

Abstract
Cocaine self-administration increases expression of GluA1 subunits in ventral tegmental area (VTA) dopamine neurons, which subsequently enhance the motivation for cocaine. This increase in GluA1 may be dependent on concomitant NMDA receptor (NMDAR) activation during self-administration, similar to cocaine-induced long-term potentiation in the VTA. In this study, we used viral-mediated expression of a dominant-negative GluN1 subunit (HSV-dnGluN1) in VTA neurons to study the effect of transient NMDAR inactivation on the GluA1 increases induced by chronic cocaine self-administration in male rats. We found that dnGluN1 expression in the VTA limited to the 3 weeks of cocaine self-administration prevents the subsequent increase in tissue GluA1 levels when compared with control infusions of HSV-LacZ. Surprisingly, dnGluN1 expression led to an enhancement in the motivation to self-administer cocaine as measured using a progressive ratio reinforcement schedule and to enhanced cocaine seeking measured in extinction/reinstatement tests following an extended 3 week withdrawal period. Despite blocking tissue GluA1 increases in cocaine self-administering animals, the HSV-dnGluN1 treatment resulted in increased membrane levels of GluA1 and GluN2B, along with markedly higher locomotor responses to intra-VTA infusions of AMPA, suggesting a paradoxical increase in VTA AMPA receptor responsiveness. Together, these data suggest that NMDARs mediate cocaine-induced increases in VTA GluA1 expression, but such transient NMDAR inactivation also leads to compensatory scaling of synaptic AMPA receptors that enhance the motivational for cocaine.
SIGNIFICANCE STATEMENT Dopamine neurons in the ventral tegmental area (VTA) are critical substrates of drug rewards. Animal models indicate that chronic cocaine use enhances excitatory glutamatergic input to these neurons, making them more susceptible to environmental stimuli that trigger drug craving and relapse. We previously found that self-administration of cocaine increases AMPA glutamate receptors in the VTA, and this effect enhances motivation for cocaine. Here we report that the mechanism for this upregulation involves NMDA receptor activity during cocaine use. While interference with NMDA receptor function blocks AMPA receptor upregulation, it also produces a paradoxical enhancement in membrane AMPA receptor subunits, AMPA responsiveness, and the motivation for cocaine. Thus, pharmacotherapy targeting NMDA receptors may inadvertently produce substantial adverse consequences for cocaine addiction.
Keywords: cocaine, GluA1, GluN1, self-administration, VTA
Introduction
Animal models of cocaine addiction are characterized by compulsive drug-seeking and drug-taking even after extended periods of abstinence (Ahmed and Koob, 1998; Grimm et al., 2001; Deroche-Gamonet et al., 2004). A major hypothesis is that these addictive phenotypes reflect enhanced incentive motivation for the drug and associated stimuli (Robinson and Berridge, 1993) mediated by a potentiation of glutamatergic synapses on ventral tegmental area (VTA) dopamine neurons (Kauer and Malenka, 2007; Thomas et al., 2008). Indeed, VTA dopamine neurons exhibit transient NMDA receptor (NMDAR)-dependent increases in AMPA receptor (AMPAR)-mediated currents following either single or repeated cocaine injections (Ungless et al., 2001; Saal et al., 2003; Borgland et al., 2004). Similarly, cocaine has been shown to facilitate induction of long-term potentiation (LTP) and enhance responsiveness of dopamine neurons to AMPA application (White et al., 1995; Zhang et al., 1997; Liu et al., 2005). One potential mechanism may involve enhanced expression of the AMPAR subunits in the VTA. Such enhanced expression has been shown to occur following repeated cocaine exposure (Fitzgerald et al., 1996; Churchill et al., 1999; but see Lu et al., 2002) or through incorporation of calcium-permeable AMPARs (Bellone and Lüscher, 2006).
While passive (experimenter-delivered) cocaine administration has been found to produce transient enhancement in glutamatergic input to VTA dopamine neurons, persistent LTP of VTA excitatory synapses has been found following volitional cocaine self-administration (SA) in rats, lasting for up to 3 months after cessation of cocaine SA (Chen et al. (2008)). This effect was not found in control rats receiving an equivalent number and pattern of cocaine injections by passive yoked infusion, indicating a reinforcement-related induction of LTP in VTA dopamine neurons. Similarly, we previously reported reinforcement-related upregulation of both phosphorylated and total GluA1 protein levels in the VTA in cocaine-SA rats that is not evident in yoked controls (Choi et al., 2011). Furthermore, viral-mediated GluA1 overexpression in the VTA dramatically increases the level of effort rats would exert to SA the cocaine injections on a progressive ratio (PR) reinforcement schedule, functionally linking increased AMPA-mediated input to VTA with enhanced motivation for cocaine (Choi et al., 2011).
Cocaine-induced LTP in VTA dopamine neurons is blocked by local NMDAR antagonists (Ungless et al., 2001), and studies also have found that selective GluN1 deletion in dopamine neurons blocks cocaine-induced synaptic plasticity (Engblom et al., 2008; Zweifel et al., 2008; Mameli et al., 2009). However, this GluN1 deletion may occlude rather than block LTP due to compensatory scaling up of AMPA-mediated synaptic currents, and consequently induce sensitized responses to initial cocaine exposure. Thus, a potential drawback to the GluN1-deletion approach is that homeostatic alterations exist not only during initial cocaine exposure, but also during subsequent behavioral tests. Thus, one cannot dissociate mechanisms of induction from expression of addictive behavior. We sought to circumvent this issue by using inducible and transient viral-mediated expression of a dominant-negative mutant of GluN1. We hypothesized that such transient inactivation of NMDARs in the VTA limited to cocaine-SA training would (1) prevent cocaine-induced upregulation of VTA GluA1 levels and (2) subsequently reduce cocaine-seeking behavior when NMDAR activity in the VTA is normalized.
Materials and Methods
Experimental animals.
Male Sprague Dawley rats, weighing 300–350 g on arrival (Charles River), were housed individually in a climate-controlled environment (21°C) on a 12 h light/dark cycle (lights on at 6:00 A.M.). All rats were maintained in accordance to the guidelines of the National Institutes of Health and approved by the Institutional Animal Care and Use Committee of the University of Texas Southwestern Medical Center. To facilitate acquisition of cocaine SA, animals were temporarily maintained on a restricted diet of laboratory chow at 85% of their original body weight and trained to lever press for ≤100 sucrose pellets (45 mg) per session on a fixed ratio 1 (FR1) reinforcement schedule for 3 consecutive days. Animals were then fed ad libitum for ≥2 d before surgical procedures.
Intravenous catheterization and intracranial cannulation.
Before surgery, rats were given atropine (0.10 ml, s.c.) to facilitate breathing, anesthetized with a ketamine/xylazine (100/10 mg/kg, i.p.) mixture, supplemented with isoflurane gas (0.5–1%) as needed, and implanted with a chronic in-dwelling Silastic intravenous catheter as described previously (Edwards et al., 2007). Rats also received stereotaxic surgery to implant 26 gauge bilateral guide cannulae (Plastics One) aimed 2.0 mm above the VTA, −5.6 mm posterior to bregma, ±0.8 mm lateral to bregma, and −6.0 mm ventral to dura (Paxinos and Watson, 1998). Dummy cannulae (33 gauge) were left in place throughout the experiment to prevent cannula obstruction. Upon surgery completion, rats were administered daily ketofen injections (5 mg/kg, s.c.) for ≤3 d after surgery to reduce pain and discomfort and 2.27% enrofloxacin (0.05 ml, i.v.) for to 10 d after surgery to curb infections. Catheters were flushed daily with 0.2 ml of saline containing heparin (20 U/ml) and gentamycin sulfate (0.33 mg/ml) throughout the experiment to help maintain catheter patency.
HSV vector construction.
HSV-LacZ expressing the benign Escherichia coli LacZ gene served as a control and was constructed as described previously (Neve et al., 1997). For HSV-dnGluN1 construction, we obtained a pRK5 vector with mutant GluN1a cDNA (generous gift from Richard L. Huganir, Johns Hopkins University, School of Medicine) that contained serine-to-alanine mutations at residues 896 and 897 (Ehlers et al., 1995). The mutant GluN1a was inserted into the HSV-PrpUC plasmid, packaged using 5dl1.2 helper virus, and then purified on a sucrose gradient. The two-point mutations were confirmed by DNA sequencing. For immunohistochemistry and electrophysiology experiments, we generated HSV-dnGluN1-GFP by subcloning the mutant GluN1a cDNA into the bi-cistronic HSV-p1005+ vector that coexpresses GFP (Clark et al., 2002; Russo et al., 2009). In this vector, a CMV promoter drives GFP expression, while the HSV immediate early gene IE4/5 promoter drives mutant GluN1a expression. An unaltered HSV-p1005+ vector served as a control. Previous reports have shown that transgene expression with HSV vectors is transient, with peak expression occurring within 1–3 d after transfection and dissipating to zero by days 6–7 (Carlezon et al., 1997; Barrot et al., 2002). Vector titers were 4.0 × 107 infectious U/ml.
Immunohistochemistry.
Rats were given acute bilateral (1.0 μl/per side) infusions of HSV-dnGluN1-GFP into the VTA at 5.6 mm posterior to bregma, 0.8 mm lateral to bregma, and 8.0 mm ventral to dura, using a 5 μl syringe (Hamilton) with a 30 gauge injector tip at a rate of 0.1 μl/min. Injector tips were left in place for an additional 5 min to allow for diffusion. Two, 4, or 6 d after HSV infusions, rats were deeply anesthetized with chloral hydrate (400 mg/kg, i.p.) and perfused with PBS followed by 4% paraformaldehyde. The brains were removed and stored in 4% paraformaldehyde overnight, then cryoprotected in 30% sucrose in PBS at 4°C for ≥3 d. Coronal sections (40 μm) were taken on a cryostat and stored free floating in PBS with 0.01% sodium azide. Following three brief rinses in PBS, sections were blocked with 3% normal donkey serum and 0.3% Triton X-100 in PBS for 1 h and then incubated with chicken polyclonal anti-GFP (1:5000; Aves Labs, catalog #GFP-1010, RRID:AB_2307313) and rabbit polyclonal anti-tyrosine hydroxylase (TH; 1:1000; Millipore, catalog #AB152, RRID:AB_390204) in 3% normal donkey serum (Jackson ImmunoResearch Laboratories, catalog #017-000-121, RRID:AB_2337258) and 0.3% Triton X-100 for 18 h. Sections were then incubated with fluorescent-tagged secondary antibodies, Alexa Fluor 488 donkey anti-chicken for GFP (1:250; Jackson ImmunoResearch Laboratories, catalog #703-545-155, RRID:AB_2340375), and CY3 goat anti-rabbit for TH (1:125; Millipore, catalog #AP132C, RRID:AB_92489) for 1 h.
Electrophysiological assessment of HSV-dnGluN1-GFP.
In vitro VTA slices were prepared from 6–7-week-old Sprague Dawley rats as described previously (Krishnan et al., 2007, 2008; Iñiguez et al., 2010; Choi et al., 2011). Slices were prepared 1–2 d following in vivo viral infection of the VTA with HSV-GFP (1.0 μl) or HSV-dnGluN1-GFP (1.0 μl). A tissue block containing midbrain was taken and sliced in ice-cold solution containing the following (in mm): 254 sucrose, 3 KCl, 1.25 NaH2PO4, 10 d-glucose, 24 NaHCO3, 2 CaCl2, and 2 MgSO4. Slices (250 μm thick) were transferred to a holding chamber in 34°C containing artificial CSF containing the following (in mm): 128 NaCl, 3 KCl, 1.25 NaH2PO4, 10 d-glucose, 24 NaHCO3, 2 CaCl2, and 2 MgSO4, pH 7.35, 295–305 mOsm. After 45–60 min recovery, whole-cell voltage-clamp recordings were performed under blind experimental conditions (MultiClamp 700B, Molecular Devices). Recording electrodes (2–4 MΩ) were filled with pipette solution containing the following (in mm): 115 potassium gluconate, 20 KCl, 1.5 MgCl2, 10 phosphocreatine, 10 HEPES, 2 ATP-Mg, and 0.5 GTP, pH 7.2, 285 mOsm. GFP-positive cells were visualized with an upright fluorescence microscope using infrared differential interference contrast illumination. In these experiments, putative dopamine neurons in the VTA were identified by large hyperpolarization-activated currents (Ih) as described previously (Ungless et al., 2003; Cao et al., 2010; Iñiguez et al., 2010). Ih current was evoked by a family of 10 mV steps (duration 600 ms) from −60 to −140 mV holding potentials. Following identification, putative dopamine neurons were clamped to +40 mV to allow development of NMDAR currents, in response to bath application of 100 μm NMDA, which were measured in solution containing the following: 40 μm 6-cyano-7-nitroquinoxaline-2,3-dione, 1 μm tetrodotoxin, 200 μm cadmium chloride, and 100 μm picrotoxin. Data were acquired using DigiData 1440A and pClamp 10.3 (Molecular Devices).
Biochemical characterization of HSV-dnGluN1.
For in vitro characterization of dnGluN1 effects on NMDAR signaling, primary embryonic (embryonic day 18) striatal cultures were generated from Long–Evans rats (Charles River) as previously described (Pulipparacharuvil et al., 2008). Crude striatal cells were plated at 1.3 × 106/well on poly-D-lysine (Sigma-Aldrich)-coated six-well plates (Corning) in DMEM (Life Technologies) supplemented with 10% (v/v) FBS (Life Technologies), penicillin (50 μg/ml)-streptomycin (50 U/ml; Sigma-Aldrich), and l-glutamine (4 mm; Sigma-Aldrich) and incubated at 37°C/5% CO2. Twenty-four hours later, media was changed to supplemented Neurobasal [2% B27 (v/v); Life Technologies], penicillin (50 μg/ml)-streptomycin (50 U/ml; Sigma-Aldrich), and l-glutamine (4 mm; Sigma-Aldrich). On day 7 (DIV7), cells were infected with HSV-dnGluN1 or HSV-LacZ (1 μg/well). Forty-eight hours later (DIV9), cells were stimulated with either 0, 10, or 100 μm NMDA for 10 min and harvested with 100 μl of 1× sample buffer, briefly sonicated (30% amp, 20 s, on ice), boiled at 98°C for 10 min, and centrifuged 16,100 rpm for 10 min. Cell homogenates (10 μl) were then subjected to 10% SDS-PAGE and transferred to Hybond-P PVDF membranes (GE Healthcare Life Sciences). Membranes were probed for GluN1, phosphorylated extracellular mitogen activated kinase two (pERK2), and total ERK2 expression with mouse monoclonal anti-GluN1 CT (1:500; Millipore, catalog #05-432, RRID:AB_390129), mouse monoclonal anti-pERK (1:1000; Cell Signaling Technology, catalog #9106, RRID:AB_331768), and rabbit polyclonal anti-ERK (1:2000; Cell Signaling Technology, catalog #9102, RRID:AB_330744); visualized by ECL-plus enhanced chemiluminescence; and measured density using the National Institutes of Health ImageJ (RRID:SCR_003073), as described previously (Choi et al., 2011).
For in vivo characterization of HSV-dnGluN1 effects on NMDAR signaling, rats implanted with bilateral intracranial cannulae targeting the VTA received unilateral infusions of HSV-dnGluN1 (1 μl) and HSV-LacZ (1 μl) on the contralateral side via 33 gauge infusion needles (Plastics One) extending 2 mm beyond the guide cannula over a 5 min period. Infusion needles were left in place for an additional 2 min to allow for diffusion. Either 2 or 6 d after HSV treatment, rats received intra-VTA infusions of PBS (0.5 μl), NMDA (0.5 μg/0.5 μl), or BDNF (0.25 μg/0.5 μl) via 33 gauge infusion needles over a 100 s period. The infusion needles were left in place for an additional 30 s to allow for diffusion. Following a 10 min period, rats were killed by decapitation and the brains were removed. Tissue punches of each unilateral VTA were taken with a 16 gauge punch from chilled coronal sections (0.5 mm) and immediately homogenized by sonication in lysis buffer (320 nm sucrose, 5 nm HEPES, 50 mm NaF, 1 mm EGTA, 1% SDS) containing Protease Inhibitor Cocktail I and Phosphotase Inhibitor Cocktails II and III (Sigma-Aldrich) and stored at −80°C until further analysis. After protein quantification by the Lowry method, 20 μg of protein aliquots were separated by SDS-PAGE and transferred to a PVDF membrane and probed with mouse monoclonal anti-pERK (1:1000; Cell Signaling Technology, catalog #9106, RRID:AB_331768) and rabbit polyclonal anti-ERK (1:2000; Cell Signaling Technology, catalog #9102, RRID:AB_330744).
Cocaine SA training.
SA training sessions occurred in operant chambers (Med Associates) during the light cycle (i.e., starting between 9:00 A.M. and 11:00 A.M.) as described previously (Edwards et al., 2007). Following intravenous catheterization and intra-VTA cannulation surgery, as described above, animals were allowed to recover ≥5 d before cocaine-SA training. Animals were trained to SA cocaine on a FR1 reinforcement schedule during 4 h daily sessions/trials for 5 d/week for 3 weeks, resulting in 15 total sessions. A single response on the active left lever resulted in a cocaine injection (0.5 mg/kg/0.05 ml) delivered over a 2.5 s period. During each cocaine injection, the house light was turned off and a cue light above the active (left) lever was concurrently illuminated. This was followed by a 12.5 s timeout period where the house light remained off and responses on either lever had no programmed consequences. Animals were limited to ≤162 cocaine injections during each of the 15 4 h sessions. A subset of animals underwent saline-SA training exactly as described above, but received saline-only injections (0.05 ml) instead of cocaine injections.
Effects of transient dnGluN1 expression during cocaine SA on increases in VTA GluA1.
Animals were trained on saline (controls) or cocaine SA as described above during HSV-dnGluN1 and HSV-LacZ expression in the VTA. One day before sessions 1–5 of the SA-training period, rats received a unilateral VTA infusion of HSV-dnGluN1 (1 μl) and a contralateral infusion of HSV-LacZ (1 μl) via 33 gauge infusion needles (Plastics One) extending 2 mm beyond the guide cannula over a 5 min period. Infusion needles were left in place for an additional 2 min to allow for diffusion. Rats received the same viral infusions 1 d before cocaine-SA training sessions 6–10 and 11–15. Thus, each rat received three total viral infusions during the 3 weeks of SA training. One day following the final SA session (6 d after the HSV infusion), tissue punches of each unilateral VTA were dissected, homogenized, and stored as described above. Ten micrograms of protein aliquots were separated by SDS-PAGE and transferred to a PVDF membrane and probed with rabbit polyclonal anti-GluR1 (1:500; Millipore, catalog #07-660, RRID:AB_390157) and mouse monoclonal anti-β-tubulin (1:200,000; Millipore, catalog #05-661, RRID:AB_309885) as an internal standard.
Effects of transient dnGluN1 expression during cocaine SA on cocaine-seeking behavior (i.e., PR, extinction, and reinstatement).
Rats received three weekly infusions of HSV during cocaine SA as described above. One day following the last cocaine SA session (6 d after the last HSV infusion), animals were subjected to a single PR test for cocaine reinforcement. During PR testing, the response requirement (i.e., lever presses) for each successive cocaine injection (1.0 mg/kg/0.1 ml over a 5 s period) increased exponentially according to the following formula: (5e(inj. # × 0.2)) − 5, as described previously (Richardson and Roberts, 1996). The break point was defined as the highest ratio of responses per injection completed before a 1 h period elapsed without earning another injection. After the PR test, rats remained in their home cages for 3 weeks of forced abstinence, followed by extinction tests consisting of five daily 4 h extinction sessions where responding at the drug-paired lever was recorded but had no programmed consequence. After extinction training, animals were tested for reinstatement of cocaine seeking in four daily sessions. During each reinstatement session, rats were placed in the SA operant chambers for 3 h of additional extinction conditions, followed by the presentation of a priming stimulus when reinstatement of drug-paired lever responses was measured for an additional 1 h period. Priming stimuli consisted of noncontingent presentation of the drug-associated cue light every 2 min (i.e., 30 presentations during the 1 h) on day 1. During days 2–4, priming stimuli consisted of experimenter-delivered intraperitoneal saline or cocaine injections (0, 5, or 10 mg/kg) in counterbalanced order.
Following completion of behavioral testing, rats were deeply anesthetized with chloral hydrate (400 mg/kg, i.p.) and received bilateral intra-VTA infusions of cresyl violet (0.5 μl) through the intracranial guide cannula. Rats were then decapitated, their brains were removed, and infusion sites identified in 0.5 mm coronal sections. Animals determined to have incorrect infusion sites were not included in the data analysis.
Effects of transient dnGluN1 expression on AMPA-mediated locomotor behavior.
Locomotor behavior was measured in the dark using circular test chambers equipped with four pairs of infrared photo emitters–detectors, as described previously (Choi et al., 2011). Drug-naive rats received a single bilateral infusion of HSV-dnGluN1 or HSV-LacZ (1 μl/per side) via VTA guide cannulae as described above. Six and 7 d after HSV infusions, rats were placed in the locomotor chambers for a 2 h habituation period, and then received intra-VTA infusions of AMPA (day 6: 50 ng/ 0.5 μl) and PBS (day 7: 0.5 μl) over a 100 s period. Rats were returned to the test chambers where locomotor activity was assessed for an additional 2 h. A subset of drug-naive rats received a total of three weekly HSV infusions to emulate the cocaine-SA protocol before locomotor testing, and received similar locomotor testing following AMPA and PBS infusions 6 and 7 d after the last HSV infusion. Histological verification of infusion sites was performed as described above.
Effects of transient dnGluN1 expression on membrane and cytosol expression of glutamate receptor subunits in the VTA.
Drug-naive rats received a single or a total of three weekly bilateral infusions of HSV-dnGluN1 or HSV-LacZ (1 μl/per side) via VTA guide cannulae as described above. Six days after the final HSV infusion, VTA tissue punches were collected as described above and processed using a membrane fractionation protocol. Under chilled conditions, samples were homogenized via low amplitude (20%) sonication in 160 μl of Syn-PER synaptic protein extraction reagent buffer (https://www.thermofisher.com/order/catalog/product/87793) containing Protease Inhibitor Cocktail I and Phosphotase Inhibitor Cocktails II and III (Sigma-Aldrich). Following sonication, samples were pooled (two animals per sample) to harvest enough tissue for membrane fractionation for a total of 12 samples from 24 rats. Samples were centrifuged at 3,000 rpm for 10 min at 4°C. The supernatant (S1) was added to a new tube and the pellet was discarded. The tubes with the S1 were centrifuged at 12,000 rpm for 30 min at 4°C. The supernatant (S2) was added to a new tube (cytoplasmic fraction) and the pellet (P2) was resuspended with Syn-PER buffer (membrane fraction), sonicated briefly, and set aside for the Lowry method as described above. Ten micrograms of protein aliquots of either membrane of cytosol fractions were separated by SDS-PAGE and transferred to a PVDF membrane and probed with rabbit polyclonal anti-GluA1 (1:500; Millipore, catalog #07-660, RRID:AB_390157), rabbit polycolonal anti-GluA2 (1:2000; Millipore, catalog #AB1768, RRID:AB_2313802), rabbit polyclonal anti-GluN2B (1:500; Millipore, catalog #06-600, RRID:AB_310193), rabbit polyclonal anti-GluN3A (1:500; Millipore, catalog #07-356, RRID:AB_2112620), and mouse monoclonal anti-pan cadherin (1:2000; Sigma-Aldrich, catalog #C1821, RRID:AB_476826) as an internal standard for membrane fractions, or mouse monoclonal anti-β-tubulin (1:100,000; Millipore, catalog #05-661, RRID:AB_309885), as an internal standard for cytosol fractions.
Experimental design and statistical analysis.
Electrophysiological data were analyzed using an unpaired t test. Each group consisted of n = 10 cells from four animals (2–3 cells/per animal). For whole-tissue Western blot analysis, PBS-treated and saline-treated groups were pooled and served as controls. The data were analyzed by one-way between-subjects ANOVA followed by post hoc comparisons with Fisher's least significant difference (LSD) test. Membrane fractionation Western blot data were analyzed using a two-way between-subjects ANOVA, followed by post hoc comparisons with Fisher's LSD test, with both group and subunit as nonrepeated measures. SA data were analyzed by two-way mixed ANOVA, with trial as a repeated measure, and group as a nonrepeated measure. PR results were analyzed by unpaired t test. Drug prime reinstatement data were analyzed using a two-way mixed ANOVA, followed by post hoc comparisons with Fisher's LSD test, with group as a nonrepeated measure and dose as a repeated measure. Extinction, cue reinstatement, and locomotor data were analyzed by two-way mixed ANOVA followed by post hoc comparisons with Fisher's LSD test, with group as a nonrepeated measure and trial, test, or drug as repeated measures. Statistical significance was preset at p < 0.05.
Results
HSV-dnGluN1 expression in the VTA decreases NMDAR function in vitro and in vivo
We generated an HSV vector that expresses the GluN1 subunit containing two-point mutations (S896A/S897A) with putative dominant-negative NMDAR properties. To confirm HSV-dnGluN1-GFP transient expression, we probed for GFP following 2, 4, and 6 d after HSV infusion. Figure 1a shows HSV-mediated expression of dnGluN1-GFP in VTA coronal sections (top three panels) with the bottom three panels showing the magnified portion (white box). HSV-mediated expression is evident 2 or 4 d after HSV infusion and generally confined to the VTA region. In contrast, expression is absent after 6 d, consistent with the transient profile of HSV expression in a previous study (Barrot et al., 2002). Figure 1b shows HSV-mediated expression of dnGluN1-GFP in TH-positive dopamine neurons in the VTA. We reported previously that ∼75% of HSV-infected cells within this VTA region are TH-positive, and that HSV infects ∼25% of the local TH-positive population of neurons (Choi et al., 2011). Putative dopamine neurons in in vitro slices of HSV-infused VTA were identified by large Ih currents as described previously (Ungless et al., 2003) in GFP-positive cells. Figure 1c shows that NMDAR-mediated currents induced by bath application of NMDA (100 μm) are substantially attenuated by dnGluN1-GFP expression compared with GFP-expressing controls (t(18) = 4.75, p < 0.005). Similarly, expression of dnGluN1 substantially reduces both basal and NMDA-stimulated ERK phosphorylation in Western blots of rat primary striatal cultures compared with HSV-LacZ-infected cultures (Fig. 1d), thereby confirming a NMDAR dominant-negative profile in infected neurons in vitro.
Figure 1.
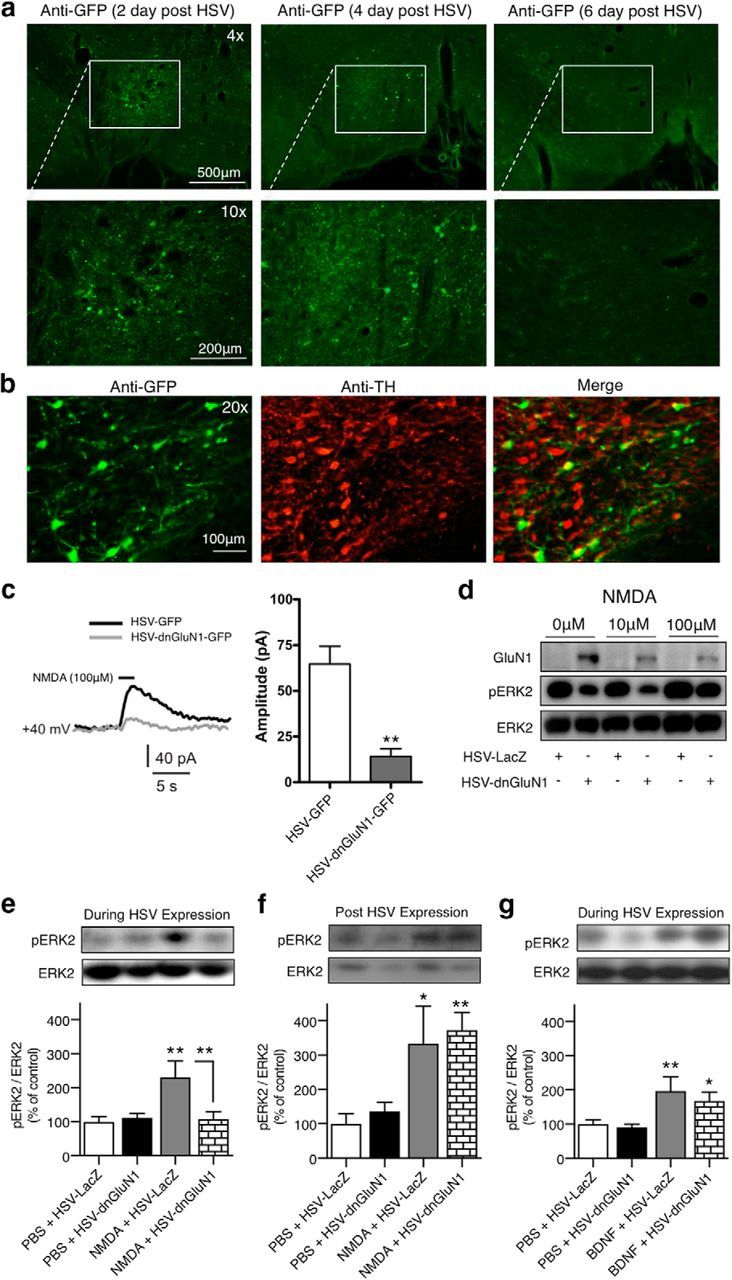
Characterization of HSV-dnGluN1 in vitro and in vivo. a, Expression of GFP in VTA sections 2, 4, and 6 d after intra-VTA HSV-dnGluN1-GFP infusions in rats. b, Colocalized expression (yellow) of mutant dnGluN1-GFP (green) in VTA dopamine neurons (red) after intra-VTA HSV-dnGluN1-GFP infusions in rats. c, Responses to bath application of NMDA in VTA dopamine neurons infected with HSV-GFP or HSV-dnGluN1-GFP. Representative traces and averaged data of whole-cell recordings show a significantly reduced response to bath application of 100 μm NMDA (horizontal black bar above trace) in HSV-dnGluN1-GFP-expressing VTA neurons compared with HSV-GFP-expressing VTA neurons held at +40 mV (n = 10 cells/group; n = 2–3 cells/4 animals). d, HSV-dnGluN1 infection led to overexpression of GluN1 protein compared with HSV-LacZ infection in primary neuronal cultures. NMDA application (100 μm) increased pERK2 levels in HSV-LacZ-infected neurons, but not HSV-dnGluN1-infected neurons. e, In vivo intra-VTA NMDA infusions (0.5 μg/0.5 μl) increased VTA pERK2 levels during HSV-LacZ expression (n = 11) compared with pooled PBS-infused (0.5 μl) groups (n = 22), an effect blocked by HSV-dnGluN1 expression (n = 11). f, In vivo intra-VTA NMDA infusions (0.5 μg/0.5 μl) increased VTA pERK2 levels in both HSV-LacZ-treated (n = 7) and HSV-dnGluN1-treated (n = 8) groups compared with pooled PBS-infused groups (n = 14) when tested 6 d after HSV expression. g, In vivo intra-VTA BDNF infusions (0.25 μg/0.5 μl) increased VTA pERK2 levels during HSV-LacZ (n = 14) and HSV-dnGluN1 (n = 14) expression compared with pooled PBS-infused groups (n = 28). Data are expressed as mean ± SEM, *p < 0.05, **p < 0.005 (significant from controls or designated group using t test or Fisher's LSD post hoc test).
We next examined the effects of HSV-dnGluN1 expression on NMDAR signaling in vivo. Awake, freely moving rats received intra-VTA infusions of NMDA (0.5 μg/0.5 μl) or the PBS vehicle 2 d after HSV infusions when HSV-mediated expression is high (Barrot et al., 2002). Figure 1e shows that NMDA infusions increase ERK phosphorylation by >130% in VTA tissue dissections compared with vehicle infusions, but this effect is abolished by expression of dnGluN1 in the VTA (F(2,41) = 7.443; p = 0.0017). There is no effect of dnGluN1 on basal ERK phosphorylation following intra-VTA PBS vehicle infusions. We determined whether attenuation of NMDAR signaling persists after 6 d (Fig. 1f) when this HSV vector no longer expresses detectable protein, and found that NMDA-induced ERK phosphorylation completely recovered to normal levels (F(2,26) = 7.55; p = 0.003). Finally, we tested the specificity of NMDAR blockade by examining the effects of dnGluN1 expression on BDNF-TrkB receptor-mediated ERK phosphorylation 2 d after HSV treatment. In contrast to attenuation of NMDA-induced ERK phosphorylation, ERK phosphorylation induced by intra-VTA infusions of BDNF is not altered by dnGluN1 expression (Fig. 1g; F(2,53) = 6.20; p = 0.004). Thus, HSV-mediated dnGluN1 expression exhibits a selective dominant-negative attenuation of NMDAR signaling that is temporally limited to dnGluN1 expression.
Transient HSV-dnGluN1 expression in VTA blocks cocaine-SA-induced increases in GluA1 expression
We reported previously that cocaine SA leads to a transient and reinforcement-specific upregulation in VTA GluA1 protein levels 1 d after withdrawal from cocaine SA (Choi et al., 2011). Given that cocaine exposure can enhance excitatory synaptic input to VTA dopamine neurons in an NMDAR-dependent manner, we investigated whether NMDAR activity is necessary for upregulation in VTA GluA1 levels induced by chronic cocaine SA. Figure 2a shows the timeline depicting HSV infusions during cocaine-SA training on a FR1 reinforcement schedule. Rats readily learn to press a lever for intravenous cocaine (0.5 mg/kg/0.05 ml) and show robust SA behavior compared with animals receiving only saline injections (Fig. 2b; group effect: F(1,15) = 71.78; p < 0.0001; trial effect: F(14,210) = 3.257 p = 0.0001; group × trial interaction; F(14,210) = 2.632; p = 0.0015). After 3 weeks of cocaine SA, GluA1 levels significantly increased in unilateral VTA receiving HSV-LacZ infusions compared with VTA from saline-SA rats that received similar HSV-LacZ infusions (F(3,28) = 2.98; p = 0.048), replicating our previous findings (Fig. 2c). However, this GluA1 upregulation is completely attenuated by transient dnGluN1 expression in the contralateral VTA of cocaine-SA animals, as they show no GluA1 increase relative to either LacZ or dnGluN1 expression in saline-SA controls. Thus, dnGluN1 expression in the VTA blocks the ability of cocaine reinforcement to upregulate GluA1 expression, presumably by blunting NMDAR activity during the cocaine-SA training period.
Figure 2.

Increased GluA1 expression in the VTA following cocaine SA is blocked by transient HSV-dnGluN1 expression. a, Timeline depiction of HSV treatment regimens for saline-SA (n = 10) and cocaine-SA (n = 7) animals. b, Number of infusions per session in saline-SA and cocaine-SA (0.5 mg/kg/infusion) animals. c, Example immunoblots of total GluA1 and β-tubulin in the VTA after 1 d withdrawal (WD) from saline and cocaine SA. Cocaine-SA animals exhibit elevated total GluR1 expression in animals treated with HSV-LacZ (n = 6), but not when treated with HSV-dnGluN1 (n = 7), in VTA homogenates, compared with pooled (n = 19) saline-SA animals. Data are expressed as mean ± SEM, *p < 0.05 (significant from controls using Fisher's LSD post hoc test).
Transient HSV-dnGluN1 expression in VTA leads to a paradoxical increase in cocaine-seeking behavior
We previously found that rats with viral-mediated GluA1 overexpression in the VTA exhibit significantly higher break points before cessation of cocaine SA on a PR test, demonstrating a functional link between enhanced VTA GluA1 levels and increased motivation for cocaine (Choi et al., 2011). Given that transient dnGluN1 expression in the VTA prevents GluA1 upregulation in cocaine-SA rats, we hypothesized that animals receiving similar intra-VTA HSV-dnGluN1 infusions during cocaine SA would exhibit decreased motivation for cocaine compared with cocaine-SA rats treated with HSV-LacZ that show GluA1 upregulation. Figure 3a shows the timeline for HSV infusions during cocaine-SA training followed by PR, extinction, and reinstatement tests conducted during weeks 4 and 5 after withdrawal from cocaine SA. Figure 3b shows that bilateral expression of HSV-dnGluN1 in the VTA does not affect cocaine SA on a low-demand FR1 reinforcement schedule compared with HSV-LacZ-infused controls (group effect: F(1,21) = 2.94; p = 0.1012; trial effect: F(14,294) = 2.285; p = 0.0056; group × trial interaction: F(14,294) = 1.507; p = 0.1071).
Figure 3.

Paradoxical increase in motivation for cocaine following transient HSV-dnGluN1-mediated inactivation of NMDARs in the VTA. a, Timeline depiction of HSV treatment regimens for cocaine-SA animals and subsequent PR, extinction, and reinstatement testing. b, Number of infusions per session in HSV-LacZ-treated (n = 12) and HSV-dnGluN1-treated (n = 11) cocaine-SA animals. c, Transient HSV-dnGluN1 expression in the VTA during cocaine SA increases break points (left), and total number of infusions earned (right), at a dose of 1 mg/kg/infusion compared with HSV-LacZ after 1 d withdrawal (WD). d, Transient HSV-dnGluN1 expression in the VTA during cocaine SA enhances drug-paired lever responding during initial extinction trial. e, Transient HSV-dnGluN1 expression in the VTA during cocaine SA has no effect on cue-primed reinstatement of drug-paired lever responding. f, Transient HSV-dnGluN1 expression in the VTA during cocaine SA increases cocaine-primed reinstatement of drug-paired lever responding (pooled 5 and 10 mg/kg i.p. cocaine). Data are expressed as mean ± SEM, *p < 0.05, **p < 0.005 (significant from controls using Fisher's LSD post hoc test).
Following cocaine-SA training, HSV-dnGluN1-treated animals show an unexpected increase in motivation for cocaine as indicated by a significant elevation in break points for cocaine SA on the more demanding PR reinforcement schedule (t(21) = 2.33; p = 0.029) and a greater total number of cocaine infusions earned (t(21) = 2.22; p = 0.037) compared with HSV-LacZ-treated controls (Fig. 3c). Similarly, HSV-dnGluN1-treated rats show greater nonreinforced responding at the drug-paired lever during initial extinction tests (Fig. 3d), despite the fact that these tests are conducted 3 weeks after cocaine SA and 4 weeks after the last HSV-dnGluN1 infusion (group effect: F(1,21) = 4.80; p = 0.039; trial effect: F(4,84) = 24.23; p < 0.0001; group × trial interaction: F(4,84) = 6.151; p = 0.0002). This enhancement in cocaine-seeking behavior in the absence of cocaine reinforcement is consistent with enhanced motivation for cocaine observed during the PR tests. Figure 3e shows that cues associated with prior cocaine infusions effectively reinstate drug-paired lever responding compared with extinction baselines (test effect: F(1,21) = 12.64; p = 0.0019), but no significant group differences are found (group effect: F(1,21) = 0.3867; p = 0.5407; group × test interaction: F(1,21) = 0.1288; p = 0.7232). Following cue-induced reinstatement testing, the ability of noncontingent priming injections of cocaine to reinstate cocaine seeking also is elevated in rats receiving prior HSV-dnGluN1 infusions (saline vs pooled 5 and 10 mg/kg cocaine doses), although both groups show low reinstatement in response to priming with saline infusions, thereby negating a generalized increase in lever-press behavior (Fig. 3f; group effect: F(1,21) = 2.57; p = 0.1241; dose effect: F(1,21) = 36.63; p < 0.0001; group × dose interaction: F(1,21) = 3.442; p = 0.0776). Together, these data indicate that prior inactivation of NMDAR function during cocaine SA leads to prolonged increases in cocaine-seeking behavior elicited by the cocaine-associated environmental context or cocaine itself, but not discrete cues associated with cocaine SA.
Transient HSV-dnGluN1 expression in VTA facilitates AMPAR-mediated locomotor activity and membrane expression
Based on the finding that dnGluN1 expression in VTA neurons increases the motivation for cocaine despite attenuating cocaine-induced upregulation in VTA GluA1 levels, we hypothesized that even transient dnGluN1-mediated attenuation of NMDAR function may lead to compensatory increases in functional (cell surface) AMPARs, a phenomenon referred to as “scaling” (Turrigiano, 2008). To determine whether transient HSV-dnGluN1 expression affected subsequent AMPAR function in the VTA, animals received a single infusion of HSV-LacZ or HSV-dnGluN1 and then were tested for locomotion in response to a subthreshold dose of intra-VTA AMPA (50 ng/ 0.5 μl) or PBS vehicle (0.5 μl) 6 d after the HSV infusion (Fig. 4a). A single infusion of HSV-dnGluN1 in VTA produced no effect on AMPA-mediated locomotor responses compared with HSV-LacZ-treated rats (group effect: F(1,7) = 0.2850; p = 0.61; drug effect: F(1,7) = 1.627; p = 0.2428; group × drug interaction: F(1,7) = 0.0072; p = 0.9344), and responses in both groups fail to differ from PBS vehicle infusions (Fig. 4b,c). We next determined whether three weekly VTA HSV infusions, similar to HSV treatment in the cocaine-SA protocol (Fig. 4d), would alter AMPA-mediated behavior. In contrast to a single VTA infusion, three weekly HSV-dnGluN1 infusions substantially enhanced the locomotor response to subthreshold AMPAR challenge (Fig. 4e,f; group effect: F(1,8) = 6.635; p = 0.0328; drug effect: F(1,8) = 14.37; p = 0.0053; group × drug interaction: F(1,8) = 5.513; p = 0.0468). This enhanced AMPA locomotor response occurs at the same time after HSV-dnGluN1 treatments when motivation for cocaine is increased in cocaine-SA rats (but tissue GluA1 levels are not), and is consistent with the hypothesis that NMDAR blockade leads to increased cell-surface AMPAR expression. Thus, prolonged attenuation of NMDAR activity over 3 weeks, but not 1 week, of dnGluN1 expression is required to induce compensatory increases in VTA AMPAR function. To test this hypothesis, we measured membrane and cytosol levels of glutamate receptor subunits in the VTA after the three weekly HSV-dnGluN1 treatments. Figure 4g shows that 3 weeks of dnGluN1 expression significantly increased levels of GluA1 and GluN2B (group effect: F(1,36) = 11.99; p = 0.0014), but failed to alter GluA2 or GluN3A (subunit effect: F(3,36) = 0.6332; p = 0.5985; group × subunit interaction: F(3,36) = 0.6332; p = 0.5985). No differences were observed in the cytosol fraction (group effect: F(1,36) = 0.0267; p = 0.8709; subunit effect: F(3,36) = 0.4447; p = 0.7225; group × subunit interaction: F(3,36) = 0.4447; p = 0.7225). Thus, sustained inactivation of NMDAR function leads to increased membrane levels of both AMPAR and NMDAR subunits, consistent with a homeostatic scaling response. Figure 5a,b depicts the location of HSV infusion sites in the VTA for all the animals included in behavioral studies based on histological analyses.
Figure 4.

Transient NMDAR inactivation in the VTA enhances AMPAR-mediated locomotor activity and membrane expression of glutamate receptor subunits. a, d, Timeline depiction of HSV-treatment regimens and subsequent locomotor testing. b, e, Locomotor counts (20 min bins) for intra-VTA HSV-LacZ-treated (n = 4–5) and HSV-dnGluN1-treated (n = 4–6) animals during habituation and following intra-VTA PBS (0.5 μl) or AMPA (50 ng/0.5 μl) infusions. c, f, Locomotor counts (2 h) for intra-VTA HSV-LacZ-treated (n = 4–5) and HSV-dnGluN1-treated (n = 4–6) animals during habituation and following intra-VTA PBS (0.5 μl) or AMPA (50 ng/0.5 μl) infusions. A single intra-VTA HSV-dnGluN1 or HSV-LacZ infusion has no significant effect on intra-VTA AMPA-mediated locomotor activity. In contrast, animals that received three intra-VTA HSV-dnGluN1 infusions over 3 weeks exhibit significantly increased levels of AMPA locomotor activity compared with controls that received three intra-VTA HSV-LacZ-treatments. Data are expressed as mean ± SEM, **p < 0.005 (significant from all groups using Fisher's LSD post hoc test). g, Example immunoblots of glutamate receptor subunits in membrane and cytosolic fractions of VTA tissue after three weekly intra-VTA HSV-dnGluN1 (n = 6) or HSV-LacZ (n = 6) treatments. Animals infused with HSV-dnGluN1 show increased membrane levels of GluA1 and GluN2b 6 d following the last HSV infusion. No differences were found in the cytosol fraction. Data are expressed as mean ± SEM, *p < 0.05 (significant from controls using Fisher's LSD post hoc test).
Figure 5.

a, b, Histological localization of HSV-LacZ and HSV-dnGluN1 infusion sites in the VTA for (a) cocaine-SA animals tested in progressive ratio/extinction/reinstatement and (b) animals tested in AMPA-mediated locomotor activity.
Discussion
Dominant-negative effect of GluN1 S896A/S897A phosphorylation-resistant mutation
We report here evidence for a novel, inducible, and transient method to downregulate NMDAR function in adult neurons via viral-mediated expression of a mutant GluN1 subunit resistant to phosphorylation by both PKA (S897) and PKC (S896; Tingley et al., 1997). We found dominant-negative actions at NMDARs in three tests. First, we show that NMDAR-mediated currents are prominently decreased in putative VTA dopamine neurons expressing dnGluN1. Second, given that ERK2 phosphorylation is a well established component of downstream NMDAR-Ca2+-dependent signaling cascades (Thomas and Huganir, 2004; Haddad, 2005; Wang et al., 2007), we show that dnGluN1 expression attenuates NMDAR-mediated ERK phosphorylation in both primary striatal cultures and following intra-VTA infusions of NMDA in vivo. Results with in vivo NMDA infusions are particularly compelling as they demonstrate that dominant-negative efficacy is contiguous with the time frame of HSV-mediated expression (<6 d) and specific to activated NMDARs in awake freely moving animals. Thus, the HSV-dnGluN1 vector can be used to induce continuous silencing of NMDAR function for relatively short time frames or phases of experimentation with complete reversion to normal NMDAR function after HSV-mediated expression declines. Earlier studies have shown that mutation of serine sites 896/897 to alanine on GluN1 suppresses trafficking of NMDARs from the endoplasmic reticulum (ER) to the cell-surface membrane (Ehlers et al., 1995; Scott et al., 2001), while mutations that mimic constitutive active phosphorylation at these sites increases cell-surface expression (Scott et al., 2001, 2003). Since NMDARs typically are composed of obligatory GluN1 subunits that coassemble with GluN2 and GluN3 subunits in the ER (McIlhinney et al., 1998; Perez-Otano et al., 2001), we speculate that dnGluN1 subunits coassemble with endogenous GluN2 and GluN3 subunits, thereby reducing the available pool of functional NMDARs that can be trafficked to the surface, resulting in the dominant-negative effects on NMDAR activity during dnGluN1 expression.
Cocaine-SA-induced increases in VTA GluA1 levels are mediated by NMDAR activity
Chronic cocaine SA induces a reinforcement-related upregulation in GluA1 subunits in the VTA, along with increased PKA-mediated GluA1 phosphorylation 1 d after cessation of daily SA sessions (Choi et al., 2011). Here we show that this GluA1 upregulation is blocked by sustained dnGluN1 expression in the VTA during the SA-training phase, indicating that reinforcement-related upregulation of GluA1 is NMDAR-dependent. These results concur with those of previous studies showing that cocaine-evoked LTP of excitatory inputs to VTA dopamine neurons also requires NMDAR activation. For example, pretreatment with the NMDAR antagonist MK-801 blocks the increase in AMPA/NMDA ratio observed following cocaine exposure (Ungless et al., 2001). Similarly, genetic ablation of the GluN1 subunit in dopamine neurons also prevents the cocaine-induced augmentation of AMPA inputs onto VTA dopamine neurons. However, the prevention of this augmentation is primarily due to compensatory increases in AMPAR input that occlude further potentiation (Engblom et al., 2008; Zweifel et al., 2008). Importantly, the inducible and reversible approach for NMDAR inactivation produced by dnGluN1 expression fails to cause similar compensatory increases in GluA1 protein levels in saline-SA controls. Thus, an NMDAR-dependent mechanism for cocaine-SA-induced GluA1 upregulation is more clearly indicated by our approach.
Given that chronic cocaine SA also increases GluA1 mRNA expression in the VTA, which could drive an increase in GluA1 protein (Choi et al., 2011), one potential mechanism may involve glutamate release and an NMDAR signaling cascade that stimulate GluA1 gene expression. Indeed, similar to GluA1 upregulation (Choi et al., 2011), increases in extracellular VTA glutamate levels are reported to occur only in cocaine-reinforced SA animals, but not in yoked controls that receive cocaine injection by passive infusion (You et al., 2007). Elevated glutamate activation of NMDARs during daily cocaine-SA sessions, and subsequent NMDAR-mediated Ca2+ influx, could stimulate BDNF secretion as has been shown in cultured neurons (Marini et al., 1998; Hartmann et al., 2001; Matsuda et al., 2009). BDNF-TrkB signaling is known to induce GluA1 transcription and translation in hippocampal neurons (Caldeira et al., 2007), and to increase synaptic expression of GluA1 (Gao et al., 2016). In this regard, a similar NMDAR-BDNF-TrkB signaling cascade mediates LTP of excitatory AMPAR input to VTA dopamine neurons following repeated systemic cocaine injections (Pu et al., 2006). Thus, by attenuating VTA NMDAR signaling with HSV-dnGluN1, such BDNF-mediated increases in VTA GluA1 gene expression may be prevented.
Transient HSV-dnGluN1 expression in VTA results in a paradoxical increase in cocaine-seeking behavior
Since GluA1 overexpression in VTA neurons directly increases motivation for cocaine in SA animals (Choi et al., 2011), we hypothesized that the blockade of endogenous GluA1 upregulation in animals treated with HSV-dnGluN1 would reduce motivation for cocaine. Surprisingly, animals receiving intra-VTA infusions of HSV-dnGluN1 during cocaine SA show increased motivation for cocaine in a PR test conducted when GluA1 upregulation is attenuated. Importantly, cocaine seeking elicited in both extinction and reinstatement (cocaine-primed) remains elevated following 3–5 weeks of protracted withdrawal. Thus, transient inactivation of VTA NMDAR signaling during cocaine SA leads to a paradoxical and enduring enhancement in cocaine-seeking behavior. In contrast, permanent deletion of GluN1 from dopamine neurons abolishes cocaine-primed reinstatement of cocaine-conditioned place preference (Engblom et al., 2008), and a cue-induced lever pressing is reduced in cocaine-SA mice (Mameli et al., 2009), although either effect may reflect the loss of NMDAR input to dopamine neurons during testing rather than attenuation of cocaine-induced neuroplasticity. Indeed, acute pharmacological blockade of VTA glutamate receptors (AMPARs or NMDARs) reduces cocaine-seeking behavior (Sun et al., 2005; You et al., 2007).
Transient HSV-dnGluN1 expression in VTA increases AMPAR-mediated locomotor activity and membrane expression of glutamate receptor subunits
Since permanent deletion of GluN1 in dopamine neurons can induce compensatory increases in AMPAR activity (Turrigiano, 2008), we determined whether increased cocaine-seeking behavior following transient dnGluN1 expression also relates to increased AMPAR function in the VTA. Consistent with this hypothesis, rats subjected to the 3 week protocol of dnGluN1 expression used in cocaine-SA experiments show strong facilitation of AMPAR-mediated locomotor responses in the VTA. Furthermore, a similar treatment of three HSV-dnGluN1 infusions increases membrane (but not whole-tissue or cytosolic) expression of both GluA1 AMPAR and GluN2B NMDAR subunits, providing biochemical evidence of homeostatic scaling following prolonged GluN1 inactivation in the VTA, similar to synaptic scaling observed with continuous knock-out of GluN1 in VTA dopamine neurons or in ventral spinal neurons following sustained pharmacological inactivation of NMDARs (Rosen et al., 2007; Engblom et al., 2008; Zweifel et al., 2008). Moreover, it's been reported that cocaine administration promotes excitatory synaptic plasticity in VTA neurons mediated in part by increased expression of GluN2b and their redistribution to the membranes (Schilström et al., 2006). NMDARs containing GluN2b have slower decay properties that would promote excitatory currents (Paoletti et al., 2013). Conversely, Schumann et al. (2009), found that inhibiting GluN2b-containing receptors with ifenprodil blocked cocaine-induced increase in AMPAR/NMDAR current ratios. Thus, the increase in motivation for cocaine, despite blockade of upregulation in GluA1 protein levels, is likely caused by a counter-adaptive increase in functional cell-surface AMPARs and NMDARs in response to prolonged GluN1 inactivation in the VTA. The augmentation in AMPAR function is not evident following only one HSV-dnGlu1 infusion, suggesting that a longer period of GluN1 inactivation is necessary to induce compensation in AMPAR responses.
In summary, dnGluN1 is a selective and effective approach to reduce NMDAR function in an inducible, continuous, and reversible manner. When coupled with short-acting viral vectors, such as HSV, which generally expresses detectable protein for only 1–5 d, dnGluN1 could be useful to dissociate NMDAR effects on neuroplasticity relating to drug treatment, learning, or other experience-dependent processes from the prominent role NMDARs have in expression of neurophysiological and behavioral responses. These findings also illustrate the potential for adverse consequences with prolonged experimental or therapeutic manipulation of NMDARs, although one could envision potential applications where long-term homeostatic compensation following relatively brief attenuation of NMDAR function would be advantageous (e.g., ketamine in treatment-resistant depression; Kavalali and Monteggia, 2012). However, the ability of even transient loss of NMDAR function to enhance excitatory transmission in the VTA, together with long-lasting facilitation of addictive behavior, suggests that VTA neurons would be hyper-responsive to conditioned glutamate release that increases the incentive motivation for cocaine.
Footnotes
This work was supported by National Institute on Drug Abuse Grants DA18743 (to D.W.S.), T32DA7290 (to D.G.), F31DA035073 (to M.B.C.), and DA032708 (to C.W.C.); and National Institute on Alcohol Abuse and Alcoholism Grant AA022445 (to M.-H.H.).
The authors declare no competing financial interests.
References
- Ahmed SH, Koob GF (1998) Transition from moderate to excessive drug intake: change in hedonic set point. Science 282:298–300. 10.1126/science.282.5387.298 [DOI] [PubMed] [Google Scholar]
- Barrot M, Olivier JD, Perrotti LI, DiLeone RJ, Berton O, Eisch AJ, Impey S, Storm DR, Neve RL, Yin JC, Zachariou V, Nestler EJ (2002) CREB activity in the nucleus accumbens shell controls gating of behavioral responses to emotional stimuli. Proc Natl Acad Sci U S A 99:11435–11440. 10.1073/pnas.172091899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellone C, Lüscher C (2006) Cocaine triggered AMPA receptor redistribution is reversed in vivo by mGluR-dependent long-term depression. Nat Neurosci 9:636–641. 10.1038/nn1682 [DOI] [PubMed] [Google Scholar]
- Borgland SL, Malenka RC, Bonci A (2004) Acute and chronic cocaine-induced potentiation of synaptic strength in the ventral tegmental area: electrophysiological and behavioral correlates in individual rats. J Neurosci 24:7482–7490. 10.1523/JNEUROSCI.1312-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldeira MV, Melo CV, Pereira DB, Carvalho R, Correia SS, Backos DS, Carvalho AL, Esteban JA, Duarte CB (2007) Brain-derived neurotrophic factor regulates the expression and synaptic delivery of alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptor subunits in hippocampal neurons. J Biol Chem 282:12619–12628. 10.1074/jbc.M700607200 [DOI] [PubMed] [Google Scholar]
- Cao JL, Covington HE 3rd, Friedman AK, Wilkinson MB, Walsh JJ, Cooper DC, Nestler EJ, Han MH (2010) Mesolimbic dopamine neurons in the brain reward circuit mediate susceptibility to social defeat and antidepressant action. J Neurosci 30:16453–16458. 10.1523/JNEUROSCI.3177-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlezon WA Jr, Boundy VA, Haile CN, Lane SB, Kalb RG, Neve RL, Nestler EJ (1997) Sensitization to morphine induced by viral-mediated gene transfer. Science 277:812–814. 10.1126/science.277.5327.812 [DOI] [PubMed] [Google Scholar]
- Chen BT, Bowers MS, Martin M, Hopf FW, Guillory AM, Carelli RM, Chou JK, Bonci A (2008) Cocaine but not natural reward self-administration nor passive cocaine infusion produces persistent LTP in the VTA. Neuron 59:288–297. 10.1016/j.neuron.2008.05.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi KH, Edwards S, Graham DL, Larson EB, Whisler KN, Simmons D, Friedman AK, Walsh JJ, Rahman Z, Monteggia LM, Eisch AJ, Neve RL, Nestler EJ, Han MH, Self DW (2011) Reinforcement-related regulation of AMPA glutamate receptor subunits in the ventral tegmental area enhances motivation for cocaine. J Neurosci 31:7927–7937. 10.1523/JNEUROSCI.6014-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churchill L, Swanson CJ, Urbina M, Kalivas PW (1999) Repeated cocaine alters glutamate receptor subunit levels in the nucleus accumbens and ventral tegmental area of rats that develop behavioral sensitization. J Neurochem 72:2397–2403. [DOI] [PubMed] [Google Scholar]
- Clark MS, Sexton TJ, McClain M, Root D, Kohen R, Neumaier JF (2002) Overexpression of 5-HT1B receptor in dorsal raphe nucleus using herpes simplex virus gene transfer increases anxiety behavior after inescapable stress. J Neurosci 22:4550–4562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deroche-Gamonet V, Belin D, Piazza PV (2004) Evidence for addiction-like behavior in the rat. Science 305:1014–1017. 10.1126/science.1099020 [DOI] [PubMed] [Google Scholar]
- Edwards S, Whisler KN, Fuller DC, Orsulak PJ, Self DW (2007) Addiction-related alterations in D1 and D2 dopamine receptor behavioral responses following chronic cocaine self-administration. Neuropsychopharmacology 32:354–366. 10.1038/sj.npp.1301062 [DOI] [PubMed] [Google Scholar]
- Ehlers MD, Tingley WG, Huganir RL (1995) Regulated subcellular distribution of the NR1 subunit of the NMDA receptor. Science 269:1734–1737. 10.1126/science.7569904 [DOI] [PubMed] [Google Scholar]
- Engblom D, Bilbao A, Sanchis-Segura C, Dahan L, Perreau-Lenz S, Balland B, Parkitna JR, Luján R, Halbout B, Mameli M, Parlato R, Sprengel R, Lüscher C, Schütz G, Spanagel R (2008) Glutamate receptors on dopamine neurons control the persistence of cocaine seeking. Neuron 59:497–508. 10.1016/j.neuron.2008.07.010 [DOI] [PubMed] [Google Scholar]
- Fitzgerald LW, Ortiz J, Hamedani AG, Nestler EJ (1996) Drugs of abuse and stress increase the expression of GluR1 and NMDAR1 glutamate receptor subunits in the rat ventral tegmental area: common adaptations among cross-sensitizing agents. J Neurosci 16:274–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao L, Tian M, Zhao HY, Xu QQ, Huang YM, Si QC, Tian Q, Wu QM, Hu XM, Sun LB, McClintock SM, Zeng Y (2016) TrkB activation by 7, 8-dihydroxyflavone increases synapse AMPA subunits and ameliorates spatial memory deficits in a mouse model of Alzheimer's disease. J Neurochem 136:620–636. 10.1111/jnc.13432 [DOI] [PubMed] [Google Scholar]
- Grimm JW, Hope BT, Wise RA, Shaham Y (2001) Neuroadaptation. Incubation of cocaine craving after withdrawal. Nature 412:141–142. 10.1038/35084134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haddad JJ. (2005) N-methyl-d-aspartate (NMDA) and the regulation of mitogen-activated protein kinase (MAPK) signaling pathways: a revolving neurochemical axis for therapeutic intervention? Prog Neurobiol 77:252–282. 10.1016/j.pneurobio.2005.10.008 [DOI] [PubMed] [Google Scholar]
- Hartmann M, Heumann R, Lessmann V (2001) Synaptic secretion of BDNF after high-frequency stimulation of glutamatergic synapses. EMBO J 20:5887–5897. 10.1093/emboj/20.21.5887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iñiguez SD, Vialou V, Warren BL, Cao JL, Alcantara LF, Davis LC, Manojlovic Z, Neve RL, Russo SJ, Han MH, Nestler EJ, Bolaños-Guzmán CA (2010) Extracellular signal-regulated kinase-2 within the ventral tegmental area regulates responses to stress. J Neurosci 30:7652–7663. 10.1523/JNEUROSCI.0951-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauer JA, Malenka RC (2007) Synaptic plasticity and addiction. Nat Rev Neurosci 8:844–858. 10.1038/nrn2234 [DOI] [PubMed] [Google Scholar]
- Kavalali ET, Monteggia LM (2012) Synaptic mechanisms underlying rapid antidepressant action of ketamine. Am J Psychiatry 169:1150–1156. 10.1176/appi.ajp.2012.12040531 [DOI] [PubMed] [Google Scholar]
- Krishnan V, Han MH, Graham DL, Berton O, Renthal W, Russo SJ, Laplant Q, Graham A, Lutter M, Lagace DC, Ghose S, Reister R, Tannous P, Green TA, Neve RL, Chakravarty S, Kumar A, Eisch AJ, Self DW, Lee FS, et al. (2007) Molecular adaptations underlying susceptibility and resistance to social defeat in brain reward regions. Cell 131:391–404. 10.1016/j.cell.2007.09.018 [DOI] [PubMed] [Google Scholar]
- Krishnan V, Han MH, Mazei-Robison M, Iñiguez SD, Ables JL, Vialou V, Berton O, Ghose S, Covington HE 3rd, Wiley MD, Henderson RP, Neve RL, Eisch AJ, Tamminga CA, Russo SJ, Bolaños CA, Nestler EJ (2008) AKT signaling within the ventral tegmental area regulates cellular and behavioral responses to stressful stimuli. Biol Psychiatry 64:691–700. 10.1016/j.biopsych.2008.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu QS, Pu L, Poo MM (2005) Repeated cocaine exposure in vivo facilitates LTP induction in midbrain dopamine neurons. Nature 437:1027–1031. 10.1038/nature04050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Monteggia LM, Wolf ME (2002) Repeated administration of amphetamine or cocaine does not alter AMPA receptor subunit expression in the rat midbrain. Neuropsychopharmacology 26:1–13. 10.1016/S0893-133X(01)00272-X [DOI] [PubMed] [Google Scholar]
- Mameli M, Halbout B, Creton C, Engblom D, Parkitna JR, Spanagel R, Lüscher C (2009) Cocaine-evoked synaptic plasticity: persistence in the VTA triggers adaptations in the NAc. Nat Neurosci 12:1036–1041. 10.1038/nn.2367 [DOI] [PubMed] [Google Scholar]
- Marini AM, Rabin SJ, Lipsky RH, Mocchetti I (1998) Activity-dependent release of brain-derived neurotrophic factor underlies the neuroprotective effect of N-methyl-d-aspartate. J Biol Chem 273:29394–29399. 10.1074/jbc.273.45.29394 [DOI] [PubMed] [Google Scholar]
- Matsuda N, Lu H, Fukata Y, Noritake J, Gao H, Mukherjee S, Nemoto T, Fukata M, Poo MM (2009) Differential activity-dependent secretion of brain-derived neurotrophic factor from axon and dendrite. J Neurosci 29:14185–14198. 10.1523/JNEUROSCI.1863-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIlhinney RA, Le Bourdellès B, Molnár E, Tricaud N, Streit P, Whiting PJ (1998) Assembly intracellular targeting and cell surface expression of the human N-methyl-d-aspartate receptor subunits NR1a and NR2A in transfected cells. Neuropharmacology 37:1355–1367. 10.1016/S0028-3908(98)00121-X [DOI] [PubMed] [Google Scholar]
- Neve RL, Howe JR, Hong S, Kalb RG (1997) Introduction of the glutamate receptor subunit 1 into motor neurons in vitro and in vivo using a recombinant herpes simplex virus. Neuroscience 79:435–447. 10.1016/S0306-4522(96)00645-8 [DOI] [PubMed] [Google Scholar]
- Paoletti P, Bellone C, Zhou Q (2013) NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat Rev Neurosci 14:383–400. 10.1038/nrn3504 [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C (1998) The rat brain in stereotaxic coordinates. San Diego: Academic. [DOI] [PubMed] [Google Scholar]
- Perez-Otano I, Schulteis CT, Contractor A, Lipton SA, Trimmer JS, Sucher NJ, Heinemann SF (2001) Assembly with the NR1 subunit is required for surface expression of NR3A-containing NMDA receptors. J Neurosci 21:1228–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pu L, Liu QS, Poo MM (2006) BDNF-dependent synaptic sensitization in midbrain dopamine neurons after cocaine withdrawal. Nat Neurosci 9:605–607. 10.1038/nn1687 [DOI] [PubMed] [Google Scholar]
- Pulipparacharuvil S, Renthal W, Hale CF, Taniguchi M, Xiao G, Kumar A, Russo SJ, Sikder D, Dewey CM, Davis MM, Greengard P, Nairn AC, Nestler EJ, Cowan CW (2008) Cocaine regulates MEF2 to control synaptic and behavioral plasticity. Neuron 59:621–633. 10.1016/j.neuron.2008.06.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson NR, Roberts DC (1996) Progressive ratio schedules in drug self-administration studies in rats: a method to evaluate reinforcing efficacy. J Neurosci Methods 66:1–11. 10.1016/0165-0270(95)00153-0 [DOI] [PubMed] [Google Scholar]
- Robinson TE, Berridge KC (1993) The neural basis of drug craving: an incentive-sensitization theory of addiction. Brain Res Brain Res Rev 18:247–291. 10.1016/0165-0173(93)90013-P [DOI] [PubMed] [Google Scholar]
- Rosen KM, Moghekar A, O'Brien RJ (2007) Activity dependent localization of synaptic NMDA receptors in spinal neurons. Mol Cell Neurosci 34:578–591. 10.1016/j.mcn.2006.12.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo SJ, Wilkinson MB, Mazei-Robison MS, Dietz DM, Maze I, Krishnan V, Renthal W, Graham A, Birnbaum SG, Green TA, Robison B, Lesselyong A, Perrotti LI, Bolaños CA, Kumar A, Clark MS, Neumaier JF, Neve RL, Bhakar AL, Barker PA, et al. (2009) Nuclear factor κB signaling regulates neuronal morphology and cocaine reward. J Neurosci 29:3529–3537. 10.1523/JNEUROSCI.6173-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saal D, Dong Y, Bonci A, Malenka RC (2003) Drugs of abuse and stress trigger a common synaptic adaptation in dopamine neurons. Neuron 37:577–582. 10.1016/S0896-6273(03)00021-7 [DOI] [PubMed] [Google Scholar]
- Schilström B, Yaka R, Argilli E, Suvarna N, Schumann J, Chen BT, Carman M, Singh V, Mailliard WS, Ron D, Bonci A (2006) Cocaine enhances NMDA receptor-mediated currents in ventral tegmental area cells via dopamine D5 receptor-dependent redistribution of NMDA receptors. J Neurosci 26:8549–8558. 10.1523/JNEUROSCI.5179-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumann J, Matzner H, Michaeli A, Yaka R (2009) NR2A/B-containing NMDA receptors mediate cocaine-induced synaptic plasticity in the VTA and cocaine psychomotor sensitization. Neurosci Lett 461:159–162. 10.1016/j.neulet.2009.06.002 [DOI] [PubMed] [Google Scholar]
- Scott DB, Blanpied TA, Swanson GT, Zhang C, Ehlers MD (2001) An NMDA receptor ER retention signal regulated by phosphorylation and alternative splicing. J Neurosci 21:3063–3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott DB, Blanpied TA, Ehlers MD (2003) Coordinated PKA and PKC phosphorylation suppresses RXR-mediated ER retention and regulates the surface delivery of NMDA receptors. Neuropharmacology 45:755–767. 10.1016/S0028-3908(03)00250-8 [DOI] [PubMed] [Google Scholar]
- Sun W, Akins CK, Mattingly AE, Rebec GV (2005) Ionotropic glutamate receptors in the ventral tegmental area regulate cocaine-seeking behavior in rats. Neuropsychopharmacology 30:2073–2081. 10.1038/sj.npp.1300744 [DOI] [PubMed] [Google Scholar]
- Thomas GM, Huganir RL (2004) MAPK cascade signalling and synaptic plasticity. Nat Rev Neurosci 5:173–183. 10.1038/nrn1346 [DOI] [PubMed] [Google Scholar]
- Thomas MJ, Kalivas PW, Shaham Y (2008) Neuroplasticity in the mesolimbic dopamine system and cocaine addiction. Br J Pharmacol 154:327–342. 10.1038/bjp.2008.77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tingley WG, Ehlers MD, Kameyama K, Doherty C, Ptak JB, Riley CT, Huganir RL (1997) Characterization of protein kinase A and protein kinase C phosphorylation of the N-methyl-d-aspartate receptor NR1 subunit using phosphorylation site-specific antibodies. J Biol Chem 272:5157–5166. 10.1074/jbc.272.8.5157 [DOI] [PubMed] [Google Scholar]
- Turrigiano GG. (2008) The self-tuning neuron: synaptic scaling of excitatory synapses. Cell 135:422–435. 10.1016/j.cell.2008.10.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungless MA, Whistler JL, Malenka RC, Bonci A (2001) Single cocaine exposure in vivo induces long-term potentiation in dopamine neurons. Nature 411:583–587. 10.1038/35079077 [DOI] [PubMed] [Google Scholar]
- Ungless MA, Singh V, Crowder TL, Yaka R, Ron D, Bonci A (2003) Corticotropin-releasing factor requires CRF binding protein to potentiate NMDA receptors via CRF receptor 2 in dopamine neurons. Neuron 39:401–407. 10.1016/S0896-6273(03)00461-6 [DOI] [PubMed] [Google Scholar]
- Wang JQ, Fibuch EE, Mao L (2007) Regulation of mitogen-activated protein kinases by glutamate receptors. J Neurochem 100:1–11. 10.1111/j.1471-4159.2006.04208.x [DOI] [PubMed] [Google Scholar]
- White FJ, Hu XT, Zhang XF, Wolf ME (1995) Repeated administration of cocaine or amphetamine alters neuronal responses to glutamate in the mesoaccumbens dopamine system. J Pharmacol Exp Ther 273:445–454. [PubMed] [Google Scholar]
- You ZB, Wang B, Zitzman D, Azari S, Wise RA (2007) A role for conditioned ventral tegmental glutamate release in cocaine seeking. J Neurosci 27:10546–10555. 10.1523/JNEUROSCI.2967-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XF, Hu XT, White FJ, Wolf ME (1997) Increased responsiveness of ventral tegmental area dopamine neurons to glutamate after repeated administration of cocaine or amphetamine is transient and selectively involves AMPA receptors. J Pharmacol Exp Ther 281:699–706. [PubMed] [Google Scholar]
- Zweifel LS, Argilli E, Bonci A, Palmiter RD (2008) Role of NMDA receptors in dopamine neurons for plasticity and addictive behaviors. Neuron 59:486–496. 10.1016/j.neuron.2008.05.028 [DOI] [PMC free article] [PubMed] [Google Scholar]