indicating globularity, particularly in the PAK4FL-CDC42 complex (21) (Fig. 2D). We next calculated asymmetric P(r) functions. If PAK4FL-CDC42 is organized as two distinct domains separated by a flexible linker, one would expect to observe two clearly distinguishable peaks in the P(r) functions (22, 23). This is not the case for PAK4FL-CDC42, where a clear shoulder appears compared with PAK4FL alone (Fig. 2E) but where no obviously distinguishable second peak is visible. We interpret these data to disagree with the two-distinct-domains hypothesis and instead to indicate that PAK4FL and CDC42 form a more compact unit.
Fig. 2.

SAXS analysis for PAK4FL and PAK4FL-CDC42 reveals a compact heterodimer. SAXS analysis of three concentrations of both PAK4FL (Upper) and PAK4FL-CDC42 (Lower). PAK4FL concentrations: 11.0 mg/mL (blue), 5.6 mg/mL (red), and 2.8 mg/mL (green). PAK4FL-CDC42 concentrations: 6.2 mg/mL (blue), 3.1 mg/mL (red), and 1.5 mg/mL (green). (A) SAXS intensity profiles (logarithmic). (B) Guinier plots for each concentration. (C) Dimensionless Kratky plots. (D) Porod–Debye plots indicate a more globular shape for PAK4FL-CDC42. (E) Normalized pair distribution functions P(r) for each concentration. PAK4FL-CDC42 displays a shoulder (at ∼60 Å).
The results from our SAXS study suggested that the PAK4FL-CDC42 complex might be a cocrystallization target. We therefore set trays with size-exclusion copurified samples (Fig. S1B) and obtained cocrystals that diffracted to 3.0 Å resolution in space group P63 (Table S2). We obtained a strong molecular replacement solution containing one copy of PAK4 kinase domain and one copy of CDC42 (Fig. 3A and Fig. S2 A and B). There are two striking features in this structure. First, CDC42 is engaged in a single protein–protein contact in the crystal, with the C-lobe of the PAK4 kinase domain (Fig. S2 C and D), and second, unsatisfied electron density is located in the substrate binding site of PAK4 which seems to connect to the expected location of the PAK4 CRIB bound to CDC42 (Fig. S3E). Unfortunately, however, although yielding a good molecular replacement solution, this 3.0-Å structure also displays high B-factors for CDC42 (>200 Å2) (Table S2). This is may be due to the lack of additional crystal contacts to stabilize CDC42, conformational flexibility of the CDC42, or low occupancy as this crystal form does not require CDC42 to form (10, 24), and therefore we are not able to refine or analyze the structure in detail. Nonetheless, the observation that CDC42 contacts the PAK4 kinase domain, specifically the C-lobe, in this crystal structure was unexpected and warranted further investigation.
Fig. 3.

Overall structure of PAK4 in complex with CDC42. (A) Ribbon diagram showing the structure of PAK4FL-CDC42. (B) Ribbon diagram showing the ternary structure of PAK4cat-PAK4N45-CDC42. AMP-PNP and GMP-PNP indicate nucleotide analog. (C) Same orientation as B showing the surfaces of PAK4cat and CDC42. PAK4N45 is shown in stick format. Location of the inset is shown with a box. (Inset) A rotation and zoom of the region boxed in red. (D) Schematic of the interaction. PB indicates polybasic region. (E) Close-up of interaction between the N terminus of PAK4N45 and the PAK4 substrate binding site. The β and γ phosphates of AMP-PNP are not visible in the electron density. (F) Close-up of the interaction between CDC42 and PAK4 showing the extended interface. Secondary structure elements are labeled.
To obtain better-resolution crystals we used an alternative approach. We incubated active CDC42, PAK4 kinase domain (PAK4cat), and the N terminus of PAK4 containing the CRIB domain (residues 1–45, PAK4N45) (Fig. 1) in a 1:1:1 mixture and set crystallization trays. We obtained heterotrimeric cocrystals that diffracted to 2.4-Å resolution in space group P21 (Table S2), a space group different from that of the PAK4FL-CDC42 cocrystals. Molecular replacement easily located a single kinase domain of PAK4 and a single small GTPase domain of CDC42. An extended segment of unambiguous electron density is also visible which we attribute to the third polypeptide chain, PAK4N45 (Fig. 3B and Fig. S3). We find that PAK4N45 extends from the substrate binding site of PAK4 to the effector binding site of CDC42 (Fig. 3 C and D and Fig. S3 C and D). Superposition of the independently determined molecular replacement solution for PAK4FL-CDC42 and refined structure for PAK4cat-PAK4N45-CDC42 shows nearly identical arrangement of CDC42 with respect to the kinase domain of PAK4, (rmsd is 0.6 Å over 462 carbon alphas for the complexes), confirming that the interaction of CDC42 with PAK4 is conserved in both structures (Fig. S4).
A more detailed analysis of the high-resolution structure indicates that the PAK4 kinase domain adopts the DFG-in conformation with the activation loop residue S474 phosphorylated and CDC42 that is in the active (GTP-loaded) state (GMP-PNP is fully visible bound to CDC42). We find that although CDC42 interacts with the PAK4 CRIB domain in a canonical fashion (Fig. 3 B and C), an additional N-terminal extension from the CRIB domain, including residues Lys4, Arg5, Lys6, Lys7, and Arg8 of the polybasic motif (19), contacts the substrate-binding site of the PAK4 kinase domain. Particularly prominent are salt bridges and polar contacts between Arg5 and four residues, Asp444, Ser443, Glu507, and Thr404, and between Arg8 and Asp440 (Fig. 3E). We also find that extensive interactions occur between the α5 helix of CDC42 and both the N terminus and C-lobe of PAK4 (activation segment and helices αEF, αG). This three-component interaction includes a salt bridge between PAK4 Arg489 and CDC42 Glu171 and hydrophobic interactions between the C terminus of CDC42 helix α5 and a hydrophobic patch centered around PAK4 kinase domain residues Leu475, Leu521, Ile487, Leu521, and Met524, and N-terminal residues Val9 and Ile11 (Fig. 3F and Fig. S2F). Both of the crystal structures therefore indicate that a compact conformation occurs between PAK4 and CDC42, in two crystal forms and using two sets of constructs.
As the crystal structures suggested a compact conformation we continued our SAXS study and using the SAXS P(r) functions (Fig. 2E) we calculated molecular envelopes (Fig. 4 and Fig. S5). We generated 20 ab initio molecular envelopes for PAK4FL-CDC42, the averaged envelope of which does not indicate two distinct domains separated by a linker, but instead points to a compact interaction between PAK4 kinase domain and CDC42 (Fig. 4B and Table S1), similar to the PAK4cat-PAK4N45-CDC42 crystal structure (Fig. 4A). To test this, we conducted rigid body modeling. Based on the two-distinct-domains hypothesis we used two rigid “domains,” PAK4cat and PAK4N45-CDC42. The modeling used coordinates from our PAK4cat-PAK4N45-CDC42 crystal structure and added unmodeled amino acids (32 at the N terminus of PAK4, 9 at the C terminus of CDC42, and 92 linking PAK4N45 and PAK4cat). Comparison of the best rigid body model with the ab initio molecular envelope yields a good fit (Fig. 4C) and compares well to the experimental data, with a χ2 value of 0.83 (Fig. 4D). The shape of the best model indicates a compact conformation between PAK4 and CDC42, with a closest distance of ∼12 Å.
Fig. 4.

SAXS structural modeling and flexibility analysis. (A) Crystal structure of PAK4cat-PAK4N45-CDC42 showing PAK4cat in blue, PAK4N45 in yellow, and CDC42 in brown. (B) Averaged SAXS molecular envelope generated using DAMMIF (DAMFILT envelopes shown) for PAK4FL-CDC42. (C) Rigid body analysis. CORAL model of PAK4FL-CDC42 with the best statistical fit to the experimental data. Ribbons are colored as in A and interdomain dummy atoms are shown as pink spheres. Superposition with the DAMMIF envelope is shown in the central pair. (D) Rigid body analysis. Experimental SAXS data (logarithmic scale) are shown fitted to the theoretical scattering profile of the best rigid body (red line). Residuals are shown below. (E) EOM analysis. Comparison of the theoretical scattering profile of the selected ensemble (containing three PDB models) is shown in red, with the experimental SAXS data (blue). Logarithmic scale. Residuals are shown below. (F) EOM analysis. Distribution of Rg values for the pool of 10,000 models (black) compared with the selected ensemble. Dashed line indicates average Rg value for the selected ensemble of 37.8 Å. (G) Most representative EOM model of PAK4FL-CDC42 superposed onto the DAMMIF molecular envelope. Interdomain dummy atoms are shown as pink spheres. Superpositions were conducted using SUPCOMB (33).
To further probe the flexibility of the PAK4-CDC42 complex in solution we analyzed the SAXS data using the ensemble optimization method (EOM) (25), using two rigid “domains,” PAK4cat and PAK4N45-CDC42, which were connected by a flexible linker of 92 dummy residues. Following generation of 10,000 individual PDB models, each encompassing a different random conformation for the linker, an optimized ensemble was assembled to best fit the experimental data. The best ensemble fits the experimental scattering data with a χ2 value of 0.13 (Fig. 4E). The Rg values for the random pool are much broader than for the ensemble, and the average Rg for the ensemble is 37.8 Å, very similar to the real and reciprocal space values that range between 37.4 Å and 39.6 Å (Table S1). We find that the ensemble Rg values indicate two populations, one with Rg = ∼32 Å and a second, smaller group with a broader peak at Rg = ∼52 Å. These populations are suggestive of a major PAK4-CDC42 population with a compact interaction and a second, smaller group with an extended conformation; indeed, the best-fitting ensemble is composed of three separate PDB models, the major component of which is compact (Fig. 4G) and the two minor components with a two-distinct-domains appearance (Fig. S5). Although the nature of EOM analysis is not to define specific conformational states, the technique clearly indicates that in solution the predominant form of PAK4-CDC42 is compact, but that conformational flexibility also occurs. Our structural studies therefore indicate that PAK4 with CDC42 form a compact conformation with an extended interaction surface which occludes the substrate binding site of the kinase. This raises two interesting possibilities, that the extended surface might impact kinase–small GTPase binding and that the interaction may suppress kinase activity. We therefore tested these possibilities.
Previous studies suggest that the interaction of PAK4 with small GTPases is dominated by the canonical CRIB binding site (26–28) and have shown that type II PAKs are specific for CDC42 over RAC1 (26, 27, 29–32), but our structural studies imply that the extended interaction surface might also have an impact on binding. Indeed, whereas previously determined structures of either RAC3 with PAK4 CRIB domain (PDB ID code 2OV2) or CDC42 with PAK6 CRIB domain (PDB ID code 2ODB) bury ∼2,070 Å2, we find that the complete PAK4-CDC42 interaction buries ∼3,150 Å2. Our structures therefore suggest two unpredicted interactions in the PAK4-CDC42 complex: between the PAK4 substrate binding site and the N terminus of PAK4 and between CDC42 and the C-lobe of PAK4. We wondered whether this extended interface might impact binding between PAK4 and RHO-family GTPases, and using isothermal titration calorimetry (ITC) we find a Kd of 300 nM for the PAK4FL -CDC42 interaction but observe approximately ninefold weaker binding to RAC1 (Table 1 and Table S3). This correlates well with our observation that RAC1 has limited copurification with PAK4FL by size-exclusion chromatography (Fig. S1C). We next introduced point mutations into CDC42 α5 helix (E171R, L174R, and E178R) (CDC42mutELE-RRR) and PAK4 αEF helix (R489A) (PAK4FL-R489A). Both sets of mutation are predicted to interrupt the noncanonical PAK4-CDC42 interaction, and we observe an approximately three- to fourfold reduction in binding affinity for both, compared with wild type. Although we observe no binding between PAK4cat and PAK4N45 by ITC, we can measure a heat of interaction between PAK4cat and previously complexed PAK4N45-CDC42 (Table 1 and Table S3), confirming the extended interaction interface. These results provide an improved understanding of PAK4 interactions with small GTPases.
Table 1.
Thermodynamic properties of the interaction between PAK4 and small GTPases RAC1 and CDC42
| Sample cell | Syringe | Kd, μM | N | ΔH, kJ/mol | ΔS, J/mol⋅K | ΔG, kJ/mol |
| PAK4FL | CDC42 | 0.3 ± 0.1 | 0.9 ± 0.02 | −81.4 ± 0.2 | −148.8 ± 3.7 | −37.1 ± 0.9 |
| PAK4FL | CDC42ELE-RRR | 1.1 ± 0.1 | 0.9 ± 0.02 | −95.6 ± 4.4 | −201.3 ± 10.2 | −35.6 ± 1.3 |
| PAK4FL-R489A | CDC42 | 1.3 ± 0.3 | 1.0 ± 0.2 | −77.3 ± 19.5 | −145.9 ± 66.7 | −33.8 ± 0.7 |
| PAK4FL | RAC1 | 3.0 ± 0.2 | 1.0 ± 0.3 | −39.7 ± 1.2 | −27.4 ± 3.3 | −31.5 ± 0.2 |
| CDC42 | PAK4N45 | 1.3 ± 0.2 | 0.6 ± 0.02 | 100.9 ± 2.3 | −255.6 ± 6.9 | −33.7 ± 0.4 |
| RAC1 | PAK4N45 | 10.1 ± 2.0 | 0.9 ± 0.03 | −106.2 ± 4.0 | −260.2 ± 15.1 | −28.6 ± 0.5 |
| PAK4cat | PAK4N45:CDC42 | 104.0 ± 38.0 | 1.3 ± 0.2 | 13.4 ± 1.0 | 121.7 ± 0.1 | −22.8 ± 0.9 |
| PAK4cat | PAK4N45 | No heat | No heat | No heat | No heat | No heat |
Data are averages from two independent ITC experiments. Experiments conducted at 298 K. No heat indicates no measurable heat was observed from the titration.
The suppression of kinase activity for PAK4 has previously been found to be predominantly driven by a pseudosubstrate interaction between the kinase domain and an N-terminal region outside of the CRIB domain, centered on proline-52 (10–12). Based on our structures, the extended PAK4-CDC42 interaction should also inhibit kinase activity in a pseudosubstrate manner. To test this, we conducted kinase activity assays for PAK4FL and PAK4FL mutated in its polybasic motif (RR5/8AA) to disrupt potential autoinhibitory ionic interactions in the substrate binding site (PAK4FL-PB). Addition of either CDC42 or CDC42 mutated to disrupt the extended binding site with PAK4, CDC42mutELE-RRR (Table 1), results in increased kinase activity, with larger increases occurring when the small GTPases are added to PAK4FL-PB (Fig. 5 and Fig. S7). The highest activity we observe (approximately fourfold more than PAK4FL) is for CDC42mutELE-RRR with PAK4FL-PB. We interpret this to indicate that canonical (Pro-52) pseudosubstrate autoinhibition is partially disrupted by CDC42 binding, but that the extended CDC42-PAK4 interaction stabilizes a second autoinhibitory pseudosubstrate interaction that incorporates residues of the polybasic motif.
Fig. 5.
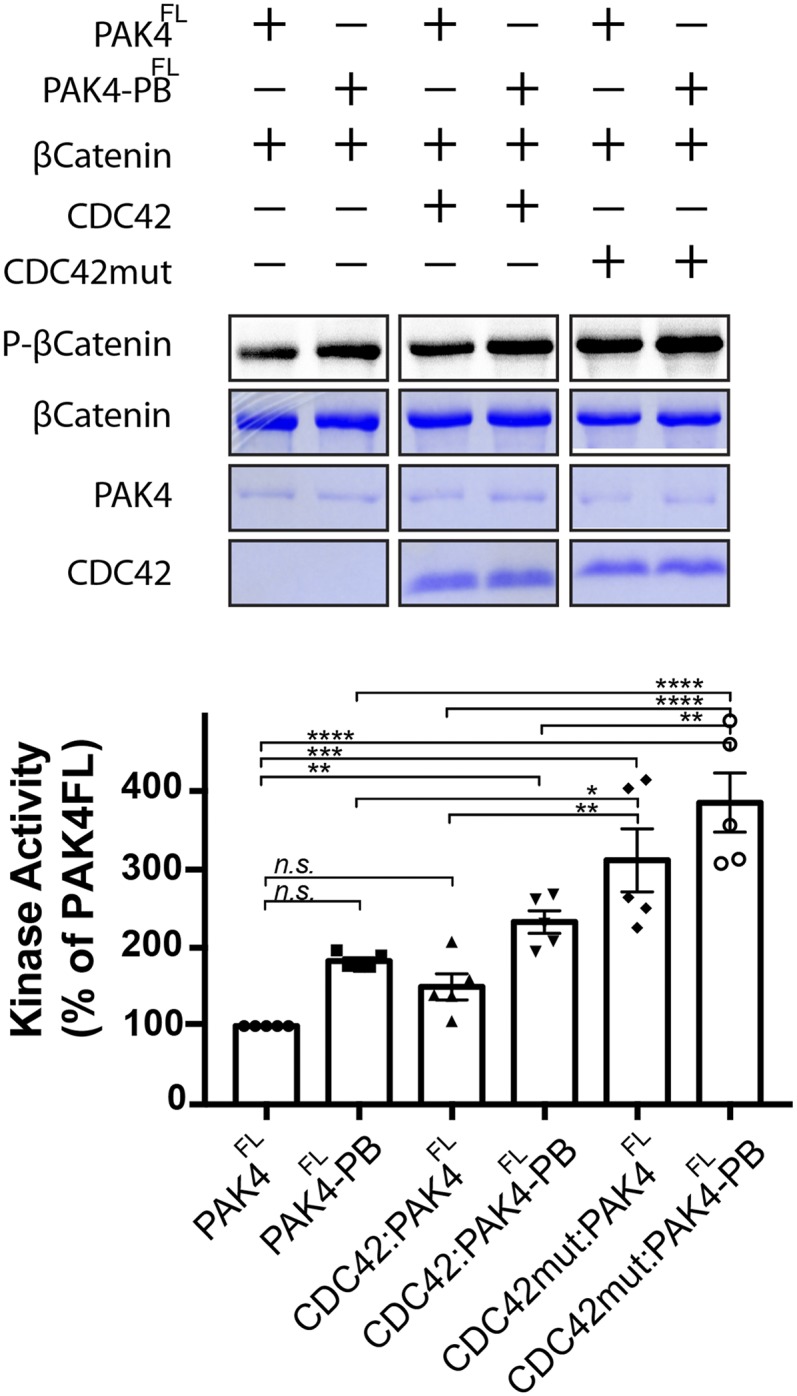
In vitro kinase assay. In vitro kinase assay using purified PAK4FL or PAK4FL mutated in its polybasic region (R5A/R8A), PAK4FL-PB. Assays were incubated with either CDC42 (Q61L, constitutively active mutant) or CDC42mut (Q61L, E171R, L174R, and E178R). Full-length β-catenin was added as the substrate, and kinase activity was monitored by autoradiography. Time point monitored is 30 min. n = 5. Bar charts show activities as a percentage of PAK4FL activity. ****P < 0.0001, ***P < 0.002, **P < 0.02, *P < 0.03, and n.s. means not significant.
Discussion
The two groups of PAKs are both considered effectors of RHO GTPases; however, small GTPase binding results in significant functional differences between the groups (3–6). Type I PAKs are considered classical GTPase effector kinases, but the type II PAKs are atypical, with GTPase binding probably important for correct subcellular localization and kinase activation controlled by other factors (10, 13, 14). Although extensive studies have investigated the interactions of the CRIB domains of type II PAKs with small GTPases, we wondered whether further insight could be gained by probing the interactions of a full-length type II PAK, PAK4, with its small GTPase, CDC42. Surprisingly, our structural studies revealed a completely unexpected overall organization of the PAK4-CDC42 complex. We had predicted an extended organization of two distinct domains connected by a flexible linker; however, SAXS very clearly indicates a compact kinase-GTPase unit. Furthermore, X-ray crystallography of the full-length PAK4-CDC42 complex and the PAK4cat-PAK4N45-CDC42 also indicates a compact complex. Although the resolution of the SAXS prevents direct comparison of the relative orientations of molecules in solution versus in crystal, our data strongly indicate that our original assumption of an extended organization was incorrect and that PAK4-CDC42 predominantly exists as a compact complex.
The crystallographically defined compact complex shows a large increase in surface area between PAK4 and CDC42 compared with previous structural studies that crystallized the small GTPase with the kinase CRIB domain alone. This extended surface seems to be important for achieving increased binding affinity, and we find that CDC42 binds full-length PAK4 with an order of magnitude tighter affinity than the kinase’s CRIB domain alone (Table 1). We find that introduction of point mutations at residues important for the extended PAK4-CDC42 interaction significantly reduce binding affinity to the equivalent of a PAK4 CRIB-CDC42 interaction. This study probes the binding affinities for a full-length type II PAK with small GTPases and determines a structure of a full-length protein kinase in complex with its GTPase. Our affinity measurements and structural studies combined, therefore, indicate that small GTPase binding and specificity may not be solely controlled by CRIB domain interactions and may mirror other systems (18) where unexpected extended interfaces play roles in achieving functionally significant outcomes, such as specificity.
Kinase activity of type II PAKs is thought to be predominantly autoregulated by a pseudosubstrate motif centered on residue Pro-52 (10–12). Point mutations to disrupt this inhibition result in significant increases in kinase activity (e.g., ∼24-fold in ref. 10). In contrast, similar to other groups (13) we observe a slight increase in kinase activity on addition of CDC42. Incorporation of mutations to disrupt the crystallographically observed extended PAK4-CDC42 interaction result in an approximately fourfold increase in activity for PAK4FL-PB with CDC42mutELE-RRR. Together, these data support a model where the Pro-52 pseudosubstrate dominates autoregulation but is partially disrupted by CDC42 binding. Thus, a secondary interaction is required to fully suppress kinase activity.
Materials and Methods
PAK4FL (residues 1–426, isoform 2), PAK4cat (residues 286–591), PAK4N45 (residues 1–45), and CDC42 were each expressed in Escherichia coli. Crystals were obtained using hanging-drop methodology at room temperature. SAXS, kinase assays, and ITC were performed using purified samples. Detailed materials and methods are described in SI Materials and Methods. X-ray diffraction images are available online at SBGrid Data Bank (34) [https://data.sbgrid.org/dataset/533/ (5UPK) and https://data.sbgrid.org/dataset/532/ (5UPL)].
Supplementary Material
Acknowledgments
We thank Ben Turk, Youngchang Kim, Lin Yang, Shirish Chodankar, Amy Stiegler, Ya Ha, Ruth Halaban, and Joseph Schlessinger. APS beamline NE-CAT (24-ID-E and -C) is thanked and is funded by NIH Grants GM103403 and RR029205. The LiX beamline is part of the Life Science Biomedical Technology Research resource, primarily supported by the National Institute of Health, National Institute of General Medical Sciences under Grant P41 GM111244, and by the Department of Energy Office of Biological and Environmental Research under Grant KP1605010, with additional support from NIH Grant S10 OD012331. As a National Synchrotron Light Source II facility resource at Brookhaven National Laboratory, work performed at Life Science and Biomedical Technology Research is supported in part by the US Department of Energy, Office of Basic Energy Sciences Program under Contract DE-SC0012704. This work was funded by National Institutes of Health Grants R01GM102262 and S10OD018007.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The crystallography, atomic coordinates, and structure factors have been deposited in the Protein Data Bank, www.wwpdb.org (PDB ID codes 5UPK and 5UPL). X-ray diffraction images are available at the SBGrid Data Bank, https://data.sbgrid.org/dataset/533/ (for 5UPK) and https://data.sbgrid.org/dataset/532/ (for 5UPL).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1717437115/-/DCSupplemental.
References
- 1.Bishop AL, Hall A. Rho GTPases and their effector proteins. Biochem J. 2000;348:241–255. [PMC free article] [PubMed] [Google Scholar]
- 2.Wennerberg K, Rossman KL, Der CJ. The Ras superfamily at a glance. J Cell Sci. 2005;118:843–846. doi: 10.1242/jcs.01660. [DOI] [PubMed] [Google Scholar]
- 3.Eswaran J, Soundararajan M, Kumar R, Knapp S. UnPAKing the class differences among p21-activated kinases. Trends Biochem Sci. 2008;33:394–403. doi: 10.1016/j.tibs.2008.06.002. [DOI] [PubMed] [Google Scholar]
- 4.Arias-Romero LE, Chernoff J. A tale of two Paks. Biol Cell. 2008;100:97–108. doi: 10.1042/BC20070109. [DOI] [PubMed] [Google Scholar]
- 5.Rane CK, Minden A. P21 activated kinases: Structure, regulation, and functions. Small GTPases. 2014;5:e28003. doi: 10.4161/sgtp.28003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kumar R, Sanawar R, Li X, Li F. Structure, biochemistry, and biology of PAK kinases. Gene. 2017;605:20–31. doi: 10.1016/j.gene.2016.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lu H, et al. PAK signalling drives acquired drug resistance to MAPK inhibitors in BRAF-mutant melanomas. Nature. 2017;550:133–136. doi: 10.1038/nature24040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Parrini MC, Lei M, Harrison SC, Mayer BJ. Pak1 kinase homodimers are autoinhibited in trans and dissociated upon activation by Cdc42 and Rac1. Mol Cell. 2002;9:73–83. doi: 10.1016/s1097-2765(01)00428-2. [DOI] [PubMed] [Google Scholar]
- 9.Ha BH, Morse EM, Turk BE, Boggon TJ. Signaling, regulation, and specificity of the type II p21-activated kinases. J Biol Chem. 2015;290:12975–12983. doi: 10.1074/jbc.R115.650416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ha BH, et al. Type II p21-activated kinases (PAKs) are regulated by an autoinhibitory pseudosubstrate. Proc Natl Acad Sci USA. 2012;109:16107–16112. doi: 10.1073/pnas.1214447109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang W, Lim L, Baskaran Y, Manser E, Song J. NMR binding and crystal structure reveal that intrinsically-unstructured regulatory domain auto-inhibits PAK4 by a mechanism different from that of PAK1. Biochem Biophys Res Commun. 2013;438:169–174. doi: 10.1016/j.bbrc.2013.07.047. [DOI] [PubMed] [Google Scholar]
- 12.Gao J, et al. Substrate and inhibitor specificity of the type II p21-activated kinase, PAK6. PLoS One. 2013;8:e77818. doi: 10.1371/journal.pone.0077818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baskaran Y, Ng YW, Selamat W, Ling FT, Manser E. Group I and II mammalian PAKs have different modes of activation by Cdc42. EMBO Rep. 2012;13:653–659. doi: 10.1038/embor.2012.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tabanifar B, Zhao Z, Manser E. PAK5 is auto-activated by a central domain that promotes kinase oligomerization. Biochem J. 2016;473:1777–1789. doi: 10.1042/BCJ20160132. [DOI] [PubMed] [Google Scholar]
- 15.Lavoie H, Therrien M. Regulation of RAF protein kinases in ERK signalling. Nat Rev Mol Cell Biol. 2015;16:281–298. doi: 10.1038/nrm3979. [DOI] [PubMed] [Google Scholar]
- 16.Dvorsky R, Blumenstein L, Vetter IR, Ahmadian MR. Structural insights into the interaction of ROCKI with the switch regions of RhoA. J Biol Chem. 2004;279:7098–7104. doi: 10.1074/jbc.M311911200. [DOI] [PubMed] [Google Scholar]
- 17.Truebestein L, Elsner DJ, Leonard TA. Made to measure–Keeping Rho kinase at a distance. Small GTPases. 2016;7:82–92. doi: 10.1080/21541248.2016.1173770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bae JH, et al. The selectivity of receptor tyrosine kinase signaling is controlled by a secondary SH2 domain binding site. Cell. 2009;138:514–524. doi: 10.1016/j.cell.2009.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morse EM, et al. PAK6 targets to cell-cell adhesions through its N-terminus in a Cdc42-dependent manner to drive epithelial colony escape. J Cell Sci. 2016;129:380–393. doi: 10.1242/jcs.177493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Morreale A, et al. Structure of Cdc42 bound to the GTPase binding domain of PAK. Nat Struct Biol. 2000;7:384–388. doi: 10.1038/75158. [DOI] [PubMed] [Google Scholar]
- 21.Rambo RP, Tainer JA. Characterizing flexible and intrinsically unstructured biological macromolecules by SAS using the Porod-Debye law. Biopolymers. 2011;95:559–571. doi: 10.1002/bip.21638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Svergun DI, Koch MHJ. Small-angle scattering studies of biological macromolecules in solution. Rep Prog Phys. 2003;66:1735–1782. [Google Scholar]
- 23.Sethi R, Ylänne J. Small-angle X-ray scattering reveals compact domain-domain interactions in the N-terminal region of filamin C. PLoS One. 2014;9:e107457. doi: 10.1371/journal.pone.0107457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baskaran Y, et al. An in cellulo-derived structure of PAK4 in complex with its inhibitor Inka1. Nat Commun. 2015;6:8681. doi: 10.1038/ncomms9681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bernadó P, Mylonas E, Petoukhov MV, Blackledge M, Svergun DI. Structural characterization of flexible proteins using small-angle X-ray scattering. J Am Chem Soc. 2007;129:5656–5664. doi: 10.1021/ja069124n. [DOI] [PubMed] [Google Scholar]
- 26.Abo A, et al. PAK4, a novel effector for Cdc42Hs, is implicated in the reorganization of the actin cytoskeleton and in the formation of filopodia. EMBO J. 1998;17:6527–6540. doi: 10.1093/emboj/17.22.6527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dan C, Nath N, Liberto M, Minden A. PAK5, a new brain-specific kinase, promotes neurite outgrowth in N1E-115 cells. Mol Cell Biol. 2002;22:567–577. doi: 10.1128/MCB.22.2.567-577.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wells CM, Abo A, Ridley AJ. PAK4 is activated via PI3K in HGF-stimulated epithelial cells. J Cell Sci. 2002;115:3947–3956. doi: 10.1242/jcs.00080. [DOI] [PubMed] [Google Scholar]
- 29.Cotteret S, Jaffer ZM, Beeser A, Chernoff J. p21-activated kinase 5 (Pak5) localizes to mitochondria and inhibits apoptosis by phosphorylating BAD. Mol Cell Biol. 2003;23:5526–5539. doi: 10.1128/MCB.23.16.5526-5539.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pandey A, et al. Cloning and characterization of PAK5, a novel member of mammalian p21-activated kinase-II subfamily that is predominantly expressed in brain. Oncogene. 2002;21:3939–3948. doi: 10.1038/sj.onc.1205478. [DOI] [PubMed] [Google Scholar]
- 31.Wu X, Frost JA. Multiple Rho proteins regulate the subcellular targeting of PAK5. Biochem Biophys Res Commun. 2006;351:328–335. doi: 10.1016/j.bbrc.2006.09.172. [DOI] [PubMed] [Google Scholar]
- 32.Lee SR, et al. AR and ER interaction with a p21-activated kinase (PAK6) Mol Endocrinol. 2002;16:85–99. doi: 10.1210/mend.16.1.0753. [DOI] [PubMed] [Google Scholar]
- 33.Kozin M, Svergun DI. Automated matching of high- and low-resolution structural models. J Appl Cryst. 2000;34:33–41. [Google Scholar]
- 34.Meyer PA, et al. Data publication with the structural biology data grid supports live analysis. Nat Commun. 2016;7:10882. doi: 10.1038/ncomms10882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang L. Using an in-vacuum CCD detector for simultaneous small- and wide-angle scattering at beamline X9. J Synchrotron Radiat. 2013;20:211–218. doi: 10.1107/S0909049512048984. [DOI] [PubMed] [Google Scholar]
- 36.Konarev PV, Volkov VV, Sololova AV, Koch MHJ, Svergun DI. PRIMUS: A windows PC-based system for small-angle scattering data analysis. J Appl Cryst. 2003;36:1277–1282. [Google Scholar]
- 37.Petoukhov MV, Konarev PV, Kikhney AG, Svergun DI. ATSAS 2.1–Towards automated and web-supported small-angle scattering data analysis. J Appl Cryst. 2007;40(Suppl 1):S223–S228. [Google Scholar]
- 38.Franke D, Svergun DI. DAMMIF, a program for rapid ab-initio shape determination in small-angle scattering. J Appl Cryst. 2009;42:342–346. doi: 10.1107/S0021889809000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Durand D, et al. NADPH oxidase activator p67(phox) behaves in solution as a multidomain protein with semi-flexible linkers. J Struct Biol. 2010;169:45–53. doi: 10.1016/j.jsb.2009.08.009. [DOI] [PubMed] [Google Scholar]
- 40.Petoukhov MV, Svergun DI. Global rigid body modeling of macromolecular complexes against small-angle scattering data. Biophys J. 2005;89:1237–1250. doi: 10.1529/biophysj.105.064154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 42.McCoy AJ. Solving structures of protein complexes by molecular replacement with Phaser. Acta Crystallogr D Biol Crystallogr. 2007;63:32–41. doi: 10.1107/S0907444906045975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Adams PD, et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen VB, et al. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Murshudov GN, et al. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr D Biol Crystallogr. 2011;67:355–367. doi: 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Morin A, et al. Collaboration gets the most out of software. Elife. 2013;2:e01456. doi: 10.7554/eLife.01456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McNicholas S, Potterton E, Wilson KS, Noble ME. Presenting your structures: The CCP4mg molecular-graphics software. Acta Crystallogr D Biol Crystallogr. 2011;67:386–394. doi: 10.1107/S0907444911007281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.