

Abstract
The MAPK/ERK pathway has a critical role in PNS development. It is required for Schwann cell (SC) differentiation and myelination; sustained embryonic MAPK/ERK activation in SCs enhances myelin growth overcoming signals that normally end myelination. Excess activation of this pathway can be maladaptive as in adulthood acute strong activation of MAPK/ERK has been shown to cause SC dedifferentiation and demyelination. We used a mouse model (including male and female animals) in which the gain-of-function MEK1DD allele produces sustained MAPK/ERK activation in adult SCs, and we determined the impact of such activation on nerve repair. In the uninjured nerve, MAPK/ERK activation neither impaired myelin nor reactivated myelination. However, in the injured nerve it was detrimental and resulted in delayed repair and functional recovery. In the early phase of injury, the rate of myelin clearance was faster. Four weeks following injury, when nerve repair is normally advanced, myelinated axons of MEK1DD mutants demonstrated higher rates of myelin decompaction, a reduced number of Cajal bands. and decreased internodal length. We noted the presence of abnormal Remak bundles with long SCs processes and reduced numbers of C-fibers/Remak bundle. Both the total number of regenerating axons and the intraepidermal nerve fiber density in the skin were reduced. Sustained activation of MAPK/ERK in adult SCs is therefore deleterious to successful nerve repair, emphasizing the differences in the signaling processes coordinating nerve development and repair. Our results also underline the key role of SCs in axon regeneration and successful target reinnervation.
SIGNIFICANCE STATEMENT The MAPK/ERK pathway promotes developmental myelination and its sustained activation in SCs induced continuous myelin growth, compensating for the absence of essential myelination signals. However, the strength of activation is fundamental because acute strong induction of MAPK/ERK in adulthood induces demyelination. What has been unknown is the effect of a mild but sustained MAPK/ERK activation in SCs on nerve repair in adulthood. This promoted myelin clearance but led to abnormalities in nonmyelinating and myelinating SCs in the later phases of nerve repair, resulting in slowed axon regeneration, cutaneous reinnervation, and functional recovery. Our results emphasize the distinct role of the MAPK/ERK pathway in developmental myelination versus remyelination and the importance of signaling between SCs and axons for successful axon regeneration.
Keywords: MAPK/ERK, nerve injury, nerve repair, regeneration, remyelination, Schwann cells
Introduction
Peripheral nerve regeneration after injury is a complex process requiring signaling among injured axons, Schwann cells (SCs), and immune cells. During regeneration, a repair program is induced in myelinating and nonmyelinating SCs that supports regeneration (Arthur-Farraj et al., 2012; Jessen and Mirsky, 2016). The first phase of the injury response is Wallerian degeneration (WD), which involves axon destruction, dedifferentiation of SCs, activation of an inflammatory response, and phagocytosis of myelin debris (Chen et al., 2007; Jessen and Mirsky, 2016). Schwann cells start to proliferate, downregulate myelin gene expression, and adopt a repair phenotype. Subsequently, repair SCs differentiate again into mature SCs to generate myelin-forming and non-myelin-forming (Remak) cells (Arthur-Farraj et al., 2012). Although regeneration happens spontaneously in the peripheral nervous system (PNS), unfortunately, it is imperfect: large-diameter axons are remyelinated, but such myelin is thinner compared with the naive state (Schröder, 1972); target reinnervation occurs but can be slow and misdirected; and, as a consequence, functional outcomes in patients are often poor (Palispis and Gupta, 2017).
Mitogen-activated protein kinase (MAPK) is a signal transduction pathway involved in many physiological processes such as gene expression, metabolism, survival, proliferation, and cell differentiation (Cargnello and Roux, 2011; Pearson et al., 2001). This family of kinases is regulated by a phosphorylation cascade and requires an extracellular stimulus for activation. The ERK1/2 cascade was the first MAPK pathway described (Seger and Krebs, 1995) in which phosphorylation of ERK1/2 by the MAPK kinase 1 (MEK1) activates MAPK signaling. The role of this pathway in developmental myelination both in the CNS and PNS has been widely investigated. In CNS, ERK1/2 has been shown to be critical for myelination during development (Ishii et al., 2012) and for myelin maintenance (Ishii et al., 2014). In the PNS, the ablation of ERK1/2 in SCs arrested their differentiation, resulting in dramatic hypomyelination and suggesting a positive role of ERK1/2 in the regulation of myelination (Newbern et al., 2011; Ishii et al., 2014). When sustained MAPK/ERK activation was induced in SCs during embryonic development, this resulted in increased myelin thickness (Ishii et al., 2012; Sheean et al., 2014) due to continuous myelin growth. Such sustained MAPK/ERK activation was able to replace the ablation of essential myelinating genes such as ErbB3 and Shp2 (Sheean et al., 2014).
It is known that after nerve injury ERK1/2 signaling is rapidly activated and contributes to SC dedifferentiation phenotype of WD (Sheu et al., 2000; Harrisingh et al., 2004). Napoli et al. (2012) demonstrated that a rapid and transient ERK1/2 activation in SCs in the absence of axonal injury was sufficient to induce demyelination and recruitment of inflammatory cells and was followed by remyelination as ERK1/2 levels normalized. The strength and temporal profile of MAPK/ERK induction is a key factor in the regulation of the signaling pathway, and our aim was to use a mutant mouse expressing a MEK1DD gain-of-function allele in SCs, which has been shown to promote developmental myelination, to determine whether sustained MAPK/ERK activation in adult SCs could enhance myelin production and nerve repair (Sheean et al., 2014).
By using this model, we observed that this MAPK/ERK activation did not have any effect on myelin stability in the naive state up to 6 weeks. After nerve crush, mutant mice showed signs of more rapid clearance of myelin debris. However, at later stages of the injury response, functional recovery was delayed with morphological defects in both myelinated and nonmyelinated fibers and a reduced number of regenerating axons. MAPK/ERK activation decreased myelin stability and reduced internodal length during regeneration without affecting myelin thickness. In unmyelinated fibers, it resulted in elongated SCs processes, abnormal Remak bundle formation, loss of axons, and decreased small-fiber regeneration. Together, these results show that the MAPK/ERK pathway has distinct roles in development and after injury and that enhanced activation of this pathway during regeneration has a negative impact on nerve repair.
Materials and Methods
Transgenic animals
All work performed was conform to UK Home Office legislation (Scientific Procedure Act 1986). PLPCre-ERT2;MEK1DDfl/fl mice were bred by crossing PLPCre-ERT2 mice (The Jackson Laboratory) with R26StopFLMEK1DDfl/fl mice (provided by K. Rajewsky, generated at the Boston Children's Hospital, Boston, MA); both colonies were on a C57BL/6 background. Generation of PLPCre-ERT2 mice has been previously described (Doerflinger et al., 2003) to obtain an inducible, site-specific recombination under transcriptional control of the myelin proteolipid protein gene (Plp) expressed in oligodendrocytes and SCs. Mice homozygous for the R26StopFLMEK1DD conditional allele contain a loxP-flanked Neo-STOP cassette (for details, see Srinivasan et al., 2009; Sheean et al., 2014). In PLPCre-ERT2;MEK1DDfl/fl transgenic mice, Cre-mediated excision of the STOP cassette leads to the expression of active MEK1DD signaling molecule in Plp-expressing cells. Conditional MEK1DD mice were generated by the administration of tamoxifen (0.25 mg/g body weight in corn oil; catalog #T5648, Sigma-Aldrich) by oral gavage for 5 consecutive days to PLPCre-ERT2;MEK1DDfl/fl mice that were 8 weeks of age. Littermate PLPCre-negative mice were used as controls. Wherever possible, an equal number of animals of each gender was used in the experimental groups.
Immunoblotting
Sciatic nerves were homogenized in NP-40 lysis buffer and protein concentration was determined using a BCA Protein Assay Kit (Thermo Fisher Scientific). Proteins (20–40 μg) were mixed with SDS gel sample buffer, electrophoresed in SDS-PAGE precasted gels (Bio-Rad), transferred to membranes, blocked for 30 min with 5% BSA in TBS-T; and were immunoblotted with antibodies against αRb phosphorylated (p)-ERK (1:5000; Cell Signaling Technology), p-AKT (1:1000; Cell Signaling Technology), ERK1/2 (1:5000; Cell Signaling Technology), αRat myelin basic protein (MBP; 1:300; Abcam), αRb dystrophin-related protein 2 (DRP2; 1:3000), and αRb periaxin N terminal (1:20,000), both of which were donated by the Brophy laboratory (University of Edinburgh, UK). αRb calnexin was used as a control (1:1000; Enzo Life Sciences). Second antibodies were ECL anti-Rabbit IgG (GE Healthcare) and polyclonal Rb anti-Rat Igs/HRP (DAKO). ECL prime Western blotting detection system (GE Healthcare) was used to detect the immunoreactive bands on chemoluminescence films. To measure signal intensities ImageJ was used.
Immunofluorescence microscopy
Mice were deeply anesthetized with pentobarbital and transcardially perfused with 5 ml of saline followed by 10 ml of paraformaldehyde [PFA; 4% in 0.1% phosphate buffer (PB)]. Tissues were cut on a cryostat and sections blocked using 5% BSA/5% donkey serum in PBS/0.2% Triton X-100 and 0.1% sodium azide. All primary antibodies were incubated overnight and secondary for 2 h. Immunofluorescence was visualized under a Zeiss Imager Z1 microscope. Photographs were taken using an Axio Camera (Zeiss). Skin biopsy samples were taken from the hindpaw glabrous skin proximal to the last nodule and were postfixed in PFA 4% for 30 min, then were transferred to 20% sucrose overnight (4°C), successively blocked in OCT embedding compound on dry ice, and stored at −80°C. Transverse sections were cut on a cryostat at the thickness of 14 μm onto gelatin-coated Super Frost Ultra Plus slides. C-fibers in the epidermis were visualized by immunostaining with polyclonal rabbit PGP 9.5 (1:200; Zytomed). Secondary antibody used was anti-rabbit Cy3 (1:1000; Stratech Scientific). Epidermal fibers were counted at a 63× magnification live on the microscope according to the rules set out by Lauria (2005). Four sections per animal were counted with the analyst blind to genotype. The length of the epidermis was measured by ImageJ in images taken with 20× magnification, and used to calculate the density of intraepidermal fibers [intraepidermal nerve fiber density (IENFD)] as fibers per millimeter. Gastrocnemius muscles were postfixed for 2 h in PFA 4% and transferred to 20% sucrose overnight (4°C). Sections were cut at a thickness of 100 μm, put into a 24-well plate, and stored in PBS/0.1% sodium azide. Floating staining was performed using mouse anti-2H3 (1:200; Developmental Studies Hybridoma Bank), monoclonal mouse anti-SV2 (1:200; Developmental Studies Hybridoma Bank). The following secondary antibodies used: Alexa Fluor 546 goat anti-rat IgG (Life Technologies) and α-bungarotoxin tetramethylrodamine conjugate (Invitrogen). About 50 muscle terminals were analyzed per mouse.
Nerve teasing
Sciatic nerves were fixed in 4% PFA/0.1 m PB for 30 min and stored in PBS/0.1% sodium azide. Nerves were stained with 0.5% osmium tetroxide for 1 h followed by 3 washes of 5 min each with 25% EtOH, 50% EtOH, and 75% EtOH. Nerves were stored in 50% glycerol and 50% of 70% EtOH and teased in glycerol. Images were taken with the Axio Camera of the Zeiss Imager Z1 microscope. Internodal distance was quantified using ImageJ; ∼50 fibers per nerve were analyzed.
Electron microscopy
Distal sciatic nerves were postfixed with 4% PFA and 3% glutaraldehyde in 0.1 m phosphate buffer. Nerves were osmicated with 1.5% osmium/0.2 m PB for 90 min, dehydrated and embedded in epoxy resin (embedding materials, TAAB). Ultrathin sections (90 nm) were cut using a Diatome diamond knife on the Leica UC7 Ultramicrotome and mounted onto 100 mesh Cu grids coated with formvar. Sections were poststained for 5 min with lead citrate. Pictures were taken with a Thermo Fisher Scientific Tecnai T12 Transmission Electron Microscope (TEM) at the Sir William Dunn School of Pathology Bioimaging Facility (University of Oxford, Oxford, UK). For analysis of axons and macrophages, photographs of randomly chosen entire grid squares were taken, the area of which covered ∼20% of the total area of the cross section of the sciatic nerve. The axon count was normalized for the area in square millimeters (uninjured nerves and 5 d postinjury nerves) or for the total nerve area calculated in semithin sections of the same sciatic nerve (1 month after injury nerves). Analysis was performed using AxioVision LE software, release 4.2 (Zeiss).
Behavioral tests
All behavioral testing was performed with the observer blind to the genotype of the animal.
Beam test.
Proprioception was assessed using a 1 m long wooden beam in which mice were recorded meanwhile walking from a side to the other. Percentage of incorrect steps were counted successively as ratio between the mean of slips and hops and the total number of steps mice were taking during each run.
Rotarod.
Mice were placed on the rotarod (Jones and Roberts, 1968) as it was rotating at 20 rpm. The time each mouse spent on the rod was measured, with a cutoff time of 7 min. The test was performed twice for each animal with an interval of at least 15 min between each test.
Toe-spreading reflex.
Motor–muscular function was assessed by gently lifting mice by the tail in order for them to extend legs laterally and spread the digits. Toe-spreading reflex of ipsilateral paw was compared with the contralateral and graded from 0 to 2, as follows: 0, no spreading; 1, intermediate spreading; 2 normal spreading. Appearance of this reflex results from reinnervation of small muscles in the foot.
Pinprick test.
Sciatic nerve reinnervation of the skin was assessed pinching the injured side with a fine insect needle (Fine Science Tools) in specific parts of paw and toes. Score was given from 0 to 2, as follows: 0, nonreflex withdrawal response; 1, mild reflex; 2, normal reflex.
Experimental design and statistical analysis
All statistical analysis has been performed using GraphPad Prism 6 Software. Data were reported as the mean ± SEM, and the number of replicates was indicated in each figure legend. The level of significance was set at p < 0.05. The two-tailed unpaired Student's t test was used to analyze differences between two groups. To assess statistical differences in behavior experiments with multiple time points repeated-measures two-way ANOVA was performed with Sidak's multiple-comparison tests. Distribution plots have been performed with the SPSS software (IBM), and the Kolmogorov–Smirnov test has been used for statistical analysis. Statistical analysis for each panel is specified in the figure legend.
Results
MAPK/ERK activation, as a consequence of MEK1DD expression in adult Schwann cells, does not affect myelination in uninjured nerve over a 6 week time course
We used mice homozygous for the conditional MEK1DD gain of function allele (MEK1DDfl/fl) that have been crossed with PLPCre-ERT2 mice, in which the induction of Cre recombinase activity in the PNS occurs specifically in SCs after tamoxifen treatment. PLPCre-ERT2;MEK1DDfl/fl adult mice (subsequently termed MEK1DD mutant), and MEK1DDfl/fl littermates (controls) were dosed with tamoxifen for 1 week. Two weeks later, we performed nerve crush injury in the left sciatic nerve, and tissue was harvested at up to 4 weeks after injury. Tissues taken from the contralateral side to the injury (this included sciatic nerve, plantar paw skin, and gastrocnemius muscle) were used to assess the impact of MAPK/ERK signaling on uninjured peripheral nerve.
At 6 weeks post-tamoxifen dosing, p-ERK protein expression in the uninjured nerve was significantly increased in mutants compared with controls, showing that MAPK/ERK signaling had been activated (Fig. 1A; control, 1 ± 0.37, vs MEK1DD mutants, 5.45 ± 1.5; p = 0.04). No difference was found in the total ERK1/2 protein expression (Fig. 1A; control, 1 ± 0.5, vs MEK1DD mutants, 1 ± 0.05). The two groups were also comparable in the expression of p-Akt (control, 1 ± 0.2, vs MEK1DD mutants, 1.3 ± 0.03) confirming the specificity of p-ERK induction. Using ultrastructural analysis, we did not find any differences in myelinated or nonmyelinated axon number, size, or morphology when comparing the two groups 6 weeks after dosing (Fig. 1B,C). There was no significant difference in the g-ratio distribution per axon diameter (Fig. 1D). The number of small fibers counted in paw skin biopsy specimens (Fig. 1E; controls, 40.84 ± 2.2, vs MEK1DD mutants, 34.6 ± 1.9), internodal distance measured in sciatic teased fibers (Fig. 1F; controls, 540.5 ± 36.98, vs MEK1DD mutants, 584.7 ± 18.5) and neuromuscular junction (NMJ) innervation were unchanged (Fig. 1G; fully innervated: controls, 73.06 ± 1.9, vs MEK1DD mutants, 73.57 ± 4.1; partially innervated: controls, 26.94 ± 0.37, vs Mek1DD mutants, 26.43 ± 0.37). Thus, activation of the MAPK/ERK in young Mek1DD mutants prolongs developmental myelination, but MAPK/ERK activation in the adult does not suffice to reinitiate myelination.
Figure 1.

MAPK/ERK activation in adult Schwann cells does not affect myelin and axons stability. A, Immunoblotting of p-ERK, p-Akt, and ERK1/2 proteins in contralateral nerves 6 weeks after tamoxifen dosing in sciatic nerves of MEK1DD mutant mice (MEK) compared with controls (Ctr) mice. Data were normalized vs Calnexin. B, TEM micrographs of uninjured sciatic nerve 6 weeks post-tamoxifen dosing. C, Summary of the main parameters analyzed to study myelination and axon survival. Data are reported as percentages of myelinated and nonmyelinated axons normalized for the area in square millimeters. The g-ratio of individual fibers in relation to their axon diameter is shown in the scatter plot in D. E, Quantification of IENFD/millimeter from uninjured skin biopsy specimens. F, Internodal distance measured in teased fibers of sciatic nerve. G, Percentage of innervated NMJs in uninjured nerves. For all analyses, three to four mice per group were analyzed. Scale bar, 5 μm. *p < 0.05, two-tailed unpaired t test. Error bars indicate SEM.
Wallerian degeneration is accelerated by MAPK/ERK activation
MEK1DD mutants and control littermates mice were dosed for a week with tamoxifen to activate the MAPK/ERK pathway, and after 2 weeks sciatic nerve crush was performed. Mice were sacrificed 5 d postinjury, and ipsilateral sciatic nerves were analyzed. As showed by TEM analysis, in both control and mutant nerves there were clear features of WD with collapsed myelin, resulting in myelin ovoid formation (Fig. 2A). However, myelin breakdown was faster in mutant nerves, as confirmed by a significant decrease in the percentage of intact myelin sheaths in relation to the total number of myelinated axons (Fig. 2B; controls, 28.61 ± 1.81, vs MEK1DD mutants, 17.31 ± 3.45; p = 0.015). An acceleration of WD in mutants was also supported by a significant increase in the number of macrophages per square millimeter (Fig. 2C; controls, 1270.83 ± 90.81, vs MEK1DD mutants, 2416.67 ± 265.98; p = 0.0003), as assessed by TEM analysis, and a 2.4-fold decrease in MBP expression (Fig. 2D; controls, 1 ± 0.15, vs MEK1DD mutants, 0.41 ± 0.12, p = 0.037). To conclude that MAPK/ERK activation in MEK1DD mutants at the time of injury accelerates WD, increasing the number of degenerated axons and macrophage recruitment and decreasing MBP expression.
Figure 2.

Wallerian degeneration is faster in mutant mice after p-ERK induction. A, Electron micrographs of ipsilateral sciatic nerve sections 5 d postinjury. Arrows indicate intact myelin sheaths that are quantified in B. C, Macrophage count in TEM pictures reported as the number of macrophages per square millimeter. D, Western blot analysis of MBP expression in MEK1DD mutants (MEK) compared with controls (Ctr). Calnexin was used as a control. n = 3 mice/group. Scale bar, 10 μm. *p < 0.05, ***p < 0.001, two-tailed unpaired t test. Error bars indicate SEM.
Activation of MAPK/ERK results in impaired functional recovery after injury
To assess the functional consequences of MAPK/ERK activation in regeneration, we assessed behavioral measures of motor and sensory function in mice for 1 month after nerve crush. There was no difference in baseline (preinjury) values comparing MEK1DD mutants and littermate controls. MEK1DD mutants showed an impaired recovery in motor coordination and balance, as assessed by the beam test, that were significantly slower at days 13 and 17 postinjury and still significantly different from controls at day 32, the end point of the experiment (Fig. 3A). Furthermore, the rotarod test confirmed decreased motor performance in mutant mice 1 month after injury (Fig. 3B; controls, 136.7 ± 47.38 s, vs MEK1DD mutants, 27.50 ± 13.92 s; p = 0.048). Distal sensorimotor function was assessed by the toe-spreading reflex test. This showed a significant slowing in the recovery of toe spreading in MEK1DD mutants, as shown in the representative photographs from day 13 postinjury. By day 25 postinjury, the reflex in the two groups was comparable. Sensory function was assessed by pin-prick test, and mutant mice actually showed a hypersensitivity to pin-prick at days 21 and 25 postinjury, but at day 32 postinjury the difference was no longer statistically significant (Fig. 3E). In summary, MEK1DD mutant mice showed impaired recovery of motor coordination, balance, and a hypersensitivity to pin-prick post-nerve injury.
Figure 3.
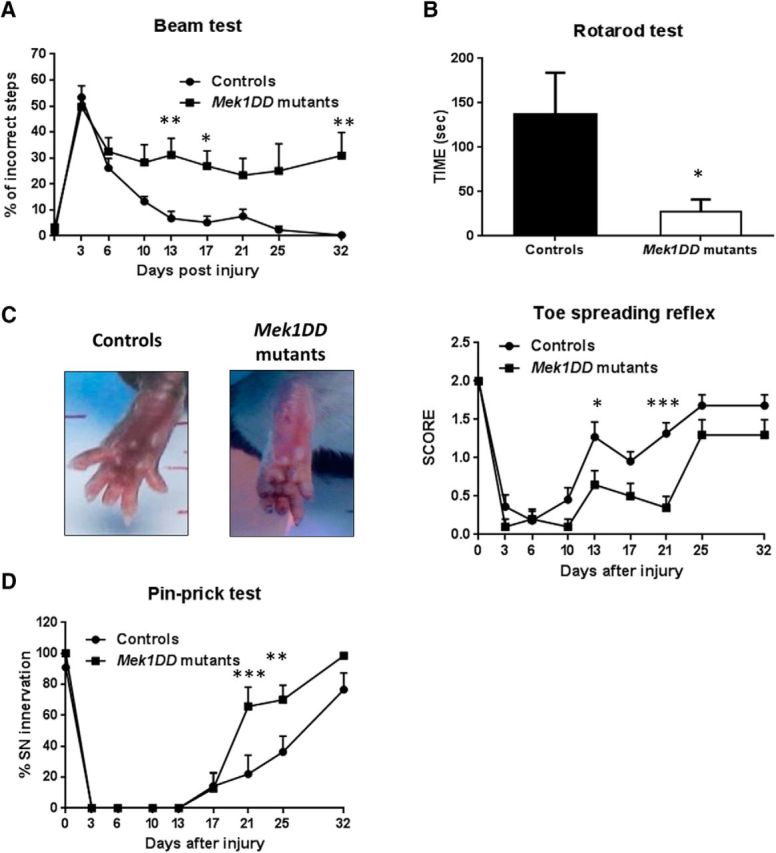
Slower recovery in MEK1DD mutant mice vs controls, 1 month postinjury. A, Beam test was used to assess balance and motor coordination after injury. B, C, Rotarod test (B) and toe-spreading reflex test (C) confirmed deficits in motor function in MEK1DD mutant mice. C, Representative photographs of the toe spreading from day 13 postinjury are shown. D, Pin-prick test was used to analyze recovery in sensory functions. n = 11 mice. All tests were analyzed with two-way repeated-measures ANOVA; Sidak's multiple-comparison test apart from the rotarod test in which two-tailed unpaired t test was used. *p < 0.05, **p < 0.005, ***p < 0.001. Error bars indicate SEM.
MAPK/ERK activation results in morphological abnormalities in myelinated fibers during regeneration
Given the differences in sensorimotor behavior in MEK1DD mutants versus controls, we undertook morphological assessment in ipsilateral sciatic nerve of control and mutant mice 1 month after injury. As showed in Table 1, no difference was found in total g-ratio measurement between the two groups, and the analysis of the g-ratio frequency of distribution confirmed this result (Fig. 4A–C). However, the distribution of the diameters of myelinated axons showed a significant shift, and smaller myelinated axons were present in greater frequency in mutants than in control animals (Fig. 4B; p = 0.02, Kolmogorov–Smirnov test). This was not due to the myelination of small axons that would not normally be myelinated, because the number of myelinated axons with <1 μm diameter between the groups was not different (controls, 0.33 ± 0.7%, vs MEK1DD mutants, 1.35 ± 0.6%; p = 0.08). Although MAPK/ERK activation did not alter myelin thickness, it did have a detrimental effect on myelin compaction and structure. We defined these myelin defects as myelinated axons showing the following: recurrent loops, infoldings, and myelin decompaction (Fig. 4E) as described by Goebbels et al. (2012). Table 1 reports the significant increase in the frequency of aberrant myelinated axons per nerve that was almost twice as high in mutants as in controls. We also analyzed macrophage number in ipsilateral nerves at the end point of the experiment to check whether the activation of MAPK/ERK pathway could cause a chronic macrophage infiltration. However, we did not find any difference on their number between control and mutant mice (controls, 1475 ± 145.2 macrophages/mm2, vs MEK1DD mutants, 1556 ± 141.2 macrophages/mm2; p = 0.15). In addition, after injury we observed an increase in the area of mutant nerves compared with the controls (controls, 0.17 ± 0.011 mm2, vs MEK1DD mutants, 0.23 ± 0.014 mm2; p = 0.02) and a reduced axonal packing. In addition, a significant reduction in the total number of axons per nerve was observed (Table 1).
Table 1.
MAPK/ERK activation does not affect myelination during regeneration but does affect nonmyelinated fiber survival and C-fiber organization in Remak bundles
| Controls | MEK1DD mutants | p value (t test) | |
|---|---|---|---|
| G-ratio | 0.66 | 0.64 | 0.501 |
| Myelinated axon/nerve | 3435.9 | 2872.7 | 0.202 |
| Nonmyelinated axon/nerve | 8032.6 | 6129.2 | 0.023 |
| Aberrant myelinated axon/nerve | 293.9 | 599.9 | 0.026 |
| Total axon number/nerve | 11762.4 | 9601.9 | 0.048 |
| C-fibers/RB | 5.4 | 2.8 | 0.001 |
The numbers of nonmyelinated fibers and total axons are decreased in mutant mice, whereas no significant differences between the two groups are observed for the numbers of myelinated fibers. Aberrant myelinated fibers (presenting with recurrent loops, outfoldings, and myelin decompaction) are increased in mutants. The number of C-fibers/Remak bundles (RBs) shows a dramatic decrease following MAPK activation due to a lack of normal Remak bundle formation. The axon count was normalized for the total nerve area calculated in semithin sections of the same sciatic nerve. n = 3–4 mice/group. Numbers in italics indicate a significant difference between the two groups; two-tailed unpaired t test.
Figure 4.

MAPK/ERK activation in Schwann cells does not affect myelin thickness, but does affect myelin stability. A–C, Morphological analysis of MEK1DD mutant vs control sciatic nerve at 4 weeks postinjury was undertaken to determine the following: frequency distribution of g-ratio (A); frequency distribution of axon diameter (B); distribution graph of g-ratio in relation to axon diameter (C). D, TEM pictures of ipsilateral sciatic nerves from control and MEK1DD mutant mice, which illustrate that although myelin thickness does not change there are myelin defects shown in E: asterisks illustrate axons with myelin decompaction; arrows point to invaginating recurrent loops; arrowheads indicate supernumerary SC processes surrounding a myelinated axon with poorly compacted myelin that have the appearance of an early “onion bulb” structure. Error bars indicate SEM. n = 3 mice/group. Scale bars: D, 10 μm; E, top left, 5 μm; E, all other panels, 2 μm. Kolmogorov–Smirnov test performed.
A further dramatic effect of Mek1 induction in remyelinating SCs was the formation of Cajal bands in regenerated myelinated fibers. In control mice (Fig. 5A), the abaxonal surface of the myelin sheath apposes and adheres with the SC plasma membrane forming the so-called appositions that flank cytoplasmic channels known as Cajal bands (Court et al., 2004). However, mutant mice presented a loss of appositions (Fig. 5A, arrows in the insert) with consequent disruption of Cajal band formation (Fig. 5A, asterisk in control mice). Quantification showed a significant increase in the number of myelinated axons with no or only one apposition and a significant decrease of axons with three appositions in MEK1DD mutant mice versus controls (Fig. 5B). No differences in Cajal bands were found in contralateral nerves (data not shown). It is known that appositions contain complexes formed by DRP2, periaxin, and dystroglycan (Gillespie et al., 1994), with DRP2 considered a marker for these appositions. In MEK1DD mutant mice, the expression of DRP2 was significantly decreased, as assessed by Western blot analysis (Fig. 5C; controls, 1 ± 0.2, vs MEK1DD mutants, 0.45 ± 0.09; p = 0.017), while periaxin protein expression did not differ (Fig. 5C; controls, 1 ± 0.13, vs MEK1DD mutants, 1.15 ± 0.16). Furthermore, elongation of SCs was compromised in mutants after injury, as seen by decreased internodal distance measured in ipsilateral teased fibers (Fig. 5D; controls, 280.2 ± 0.87 μm, vs MEK1DD mutants, 253.8 ± 0.39 μm; p = 0.036).
Figure 5.

Cajal band disruption and DRP2 impairment in mutant ipsilateral nerves after injury. A, TEM pictures showing a lack of appositions between SC plasma membrane and the abaxonal layer of the myelin sheath in mutant mice. Scale bars: 2 μm; insert, 1 μm. Asterisk shows Cajal bands location; arrow points to the apposition. B, Quantification. C, Periaxin and DRP2 were quantified by Western blot 1 month after injury in sciatic nerves of MEK1DD mutant mice (MEK) compared with control (Ctr) mice; calnexin was used as a loading control. D, Teased fibers from ipsilateral sciatic nerves showed decreased internodal distance in mutant mice. n = 3 mice/group. *p < 0.05, ***p < 0.001, two-tailed unpaired t test. Holm–Sidak test was performed in B. Error bars indicate SEM.
These results suggest that MAPK/ERK activation during regeneration did not promote remyelination as the g-ratio was unchanged. Instead, it led to a higher rate of axons with myelin defects. Furthermore, MAPK/ERK activation in myelinating SCs resulted in the reduced number of Cajal bands and shortened internodal distances. Despite these structural changes in myelinating SCs, motoneurons still reinnervate their targets at 4 weeks after injury, as assessed by quantification of gastrocnemius muscle innervation, which was not significantly different in controls and mutants (Fig. 6; fully innervated neuromuscular junctions: control mice, 87.46 ± 3.4%; MEK1DD mutants, 82.57 ± 1.1%; partially innervated neuromuscular junctions: controls, 13.83 ± 2.3%, vs MEK1DD mutants, 17.42 ± 1.1%).
Figure 6.
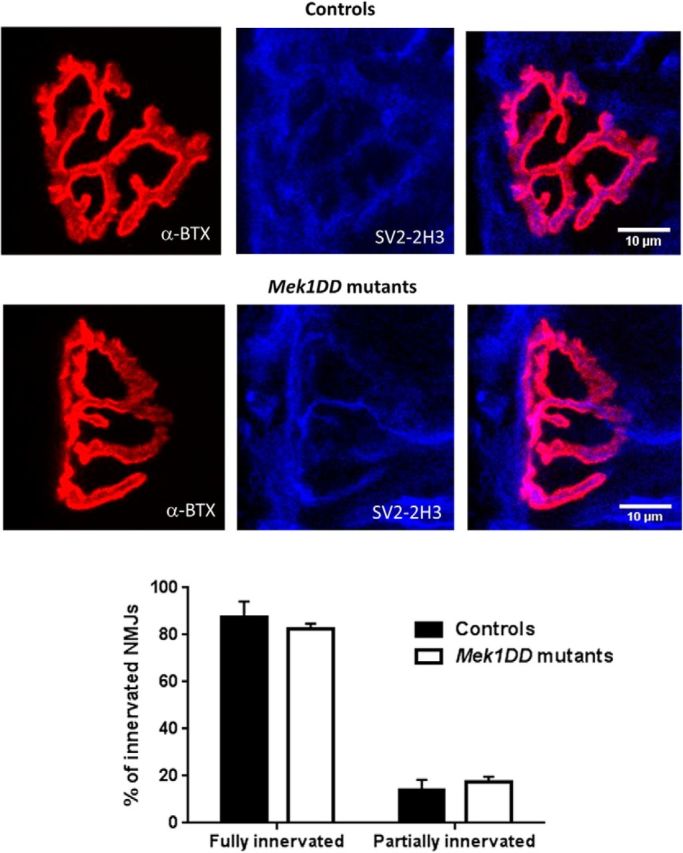
MAPK/ERK activation does not affect reinnervation of the gastrocnemius muscle at 4 weeks postinjury. Muscle innervation has been calculated as the number of reinnervated neuromuscular junctions. Gastrocnemius muscle sections of 100 μm were stained for a-bungarotoxin (α-BTX) as a marker of the postsynaptic NMJ, and for synaptic vesicle protein 2 (SV2) and neurofilament (2H3) antibodies for presynaptic terminal markers. n = 3 mice/group. No significant difference in two-tailed unpaired t test. Scale bar, 10 μm. Error bars indicate SEM.
MAPK/ERK activation impairs Remak bundle formation and unmyelinated axon regeneration after injury
Interestingly, we found that regeneration of small-diameter axons was more dramatically affected by MAPK/ERK activation. As shown in Table 1, the number of nonmyelinated axons 2 mm distal to a nerve crush was significantly decreased at 4 weeks postinjury in MEK1DD mutants compared with controls. In mutant nerves following injury, we noted the abnormalities of nonmyelinating SCs. There were supernumerary SCs surrounding myelinated axons (Fig. 7A, arrowheads). Furthermore, in mutant nerves the morphology of the Remak bundles was altered. Specifically, the SCs demonstrated longer processes and the number of C-fibers per Remak bundle was significantly reduced (Table 1, Fig. 7A).
Figure 7.
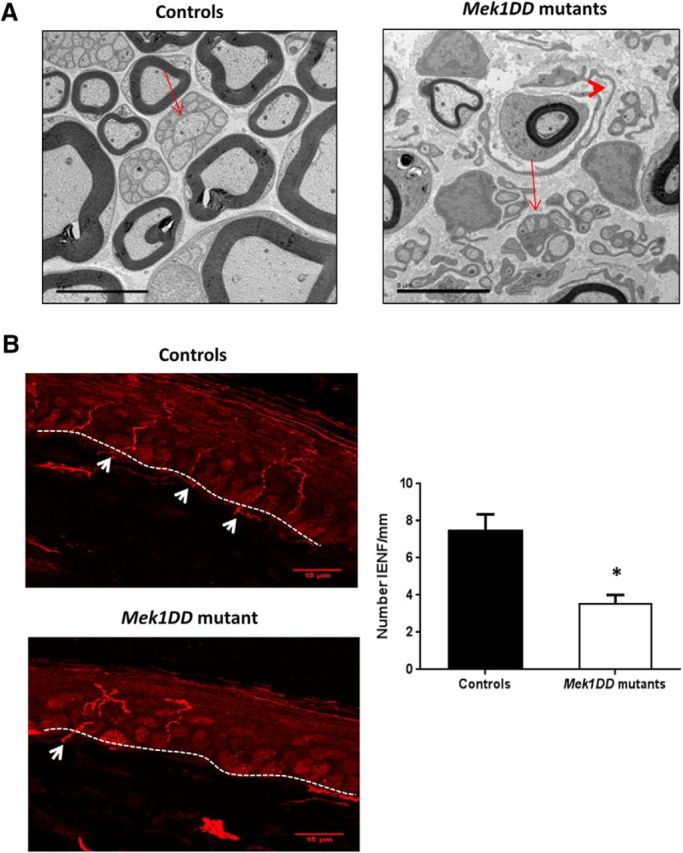
MAPK/ERK induction disrupts Remak bundle formation and small-fiber reinnervation after injury. A, Electron micrograph showing disruption of Remak bundle formation with decreased C-fiber numbers per Remak bundle. Arrows show Remak bundles in controls and mutants. Visible formation of elongated and supernumerary SCs failing to assemble into normal Remak bundles (arrowhead). Scale bar, 5 μm. B, Photomicrographs of transverse sections of ipsilateral skin biopsy specimens stained with PGP9.5. Quantification of epidermal reinnervation, in IENF per millimeter, showed a decrease in mutant mice. n = 3 mice/group. Arrows point to nerve fibers crossing the epidermis. *p < 0.05, two-tailed unpaired t test. Scale bar, 15 μm. Error bars indicate SEM.
We also analyzed reinnervation of the skin, the targets of small-diameter fibers. We collected skin biopsy specimens 3 weeks after injury from the paws of mutant mice, and we stained them with the axonal marker PGP9.5 to measure intraepidermal nerve fiber density. Skin ipsilateral to injury in mutant mice showed a significant decrease in intraepidermal nerve fiber density compared with skin ipsilateral to injury in controls, which is consistent with impaired target reinnervation by sensory fibers (Fig. 7B; controls, 7.47 ± 0.0001, vs MEK1DD mutants, 3.52 ± 0.0005; p = 0.017).
Discussion
We have used a mouse model in which sustained MAPK/ERK activation was induced in adult SCs (i.e., after developmental myelination was completed). Sheean et al. (2014) have previously shown that MAPK/ERK activation in SCs during development induced continuous myelination and rescued the myelination deficit caused by ablation of Shp2 and ErbB3, two signaling molecules essential for myelination. Given the developmental effects of MAPK/ERK activation, we hypothesized that it may promote remyelination. Although remyelination is relatively efficient in the PNS, there are lasting morphological alterations in remyelinated axons after nerve injury, including an increase in the g-ratio and shorter internodal length (Beuche and Friede, 1985). However, we found that MAPK/ERK activation in SCs leads to impaired peripheral nerve repair, profoundly affecting both nonmyelinating and myelinating SCs, axon regeneration, and functional recovery.
Before studying the role of MAPK/ERK in nerve repair, we confirmed that the sustained induction of the pathway did not interfere with myelin stability in uninjured adult nerve over this time course. In contrast to SCs, sustained activation of ERK1/2 in oligodendrocytes of adult mice had already increased myelin thickness at 21 d after activation (Jeffries et al., 2016), although a direct comparison of the degree of ERK activation achieved by this allele in oligodendrocytes versus SCs has not been undertaken. During WD, the first step of nerve regeneration after injury, we found a higher inflammatory response in the form of increased macrophage recruitment and an increased myelin clearance in mutants (Fig. 2A–D). This was in line with the findings of previous studies showing that the MAPK/ERK pathway is directly involved in WD and p-ERK is induced in SCs following injury (Harrisingh et al., 2004; Napoli et al., 2012). Napoli et al. (2012) showed that a transient and strong activation of the Raf/MEK/ERK pathway was able to induce a recruitment of inflammatory cells (macrophages, mast cells, neutrophils, and T cells) even in the absence of injury. SCs and macrophages participate in myelin phagocytosis following injury and may be contributing to the increased myelin clearance we observed after MAPK/ERK activation (Gomez-Sanchez et al., 2015).
Following the initial phase of WD, axons begin to regenerate, re-establishing their close relationship with SCs, which provide critical guidance and growth factors for axons. The SCs then redifferentiate and remyelinate axons (DeFrancesco-Lisowitz et al., 2015). Ultimately, axons will reinnervate their targets, leading to restoration of sensorimotor function, although the degree of recovery is highly dependent on factors such as type and duration of injury (He et al., 2014). MEK1DD mutant mice demonstrated delayed recovery of motor function compared with controls. An unexpected finding was that mutants showed a more rapid recovery of sensory response to pin-prick application to the paw than control littermates (Fig. 3E), although the two groups did not differ at the end point of 4 weeks. This contrasted with the fact that we noted reduced intraepidermal nerve fiber density in MEK1DD mutants at 3 weeks postinjury (i.e., at a time point when a clear behavioral difference was observed). This is likely to be due to the fact that mechanical pain thresholds are well known to be reduced by inflammatory mediators, which cause a sensitization of these afferents and therefore do not simply reflect the number of sensory terminals (Petho and Reeh, 2012).
Given these differences in functional recovery, we investigated morphological changes in both myelinating and nonmyelinating SCs in MEK1DD mutant mice. MAPK/ERK induction in adult SCs induced an enlargement of the nerve area, resulting in a reduced axonal packing as already shown during development (Sheean et al., 2014; Ishii et al., 2016). However, MAPK/ERK induction did not enhance myelin thickness in regenerating myelinated fibers as it does during development (Sheean et al., 2014; Ishii et al., 2016). In fact, 1 month after injury, we found deficits in myelin compaction and other abnormalities such as recurrent myelin loops and infoldings. Similar myelin defects were found as a consequence of MAPK/ERK activation in SCs following MAPK/ERK induction in the postnatal period (Sheean et al., 2014; Ishii et al., 2016). Nerve injury seems to accelerate myelin abnormalities associated with p-ERK activation since in the context of regeneration they are abundant already 30 d after injury.
There are differences in the regulation of developmental myelination and remyelination. For example, during development Neuregulin-1 signaling is essential for the initiation of myelination (Garratt et al., 2000; Taveggia et al., 2005; Birchmeier and Nave, 2008; Birchmeier and Bennett, 2016), whereas during nerve repair Neuregulin-1 determines the rate of remyelination (Fricker et al., 2011). Neuregulin-1 is dispensable at later stages of repair and, in particular, does not determine myelination fate (Fricker et al., 2013). The MAPK/ERK pathway is activated by Neuregulin-1 and during development promotes myelination, but excess activation of MAPK/ERK by Neuregulin-1 can inhibit myelination in vitro (Syed et al., 2010). In the context of animal models of Charcot-Marie-Tooth (CMT) disease, the type I isoform of Neuregulin 1 helps to restore the imbalance between MAPK/ERK and phosphoinositide-3 kinase–AKT signaling pathways and promotes SC differentiation and axonal survival (Fledrich et al., 2014).
A further unexpected finding in MEK1DD mutant mice was a loss of appositions between the abaxonal myelin surface and the SC plasma membrane (Fig. 5A). Appositions in myelinating SCs are generated by the formation of the so-called PDG complex which is composed of periaxin, DRP2, and dystroglycan (Gillespie et al., 1994, 2000; Sherman et al., 2001). These appositions define the border of Cajal bands, which are cytoplasmic channels first described by Ramon y Cajal (1912) and contain microtubules that may serve a nutritive function (Court et al., 2004). We found that DRP2 expression was decreased in mutants, whereas periaxin levels were unchanged (Fig. 5C). To our knowledge, this is the first evidence of a link among activation of the MAPK/ERK pathway, DRP2 loss, and Cajal band disruption. Microtubule-based transport in Cajal bands is used to incorporate new myelin, and gene ablation studies of periaxin or DRP2 suggest that the PDG complex stabilizes and limits radial growth of myelin (Ainger et al., 1997; Carson et al., 1997; Sherman and Brophy, 2005). The reduction in the number of Cajal bands and DRP2 expression could therefore have relevance to the aberrant myelin we found in MEK1DD mutants. We also found a decrease in internodal length, suggesting impaired SC elongation in MEK1DD mutants (Fig. 5D). This reduction in internodal length is not necessarily caused by a change in the PDG complex, given that internodal length is not changed in SCs lacking DRP2. However, periaxin loss results in a shorter internodal length, indicating that periaxin can have DRP2-independent functions (Sherman et al., 2012).
MAPK/ERK activation in SCs resulted in dramatic changes in small-fiber regeneration. Activation of MAPK/ERK led to disruption of Remak bundle formation. In particular, we noted elongated processes of the nonmyelinating SCs and a significant decrease in the number of unmyelinated axons per Remak bundle. These changes were associated with deficits in reinnervation of cutaneous targets by unmyelinated axons. Previous literature has focused on the effect of the MAPK/ERK pathway in myelinating but not in nonmyelinating glia (Ishii et al., 2013; Sheean et al., 2014). However, similar abnormalities in nonmyelinating axons have been described in animal models of the hereditary neuropathies CMT1A and CMT4C (Robertson et al., 2002; Houlden et al., 2009). Interestingly, activated MEK/ERK has been reported in such models before axonal damage (Kohl et al., 2010; Fledrich et al., 2014).
Neurofibromin (NF1) is a Ras-GTPase, which negatively regulates Ras signaling, and Schwann cells deficient in NF1 develop dysregulated ERK signaling (Harrisingh and Lloyd, 2004; Jessen et al., 2013). Interestingly, analysis of the peripheral nerves of mice in which NF1 is deficient within SCs also demonstrate abnormalities of Remak bundles, including a reduced number of axons per Remak bundle and elongated SC processes (Zhu et al., 2002; Zheng et al., 2008; Mayes et al., 2011).
Schwann cell processes have been shown to guide regenerating axons, and it is possible that the impaired regeneration of unmyelinated axons that we observed is due to deficient physical interaction between axons and the elongated SC processes (Son and Thompson, 1995; Höke et al., 2006; Jessen and Mirsky, 2016). It is now recognized that the activation of a transcriptional repair program in SCs is a critical determinant of the success of axon regeneration (Painter et al., 2014; Jessen and Mirsky, 2016; Mindos et al., 2017). It is possible that uncoordinated MAPK activation leads to reduced trophic factor production by nonmyelinating SCs, leading to reduced reinnervation by sensory fibers. Furthermore, the basal membrane might not be correctly formed by nonmyelinating SCs, a further determinant of successful reinnervation (Frostick et al., 1998; Toy and Namgung, 2013).
In summary, we have investigated the effects of MAPK/ERK induction in adult SCs on the regenerative response to nerve injury. At early time points, this activation produced an increased inflammatory response and a faster myelin clearance. Despite our initial hypothesis that MAPK/ERK activation in SCs may promote remyelination, it was actually detrimental to functional nerve repair. MAPK/ERK induction had no effect on myelin thickness but did result in higher rates of myelin decompaction, a reduced number of Cajal bands, and decreased internodal length. We also noted abnormal Remak bundle structures with elongated SC processes, reduced number of C-fibers per Remak bundle, and reduced reinnervation by sensory fibers. Persistent activation of MAPK/ERK in SCs is deleterious for nerve repair; however, we cannot exclude the possibility that transient p-ERK activation (which we are unable to achieve using this model) could have positive effects, for instance through enhanced myelin clearance in the early phase of the injury response. These findings emphasize the differences in the signaling processes that control myelin development and repair and the importance of axoglial interactions in coordinating effective axon regeneration.
Footnotes
This work was supported by Wellcome Clinical Scientist Grant 202747/Z/16/Z and Action Medical Research Grant GN2526. We thank Professor Peter Brophy and Dr. Diane Sherman (University of Edinburgh, Edinburgh, UK) for providing DRP2 and periaxin antibodies and for their valuable comments of the manuscript. We also thank Dr. Errin Johnson and Anna Pielach from the Electron Microscopy Facility (University of Oxford, Oxford, UK) for their helpful assistance with the electron microscopy; and Dr. Klaus Rajewsky for providing R26StopFLMEK1DDfl/fl mice.
The authors declare no competing financial interests.
References
- Ainger K, Avossa D, Diana AS, Barry C, Barbarese E, Carson JH (1997) Transport and localization elements in myelin basic protein mRNA. J Cell Biol 138:1077–1087. 10.1083/jcb.138.5.1077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arthur-Farraj PJ, Latouche M, Wilton DK, Quintes S, Chabrol E, Banerjee A, Woodhoo A, Jenkins B, Rahman M, Turmaine M, Wicher GK, Mitter R, Greensmith L, Behrens A, Raivich G, Mirsky R, Jessen KR (2012) c-Jun reprograms Schwann cells of injured nerves to generate a repair cell essential for regeneration. Neuron 75:633–647. 10.1016/j.neuron.2012.06.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beuche W, Friede RL (1985) A new approach toward analyzing peripheral nerve fiber populations. II. Foreshortening of regenerated internodes corresponds to reduced sheath thickness. J Neuropathol Exp Neurol 44:73–84. 10.1097/00005072-198501000-00006 [DOI] [PubMed] [Google Scholar]
- Birchmeier C, Bennett DL (2016) Neuregulin/ErbB signaling in developmental myelin formation and nerve repair. Curr Top Dev Biol 116:45–64. 10.1016/bs.ctdb.2015.11.009 [DOI] [PubMed] [Google Scholar]
- Birchmeier C, Nave KA (2008) Neuregulin-1, a key axonal signal that drives Schwann cell growth and differentiation. Glia 56:1491–1497. 10.1002/glia.20753 [DOI] [PubMed] [Google Scholar]
- Cargnello M, Roux PP (2011) Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol Mol Biol Rev 75:50–83. 10.1128/MMBR.00031-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carson JH, Worboys K, Ainger K, Barbarese E (1997) Translocation of myelin basic protein mRNA in oligodendrocytes requires microtubules and kinesin. Cell Motil Cytoskeleton 38:318–328. 10.1002/(SICI)1097-0169(1997)38:4%3C318::AID-CM2%3E3.0.CO;2-%23 [DOI] [PubMed] [Google Scholar]
- Chen ZL, Yu WM, Strickland S (2007) Peripheral regeneration. Annu Rev Neurosci 30:209–233. 10.1146/annurev.neuro.30.051606.094337 [DOI] [PubMed] [Google Scholar]
- Court FA, Sherman DL, Pratt T, Garry EM, Ribchester RR, Cottrell DF, Fleetwood-Walker SM, Brophy PJ (2004) Restricted growth of Schwann cells lacking Cajal bands slows conduction in myelinated nerves. Nature 431:191–195. 10.1038/nature02841 [DOI] [PubMed] [Google Scholar]
- DeFrancesco-Lisowitz A, Lindborg JA, Niemi JP, Zigmond RE (2015) The neuroimmunology of degeneration and regeneration in the peripheral nervous system. Neuroscience 302:174–203. 10.1016/j.neuroscience.2014.09.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doerflinger NH, Macklin WB, Popko B (2003) Inducible site-specific recombination in myelinating cells. Genesis 35:63–72. 10.1002/gene.10154 [DOI] [PubMed] [Google Scholar]
- Fledrich R, Stassart RM, Klink A, Rasch LM, Prukop T, Haag L, Czesnik D, Kungl T, Abdelaal TA, Keric N, Stadelmann C, Brück W, Nave KA, Sereda MW (2014) Soluble neuregulin-1 modulates disease pathogenesis in rodent models of Charcot-Marie-Tooth disease 1A. Nat Med 20:1055–1061. 10.1038/nm.3664 [DOI] [PubMed] [Google Scholar]
- Fricker FR, Lago N, Balarajah S, Tsantoulas C, Tanna S, Zhu N, Fageiry SK, Jenkins M, Garratt AN, Birchmeier C, Bennett DL (2011) Axonally derived neuregulin-1 is required for remyelination and regeneration after nerve injury in adulthood. J Neurosci 31:3225–3233. 10.1523/JNEUROSCI.2568-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fricker FR, Antunes-Martins A, Galino J, Paramsothy R, La Russa F, Perkins J, Goldberg R, Brelstaff J, Zhu N, McMahon SB, Orengo C, Garratt AN, Birchmeier C, Bennett DL (2013) Axonal neuregulin 1 is a rate limiting but not essential factor for nerve remyelination. Brain 136:2279–2297. 10.1093/brain/awt148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frostick SP, Yin Q, Kemp GJ (1998) Schwann cells, neurotrophic factors, and peripheral nerve regeneration. Microsurgery 18:397–405. 10.1002/(SICI)1098-2752(1998)18:7%3C397::AID-MICR2%3E3.0.CO;2-F [DOI] [PubMed] [Google Scholar]
- Garratt AN, Voiculescu O, Topilko P, Charnay P, Birchmeier C (2000) A dual role of erbB2 in myelination and in expansion of the schwann cell precursor pool. J Cell Biol 148:1035–1046. 10.1083/jcb.148.5.1035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillespie CS, Sherman DL, Blair GE, Brophy PJ (1994) Periaxin, a novel protein of myelinating Schwann cells with a possible role in axonal ensheathment. Neuron 12:497–508. 10.1016/0896-6273(94)90208-9 [DOI] [PubMed] [Google Scholar]
- Gillespie CS, Sherman DL, Fleetwood-Walker SM, Cottrell DF, Tait S, Garry EM, Wallace VC, Ure J, Griffiths IR, Smith A, Brophy PJ (2000) Peripheral demyelination and neuropathic pain behavior in periaxin-deficient mice. Neuron 26:523–531. 10.1016/S0896-6273(00)81184-8 [DOI] [PubMed] [Google Scholar]
- Goebbels S, Oltrogge JH, Wolfer S, Wieser GL, Nientiedt T, Pieper A, Ruhwedel T, Groszer M, Sereda MW, Nave KA (2012) Genetic disruption of Pten in a novel mouse model of tomaculous neuropathy. EMBO Mol Med 4:486–499. 10.1002/emmm.201200227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Sanchez JA, Carty L, Iruarrizaga-Lejarreta M, Palomo-Irigoyen M, Varela-Rey M, Griffith M, Hantke J, Macias-Camara N, Azkargorta M, Aurrekoetxea I, De Juan VG, Jefferies HB, Aspichueta P, Elortza F, Aransay AM, Martínez-Chantar ML, Baas F, Mato JM, Mirsky R, Woodhoo A, et al. (2015) Schwann cell autophagy, myelinophagy, initiates myelin clearance from injured nerves. J Cell Biol 210:153–168. 10.1083/jcb.201503019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrisingh MC, Lloyd AC (2004) Ras/Raf/ERK signalling and NF1. Cell Cycle 3:1255–1258. 10.4161/cc.3.10.1182 [DOI] [PubMed] [Google Scholar]
- Harrisingh MC, Perez-Nadales E, Parkinson DB, Malcolm DS, Mudge AW, Lloyd AC (2004) The Ras/Raf/ERK signalling pathway drives Schwann cell dedifferentiation. EMBO J 23:3061–3071. 10.1038/sj.emboj.7600309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He B, Zhu Z, Zhu Q, Zhou X, Zheng C, Li P, Zhu S, Liu X, Zhu J (2014) Factors predicting sensory and motor recovery after the repair of upper limb peripheral nerve injuries. Neural Regen Res 9:661–672. 10.4103/1673-5374.130094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Höke A, Redett R, Hameed H, Jari R, Zhou C, Li ZB, Griffin JW, Brushart TM (2006) Schwann cells express motor and sensory phenotypes that regulate axon regeneration. J Neurosci 26:9646–9655. 10.1523/JNEUROSCI.1620-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houlden H, Laura M, Ginsberg L, Jungbluth H, Robb SA, Blake J, Robinson S, King RH, Reilly MM (2009) The phenotype of Charcot-Marie-Tooth disease type 4C due to SH3TC2 mutations and possible predisposition to an inflammatory neuropathy. Neuromuscul Disord 19:264–269. 10.1016/j.nmd.2009.01.006 [DOI] [PubMed] [Google Scholar]
- Ishii A, Fyffe-Maricich SL, Furusho M, Miller RH, Bansal R (2012) ERK1/ERK2 MAPK signaling is required to increase myelin thickness independent of oligodendrocyte differentiation and initiation of myelination. J Neurosci 32:8855–8864. 10.1523/JNEUROSCI.0137-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii A, Furusho M, Bansal R (2013) Sustained activation of ERK1/2 MAPK in oligodendrocytes and Schwann cells enhances myelin growth and stimulates oligodendrocyte progenitor expansion. J Neurosci 33:175–186. 10.1523/JNEUROSCI.4403-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii A, Furusho M, Dupree JL, Bansal R (2014) Role of ERK1/2 MAPK signaling in the maintenance of myelin and axonal integrity in the adult CNS. J Neurosci 34:16031–16045. 10.1523/JNEUROSCI.3360-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii A, Furusho M, Dupree JL, Bansal R (2016) Strength of ERK1/2 MAPK activation determines its effect on myelin and axonal integrity in the adult CNS. J Neurosci 36:6471–6487. 10.1523/JNEUROSCI.0299-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffries MA, Urbanek K, Torres L, Wendell SG, Rubio ME, Fyffe-Maricich SL (2016) ERK1/2 Activation in preexisting oligodendrocytes of adult mice drives new myelin synthesis and enhanced CNS function. J Neurosci 36:9186–9200. 10.1523/JNEUROSCI.1444-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessen KR, Mirsky R (2016) The repair Schwann cell and its function in regenerating nerves. J Physiol 594:3521–3531. 10.1113/JP270874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessen WJ, Miller SJ, Jousma E, Wu J, Rizvi TA, Brundage ME, Eaves D, Widemann B, Kim MO, Dombi E, Sabo J, Hardiman Dudley A, Niwa-Kawakita M, Page GP, Giovannini M, Aronow BJ, Cripe TP, Ratner N (2013) MEK inhibition exhibits efficacy in human and mouse neurofibromatosis tumors. J Clin Invest 123:340–347. 10.1172/JCI60578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones BJ, Roberts DJ (1968) The quantiative measurement of motor inco-ordination in naive mice using an acelerating rotarod. J Pharm Pharmacol 20:302–304. 10.1111/j.2042-7158.1968.tb09743.x [DOI] [PubMed] [Google Scholar]
- Kohl B, Fischer S, Groh J, Wessig C, Martini R (2010) MCP-1/CCL2 modifies axon properties in a PMP22-overexpressing mouse model for Charcot-Marie-tooth 1A neuropathy. Am J Pathol 176:1390–1399. 10.2353/ajpath.2010.090694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauria G. (2005) Small fibre neuropathies. Curr Opin Neurol 18:591–597. 10.1097/01.wco.0000177330.35147.70 [DOI] [PubMed] [Google Scholar]
- Mayes DA, Rizvi TA, Cancelas JA, Kolasinski NT, Ciraolo GM, Stemmer-Rachamimov AO, Ratner N (2011) Perinatal or adult Nf1 inactivation using tamoxifen-inducible PlpCre each cause neurofibroma formation. Cancer Res 71:4675–4685. 10.1158/0008-5472.CAN-10-4558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mindos T, Dun XP, North K, Doddrell RD, Schulz A, Edwards P, Russell J, Gray B, Roberts SL, Shivane A, Mortimer G, Pirie M, Zhang N, Pan D, Morrison H, Parkinson DB (2017) Merlin controls the repair capacity of Schwann cells after injury by regulating Hippo/YAP activity. J Cell Biol 216:495–510. 10.1083/jcb.201606052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napoli I, Noon LA, Ribeiro S, Kerai AP, Parrinello S, Rosenberg LH, Collins MJ, Harrisingh MC, White IJ, Woodhoo A, Lloyd AC (2012) A central role for the ERK-signaling pathway in controlling Schwann cell plasticity and peripheral nerve regeneration in vivo. Neuron 73:729–742. 10.1016/j.neuron.2011.11.031 [DOI] [PubMed] [Google Scholar]
- Newbern JM, Li X, Shoemaker SE, Zhou J, Zhong J, Wu Y, Bonder D, Hollenback S, Coppola G, Geschwind DH, Landreth GE, Snider WD (2011) Specific functions for ERK/MAPK signaling during PNS development. Neuron 69:91–105. 10.1016/j.neuron.2010.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Painter MW, Brosius Lutz A, Cheng YC, Latremoliere A, Duong K, Miller CM, Posada S, Cobos EJ, Zhang AX, Wagers AJ, Havton LA, Barres B, Omura T, Woolf CJ (2014) Diminished Schwann cell repair responses underlie age-associated impaired axonal regeneration. Neuron 83:331–343. 10.1016/j.neuron.2014.06.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palispis WA, Gupta R (2017) Surgical repair in humans after traumatic nerve injury provides limited functional neural regeneration in adults. Exp Neurol 290:106–114. 10.1016/j.expneurol.2017.01.009 [DOI] [PubMed] [Google Scholar]
- Pearson G, Robinson F, Beers Gibson T, Xu BE, Karandikar M, Berman K, Cobb MH (2001) Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr Rev 22:153–183. 10.1210/edrv.22.2.0428 [DOI] [PubMed] [Google Scholar]
- Petho G, Reeh PW (2012) Sensory and signaling mechanisms of bradykinin, eicosanoids, platelet-activating factor, and nitric oxide in peripheral nociceptors. Physiol Rev 92:1699–1775. 10.1152/physrev.00048.2010 [DOI] [PubMed] [Google Scholar]
- Ramon y Cajal S. (1912) El aparato endocelular de Golgi de la celula de Schwann y algunas observaciones sobre la estructura de los tubos nerviosos. Trabajos del Laboratorio de Investigaciones Biologicas de la Universidad de Madrid 10:221–246. [Google Scholar]
- Robertson AM, Perea J, McGuigan A, King RH, Muddle JR, Gabreëls-Festen AA, Thomas PK, Huxley C (2002) Comparison of a new pmp22 transgenic mouse line with other mouse models and human patients with CMT1A. J Anat 200:377–390. 10.1046/j.1469-7580.2002.00039.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schröder JM. (1972) Altered ratio between axon diameter and myelin sheath thickness in regenerated nerve fibers. Brain Res 45:49–65. 10.1016/0006-8993(72)90215-6 [DOI] [PubMed] [Google Scholar]
- Seger R, Krebs EG (1995) The MAPK signaling cascade. FASEB J 9:726–735. [PubMed] [Google Scholar]
- Sheean ME, McShane E, Cheret C, Walcher J, Müller T, Wulf-Goldenberg A, Hoelper S, Garratt AN, Krüger M, Rajewsky K, Meijer D, Birchmeier W, Lewin GR, Selbach M, Birchmeier C (2014) Activation of MAPK overrides the termination of myelin growth and replaces Nrg1/ErbB3 signals during Schwann cell development and myelination. Genes Dev 28:290–303. 10.1101/gad.230045.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman DL, Brophy PJ (2005) Mechanisms of axon ensheathment and myelin growth. Nat Rev Neurosci 6:683–690. 10.1038/nrn1743 [DOI] [PubMed] [Google Scholar]
- Sherman DL, Fabrizi C, Gillespie CS, Brophy PJ (2001) Specific disruption of a schwann cell dystrophin-related protein complex in a demyelinating neuropathy. Neuron 30:677–687. 10.1016/S0896-6273(01)00327-0 [DOI] [PubMed] [Google Scholar]
- Sherman DL, Wu LM, Grove M, Gillespie CS, Brophy PJ (2012) Drp2 and periaxin form Cajal bands with dystroglycan but have distinct roles in Schwann cell growth. J Neurosci 32:9419–9428. 10.1523/JNEUROSCI.1220-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheu JY, Kulhanek DJ, Eckenstein FP (2000) Differential patterns of ERK and STAT3 phosphorylation after sciatic nerve transection in the rat. Exp Neurol 166:392–402. 10.1006/exnr.2000.7508 [DOI] [PubMed] [Google Scholar]
- Son YJ, Thompson WJ (1995) Schwann cell processes guide regeneration of peripheral axons. Neuron 14:125–132. 10.1016/0896-6273(95)90246-5 [DOI] [PubMed] [Google Scholar]
- Srinivasan L, Sasaki Y, Calado DP, Zhang B, Paik JH, DePinho RA, Kutok JL, Kearney JF, Otipoby KL, Rajewsky K (2009) PI3 kinase signals BCR-dependent mature B cell survival. Cell 139:573–586. 10.1016/j.cell.2009.08.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syed N, Reddy K, Yang DP, Taveggia C, Salzer JL, Maurel P, Kim HA (2010) Soluble neuregulin-1 has bifunctional, concentration-dependent effects on Schwann cell myelination. J Neurosci 30:6122–6131. 10.1523/JNEUROSCI.1681-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taveggia C, Zanazzi G, Petrylak A, Yano H, Rosenbluth J, Einheber S, Xu X, Esper RM, Loeb JA, Shrager P, Chao MV, Falls DL, Role L, Salzer JL (2005) Neuregulin-1 type III determines the ensheathment fate of axons. Neuron 47:681–694. 10.1016/j.neuron.2005.08.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toy D, Namgung U (2013) Role of glial cells in axonal regeneration. Exp Neurobiol 22:68–76. 10.5607/en.2013.22.2.68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H, Chang L, Patel N, Yang J, Lowe L, Burns DK, Zhu Y (2008) Induction of abnormal proliferation by nonmyelinating schwann cells triggers neurofibroma formation. Cancer Cell 13:117–128. 10.1016/j.ccr.2008.01.002 [DOI] [PubMed] [Google Scholar]
- Zhu Y, Ghosh P, Charnay P, Burns DK, Parada LF (2002) Neurofibromas in NF1: Schwann cell origin and role of tumor environment. Science 296:920–922. 10.1126/science.1068452 [DOI] [PMC free article] [PubMed] [Google Scholar]