

Abstract
The vertebrate retina has the remarkable ability to support visual function under conditions of limited illumination, including the processing of signals evoked by single photons. Dim-light vision is regulated by several adaptive mechanisms. The mechanism explored in this study is responsible for increasing the light sensitivity and operational range of rod bipolar cells, the retinal neurons operating immediately downstream of rod photoreceptors. This sensitization is achieved through the sustained dopamine-dependent GABA release from other retinal neurons. Our goals were to identify the cell type responsible for the GABA release and the site of its modulation by dopamine. Previous studies have suggested the involvement of amacrine and/or horizontal cells. We now demonstrate, using mice of both sexes, that horizontal cells do not participate in this mechanism. Instead, sustained GABA input is provided by a subpopulation of wide-field amacrine cells, which stimulate the GABAC receptors at rod bipolar cell axons. We also found that dopamine does not act directly on either of these cells. Rather, it suppresses inhibition imposed on these wide-field cells by another subpopulation of upstream GABAergic amacrine cells, thereby sustaining the GABAC receptor activation required for rod bipolar cell sensitization.
SIGNIFICANCE STATEMENT The vertebrate retina has an exquisite ability to adjust information processing to ever-changing conditions of ambient illumination, from bright sunlight to single-photon counting under dim starlight. Operation under each of these functional regimes requires an engagement of specific adaptation mechanisms. Here, we describe a mechanism optimizing the performance of the dim-light channel of vision, which consists of sensitizing rod bipolar cells by a sustained GABAergic input originating from a population of wide-field amacrine cells. Wide-field amacrine cells span large segments of the retina, making them uniquely equipped to normalize and optimize response sensitivity across distant receptive fields and preclude any bias toward local light-intensity fluctuations.
Keywords: adaptation, amacrine cell, bipolar cell, dopamine, GABA
Introduction
During the course of the day, our visual system efficiently operates over a range of light intensities spanning ∼10 orders of magnitude (Rodieck, 1998). To fulfill this task, the retina undergoes dark and light adaptation at multiple levels of information processing. At one of these levels is the rod-driven circuit mediating dim-light vision (Shapley and Enroth-Cugell, 1984; Dunn et al., 2006). In this circuit, rod photoreceptors hyperpolarize in response to light stimuli, which results in reduced glutamate release from their synaptic terminals, causing depolarization of the downstream rod bipolar cells. In higher vertebrates, rod bipolar cells pass the signal through AII amacrine to ON and OFF cone bipolar cells to reach the ganglion cells, which convey the signal to the brain. This vertical pathway through the retina is sculpted by lateral inhibition in the outer and inner retina, with the activity of rod bipolar cells being further regulated by multiple inhibitory inputs, both GABAergic and glycinergic (Tachibana and Kaneko, 1987; Vigh and von Gersdorff, 2005; Eggers and Lukasiewicz, 2006a, 2011; Chávez et al., 2010).
One mechanism optimizing the light sensitivity of rod bipolar cells involves their sensitization by GABA (Herrmann et al., 2011; Herrmann and Arshavsky, 2014; Smith et al., 2015). In this mechanism, rod bipolar cells receive a sustained GABAergic input from another type(s) of retinal neuron whose identity remains a subject of active investigation. The continuous GABA release supports a hyperpolarizing chloride current through GABAC receptor (GABACR) channels on the rod bipolar cells. This hyperpolarization mechanism coexists with the traditional mechanism driven by potassium leak from these cells. Only when both chloride and potassium currents are active do rod bipolar cells achieve the hyperpolarization level required for producing normal light-response amplitudes (Herrmann et al., 2011). This GABAergic input relies on the presence of dopamine, which stimulates D1 receptors (D1Rs) localized either on the cells responsible for the GABA release or further upstream in the circuitry. Accordingly, both response amplitude and operational range of rod bipolar cells are decreased in mice lacking either GABACRs or D1Rs (Herrmann et al., 2011).
The goal of this study was to identify the cell type(s) supporting this sensitizing mechanism. Because GABACRs are found on both dendrites and axon terminals of rod bipolar cells (Lukasiewicz et al., 2004; Herrmann et al., 2011), it is conceivable that sustained GABAergic input could be received at either location (the retinal neurons known to release GABA onto rod bipolar cells are illustrated in Fig. 1). Accordingly, sustained GABA release could potentially originate from either horizontal cells at the bipolar cell dendrites or amacrine cells forming synapses with rod bipolar cell axons. Detailed evidence for the potential involvement of each cell type is reviewed by Herrmann and Arshavsky, 2014. We assessed potential involvement of each cell type in this mechanism by combining transgenic and pharmacological approaches. We concluded that rod bipolar cell sensitivity is not regulated by horizontal cells. Rather, it is regulated by wide-field amacrine cells, whose output is further modulated by serial inhibition.
Figure 1.

Schematic representation of retinal neuron types potentially involved in regulating rod bipolar cell sensitivity.
Materials and Methods
Animals.
C57BL/6 (WT) mice were purchased from Jackson Laboratory. GABACR−/− mice, described by McCall et al., 2002, have been backcrossed onto a C57BL/6 background. The D1RlacZ mouse strain was generated by the trans-National Institutes of Health Knock-Out Mouse Project (KOMP) and obtained from the KOMP Repository (www.komp.org) and then backcrossed with C57BL/6 mice for nine generations. To obtain D1Rfl mice, the D1RlacZ line was bred with Gt(ROSA)26Sortm2(FLP*)Sor/J (Jackson Laboratory, strain #012930) to obtain offspring with complete FRT (flipase recognition target) site recombination. The resulting line with backcrossed with C57BL/6 for nine generations. Cx57Cre mice were described by Hirano et al., 2016 and VGATfl mice were obtained from the Jackson Laboratory (strain #012897). Animals were reared under a normal day/night cycle and handled according to the protocol approved by the Institutional Animal Care and Use Committees of Duke University. All experiments were performed during the day.
Immunofluorescence analysis.
Mouse eyes were enucleated and the posterior eyecups were dissected and then fixed for 1 h in 4% paraformaldehyde, rinsed in PBS, cryoprotected in 30% sucrose, and embedded in optimal cutting temperature (OCT) embedding medium (Tissue-Tek, Sakura Finetek). Retinal 16 μm cross sections were collected using a cryostat microtome (Microm HM 550, Thermo Fisher Scientific). Sections were rehydrated with PBS, treated with a solution to block nonspecific binding (5% goat or donkey serum in PBS with 0.2% Triton X-100) for 1 h, incubated in primary antibodies in the same blocking solution for 3 h, washed three times, incubated in secondary antibodies in blocking solution, and then finally washed three more times. The same general procedure but without cryoprotection and OCT embedding was used to process retinal whole mounts. The retinas were dissected away from the retinal pigment epithelium before fixation and the incubation times for primary and secondary antibodies were 3 and 1 d, respectively.
Both retinal sections and whole mounts were then mounted with Fluoromount G (Electron Microscopy Sciences) under glass coverslips. Images were acquired using a Nikon Eclipse 90i confocal microscope, CFI Plan Fluor 20× or 60× (oil) objectives, and a C1 confocal scanner controlled by EZ-C1 v 3.10 software (Nikon).
The primary antibodies were as follows: rat anti-D1R (1:500; Sigma-Aldrich, D187, clone 1-1-F11 s.E6), rabbit anti-calbindin (1:1000; Millipore, AB1778), rabbit anti-β-galactosidase (1:5000; a kind gift from Jeremy Kay, Duke University), mouse anti-PKCα H-7 (1:1000; Santa Cruz Biotechnology, sc-8393 Lot#I1306), goat anti-Bhlhb5 E-17 (1:500; Santa Cruz Biotechnology, sc-6045 Lot#K1414), and goat anti-choline acetyltransferase (ChAT; 1:400; Millipore, AB144P). Secondary goat anti-rat or anti-mouse and donkey anti-goat, anti-rabbit, or anti-mouse antibodies were Alexa Fluor 488, Alexa Fluor 594, or Alexa Fluor 647 (1:500; Invitrogen). Nuclei were stained using DAPI (Sigma-Aldrich).
Electroretinography.
Electroretinograms (ERGs) were recorded from dark-adapted mice as described by Herrmann et al., 2011, using the Espion E2 system with a ColorDome ganzfeld stimulator (Diagnosys). Six-to-12-week-old mice of both sexes were anesthetized by an intraperitoneal injection of 100 mg/kg ketamine and 10 mg/kg xylazine. Pupils were dilated with a mixture of 1% cyclopentolate-HCl and 2.5% phenylephrine. Eyes were kept lubricated during the recordings by a 1% carboxymethylellulose sodium gel. Body temperature was maintained by a heated platform. Simultaneous recordings were made from both eyes using gold contact-lens electrodes, with stainless steel needle electrodes (Ocuscience) in the mouth (reference) and at the base of the tail (ground). ERG signals were sampled at 1 kHz and recorded with 0.15 Hz low-frequency and 500 Hz high-frequency cutoffs. Responses to flashes from 0.0003 to 100 cd s/m2 with 10 to 1 trials averaged and interflash intervals of 5–180 s were recorded in the dark and then recorded at increasing background intensities (0.001, 0.01, 0.1 cd/m2) after 2 min of adaptation. In our experimental setup, the 456 nm background light-activated rhodopsin at the rate of ∼540 isomerization/rod/s at 1 cd/m2.
The data from the electroretinogram recordings were analyzed using Matlab 2016a (MathWorks). Oscillatory potentials were removed from the signals by 55 Hz FFT low-pass frequency filtering. The amplitude of the b-wave was calculated from baseline to the peak for dimmer flashes and from the bottom of the a-wave to the b-wave peak for brighter flashes. The rod bipolar cell sensitivity was then determined for each background illumination. After plotting b-wave amplitudes as a function of flash intensity at each background, the stimulus–response curves were fit with Matlab using the following equation (Eq. 1):
 |
Rmax,1 is the maximal response amplitude, n1 is the Hill coefficient, and I0.5,1 is the half-saturating flash intensity for the rod-mediated responses. The second term of Equation 1 characterizes the cone-mediated response. Sensitivity (S) for the rod system was then determined by dividing the maximal response amplitude by the half-saturating flash intensity for the set of flashes at each background-light level. The relationship between S and background light for each genotype or pharmacological manipulation can then be fit using the Weber–Fechner equation as follows (Eq. 2):
 |
Ib is the background light intensity, Ihalf is the background luminance that causes a half-maximal reduction of S, and n is again a Hill coefficient. In the text, S is referred to as rod bipolar cell sensitivity.
Intraocular injections.
Intravitreal injections were performed using a syringe with a 33 gauge, 12° beveled needle (Hamilton) under dim red light. The following compounds from Tocris Bioscience or Sigma-Aldrich were dissolved in PBS and then a volume of 1 μl was injected: 200 mm GABA (Sigma-Aldrich), 10 μm tetrodotoxin (TTX; Tocris Bioscience), 200 μm SR-95531 [2-(3-carboxypropyl)-3-amino-6-(4 methoxyphenyl)pyridazinium bromide, Sigma-Aldrich], 200 μm SKF 83566 (8-bromo-2,3,4,5-tetrahydro-3-methyl-5-phenyl-1H-3-benzazepin-7-olhydrobromide; Tocris Bioscience), and 500 μm SKF 81297 [(±)-6-chloro-2,3,4,5-tetrahydro-1-phenyl-1H-3-benzazepine hydrobromide; Tocris Bioscience). Recordings began 20 min after injection.
Experimental design and statistical analysis.
All electrophysiological experiments were performed on mice between 8 and 12 weeks old. For the experiments using genetic models, animals of both sexes were used (eight male and seven female Cx57-Cre+/− D1RWT/WT mice; four male and eight female Cx57-Cre+/− D1Rflox/flox mice; four male and three female Cx57-Cre+/− VGATWT/WT mice; two male and three female Cx57-Cre+/− VGATflox/flox mice), but for the experiments using intravitreal injections, all animals were female.
To compare sensitivities of different experimental groups, the three components of the Weber–Fechner fit (Eq. 2) were compared using either an ordinary one-way ANOVA or a two-tailed t test in GraphPad Prism version 7.00 for Windows (GraphPad Software, www.graphpad.com; Table 1).
Table 1.
Fitting parameters for rod bipolar cell sensitivity of each animal type and experimental condition and statistical analysis of the differences among selected groups
| S0 | SEM | p (ordinary one-way ANOVA or *two-tailed t test) | Ihalf | SEM | p (ordinary one-way ANOVA or *two-tailed t test) | n | SEM | p (ordinary one-way ANOVA or *two-tailed t test) | R2 | |
|---|---|---|---|---|---|---|---|---|---|---|
| Cx57-Cre+/− D1RWT/WT | 1 | 0.03397 | <0.0001 | 1.13 | 0.228 | 0.2339 | 0.7062 | 0.08374 | 0.4118 | 0.8905 |
| Cx57-Cre+/− D1Rflox/flox | 0.8989 | 0.04447 | 2.053 | 0.546 | 0.917 | 0.1493 | 0.8397 | |||
| Cx57-Cre+/− D1Rflox/flox + D1R antagonist | 0.4204 | 0.008282 | 1.413 | 0.1481 | 0.8695 | 0.05664 | 0.9967 | |||
| Cx57-Cre+/− D1RWT/WT | 1 | 0.03397 | 0.0742* | 1.13 | 0.228 | 0.124* | 0.7062 | 0.08374 | 0.2222* | 0.8905 |
| Cx57-Cre+/− D1Rflox/flox | 0.8989 | 0.04447 | 2.053 | 0.546 | 0.917 | 0.1493 | 0.8397 | |||
| Cx57-Cre+/− VGATWT/WT | 0.998 | 0.09241 | 0.8556* | 2.159 | 1.264 | 0.9561* | 0.656 | 0.2229 | 0.8956* | 0.6699 |
| Cx57-Cre+/− VGATflox/flox | 0.9697 | 0.1233 | 2.047 | 1.572 | 0.7082 | 0.3248 | 0.6253 | |||
| WT | 1 | 0.04185 | 0.0007 | 0.8998 | 0.248 | 0.531 | 0.6227 | 0.09645 | 0.7096 | 0.8374 |
| WT + TTX | 0.8633 | 0.06843 | 0.5021 | 0.212 | 0.7045 | 0.2009 | 0.7443 | |||
| GABACR−/− | 0.7055 | 0.04237 | 0.7016 | 0.3038 | 0.812 | 0.225 | 0.8527 | |||
| WT | 1 | 0.04185 | 0.0002 | 0.8998 | 0.248 | 0.4941 | 0.6227 | 0.09645 | 0.9064 | 0.8374 |
| WT + TTX | 0.8633 | 0.06843 | 0.5021 | 0.212 | 0.7045 | 0.2009 | 0.7443 | |||
| WT + TTX + GABA | 1.283 | 0.04437 | 0.759 | 0.1702 | 0.6429 | 0.08504 | 0.9634 | |||
| GABACR−/− | 0.7055 | 0.04237 | 0.8753 | 0.7016 | 0.3038 | 0.756 | 0.812 | 0.225 | 0.871 | 0.8527 |
| GABACR−/−+TTX | 0.6866 | 0.04916 | 0.6175 | 0.2095 | 0.7364 | 0.1656 | 0.6988 | |||
| GABACR−/− +TTX + GABA | 0.755 | 0.0231 | 0.1749 | 0.06759 | 0.5301 | 0.09671 | 0.9918 | |||
| WT | 1.074 | 0.06428 | 0.3063 | 0.6685 | 0.2712 | 0.7668 | 0.6217 | 0.1473 | 0.8616 | 0.8014 |
| WT + gabazine | 0.9997 | 0.02406 | 0.6328 | 0.09676 | 0.6712 | 0.06576 | 0.9826 | |||
| WT + gabazine + SKF83566 | 0.9992 | 0.05171 | 0.8813 | 0.263 | 0.7327 | 0.14 | 0.8945 | |||
| WT + gabazine + SKF81297 | 0.8796 | 0.03214 | 1.081 | 0.2127 | 0.8163 | 0.1045 | 0.9798 |
Results
Horizontal cells are not responsible for dopamine/GABA-dependent rod bipolar cell sensitization
One cell type proposed as a potential source of the dopamine/GABA-dependent rod bipolar cell sensitization is the horizontal cell. Horizontal cells contain a significant amount of GABA and produce the strongest D1R immunostaining in the retina (Guo et al., 2010; Deniz et al., 2011; Herrmann et al., 2011; Hirano et al., 2016). Additionally, the GABA content of these cells is regulated by light and this light dependence is abolished in D1R knock-out (D1R−/−) mice (Herrmann et al., 2011). Therefore, horizontal cells satisfy both criteria of a strong candidate for this mechanism: they could release GABA at the location where rod bipolar cells express GABACR and their GABAergic activity is regulated by D1Rs.
We interrogated the potential involvement of horizontal cells in providing the dopamine-dependent GABA release onto rod bipolar cell dendrites using two models of genetically manipulated mice. First, we generated a conditional knock-out mouse lacking D1Rs specifically in horizontal cells. Two LoxP sites were inserted around Exon 2 of the Drd1a gene, which contains the entire D1R coding region (Fig. 2A). After excising out the FRT-flanked gene-trapped lacZ cassette by breeding this mouse with a flp-expressing mouse, we bred this new line with the mouse expressing Cre recombinase in place of one allele of the horizontal cell-specific protein, connexin 57 (Hirano et al., 2016). The resulting Cx57-Cre+/− D1Rflox/flox genotype showed a near complete elimination of D1R immunostaining in horizontal cells with the rest of the retina being unaffected (Fig. 2B).
Figure 2.
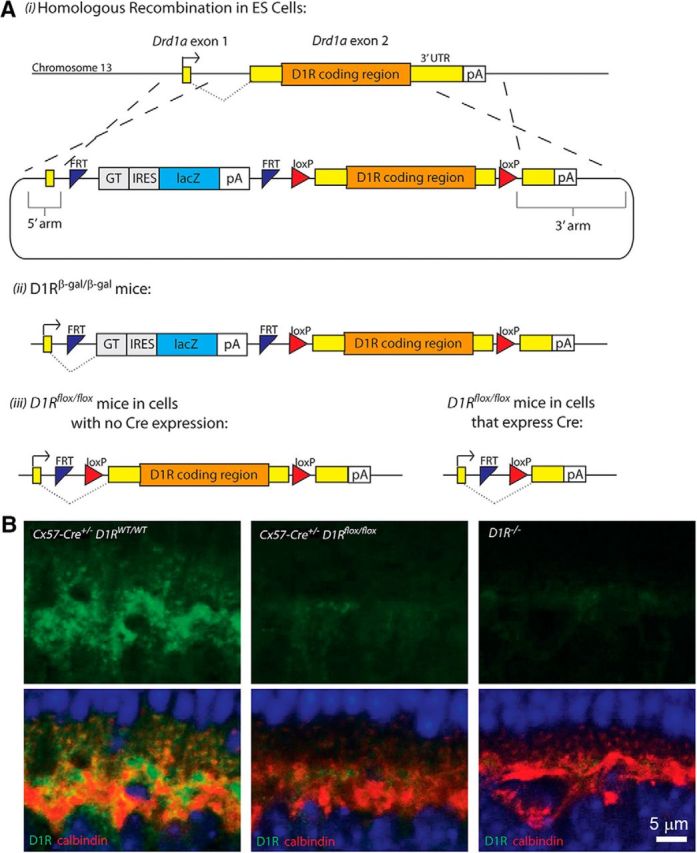
Generation of horizontal cell-specific D1R knock-out mouse. A, Strategy for generating D1Rβ-gal/β-gal and D1Rflox/flox mouse lines: (i) a DNA cassette encoding both FRT-flanked sequence of lacZ and loxP-flanked D1R coding region underwent homologous recombination in ES cells; (ii) mice derived from these cells transcribed lacZ instead of Drd1a at the D1R allele (D1Rβ-gal/β-gal mice); (iii) germline recombination at the FRT sites was achieved by breeding these mice with a flp mouse line [Gt(ROSA)26Sortm2(FLP*)Sor] to produce the D1Rflox/flox mouse, in which the Drd1a gene can be excised in the presence of Cre recombinase. Arrows indicate transcription start sites. pA, Transcription termination site; GT, splice acceptor site; IRES, internal ribosome entry site. B, The lack of D1R immunostaining in the outer plexiform layer of Cx57-Cre+/− D1Rflox/flox mice. Faint residual signal was indistinguishable from that in the global D1R−/− mouse and, therefore, was nonspecific. Horizontal cells are visualized by calbindin staining. Scale bar, 5 μm.
The effect of conditional D1R knock-out on the light sensitivity of rod bipolar cells was characterized using ERG. ERG monitors the activity of retinal neurons in living animals without disrupting their synaptic connections, surrounding neurotransmitters, or intracellular and extracellular ion concentrations (Robson and Frishman, 1998). Given the complexity of the circuit interrogated in this study, with at least some participating neurons likely located distant from their targets, the use of ERG ensures that none of this circuit's components are disrupted and all function in their natural physiological environments.
The response of rod bipolar cells to a dim flash is represented by a positive ERG deflection, the b-wave (Robson and Frishman, 1998; Robson et al., 2004). Accordingly, the sensitivity of rod bipolar cells, defined as a ratio between the maximal b-wave amplitude and the half-saturating flash intensity, can be calculated after measuring b-wave amplitudes in response to flashes of increasing intensity and subtracting the cone-driven b-wave contributions. Conducting these experiments in the dark and in the presence of various levels of background illumination, followed by data normalization to the dark-adapted sensitivity, provides a useful comparative measure of rod bipolar cell sensitivity and adaptation across genotypes and pharmacological conditions (Herrmann et al., 2011; Smith et al., 2015).
The absence of D1Rs in horizontal cells had no effect on ERG responses under any of our experimental conditions (Fig. 3A), and applying the rod bipolar cell sensitivity analysis to the conditional D1R knock-out did not reveal any appreciable changes across a wide range of background-light intensities (Fig. 3B; see Table 1 for statistical parameters from these and all subsequent ERG recordings). This was in striking contrast to the amplitude (Fig. 3A) and sensitivity (Fig. 3B) reduction in the presence of a D1R antagonist or in the global D1R knock-out (Herrmann et al., 2011).
Figure 3.

Horizontal cells do not regulate rod bipolar cell sensitivity. A, Representative traces from ERG recordings of Cx57-Cre+/− D1RWT/WT mice, Cx57-Cre+/− D1Rflox/flox mice, and Cx57-Cre+/− D1Rflox/flox mice after intravitreal injection of a D1R antagonist SCH-23390. Conditions of dark or light adaptation and flash intensities are indicated in the panels. B, Rod bipolar cell sensitivity of Cx57-Cre+/− D1Rflox/flox mice and their Cx57-Cre+/− D1RWT/WT control littermates was determined in the dark and in the presence of three background illumination levels. Each sensitivity value was calculated as described in Materials and Methods, normalized to the dark sensitivity of control littermates and plotted as a function of background light. Light sensitivity of Cx57-Cre+/− D1Rflox/flox mice was similarly analyzed following intravitreal injection of D1R antagonist SCH-23390 and included for comparison. C, Representative traces from ERG recordings of Cx57-Cre+/− VGATWT/WT, Cx57-Cre+/− Vgatflox/flox, and GABACR knock-out mice. D, Rod bipolar cell sensitivity of Cx57-Cre+/− VGATflox/flox mice and their Cx57-Cre+/− VGATWT/WT control littermates was normalized to the dark sensitivity of control littermates and plotted as a function of background light. Rod bipolar cell sensitivity of GABACR knock-out mice was normalized to the dark sensitivity of the Cx57-Cre+/− VGATWT/WT mice and included for comparison. Animal types and pharmacological interventions used in each experiment are indicated and color-coded in the panels. Data from ≥12 eyes were averaged for each condition, except for the SCH-23390 injection experiment for which three eyes were analyzed. Data are presented as mean ± SEM.
In our second conditional knock-out mouse, recently described by Hirano et al., 2016, cre-lox recombination was used to eliminate the vesicular GABA transporter (VGAT) specifically in horizontal cells, preventing vesicular GABA release from these cells. ERG recordings conducted with these mice once again showed that there is no difference in the overall ERG waveforms (Fig. 3C) or the rod bipolar cell sensitivity (Fig. 3D) between knock-out and control mice. This result is in stark contrast to the phenotype of the GABACR knock-out (Fig. 3D), which essentially phenocopies the effect of D1R knock-out (Herrmann et al., 2011). Together, these data rule out horizontal cells as a source of sustained dopamine-dependent GABA release onto rod bipolar cells.
The GABA-dependent rod bipolar cell sensitization is driven by wide-field amacrine cells
The elimination of horizontal cells as the source of GABA responsible for sensitizing rod bipolar cells implies that GABA activates GABACRs at the rod bipolar cell axons. Consistent with this idea, sustained GABACR-mediated currents have been documented in axon terminals of fish bipolar cells (Hull et al., 2006; Jones and Palmer, 2009), which receive amacrine cell inputs (Fletcher and Wässle, 1999). Similarly, experiments in rat retinal slices demonstrated that antagonists of ionotropic GABA receptors decrease the dynamic range of light responses in intact rod bipolar cells, but not in cells with severed axon terminals (Euler and Masland, 2000).
There are two types of GABAergic amacrine cells forming synapses at this location, A17 amacrine cells and unclassified wide-field amacrine cells (Chávez et al., 2006, 2010; Eggers and Lukasiewicz, 2006a; Grimes, 2012). A17 cells provide inhibitory feedback at a reciprocal synapse, which enhances the temporal resolution of rod-mediated visual signals (Eggers and Lukasiewicz, 2006b) and improves signal-to-noise characteristics of rod bipolar cell outputs (Grimes et al., 2015). This feedback is highly dynamic and is conveyed by rapidly desensitizing GABAARs and a population of GABACRs separate from those activated at nonreciprocal inhibitory synapses (Chávez et al., 2006, 2010). Additionally, chemical inactivation of A17 cells did not affect sensitivity of rod bipolar cells in dark-adapted mice (Smith et al., 2015).
On the other hand, the nonreciprocal synapses of wide-field amacrine cells act primarily via the GABACRs and are dependent on spiking activity/voltage-gated Nav channels (Shields and Lukasiewicz, 2003; Chávez et al., 2010), which makes them a plausible candidate for sensitizing rod bipolar cells through GABA release. Indeed, Smith and colleagues (2013, 2015) demonstrated that the amplitude of scotopic b-waves in WT mice is reduced by TTX. They concluded that this effect is conveyed through wide-field amacrine cells, the only TTX-sensitive GABAergic cell type acting upstream of rod bipolar cells. They also showed that this amplitude decrease is comparable to that caused by a pharmacological block of GABACRs. Our data provide further evidence supporting this hypothesis.
First, we analyzed the effect of TTX on rod bipolar cell sensitivity across a broad range of background-light intensities and observed a degree of sensitivity reduction essentially indistinguishable from the global GABACR knock-out mouse (Fig. 4A). This suggests that the sensitizing GABAergic input is strictly TTX-sensitive, consistent with GABA being released from wide-field amacrine cells. Light-evoked, TTX-sensitive input to the rod system via wide-field amacrines has also been shown in other animals (Taylor, 1999; Vigh et al., 2011).
Figure 4.

GABACR-dependent rod bipolar cell sensitization is TTX-sensitive. A, Representative traces from ERG recordings of GABACR knock-out mice and WT mice with or without injections of TTX or TTX and GABA. Conditions of dark or light adaptation and flash intensities are indicated in the panels. B, Rod bipolar cell sensitivity for the animal groups represented in A was normalized to the dark sensitivity of WT mice and plotted as a function of background light. C, Representative traces from ERG recordings of WT mice and GABACR knock-out mice with or without injections of TTX or TTX and GABA. D, Rod bipolar cell sensitivity for the groups represented in C was normalized to the dark sensitivity of WT mice and plotted as a function of background light. Between 3 and 12 eyes were analyzed for each condition; data are presented as mean ± SEM.
An important control experiment showed that an intraocular injection of GABA reverses the rod bipolar cell sensitivity reduction imposed by TTX (Fig. 4B), establishing that it is GABA release that impacts sensitivity in the absence of voltage-gated sodium channel function. Another critical control showed that TTX does not alter rod bipolar cell sensitivity in GABACR knock-out mice (Fig. 4C), which rules out a GABACR-independent effect of TTX on the light sensitivity of these cells. Consistently, intravitreal injections of exogenous GABA had no effect on light sensitivity of GABACR knock-out mice in the presence of TTX (Fig. 4D). This further demonstrates that GABACR-dependent regulation of rod bipolar cell sensitivity relies entirely on TTX-dependent GABA release and there are no off-target effects mediated, for example, via GABAARs or GABABRs somewhere else in the retinal circuitry.
Dopamine promotes sustained GABA release by modulating serial inhibition in wide-field amacrine cells
We next considered the site where dopamine is acting to modulate the GABA-dependent rod bipolar cell sensitization. A hallmark of wide-field amacrine cells is their serial inhibition, in which GABA release from upstream cells decreases GABAergic activity of downstream cells (Eggers and Lukasiewicz, 2006a, 2010, 2011). Therefore, the GABAergic input onto rod bipolar cell axons could be modulated by dopamine acting at any wide-field amacrine cell engaged in a serial inhibition circuit, not necessarily the cell providing direct GABAergic input. The difference between these cells is that serial inhibition is accomplished by activating GABAARs (Eggers and Lukasiewicz, 2011), whereas the synapse between wide-field and rod bipolar cells uses GABACRs (Fletcher and Wässle, 1999; Chávez et al., 2010). Therefore, inhibition of GABAARs allows the study of GABA-dependent rod bipolar cell sensitization in the absence of serial inhibition.
Blocking GABAARs with gabazine (SR 95531) in WT animals has no effect on rod bipolar cell sensitivity (Herrmann et al., 2011; Smith et al., 2015), suggesting that the primary wide-field cell (i.e., the cell directly responsible for releasing GABA onto rod bipolar cell; see Fig. 6) does not receive serial inhibition. However, blocking GABAARs reversed the reduction in b-wave amplitude caused by the D1R antagonist SKF83566 (Smith et al., 2015), a result reproduced in Figure 5A. The most parsimonious explanation for this puzzling observation is that the primary wide-field cell does receive serial inhibition but, under the conditions of our and their experiments, this inhibition is prevented by activated D1Rs on the secondary wide-field cell. In this case, D1R inhibition on the secondary cell allows serial inhibition to occur, which in turn suppresses the GABAergic output of the primary cell. This model is illustrated schematically in Figure 6.
Figure 6.

The circuit responsible for rod bipolar cell sensitization by the dopamine/GABA-dependent mechanism. A subset of wide-field amacrine cells (primary cell in blue) provides sustained GABA release onto GABACRs located in the rod bipolar cell axon. This GABA release can be suppressed by upstream amacrine cells (secondary cell in magenta), which release GABA onto GABAARs located at the primary amacrine cell. This serial inhibition is suppressed by dopamine acting through the D1 receptors located on the secondary cells. When these D1Rs are blocked, secondary cells inhibit primary cells through the GABAAR-dependent mechanism, which ultimately causes reduction in rod bipolar cell sensitivity.
Figure 5.

D1R activity does not affect rod bipolar cell sensitivity in the absence of serial inhibition. A–C, ERG b-wave responses of dark-adapted WT mice are plotted as a function of flash intensity and fit using a double hyperbolic function (Eq. 1). Experiments were conducted with either untreated mice or following intravitreal injections with D1R antagonist SKF83566, D1R agonist SKF81297, GABAAR antagonist gabazine, or a combination of gabazine with either D1R drug. Specific pharmacological interventions used in each experiment are indicated and color-coded in the panels. D, Rod bipolar cell sensitivity in the presence of either gabazine alone or a combination of gabazine with D1R antagonist SKF83566 or D1R agonist SKF81297 was normalized to the dark sensitivity in the presence of gabazine alone and plotted as a function of background light. Between 3 and 10 eyes were analyzed for each condition; data are presented as mean ± SEM.
The explanation provided by Smith and colleagues (2015) was more complex and presumed an additional role for D1Rs on the primary wide-field cell. This reasoning was based on two observations. First, b-wave amplitudes in their experiments were reduced by the D1R agonist SKF38393. Second, the D1R antagonist SKF83566 increased the b-wave amplitudes over WT conditions in the absence of serial inhibition (when gabazine is present). We explored this possibility further.
We first noted that SKF38393 is not a D1R agonist, but rather a partial agonist (Watts et al., 1993), which could either activate or inhibit D1R, dependent on the background dopamine level. Therefore, we tested whether a similar effect would be seen with a full agonist, SKF81297 (Spealman et al., 1997), and did not observe any inhibition (Fig. 5B). Regarding SKF38393, we did observe a slight trend toward b-wave amplitude reduction, but it did not reach statistical significance in our hands (data not shown).
We next considered the contributions of D1Rs under conditions in which serial inhibition is blocked by gabazine (Fig. 5C). As in the case of Smith et al. (2015), the D1R antagonist SKF83566 caused an increase in b-wave amplitudes. However, the D1R agonist SKF81297 increased b-wave amplitudes to an even larger degree, which is inconsistent with the idea that D1Rs suppress GABA release from the primary wide-field cell. Most importantly, neither D1R agonist nor antagonist had any effect on b-wave sensitivity (Fig. 5D), which is the hallmark of the mechanism investigated in this study. In addition, b-wave amplitudes were affected at light intensities that activated cones, whereas responses to the dimmest flashes driven exclusively by rods were essentially unchanged. This argues the following: (1) D1R-dependent regulation of b-wave amplitude observed in the presence of gabazine is conferred through alternative circuitry, (2) the primary wide-field amacrine cells in our circuit are not directly regulated by dopamine, and (3) the D1Rs critical for modulating rod bipolar cell sensitivity are located at a “secondary” amacrine cell (Fig. 6).
A subset of wide-field amacrine cells express D1Rs
One prediction from the data presented so far is that there is a pool of wide-field amacrine cells that express D1Rs and another pool that does not. A challenge in validating this prediction is that nearly all D1Rs found in the inner retina, including the D1Rs close to rod bipolar cell terminals (Herrmann et al., 2011), are localized to the dense synaptic milieu of the inner plexiform layer, making it extremely difficult to assign D1R immunoreactivity to specific cell types. Therefore, we used an alternative approach to identify D1R-expressing cells by analyzing retinas of a mouse containing a gene trap driving expression of β-galactosidase (β-gal) at the D1R locus (Fig. 2). These mice accumulate β-gal in the cytoplasm of cells normally expressing D1R, which allows clear identification of individual D1R-positive neurons.
This approach revealed prominent β-gal staining in two distinct cell populations (Fig. 7A): cone bipolar cells known to express D1Rs (Farshi et al., 2016) and a subset of amacrine cells. The juxtaposition of these amacrine cells in relation to rod bipolar cell axons (stained for protein kinase C) and starburst amacrine cells (stained for ChAT) is further illustrated in an optical section through the inner nuclear layer of a whole-mount retina (Fig. 7B). It is worth noting that the lack of β-gal staining in rod bipolar cells serves as a further independent confirmation that these cells do not express D1Rs (Herrmann and Arshavsky, 2014; Farshi et al., 2016) and the lack of β-gal staining in starburst cells illustrates that not all GABAergic retinal neurons express D1Rs.
Figure 7.
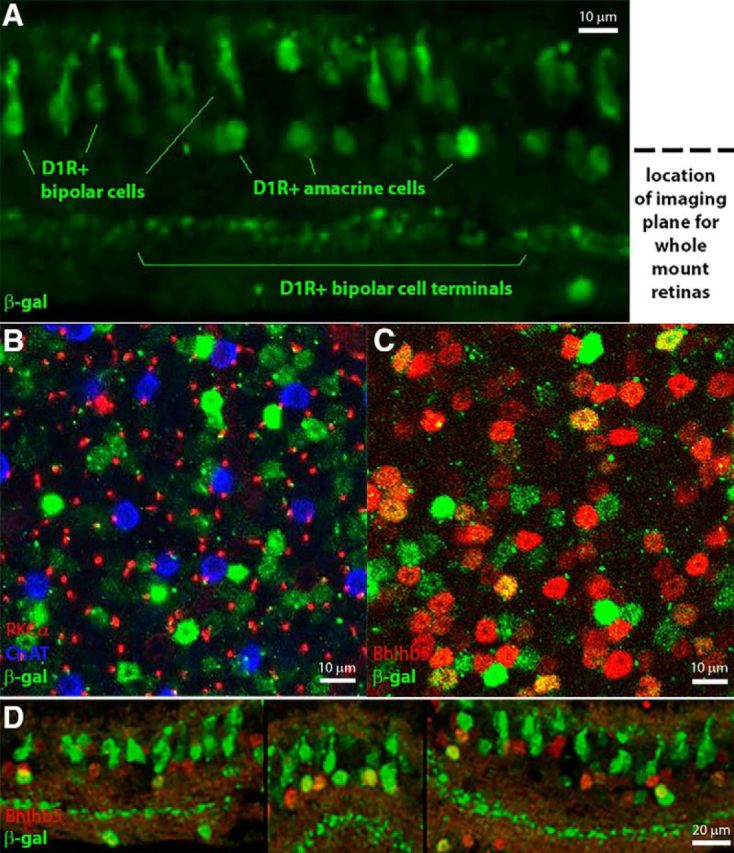
D1Rs are expressed in a subset of wide-field amacrine cells. A, β-Gal staining (green) in a tangential section from a D1Rβ-gal/β-gal mouse showing D1R expression in cone bipolar and amacrine cells. The dotted line indicates the depth at which the images in B and C were taken. B, Image taken at the level of amacrine cell bodies in a D1Rβ-gal/β-gal whole-mount retina. ChAT (blue) labels the bodies of starburst amacrine cells, PKCα (red) labels rod bipolar cell axons, and β-gal (green) labels both cell bodies and axons (diameter equivalent to the PKCα axons) of cells located in this section. C, Image taken at the level of amacrine cell bodies in a D1Rβ-gal/β-gal whole-mount retina. β-Gal staining (green) colocalizes with a subset of wide-field amacrine cell bodies labeled with Bhlhb5 (red). D, Retinal cross sections from a D1Rβ-gal/β-gal mouse showing β-gal staining (green) colocalization with a subset of wide-field amacrine cell bodies labeled with Bhlhb5 (red). Individual optical sections are shown in all panels.
We next coimmunostained β-gal with Bhlhb5, a marker expressed in a large fraction of GABAergic amacrine cells (Feng et al., 2006) and found that a distinct population of the Bhlhb5-positive cells were also β-gal-positive. This result was obtained using both retinal whole mounts (Fig. 7C) and cross sections (Fig. 7D), with location of double-stained cell bodies corresponding to that of wide-field amacrine cells. This is consistent with our model showing that the activity of a distinct subset of GABAergic amacrine cells is directly regulated by dopamine.
Discussion
In this study, we analyzed the regulation of light sensitivity within the dim-light channel of vision. Sensitivity is perhaps the most critical parameter for light perception in dim light, with behavioral assays demonstrating that sensitivity (rather than response amplitude or speed) determines visual performance under these conditions (Sarria et al., 2015). This appears to be an efficient strategy to maximize information collection when the number of absorbed photons is limited.
The circuit investigated here uses the GABACRs in rod bipolar cells to extend the voltage range of their light responses, thereby enhancing the sensitivity and expanding the operating range of these cells (Herrmann et al., 2011). This sensitization is achieved through a Cl− current carried by GABACRs, which hyperpolarizes rod bipolar cells beyond the level achieved by the conventional mechanism involving a K+ current (see Herrmann et al., 2011, their Fig. 4, for an electrical circuit illustrating the additive nature of Cl− and K+ contributions).
This GABACR activation is sustained by GABA release from other retinal neurons in a circuit further regulated by dopamine. Our goal was to establish the identity of these neurons. Previous studies suggested that they could be either horizontal or amacrine cells (Fig. 1); however, our results eliminate horizontal cells as a potential source of GABA release in this mechanism. We showed that rod bipolar cell sensitivity is unaffected by horizontal cell-specific knock-outs of either D1R or VGAT. Furthermore, D1R activity was recently shown to decrease calcium-channel currents in isolated horizontal cells (Liu et al., 2016), a property that would lead to increased rather than decreased GABA release in the absence of D1R activation. Therefore, the functional role of GABACRs in the rod bipolar cell dendrites remains a subject for future studies.
On the other hand, we provide multiple lines of evidence that the GABA driving rod bipolar cell sensitization is released from wide-field amacrine cells, consistent with earlier observations in other species (Taylor, 1999; Vigh et al., 2011) and the conclusions by Smith and colleagues (2013, 2015). Most importantly, wide-field amacrine cells, which release GABA at the GABACR synapses on rod bipolar cell axons, are sensitive to TTX and are regulated by GABAAR-dependent serial inhibition, key properties of the cell type releasing GABA in this mechanism. One caveat is that voltage-gated sodium channels could be impacting this circuit in more than one cell type. Some ON-cone bipolar cell types, the most likely candidate for depolarizing the wide-field amacrine cells (Chávez et al., 2010), have been shown to contain TTX-sensitive NaV channels (Pan and Hu, 2000; Ichinose et al., 2005; Puthussery et al., 2013), and changes in ON-cone bipolar input to the circuit could indirectly change GABAergic input to the rod bipolar cell.
We further demonstrated that a distinct fraction of wide-field amacrine cells express D1R and analyzed whether D1R-dependent regulation of this circuit takes place at the primary cell releasing GABA into rod bipolar cells and/or the secondary cell responsible for the serial inhibition of the primary cell (Fig. 7). We concluded that this regulation is confined to the secondary cell based on the lack of rod bipolar cell sensitivity regulation by D1R agonists and antagonists when serial inhibition was blocked. Notably, this contradicts the conclusions by Smith et al. (2015), who suggested that D1Rs are expressed in all wide-field amacrine cells in this circuit. As elaborated in Results, we believe that the reason for this discrepancy is twofold. First, Smith et al. drew their conclusions using a partial rather than full D1R agonist. Second, they did not discriminate the effects of dopamine drugs on rod bipolar cell sensitivity from other effects that change response amplitude, independent of changing response sensitivity. A similar phenomenon is seen with exogenous GABA, which has long been known to increase b-wave amplitudes (Naarendorp and Sieving, 1991; Robson et al., 2004) but does not increase rod bipolar cell sensitivity. In this case, GABA likely decouples the level of rod bipolar cell hyperpolarization from the regulatory mechanism interrogated in this study (Herrmann et al., 2011).
Another interesting result is that the dopamine/GABA-dependent rod bipolar cell sensitization was constitutively present under all experimental conditions explored in this study. Rod bipolar cell sensitivity remained unchanged when serial inhibition was blocked by gabazine, indicating that serial inhibition was suppressed by dopamine even in the absence of gabazine. Such a constitutive suppression of serial inhibition could be a consequence of using full-field illumination during ERG recordings, which presents identical stimuli across all visual fields, eliminating any need for lateral information transmission. This begs the question of whether serial inhibition is in fact engaged in dynamic regulation of information processing in a natural environment where light stimuli rapidly and uniquely change across the retina. In this case, wide-field amacrine cells, with their long processes, are uniquely equipped to normalize and optimize response sensitivity across distant receptive fields and preclude any bias toward local light-intensity fluctuations. Exploring this question is an exciting task for future studies.
Footnotes
This work was supported by National Institutes of Health Grants EY10336 (V.Y.A.), EY05722 (V.Y.A.), EY04067 (N.C.B.), EY15573 (N.C.B.); an unrestricted award from Duke University; a Nelson Trust Award (V.Y.A.) from Research to Prevent Blindness; and a Career Scientist Award (14F-RCS-004) from the United States Department of Veterans Affairs. N.C.B. is a U.S. Department of Veterans Affairs Career Research Scientist.
The authors declare no competing financial interests.
References
- Chávez AE, Singer JH, Diamond JS (2006) Fast neurotransmitter release triggered by Ca influx through AMPA-type glutamate receptors. Nature 443:705–708. 10.1038/nature05123 [DOI] [PubMed] [Google Scholar]
- Chávez AE, Grimes WN, Diamond JS (2010) Mechanisms underlying lateral GABAergic feedback onto rod bipolar cells in rat retina. J Neurosci 30:2330–2339. 10.1523/JNEUROSCI.5574-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deniz S, Wersinger E, Schwab Y, Mura C, Erdelyi F, Szabó G, Rendon A, Sahel JA, Picaud S, Roux MJ (2011) Mammalian retinal horizontal cells are unconventional GABAergic neurons. J Neurochem 116:350–362. 10.1111/j.1471-4159.2010.07114.x [DOI] [PubMed] [Google Scholar]
- Dunn FA, Doan T, Sampath AP, Rieke F (2006) Controlling the gain of rod-mediated signals in the mammalian retina. J Neurosci 26:3959–3970. 10.1523/JNEUROSCI.5148-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggers ED, Lukasiewicz PD (2006a) GABA(A), GABA(C) and glycine receptor-mediated inhibition differentially affects light-evoked signalling from mouse retinal rod bipolar cells. J Physiol 572:215–225. 10.1113/jphysiol.2005.103648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggers ED, Lukasiewicz PD (2006b) Receptor and transmitter release properties set the time course of retinal inhibition. J Neurosci 26:9413–9425. 10.1523/JNEUROSCI.2591-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggers ED, Lukasiewicz PD (2010) Interneuron circuits tune inhibition in retinal bipolar cells. J Neurophysiol 103:25–37. 10.1152/jn.00458.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggers ED, Lukasiewicz PD (2011) Multiple pathways of inhibition shape bipolar cell responses in the retina. Vis Neurosci 28:95–108. 10.1017/S0952523810000209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Euler T, Masland RH (2000) Light-evoked responses of bipolar cells in a mammalian retina. J Neurophysiol 83:1817–1829. [DOI] [PubMed] [Google Scholar]
- Farshi P, Fyk-Kolodziej B, Krolewski DM, Walker PD, Ichinose T (2016) Dopamine D1 receptor expression is bipolar cell type-specific in the mouse retina. J Comp Neurol 524:2059–2079. 10.1002/cne.23932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng L, Xie X, Joshi PS, Yang Z, Shibasaki K, Chow RL, Gan L (2006) Requirement for Bhlhb5 in the specification of amacrine and cone bipolar subtypes in mouse retina. Development 133:4815–4825. 10.1242/dev.02664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher EL, Wässle H (1999) Indoleamine-accumulating amacrine cells are presynaptic to rod bipolar cells through GABA(C) receptors. J Comp Neurol 413:155–167. 10.1002/(SICI)1096-9861(19991011)413:1%3C155::AID-CNE11%3E3.0.CO;2-6 [DOI] [PubMed] [Google Scholar]
- Grimes WN. (2012) Amacrine cell-mediated input to bipolar cells: variations on a common mechanistic theme. Vis Neurosci 29:41–49. 10.1017/S0952523811000241 [DOI] [PubMed] [Google Scholar]
- Grimes WN, Zhang J, Tian H, Graydon CW, Hoon M, Rieke F, Diamond JS (2015) Complex inhibitory microcircuitry regulates retinal signaling near visual threshold. J Neurophysiol 114:341–353. 10.1152/jn.00017.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo C, Hirano AA, Stella SL Jr, Bitzer M, Brecha NC (2010) Guinea pig horizontal cells express GABA, the GABA-synthesizing enzyme GAD 65, and the GABA vesicular transporter. J Comp Neurol 518:1647–1669. 10.1002/cne.22294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann R, Arshavsky VY (2014) The role of dopamine in fine-tuning cone- and rod-driven vision. In: G-protein signaling mechanisms in the retina (Martemyanov KA, Sampath AP, eds), pp 121–141. New York: Springer. [Google Scholar]
- Herrmann R, Heflin SJ, Hammond T, Lee B, Wang J, Gainetdinov RR, Caron MG, Eggers ED, Frishman LJ, McCall MA, Arshavsky VY (2011) Rod vision is controlled by dopamine-dependent sensitization of rod bipolar cells by GABA. Neuron 72:101–110. 10.1016/j.neuron.2011.07.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano AA, Liu X, Boulter J, Grove J, Perez de Sevilla Muller L, Barnes S, Brecha NC (2016) Targeted deletion of vesicular GABA transporter from retinal horizontal cells eliminates feedback modulation of photoreceptor calcium channels. eNeuro 3:pii:ENEURO.0148-15.2016. 10.1523/ENEURO.0148-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hull C, Li GL, von Gersdorff H (2006) GABA transporters regulate a standing GABAC receptor-mediated current at a retinal presynaptic terminal. J Neurosci 26:6979–6984. 10.1523/JNEUROSCI.1386-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichinose T, Shields CR, Lukasiewicz PD (2005) Sodium channels in transient retinal bipolar cells enhance visual responses in ganglion cells. J Neurosci 25:1856–1865. 10.1523/JNEUROSCI.5208-04.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones SM, Palmer MJ (2009) Activation of the tonic GABAC receptor current in retinal bipolar cell terminals by nonvesicular GABA release. J Neurophysiol 102:691–699. 10.1152/jn.00285.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Grove JC, Hirano AA, Brecha NC, Barnes S (2016) Dopamine D1 receptor modulation of calcium channel currents in horizontal cells of mouse retina. J Neurophysiol 116:686–697. 10.1152/jn.00990.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukasiewicz PD, Eggers ED, Sagdullaev BT, McCall MA (2004) GABAC receptor-mediated inhibition in the retina. Vision Res 44:3289–3296. 10.1016/j.visres.2004.07.023 [DOI] [PubMed] [Google Scholar]
- McCall MA, Lukasiewicz PD, Gregg RG, Peachey NS (2002) Elimination of the rho1 subunit abolishes GABAC receptor expression and alters visual processing in the mouse retina. J Neurosci 22:4163–4174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naarendorp F, Sieving PA (1991) The scotopic threshold response of the cat ERG is suppressed selectively by GABA and glycine. Vision Res 31:1–15. 10.1016/0042-6989(91)90068-G [DOI] [PubMed] [Google Scholar]
- Pan ZH, Hu HJ (2000) Voltage-dependent Na(+) currents in mammalian retinal cone bipolar cells. J Neurophysiol 84:2564–2571. [DOI] [PubMed] [Google Scholar]
- Puthussery T, Venkataramani S, Gayet-Primo J, Smith RG, Taylor WR (2013) NaV1.1 channels in axon initial segments of bipolar cells augment input to magnocellular visual pathways in the primate retina. J Neurosci 33:16045–16059. 10.1523/JNEUROSCI.1249-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robson JG, Frishman LJ (1998) Dissecting the dark-adapted electroretinogram. Doc Ophthalmol 95:187–215. 10.1023/A:1001891904176 [DOI] [PubMed] [Google Scholar]
- Robson JG, Maeda H, Saszik SM, Frishman LJ (2004) In vivo studies of signaling in rod pathways of the mouse using the electroretinogram. Vision Res 44:3253–3268. 10.1016/j.visres.2004.09.002 [DOI] [PubMed] [Google Scholar]
- Rodieck RW. (1998) The first steps in seeing. Sunderland, MA: Sinauer. [Google Scholar]
- Sarria I, Pahlberg J, Cao Y, Kolesnikov AV, Kefalov VJ, Sampath AP, Martemyanov KA (2015) Sensitivity and kinetics of signal transmission at the first visual synapse differentially impact visually-guided behavior. Elife 4:e06358. 10.7554/eLife.06358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapley R, Enroth-Cugell C (1984) Visual adaptation and retinal gain controls. In: Progress in retinal research (Osborne N, ed). Amsterdam: Elsevier; 10.1016/0278-4327(84)90011-7 [DOI] [Google Scholar]
- Shields CR, Lukasiewicz PD (2003) Spike-dependent GABA inputs to bipolar cell axon terminals contribute to lateral inhibition of retinal ganglion cells. J Neurophysiol 89:2449–2458. 10.1152/jn.00916.2002 [DOI] [PubMed] [Google Scholar]
- Smith BJ, Tremblay F, Côté PD (2013) Voltage-gated sodium channels contribute to the b-wave of the rodent electroretinogram by mediating input to rod bipolar cell GABA(c) receptors. Exp Eye Res 116:279–290. 10.1016/j.exer.2013.09.006 [DOI] [PubMed] [Google Scholar]
- Smith BJ, Côté PD, Tremblay F (2015) Dopamine modulation of rod pathway signaling by suppression of GABAC feedback to rod-driven depolarizing bipolar cells. Eur J Neurosci 42:2258–2270. 10.1111/ejn.12993 [DOI] [PubMed] [Google Scholar]
- Spealman RD, Bergman J, Rosenzweig-Lipson S (1997) Differential modulation of behavioral effects of cocaine by low- and high-efficacy D1 agonists. Psychopharmacology (Berl) 133:283–292. 10.1007/s002130050403 [DOI] [PubMed] [Google Scholar]
- Tachibana M, Kaneko A (1987) γ-Aminobutyric acid exerts a local inhibitory action on the axon terminal of bipolar cells: evidence for negative feedback from amacrine cells. Proc Natl Acad Sci U S A 84:3501–3505. 10.1073/pnas.84.10.3501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor WR. (1999) TTX attenuates surround inhibition in rabbit retinal ganglion cells. Vis Neurosci 16:285–290. [DOI] [PubMed] [Google Scholar]
- Vigh J, von Gersdorff H (2005) Prolonged reciprocal signaling via NMDA and GABA receptors at a retinal ribbon synapse. J Neurosci 25:11412–11423. 10.1523/JNEUROSCI.2203-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigh J, Vickers E, von Gersdorff H (2011) Light-evoked lateral GABAergic inhibition at single bipolar cell synaptic terminals is driven by distinct retinal microcircuits. J Neurosci 31:15884–15893. 10.1523/JNEUROSCI.2959-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts VJ, Lawler CP, Gilmore JH, Southerland SB, Nichols DE, Mailman RB (1993) Dopamine D1 receptors: efficacy of full (dihydrexidine) vs partial (SKF38393) agonists in primates vs rodents. Eur J Pharmacol 242:165–172. 10.1016/0014-2999(93)90076-T [DOI] [PubMed] [Google Scholar]