

Abstract
Although RBC transfusion can result in the development of anti-RBC alloantibodies that increase the probability of life-threatening hemolytic transfusion reactions, not all patients generate anti-RBC alloantibodies. However, the factors that regulate immune responsiveness to RBC transfusion remain incompletely understood. One variable that may influence alloantibody formation is RBC alloantigen density. RBC alloantigens exist at different densities on the RBC surface and likewise exhibit distinct propensities to induce RBC alloantibody formation. However, although distinct alloantigens reside on the RBC surface at different levels, most alloantigens also represent completely different structures, making it difficult to separate the potential impact of differences in Ag density from other alloantigen features that may also influence RBC alloimmunization. To address this, we generated RBCs that stably express the same Ag at different levels. Although exposure to RBCs with higher Ag levels induces a robust Ab response, RBCs bearing low Ag levels fail to induce RBC alloantibodies. However, exposure to low Ag–density RBCs is not without consequence, because recipients subsequently develop Ag-specific tolerance. Low Ag–density RBC–induced tolerance protects higher Ag–density RBCs from immune-mediated clearance, is Ag specific, and occurs through the induction of B cell unresponsiveness. These results demonstrate that Ag density can potently impact immune outcomes following RBC transfusion and suggest that RBCs with altered Ag levels may provide a unique tool to induce Ag-specific tolerance.
Red blood cell transfusion represents the most common medical intervention in modern medicine (1, 2). Although transfusion can provide a therapeutic benefit to most patients, repeat exposure to RBCs can result in the formation of alloantibodies directed against donor RBC alloantigens not expressed by transfused recipients (3, 4). Formation of RBC alloantibodies can make it difficult to find compatible blood for future transfusions and directly increases the risk for hemolytic transfusion reactions, one of the most common causes of transfusion-related mortality (5–8). However, not all patients develop RBC alloantibodies following transfusion (9, 10). Although a variety of factors likely influence the development of an immune response following RBC transfusion, previous studies suggest that RBC alloantigen density may impact immune responsiveness following RBC exposure (11, 12). However, because many distinct alloantigens reside on the RBC surface (13), studies capable of separating the potential impact of the distinct structural features of individual alloantigens from the possible influence of differences in alloantigen density on immune outcomes have been difficult to conduct.
Unlike models of transplantation, RBCs isolated from different strains of mice do not inherently express distinct alloantigens capable of inducing RBC alloantibodies observed clinically following transfusion (14, 15). As a result, although RBC transfusion–induced alloantibody formation predates transplantation and has been recognized for nearly 80 y, models to study this process have historically not been available. To address this limitation, we developed founders that express human Kell (KEL) Ag, one of the most common RBC alloantigens implicated in hemolytic transfusion reactions following RBC transfusion (4, 16, 17). By using a β-globin promoter to specifically drive KEL expression on the surface of RBCs (18), we isolated two founders that stably express different levels of the KEL alloantigen. Because previous studies suggest that RBC alloantigen density might influence RBC alloantibody formation following transfusion (11, 12), we examined the immunological consequence of transfusion of RBCs expressing distinct levels of an RBC alloantigen.
Materials and Methods
Mice
C57BL/6 (H-2b) recipients were purchased from the National Cancer Institute (Frederick, MD) or Charles River (Wilmington, MA). B cell–deficient μMT (C57BL/6-Ighmtm1Cgn/J) mice were purchased from the Jackson Laboratory (Bar Harbor, ME) and maintained as described previously (19). IL-10–GFP/Vert-x [C57BL/6(Cg)-IL10tm1.1Karp/J] and Foxp3-RFP (C57BL/6-Foxp3tm1Flv/J) recipients were generous gifts from Dr. J. Galipeau and Dr. E. Waller, respectively (20). All recipients were 6–12 wk old. C57BL/6 KEL-transgenic mice that express different densities of the human KEL glycoprotein under an RBC-specific promoter were developed as outlined below. Hen egg lysozyme (HEL), OVA fused to Duffy (HOD) (H-2q) donors expressing a fusion protein HEL, OVA, and human Duffy b under an RBC-specific promoter were developed previously (21). KEL and HOD mice were crossed to generate KEL × HOD F1 progeny expressing both transgenes. Donor mice were 8–12 wk of age. Mice were bred and housed in Emory University Department of Animal Resources facilities. All procedures were performed according to approved Institutional Care and Use Committee protocols.
Generation of KELlo mice
The KEL-transgenic mouse was developed by PCR amplification of the open reading frame of KEL from a human bone marrow cDNA library (Clontech, Mountain View, CA), as described previously (17, 22). Briefly, primers for amplification were designed to insert a BamHI site on the 5′ end and to engineer a Kozak consensus sequence upstream of the start codon (5′-GGGGGATCCTAAGCCACCATGGAAGGTGGGGACCAAAGTG-3′ and 5′-TTGGAACAGAAGCAGAAAGGAA-3′), and the open reading frame was then ligated into a vector containing μ′LCR β, followed by isolation of successful recombinants. The purified construct was subsequently submitted to the Emory Core Transgenic Facility for pronuclear injection. Using PCR and flow cytometry, two founder pups were isolated, each expressing different densities of the KEL glycoprotein, hereafter referred to as KEL and KELlo.
Characterization of KELlo mice
RBC-specific expression of KEL was assessed in the peripheral blood and spleen of KEL and KELlo mice. To detect KEL, samples were incubated with monoclonal anti-KEL Abs, Mima-8 or Mima-9, which react with the specific Jsb and Kpb epitopes, respectively, as described previously (17). The mean fluorescence intensity (MFI) of binding on RBCs expressing high levels of KEL (KEL RBCs) or RBCs expressing low levels of KEL (KELlo RBCs) was calculated by subtracting the MFI of anti-IgG alone from the binding of anti-KEL plus anti-IgG (17). Cell-specific expression was determined by costaining samples with anti-KEL mAbs and Abs to cell-specific markers to RBCs (anti-Ter119), T cells (anti-CD3), B cells (anti-CD19), or platelets (anti-CD41).
Abs for flow cytometry
Allophycocyanin rat anti-mouse CD4, PE rat anti-mouse CD25, FITC rat anti-mouse CD8α, allophycocyanin rat anti-mouse CD45R/B220, BV786 rat anti-mouse CD3ε, V500 rat anti-mouse CD4, FITC rat anti-mouse CD5, V450 rat anti-mouse CD45R/B220, PE rat anti-mouse CD1d, PerCP rat anti-mouse IgD, allophycocyanin rat anti-mouse CD25, PE rat anti-mouse CD1d, FITC polyclonal goat anti-mouse Ig, and allophycocyanin streptavidin were purchased from BD Biosciences (San Jose, CA). Alexa Fluor 700 rat anti-mouse IL-10, Alexa Fluor 700 rat IgG2b, к, PE Cy7 mouse IgG1, к, and PE Cy7 mouse anti-mouse TGF-β were purchased from eBioscience (San Diego, CA). Biotinylated rat anti-mouse C3 was obtained from CEDARLANE (Burlington, ON, Canada), and allophycocyanin goat anti-mouse IgG and FITC goat anti-mouse IgM were purchased from Jackson ImmunoResearch (West Grove, PA). LIVE/DEAD Fixable Near-IR Dead Cell Stain was bought from Life Technologies (Waltham, MA). Anti-KEL (Mima-8, Jsb epitope and Mima-9, Kpb epitope) and anti-Fy3 (Mima-29) mAbs were purchased from Bio X Cell (West Lebanon, NH).
RBC transfer, survival, and staining
Donor C57BL/6, KEL, KELlo, HOD, or HOD × KEL F1 blood was collected 1:8 into acid citrate dextrose (Vacutainer, Franklin Lakes, NJ) and washed three times with PBS. For polyinosinic-polycytidylic acid (PIC) priming, recipients received a single i.p. injection of 100 μg of PIC (GE, Chicago, IL) 2 h prior to transfusion, as done previously (17). Recipients were then transfused with RBCs once, twice, or three times at 2-wk intervals via the lateral tail vein, as done previously (17). KEL, HOD, or KEL × HOD F1 RBC challenge occurred 5 wk following KEL or KELlo RBC exposure. To facilitate detection of RBCs posttransfer, RBCs were labeled with DiO or CM-DiI (both from Molecular Probes, Eugene, OR), as described previously (22, 23). Briefly, 1 ml of packed RBCs was diluted 1:10 in PBS, followed by the addition of DiO or DiI at a 1:100 dilution. Samples were incubated for 30 min at 37°C, and unbound dye was removed by washing the samples three times in PBS. C57BL/6-DiO and KEL or KELlo-DiI RBCs were mixed at a 1:1 ratio and transfused via the lateral tail vein with 50 μl of each type of blood, as outlined previously (22, 23). At the indicated time points posttransfusion, RBC survival was measured by normalizing the percentage of KEL or KELlo-DiI RBCs to tracer C57BL/6-DiO RBCs. Ag density on surviving KELlo-DiI RBCs was evaluated by incubating samples with polyclonal sera containing anti-KEL Abs, followed by allophycocyanin goat anti-IgG (17, 22). To examine Abs directly bound to transfused KELlo RBCs, samples were stained with allophycocyanin goat anti-IgG or FITC goat anti-IgM (22). C3 fixation was additionally measured using a biotinylated anti-C3 Ab that was detected by flow cytometry using allophycocyanin streptavidin (22). All samples were run on a four-color BD FACSCalibur and analyzed using FlowJo software; MFI was used to assess the levels of Ag density, bound Abs, and C3 fixation on transfused RBCs.
Seroanalysis
To determine the level of anti-HOD or anti-KEL Ab formation at various time points following RBC challenge, we used a flow cross match approach, as described previously (17, 19). Briefly, to detect anti-KEL Abs bound to KEL RBCs, samples were next incubated with allophycocyanin goat anti-IgG or FITC goat anti-IgM diluted 1:100 in FACS buffer for 30 min at 4°C. C57BL/6 RBCs were similarly incubated with serum, followed by allophycocyanin goat anti-IgG or FITC goat anti-IgM (17). The nonspecific C57BL/6 RBC MFI was then subtracted from the KEL RBC–specific binding to determine KEL reactivity (17). Samples were run on a four-color BD FACSCalibur and analyzed using FlowJo software; MFI was used to measure the amount of Ag-specific Ab subsets present in the serum.
Regulatory T cell depletion
Following KELlo RBC exposure, nonexposed and KELlo-exposed recipients were depleted of regulatory T cells (Tregs; CD4+ CD25+) by a single i.p. injection of 500 μg of monoclonal rat anti-CD25 IgG1 (clone PC61) or 500 μg of rat IgG1 isotype-control Ab (both from Bio X Cell), as outlined previously (24), and then challenged with KEL RBCs. Treg depletion was assessed in the peripheral blood immediately prior to KEL RBC challenge. To accomplish this, a single capillary tube (∼100 μl) of whole blood was collected from treated and nontreated mice, and peripheral blood leukocytes were obtained by RBC lysis using RBC lysing buffer (Sigma). Isolated peripheral blood leukocytes were incubated with an Ab mixture containing allophycocyanin rat anti-CD4 and PE rat anti-CD25. Following a 30-min incubation at 4°C, samples were washed three times, run on a four-color BD FACSCalibur, and analyzed using FlowJo software. Subsequently, all recipients were challenged with KEL RBCs.
Adoptive transfer of immune cells
B cells and CD4+ T cells were isolated from nonexposed or KELlo RBC–exposed C57BL/6 recipients. To isolate each population, splenocytes from each group were harvested by sterile mechanical disruption of the spleen and lysis of RBCs. B cells or CD4+ T cells were then negatively enriched using a mouse Pan B Cell Isolation Kit II or a mouse CD4+ T Cell Isolation Kit (both from Miltenyi Biotec). CD4+ T cells or B cells were adoptively transferred into C57BL/6 mice, as done previously (107 CD4+ T cells or B cells into C57BL/6 recipients or 5 × 107 B cells into B cell–deficient μMT mice) (25–30), followed by KEL RBC transfusion. Following transfer, all recipients were challenged with KEL RBCs. For cellular transfer into μMT mice, recipients were conditioned with gamma radiation treatment (300 rad) 24 h prior to B cell transfer, as done previously (31).
Immunophenotyping
Foxp3-RFP, IL-10–GFP, and C57BL/6 recipients were exposed to KELlo or C57BL/6 RBCs, followed by KEL RBC challenge when indicated. At day 0 (pre-KEL RBC challenge) and at days 3 and 7 post–KEL RBC challenge, recipient splenocytes were harvested as described above and evaluated for alterations in the percentage of regulatory B cells (Bregs) and/or Tregs by directly staining with LIVE/DEAD Fixable Near-IR Dead Cell Stain + PE rat anti-mouse CD1d + FITC rat anti-mouse CD5 + PerCP rat anti-mouse IgD + BV786 rat anti-mouse CD3ε + V500 rat anti-mouse CD45R/B220 or with LIVE/DEAD Fixable Near-IR Dead Cell Stain + BV786 rat anti-mouse CD3ε + V500 rat anti-mouse CD4 + V450 rat anti-mouse CD45R/B220 + allophycocyanin rat anti-mouse CD25 + FITC rat anti-mouse CD8α, respectively. Cytokine production by Bregs and Tregs was additionally examined in splenocytes through intracellular cytokine staining. To evaluate cytokine production by Bregs, splenocytes were restimulated in vitro for 5 h at 37°C with 50 ng/ml PMA + 500 ng/ml ionomycin + 10 μg/ml LPS (Escherichia coli serotype 0111:B4; all from Sigma) + BD GolgiStop (containing monensin; BD Pharmingen, San Jose, CA), as done previously (25, 32, 33). For cytokine detection in Tregs, splenocytes were likewise restimulated in vitro for 5 h at 37°C with 50 ng/ml PMA + 500 ng/ml ionomycin + BD GolgiStop, as done previously (34, 35). To control for nonspecific affects of blocking the golgi, duplicate samples were cultured for 5 h at 37°C in BD GolgiStop alone. Following the 5-h incubation, the cells were washed, and cell surface markers were stained for 30 min at 4°C with LIVE/DEAD Fixable Near-IR Dead Cell Stain + PE rat anti-mouse CD1d + FITC rat anti-mouse CD5 + PerCP rat anti-mouse IgD + BV786 rat anti-mouse CD3ε + V500 rat anti-mouse CD45R/B220 for Bregs or with LIVE/DEAD Fixable Near-IR Dead Cell Stain + BV786 rat anti-mouse CD3ε + V500 rat anti-mouse CD4 + V450 rat anti-mouse CD45R/B220 + allophycocyanin rat anti-mouse CD25 + FITC rat anti-mouse CD8α for Tregs. Then samples were washed with FACS buffer, fixed, and permeabilized for 30 min at 4°C using BD Cytofix/Cytoperm (BD Pharmingen). The fixed and permeabilized cells were washed once in 1× BD Perm/Wash buffer and subsequently stained for 30 min at 4°C with Alexa Fluor 700 rat anti-mouse IL-10 + PE Cy7 mouse anti-mouse TGF-β diluted 1:100 in 1× BD Perm/Wash buffer, as outlined previously (25, 32–35). The samples were then washed and run on a BD LSR II at the Emory University School of Medicine Core Facility for Flow Cytometry. BD Pharmingen MICK2 cells were used as a positive cytokine-staining control.
Statistics
Statistical analysis was performed using one-way ANOVA with the Dunnett or Tukey posttest when multiple comparisons were made or using a Student t test when comparing two values. Significance was defined as p < 0.05. All data were reported as the mean ± SEM, unless otherwise indicated. Data are representative of at least two or three independent experiments.
Results
Prior to examining the potential impact of KEL Ag density on immune responsiveness following RBC transfusion, we first sought to characterize the expression of KEL in each founder. Examination of cells positive for the RBC-specific marker Ter119 demonstrated that each founder produces RBCs with distinct levels of KEL, hereafter referred to as KEL or KELlo RBCs (Fig. 1A–C). Levels of KEL expression on RBCs isolated from KEL or KELlo RBC founders remained stable following their initial characterization. Although we detected the KEL Ag on the surface of RBCs in KEL and KELlo mice, no KEL could be detected on the surface of B cells, T cells, or platelets (Fig. 1D, 1E), suggesting that KEL Ag expression in KEL or KELlo founders specifically occurs on the RBC surface. As a result, KEL and KELlo RBC founders provide a unique opportunity to examine the potential impact of immune responsiveness to the same Ag presented at different densities specifically on the RBC surface.
FIGURE 1.
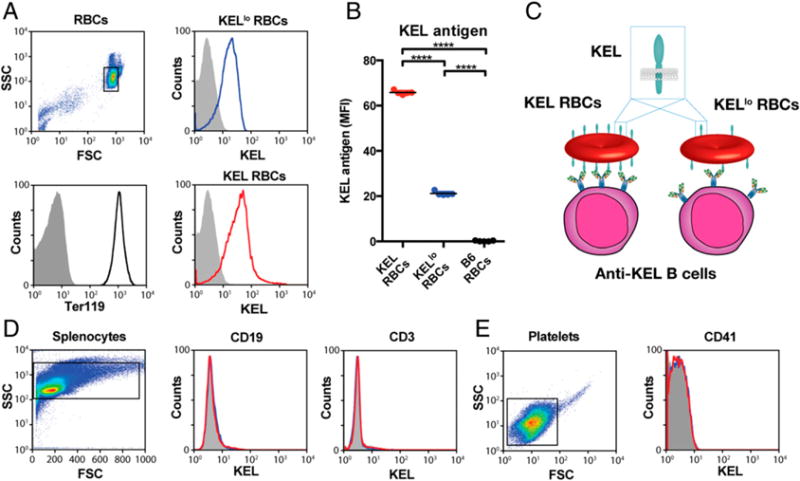
Different founders stably produce RBCs with distinct levels of the KEL Ag. (A) Flow cytometric examination of KEL Ag expression on KELlo (blue) and KEL (red) RBCs. (B) Quantitative analysis of KEL Ag expression on KEL, KELlo, and C57BL/6 (B6) KEL− RBCs. (C) Schematic of potential differences in KEL and KELlo RBC interactions with the immune system. Analysis of KEL Ag expression on CD3+ T cells and CD19+ B cells (D) and CD41+ platelets (E) from KELlo (blue) and KEL (red) RBCs. Error bars indicate SEM. ****p < 0.0001, one-way ANOVA.
To directly examine the impact of transfusing RBCs expressing KEL at different levels, C57BL/6 recipients with RBCs negative for the KEL Ag were transfused with KEL or KELlo RBCs. Recipients receiving KEL RBCs generated robust IgM anti-KEL Abs, followed by the formation of IgG anti-KEL (Fig. 2A, 2B). In contrast, following exposure to KELlo RBCs, recipients produced significantly reduced IgM or IgG anti-KEL (Fig. 2C, 2D). To determine whether the inability of KELlo RBCs to induce significant anti-KEL Abs reflects inadequate exposure, we next retransfused the same recipients with additional KEL or KELlo RBCs. Although multiple exposures to KEL RBCs resulted in an increase in anti-KEL Ab formation (Fig. 2E), repeat exposure to KELlo RBCs failed to induce a detectable increase in anti-KEL Abs (Fig. 2F), suggesting that exposure to RBCs with lower levels of the same Ag can significantly impact immune responsiveness following RBC transfusion.
FIGURE 2.
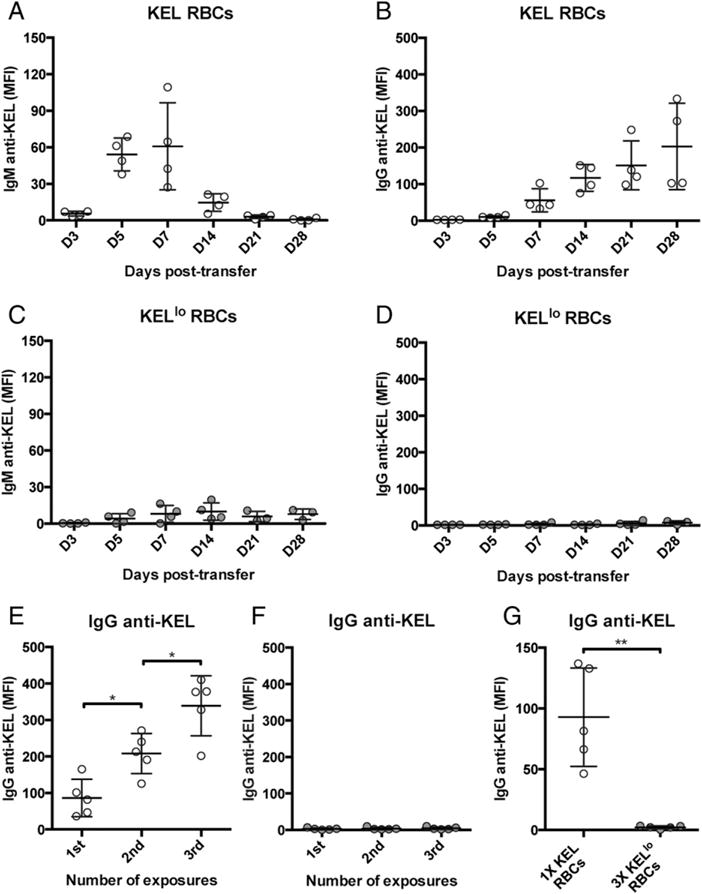
KELlo RBCs fail to induce detectable anti-KEL Abs. C57BL/6 recipients negative for the KEL Ag were transfused with KEL (A and B) or KELlo (C and D) RBCs, followed by serological detection for IgM anti-KEL or IgG anti-KEL at the time points indicated. C57BL/6 KEL− recipients were exposed to one, two, or three doses of KEL (E) or KELlo (F) RBCs, followed by examination of IgG anti-KEL Ab formation at day 14 posttransfer. (G) C57BL/6 KEL− recipients were exposed to a normal dose of KEL RBCs or three times the normal dose of KELlo RBCs, followed by evaluation of IgG anti-KEL Abs at day 14 posttransfer. Error bars indicate SEM. *p < 0.05, **p < 0.01, one-way ANOVA.
Although KEL RBCs possess the ability to readily induce anti-KEL Abs in the absence of known innate immune ligands, Ag exposure in the absence of TLR engagement can occasionally tolerize recipients to exposed Ag or otherwise prevent the development of an adequate immune response (36). To determine whether coadministration of adjuvant alters recipient immune responsiveness to KELlo RBCs, we pretreated KEL or KELlo RBC–transfused recipients with the TLR3 agonist PIC (37). Although PIC treatment enhanced the immune response to KEL RBCs, recipient priming with PIC failed to result in increased anti-KEL Ab formation following KELlo RBC exposure (Supplemental Fig. 1A, 1B). Because Ab formation may require an initial threshold of total Ag exposure, regardless of density, we next increased the dose of KELlo RBCs; however, escalating the dose of KELlo RBCs to triple that of KEL RBCs still failed to induce detectable anti-KEL Abs (Fig. 2G).
To determine whether the inability of higher doses of KELlo RBCs to induce anti-KEL Ab formation reflected the lack of an immune priming event, we similarly exposed recipients to PIC prior to higher-dose KELlo RBC transfusion. However, similar to repeat exposure to KELlo RBCs, high-dose KELlo RBCs also failed to induce detectable anti-KEL Abs, even in the presence of PIC (Supplemental Fig. 1C). Furthermore, the inability of KELlo RBCs to induce anti-KEL Abs did not appear to reflect any difference in KELlo RBC survival, because KELlo RBCs failed to be engaged by anti-KEL Abs or complement or to display any alteration in RBC clearance (Supplemental Fig. 2). In contrast, IgM, IgG, and complement were readily detected on the surface of KEL RBCs, reflecting parallel development of anti-KEL Abs and alterations in RBC survival over time (Fig. 2A, 2B; Supplemental Fig. 2). Finally, the reduced capacity of KELlo RBCs to induce anti-KEL Ab likewise did not appear to reflect instability of the KEL Ag on these cells, because no change in KEL Ag levels could be detected following transfusion over time (Supplemental Fig. 2G–I). Because KELlo RBCs failed to induce significant anti-KEL Abs, regardless of dose, number of exposures, or recipient inflammation, these results suggest that Ag density can dictate immune responsiveness following RBC transfusion.
The impact of KEL Ag density on immune responsiveness following RBC transfusion suggests that the level of KEL on KELlo RBCs may simply be below the threshold required for a productive B cell response. However, to determine whether KELlo RBC exposure impacts B cell responsiveness to the KEL Ag, we simply asked whether exposure to KELlo RBCs primes recipients for subsequent KEL RBC–induced anti-KEL Ab formation. To examine this, we challenged naive or KELlo RBC-exposed recipients with KEL RBCs. Although exposure to KEL RBCs produced anti-KEL Abs, recipients previously exposed to KELlo RBCs unexpectedly failed to generate significant anti-KEL Abs (Fig. 3A). These results suggest that, although KELlo RBCs may not possess the ability to induce significant anti-KEL Abs, KELlo RBC exposure is not without consequence, because recipients exposed to KELlo RBCs appear to be uniquely tolerized to the KEL Ag.
FIGURE 3.
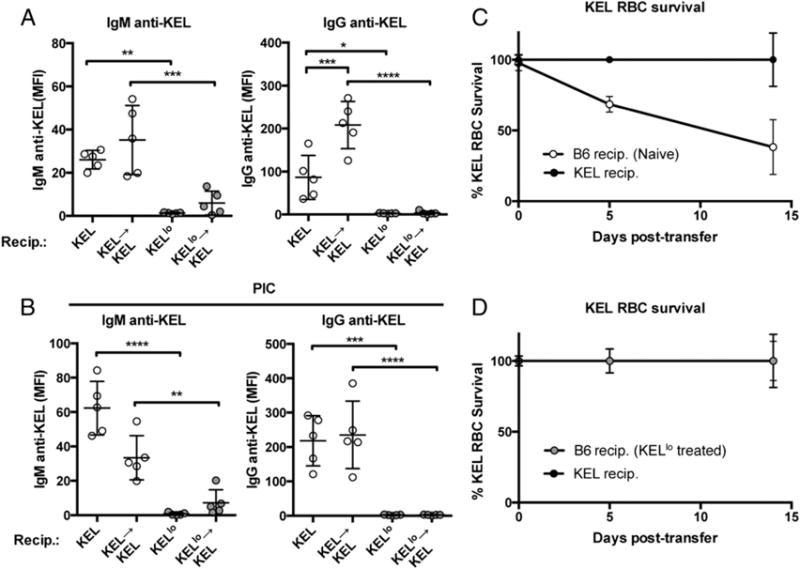
KELlo RBC exposure prevents KEL RBC–induced Ab formation. (A) C57BL/6 KEL− recipients were exposed to KEL or KELlo RBCs in the presence or absence of a secondary KEL RBC exposure, as indicated, followed by evaluation of anti-KEL Abs at day 5 (IgM) and day 14 (IgG) posttransfer. (B) C57BL/6 KEL− recipients pretreated with PIC were exposed to KEL or KELlo RBCs in the presence or absence of a secondary KEL RBC exposure as indicated, followed by evaluation of anti-KEL Abs at day 5 (IgM) and day 14 (IgG) posttransfer. Evaluation of KEL RBC survival following transfer into C57BL/6 KEL− recipients (B6 recip.) or KEL+ recipients (KEL recip.) with no previous KEL exposure (C) or previous exposure to KELlo RBCs (D) (n = 5). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, one-way ANOVA.
Because innate immune activation can occasionally break tolerance or otherwise sensitize recipients to generate an immune response following Ag exposure (38, 39), we next determined whether pretreatment with PIC could resensitize recipients transfused with KELlo RBCs to subsequent challenge with KEL RBCs. Although pretreatment with PIC enhanced naive recipient sensitivity to KEL RBCs (Supplemental Fig. 1), recipients exposed to KELlo RBCs following PIC pretreatment failed to generate a significant anti-KEL Ab response upon KEL RBC challenge (Fig. 3B). Given the ability of KELlo RBCs to prevent anti-KEL Ab formation following KEL RBC exposure, we next determined whether KELlo RBC pre-exposure protects KEL RBCs from immune-mediated clearance. Consistent with the lack of detectable anti-KEL Abs in KELlo RBC–transfused recipients, exposure to KELlo RBCs completely protected KEL RBCs from the accelerated clearance observed following transfusion into naive recipients (Fig. 3C, 3D). Taken together, these results suggest that simple exposure to KELlo RBCs possesses the unique capacity to induce immunological tolerance to KEL.
Given the unexpected ability of KELlo RBC exposure to render recipients unresponsive to KEL, we next sought to determine whether KELlo RBC–induced tolerance reflected an Ag-specific phenomenon or KELlo RBC exposure nonspecifically suppresses immune responses to other Ags. To accomplish this, we exposed naive or KELlo RBC–treated recipients to HOD RBCs, which express the model HEL, OVA, and Duffy chimeric Ag specifically on RBCs (21). Because HOD is unrelated to KEL, this provides a direct ability to determine whether KELlo RBCs suppress immune responsiveness toward a completely different Ag presented to the immune system in a similar fashion. Importantly, exposure of naive or KELlo RBC–treated recipients to HOD RBCs resulted in identical anti-HOD Ab formation (Fig. 4A–D), suggesting that KELlo RBC transfusion specifically suppresses immune responsiveness to the KEL Ag. Occasionally, generation of an immune response to one Ag can break tolerance to another Ag if that Ag is in close proximity during an immune response (40). To determine whether KELlo RBC–induced Ag-specific tolerance could be maintained following challenge with RBCs expressing KEL and HOD Ags, we crossed KEL and HOD founders to generate RBCs that express both model Ags (KEL × HOD RBCs). Exposure of naive recipients to KEL × HOD RBCs resulted in the generation of anti-KEL and anti-HOD Abs (Fig. 4E–H). In contrast, exposure of KELlo RBC–transfused recipients to KEL × HOD RBCs resulted only in significant anti-HOD Abs (Fig. 4E–H). Taken together, these results demonstrate that KELlo RBC exposure possesses the unique ability to induce Ag-specific tolerance.
FIGURE 4.
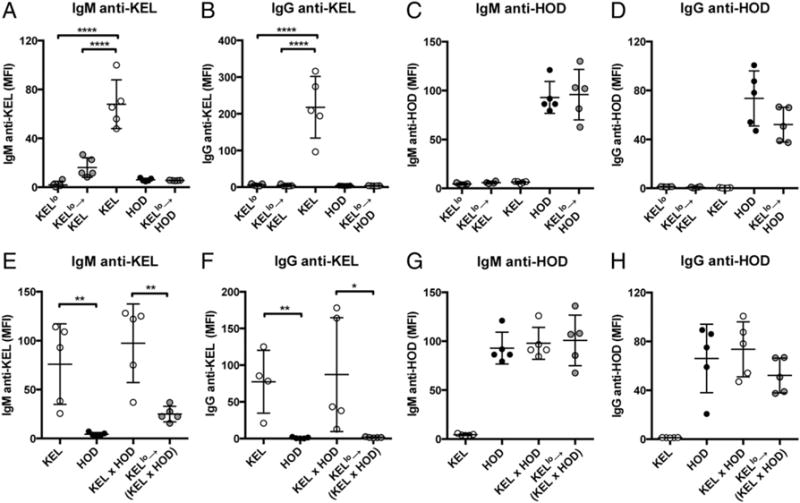
KELlo RBC exposure specifically tolerizes recipients to the KEL Ag. C57BL/6 KEL− recipients were exposed to KELlo, KEL, or HOD RBCs alone or to KELlo RBCs, followed by KEL or HOD RBCs, as indicated. IgM anti-KEL (A), IgG anti-KEL (B), IgM anti-HOD (C), or IgG anti-HOD (D) Ab formation was evaluated 5 d (IgM) or 14 d (IgG) posttransfusion. C57BL/6 KEL− recipients were exposed to KEL RBCs, HOD RBCs, RBCs expressing KEL and HOD Ags (KEL × HOD), or KELlo RBCs, followed by a KEL × HOD RBC challenge. IgM anti-KEL (E), IgG anti-KEL (F), IgM anti-HOD (G), or IgG anti-HOD (H) Ab formation was evaluated 5 d (IgM) or 14 d (IgG) days posttransfusion. Error bars indicate SEM. *p < 0.05, **p < 0.01, ****p < 0.0001, one-way ANOVA.
To explore the mechanism by which KELlo RBCs induce nonresponsiveness to KEL RBC–induced Ab formation, we first sought to determine whether KELlo RBC treatment induces any detectable changes in common immunosuppressive populations. To accomplish this, we exposed recipients to KELlo RBCs, followed by enumeration of Tregs, including CD4+ CD25+ Foxp3+ Tregs (41, 42). Transfusion of KELlo RBCs failed to result in any detectable alterations in Treg numbers (Fig. 5A, 5B). Because KELlo RBC treatment may prime Tregs to expand following subsequent KEL RBC exposure, we next tested whether any alterations in Tregs could be detected following KEL RBC challenge of KELlo RBC–transfused recipients; however, no quantitative alterations were observed in Treg populations of KELlo RBC–transfused recipients, even following subsequent KEL RBC exposure (Fig. 5A, 5B). Likewise, exposure to KELlo RBCs did not change the effector profile of Tregs (Fig. 5C, Supplemental Fig. 3), and depletion of CD4+ CD25+ Tregs following KELlo RBC transfusion failed to resensitize recipients to KEL RBC–induced Ab formation (Fig. 5D–F). Finally, because previous studies demonstrated that transfer of T cells from a tolerized recipient into naive recipients can convey Ag-specific tolerance when immunosuppressive populations play a critical role (43, 44), we transferred CD4+ T cells isolated from KELlo RBC–transfused recipients into naive recipients, as done previously (27), followed by KEL RBC challenge. However, transfer of CD4+ T cells from KELlo-treated recipients failed to yield any detectable alterations in anti-KEL Ab formation following KEL RBC exposure (Fig. 5G).
FIGURE 5.
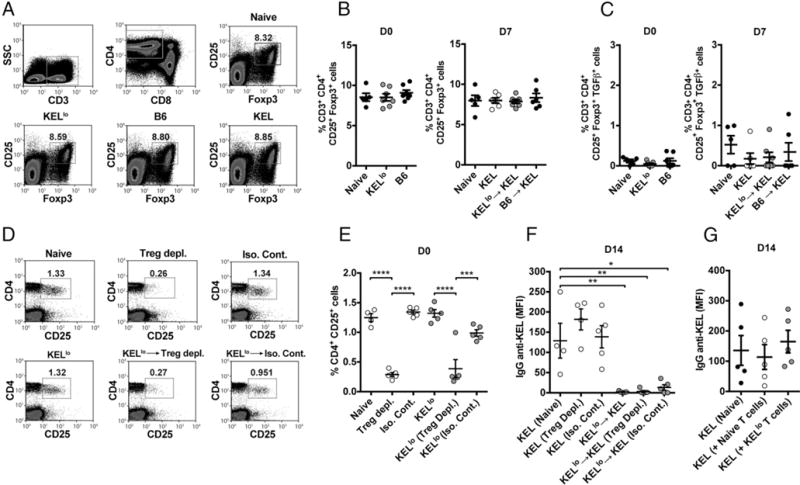
KELlo RBC exposure fails to induce detectable changes in Treg numbers or function. (A) Representative flow plots of CD25+ Foxp3+ Tregs following C57BL/6 KEL− recipient exposure to PBS (Naive), C57BL/6 (B6), KELlo, or KEL RBCs. Quantitative analysis of CD25+ Foxp3+ Tregs (B) or quantitation of TGF-β–producing CD25+ Foxp3+ Tregs (C) following exposure to C57BL/6 (B6) or KELlo RBCs prior to (D0, day 0) or following (D7, day 7 following transfer) a KEL RBC challenge. A PBS-treated control group, not exposed to KEL (Naive), was also evaluated at each time point. At D7, a group transfused with KEL only group (KEL) was included to control for endogenous Treg response to KEL RBC exposure. (D) Representative flow cytometric examination of CD4+ CD25+ T cells following injection of an anti-CD25 depleting or isotype-control Ab. (E) Quantitative analysis of CD4+ CD25+ T cells following depletion (Treg depl.) or isotype control (Iso. Cont.) injection, as indicated. (F) Examination of IgG anti-KEL Ab formation following KEL RBC transfusion (KEL) into untreated (Naive), CD4+ CD25+ T cell–depleted (Treg Depl.), or isotype control (Iso. Cont.)-treated recipients or recipients that were exposed to KELlo RBCs prior to depletion of CD4+ CD25+ T cell depletion (Treg Depl.) or isotype control (Iso. Cont.) treatment and then challenged with KEL RBCs (KELlo→KEL). (G) Evaluation of anti-KEL IgG Ab formation following KEL RBC challenge in recipients pretransfused with PBS (Naive) or adoptively transferred with 107 CD4+ T cells from C57BL/6 naive (Naive T cells) or KELlo RBC–treated (KELlo T cells) recipients. Error bars indicate SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, one-way ANOVA.
Although Tregs did not appear to play a role in the humoral nonresponsiveness of KELlo RBC–transfused recipients, we next sought to evaluate the potential impact of KELlo RBC exposure on B220+ CD5+ CD1dhi IL10+ Bregs. Similar to Tregs, no change in the number or function of Bregs could be detected following initial KELlo RBC transfusion or subsequent KEL RBC challenge of KELlo RBC–exposed recipients (Fig. 6A–C). Likewise, transfer of B cells isolated from KELlo RBC–transfused recipients into naive recipients, as done previously (28–30), failed to cause any detectable alterations in anti-KEL Ab formation following KEL RBC exposure (Fig. 6D). Taken together, these results suggest that KELlo RBC–induced tolerance does not reflect the induction of immunosuppressive populations that actively inhibit anti-KEL Ab formation following KEL RBC exposure.
FIGURE 6.
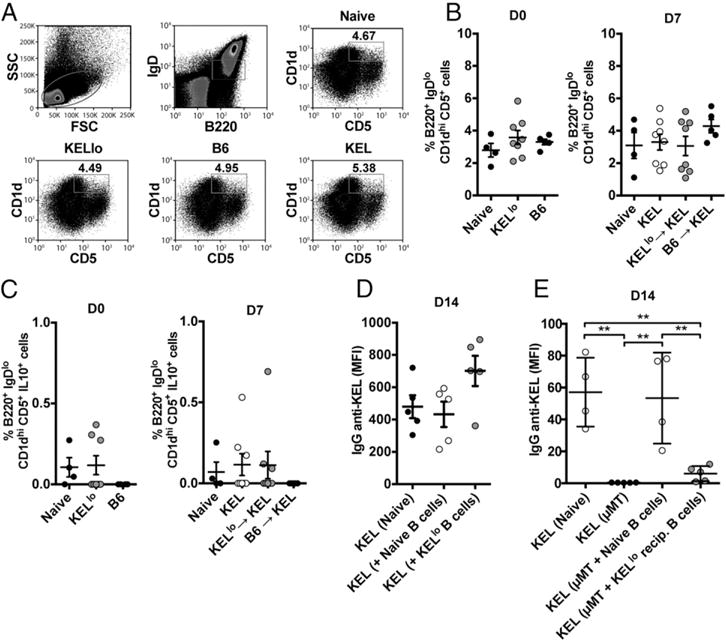
KELlo RBC exposure fails to induce detectable changes in Breg number or function, but it renders B cells unresponsive to KEL RBC challenge. (A) Representative flow cytometric examination of the percentage of CD5+ CD1dhi Bregs following C57BL/6 KEL− recipient exposure to PBS, C57BL/6 (B6), KELlo, or KEL RBCs. Quantitative analysis of CD5+ CD1dhi Bregs (B) or quantitation of IL-10–producing CD5+ CD1dhi Bregs (C) following exposure to B6 or KELlo RBCs prior to (D0, day 0) or following (D7, day 7 following transfer) a KEL RBC challenge. A PBS-treated control group, not exposed to KEL (Naive), was also evaluated at each time point. At D7, a group transfused with KEL only (KEL) was included to control for endogenous Breg response to KEL RBC exposure. (D) Evaluation of IgG anti-KEL Ab formation in KEL RBC–challenged recipients injected with PBS (Naive) or adoptively transferred with 107 CD19+ B cells from C57BL/6 naive (Naive B cells) or KELlo RBC–treated (KELlo B cells) recipients. (E) Examination of anti-KEL Ab production following KEL RBC transfusion into C57BL/6 KEL− recipients (Naive), μMT recipients (μMT), or μMT recipients adoptively transferred with 5 × 107 B cells from C57BL/6 naive (μMT + Naive B cells) recipients or 5 × 107 cells from KELlo-treated recipients (μMT + KELlo recip. B cells). Error bars indicate SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, one-way ANOVA.
Given the lack of apparent involvement of immunosuppressive T or B cell populations in KELlo RBC–induced tolerance, the ability of KELlo RBCs to induce an Ag-specific state of immunological unresponsiveness may reflect direct alterations in the activity of anti-KEL B cell clones. Indeed, although altered KEL Ag density may impact a variety of distinct immune populations, alterations in KEL Ag presentation on the RBC surface would be anticipated to most directly impact BCR engagement on the surface of B cells, which would be predicted to impact B cell function in a clone-specific fashion (45, 46). Certainly, BCR ligation would be anticipated to be the most sensitive immune detection event of altered surface Ag (45). Consistent with this, although KELlo RBC exposure rendered recipients nonresponsive to KEL, KELlo RBCs failed to impact the immune response to the unrelated HOD Ag, even when expressed on the same RBC (Fig. 4), suggesting a clone-specific phenomenon and providing further evidence against a role for Tregs or Bregs in the observed nonresponsiveness to KEL. Because the KEL and KELlo RBCs represent a new and novel system to study the impact of Ag density on immune responsiveness, typical BCR-transgenic tools to study the consequence of KEL or KELlo RBC exposure on specific B cell clone function do not exist. Therefore, to test whether differential engagement of KEL-reactive B cells by KELlo RBCs induces a state of nonresponsiveness toward the KEL Ag, we instead transferred B cells isolated from naive or KELlo RBC–transfused recipients into B cell–deficient μMT recipients, as done previously (25, 28). Exposure of μMT recipients to KEL RBCs that received B cells from naive recipients displayed significant anti-KEL Ab formation, similar to that observed in intact C57BL/6 recipients (Fig. 6E). However, μMT recipients that received B cells from KELlo RBC–exposed recipients failed to generate the same level of detectable anti-KEL Abs when evaluated in parallel (Fig. 6E). Failure to induce anti-KEL Abs in this setting did not appear to reflect B cell–mediated suppression, because μMT recipients that received a cotransfer of B cells from naive and KELlo RBC–exposed recipients generated similar levels of anti-KEL Abs (Supplemental Fig. 4). Taken together, these results demonstrate that KELlo RBC transfusion induces a state of Ag-specific tolerance, likely through the induction of B cell clone unresponsiveness.
Discussion
Our results demonstrate the effect of Ag density on immune outcomes following RBC transfusion. Although various factors likely dictate immune responsiveness following RBC transfusion, these data suggest that transfusion of RBCs expressing low Ag levels may not only fail to induce alloantibodies, but can actually render recipients unresponsive following additional challenge with RBCs expressing the same Ag at higher levels. This decreased Ab response is consistent with previous studies, suggesting that Ag density can impact the likelihood of Ab formation against RBC alloantigens, such as RhD (11, 12, 17). However, the ability of exposure to RBCs bearing lower Ag density to influence subsequent responsiveness to the same Ag presented at higher levels represents an unexpected finding and suggests that exposure to RBCs expressing varied levels of Ag may influence initial responsiveness, as well as subsequent tolerance. Thus, although a variety of factors, including recipient inflammation and MHC haplotype, may impact the likelihood of developing an alloantibody response following transfusion (9, 47–51), alloantigen density appears to be an independent and potent regulator of immunological responsiveness to RBC alloantigens.
The ability of KELlo RBCs to induce Ag-specific tolerance suggests that the temporal relationship between exposures to RBCs bearing distinct densities of an alloantigen may influence the likelihood that an individual develops an immune response following RBC transfusion. In addition, these results may provide insight into the role of pretransplant transfusions in either sensitizing or desensitizing transplant recipients. Conflicting reports of beneficial or detrimental outcomes related to transfusing patients prior to transplantation have contributed to long-standing controversy regarding the best medical practices (52); however, the factors that influence differential immune outcomes following transfusion remain unclear. Our results suggest that variation in Ag density may play an important role in influencing the likelihood of allosensitization and allograft survival in these settings, thus providing possible insight into different outcomes of transfusion on transplantation.
Although the impact of low alloantigen–density RBC exposure on subsequent immune responsiveness can be clearly observed in the KEL model system, it remains unknown whether similar alterations in response occur following exposure to other RBC alloantigens at low density. Indeed, given the structural complexity and distinct nature of different RBC alloantigens, each alloantigen may possess a unique threshold that dictates immune responsiveness following alloantigen exposure. Consistent with this, although RBCs expressing low levels of RhD (weak RhD) possess a reduced ability to induce anti-RhD alloantibodies, cases of anti-RhD Ab formation following exposure to RBCs with weak RhD expression were reported (53–55). However, it remains unknown whether weak RhD RBC exposure in individuals who fail to generate an anti-D response impacts the subsequent immune responsiveness to RhD RBCs. Previous studies demonstrated that recipient factors, such as baseline differences in inflammation and immune regulatory populations within the recipient, also impact immune responsiveness to RBC transfusion (9, 56, 57). Additionally, because this model system represents immune responsiveness to the human KEL Ag in nonhumanized C57BL/6 recipients, the xenoantigenic nature of this Ag may influence the formation and regulation of Ab production in this setting. Thus, although Ag density may influence RBC alloimmunization, a variety of donor and recipient factors likely converge to dictate the final immunological outcome of RBC transfusion.
Previous studies demonstrated that altering the affinity of Ag for a BCR directly impacts the likelihood of an Ab response following Ag engagement by impacting the ability of B cells to ligate Ag through Ag-specific BCRs. This results in an impaired ability to effectively propagate the signaling response required for Ag presentation and effective Ab production (58–60). Although the KEL Ag on KELlo and KEL RBCs appears identical, and, therefore, unlikely to possess variations that would intrinsically impact affinity, alterations in density may still impact the overall avidity of this interaction. In this way, different levels of Ag may mimic some of the affinity-based regulation observed previously (58–60). Equally important, the results of the current study demonstrate that a critical threshold can exist to generate an effective Ab response, as well as that alterations in Ag can actually inhibit responsiveness to subsequent challenge with RBCs expressing higher Ag levels. Because KELlo RBC–induced tolerance appears to occur through an Ag-specific induction of B cell unresponsiveness, this form of tolerance may reflect the ability of KELlo RBCs to induce alterations in B cell signaling that induce a state of anergy, cause apoptosis, or render cells unresponsive through some other mechanism (58, 60–63). However, although the data strongly suggest that KELlo RBC exposure impacts KEL-specific B cells directly, it remains possible that Ag engagement and uptake by APCs could also impact the subsequent immune response in such a way as to facilitate KELlo RBC induction of B cell responsiveness. Regardless of the exact mechanism of B cell unresponsiveness, these results demonstrate that simple alterations in Ag density can render a recipient unresponsive to very specific antigenic challenge. Thus, these findings provide useful insight into factors that may regulate immune responsiveness following RBC transfusion.
The unique impact of Ag density on immune responsiveness not only aids in resolving long-standing questions regarding factors that influence alloantibody formation following RBC transfusion, it also suggests that RBCs may be used as novel tools to intentionally induce Ag-specific tolerance. Although RBCs can be genetically modified, these cells uniquely extrude their nucleus, allowing RBCs to retain a desirable genetic imprint, such as altered Ag density, without conveying the same risks associated with directly introducing altered genomes into patients (64–66). Therefore, because RBC transfusion represents one of the oldest forms of cellular therapy (1, 2), genetic modification of RBCs to express altered Ag may be used to favorably manipulate immune responsiveness against specific antigenic determinants. Because current immunosuppressive approaches most often require general immunosuppression, with the accompanying risks for opportunistic infection (67–70), engineering RBCs with altered Ag levels may provide a simple and effective approach for inducing Ag-specific tolerance. Additionally, although our current results demonstrate an ability for low Ag–density RBC exposure to induce B cell unresponsiveness in naive recipients, providing a powerful tool for prophylactic treatment, future studies will investigate whether exposure to RBCs with low Ag levels can also desensitize B cells that were previously activated, which would offer a compelling potential for treatment of devastating immune-mediated conditions, such as graft rejection and active autoimmunity. Thus, understanding fundamental aspects of factors that regulate immune responsiveness to transfusion provides insight into RBC alloimmunization, as well as allows for the development of new avenues of research in the treatment, and even prevention, of a wide variety of challenging immune-mediated conditions (71, 72).
Supplementary Material
Acknowledgments
This work was supported in part by the Burroughs Wellcome Trust Career Award for Medical Scientists and National Institutes of Health Early Independence Grant DP5OD019892 (to S.R.S.).
S.R.S., C.M.A., and S.R.P. designed the research study; C.M.A. and S.R.P. carried out and analyzed experiments together with A.M., N.H.S., A.B., N.A.K., C.G.-S., and H.C.S.; critical support was provided by J.S.H., A.W., B.Y., J.C.Z., and J.E.H.; and C.M.A., S.R.P., and S.R.S. wrote the manuscript, which was edited and commented on by the other authors.
Abbreviations
- Breg
regulatory B cell
- HEL
hen egg lysozyme
- HOD
hen egg lysozyme, OVA fused to Duffy
- KEL
human Kell
- KELlo RBC
RBC expressing low levels of KEL
- KEL RBC
RBC expressing high levels of KEL
- MFI
mean fluorescence intensity
- PIC
polyinosinic-polycytidylic acid
- Treg
regulatory T cell
Footnotes
ORCIDs: 0000-0003-4909-7168 (H.C.S.); 0000-0001-9648-7922 (A.W.).
The online version of this article contains supplemental material.
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Goodnough LT, Brecher ME, Kanter MH, AuBuchon JP. Transfusion medicine. First of two parts–blood transfusion. N Engl J Med. 1999;340:438–447. doi: 10.1056/NEJM199902113400606. [DOI] [PubMed] [Google Scholar]
- 2.Landsteiner K. Zur Kenntnis der antifermativen, lytischen und agglutinierenden Wirkungendes Blutserums und der Lymphe. Zentbl Bakt Parasitkde (Abt) 1900;27:357–363. [Google Scholar]
- 3.Reali G. Forty years of anti-D immunoprophylaxis. Blood Transfus. 2007;5:3–6. doi: 10.2450/2007.0b18-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Evers D, Middelburg RA, de Haas M, Zalpuri S, de Vooght KM, van de Kerkhof D, Visser O, Péquériaux NC, Hudig F, Schonewille H, et al. Red-blood-cell alloimmunisation in relation to antigens’ exposure and their immunogenicity: a cohort study. Lancet Haematol. 2016;3:e284–e292. doi: 10.1016/S2352-3026(16)30019-9. [DOI] [PubMed] [Google Scholar]
- 5.Hillyer CD, Shaz BH, Winkler AM, Reid M. Integrating molecular technologies for red blood cell typing and compatibility testing into blood centers and transfusion services. Transfus Med Rev. 2008;22:117–132. doi: 10.1016/j.tmrv.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 6.U.S. Food and Drug Administration. Fatalities Reported to FDA Following Blood Collection and Transfusion. Annual Summary for Fiscal Year. 2015 2015 Available at: http://www.fda.gov/downloads/BiologicsBloodVaccines/SafetyAvailability/ReportaProblem/TransfusionDonationFatalities/UCM518148.pdf.
- 7.Nickel RS, Hendrickson JE, Fasano RM, Meyer EK, Winkler AM, Yee MM, Lane PA, Jones YA, Pashankar FD, New T, et al. Impact of red blood cell alloimmunization on sickle cell disease mortality: a case series. Transfusion. 2016;56:107–114. doi: 10.1111/trf.13379. [DOI] [PubMed] [Google Scholar]
- 8.Brand A. Immunological complications of blood transfusions. Presse Med. 2016;45:e313–e324. doi: 10.1016/j.lpm.2016.06.024. [DOI] [PubMed] [Google Scholar]
- 9.Higgins JM, Sloan SR. Stochastic modeling of human RBC alloimmunization: evidence for a distinct population of immunologic responders. Blood. 2008;112:2546–2553. doi: 10.1182/blood-2008-03-146415. [DOI] [PubMed] [Google Scholar]
- 10.Yazdanbakhsh K, Ware RE, Noizat-Pirenne F. Red blood cell alloimmunization in sickle cell disease: pathophysiology, risk factors, and transfusion management. Blood. 2012;120:528–537. doi: 10.1182/blood-2011-11-327361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bussel JB, Berkowitz RL, McFarland JG, Lynch L, Chitkara U. Antenatal treatment of neonatal alloimmune thrombocytopenia. N Engl J Med. 1988;319:1374–1378. doi: 10.1056/NEJM198811243192103. [DOI] [PubMed] [Google Scholar]
- 12.Chou ST, Jackson T, Vege S, Smith-Whitley K, Friedman DF, Westhoff CM. High prevalence of red blood cell alloimmunization in sickle cell disease despite transfusion from Rh-matched minority donors. Blood. 2013;122:1062–1071. doi: 10.1182/blood-2013-03-490623. [DOI] [PubMed] [Google Scholar]
- 13.Reid ME, Mohandas N. Red blood cell blood group antigens: structure and function. Semin Hematol. 2004;41:93–117. doi: 10.1053/j.seminhematol.2004.01.001. [DOI] [PubMed] [Google Scholar]
- 14.Hod EA, Arinsburg SA, Francis RO, Hendrickson JE, Zimring JC, Spitalnik SL. Use of mouse models to study the mechanisms and consequences of RBC clearance. Vox Sang. 2010;99:99–111. doi: 10.1111/j.1423-0410.2010.01327.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ryder AB, Zimring JC, Hendrickson JE. Factors influencing RBC alloimmunization: lessons learned from murine models. Transfus Med Hemother. 2014;41:406–419. doi: 10.1159/000368995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vaughan JI, Manning M, Warwick RM, Letsky EA, Murray NA, Roberts IA. Inhibition of erythroid progenitor cells by anti-Kell antibodies in fetal alloimmune anemia. N Engl J Med. 1998;338:798–803. doi: 10.1056/NEJM199803193381204. [DOI] [PubMed] [Google Scholar]
- 17.Stowell SR, Henry KL, Smith NH, Hudson KE, Halverson GR, Park JC, Bennett AM, Girard-Pierce KR, Arthur CM, Bunting ST, et al. Alloantibodies to a paternally derived RBC KEL antigen lead to hemolytic disease of the fetus/newborn in a murine model. Blood. 2013;122:1494–1504. doi: 10.1182/blood-2013-03-488874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smith NH, Henry KL, Cadwell CM, Bennett A, Hendrickson JE, Frame T, Zimring JC. Generation of transgenic mice with antithetical KEL1 and KEL2 human blood group antigens on red blood cells. Transfusion. 2012;52:2620–2630. doi: 10.1111/j.1537-2995.2012.03641.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Arthur CM, Patel SR, Sullivan HC, Winkler AM, Tormey CA, Hendrickson JE, Stowell SR. CD8+ T cells mediate antibody-independent platelet clearance in mice. Blood. 2016;127:1823–1827. doi: 10.1182/blood-2015-10-673426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wan YY, Flavell RA. Identifying Foxp3-expressing suppressor T cells with a bicistronic reporter. Proc Natl Acad Sci USA. 2005;102:5126–5131. doi: 10.1073/pnas.0501701102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Desmarets M, Cadwell CM, Peterson KR, Neades R, Zimring JC. Minor histocompatibility antigens on transfused leukoreduced units of red blood cells induce bone marrow transplant rejection in a mouse model. Blood. 2009;114:2315–2322. doi: 10.1182/blood-2009-04-214387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Girard-Pierce KR, Stowell SR, Smith NH, Arthur CM, Sullivan HC, Hendrickson JE, Zimring JC. A novel role for C3 in antibody-induced red blood cell clearance and antigen modulation. Blood. 2013;122:1793–1801. doi: 10.1182/blood-2013-06-508952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stowell SR, Liepkalns JS, Hendrickson JE, Girard-Pierce KR, Smith NH, Arthur CM, Zimring JC. Antigen modulation confers protection to red blood cells from antibody through Fcγ receptor ligation. J Immunol. 2013;191:5013–5025. doi: 10.4049/jimmunol.1300885. [DOI] [PubMed] [Google Scholar]
- 24.McHugh RS, Shevach EM. Cutting edge: depletion of CD4+CD25+ regulatory T cells is necessary, but not sufficient, for induction of organ-specific autoimmune disease. J Immunol. 2002;168:5979–5983. doi: 10.4049/jimmunol.168.12.5979. [DOI] [PubMed] [Google Scholar]
- 25.Matsushita T, Horikawa M, Iwata Y, Tedder TF. Regulatory B cells (B10 cells) and regulatory T cells have independent roles in controlling experimental autoimmune encephalomyelitis initiation and late-phase immunopathogenesis. J Immunol. 2010;185:2240–2252. doi: 10.4049/jimmunol.1001307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cabatingan MS, Schmidt MR, Sen R, Woodland RT. Naive B lymphocytes undergo homeostatic proliferation in response to B cell deficit. J Immunol. 2002;169:6795–6805. doi: 10.4049/jimmunol.169.12.6795. [DOI] [PubMed] [Google Scholar]
- 27.Lan YY, Wang Z, Raimondi G, Wu W, Colvin BL, de Creus A, Thomson AW. Alternatively activated” dendritic cells preferentially secrete IL-10, expand Foxp3+CD4+ T cells, and induce long-term organ allograft survival in combination with CTLA4-Ig. J Immunol. 2006;177:5868–5877. doi: 10.4049/jimmunol.177.9.5868. [DOI] [PubMed] [Google Scholar]
- 28.Ren X, Akiyoshi K, Dziennis S, Vandenbark AA, Herson PS, Hurn PD, Offner H. Regulatory B cells limit CNS inflammation and neurologic deficits in murine experimental stroke. J Neurosci. 2011;31:8556–8563. doi: 10.1523/JNEUROSCI.1623-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mizoguchi A, Mizoguchi E, Takedatsu H, Blumberg RS, Bhan AK. Chronic intestinal inflammatory condition generates IL-10-producing regulatory B cell subset characterized by CD1d upregulation. Immunity. 2002;16:219–230. doi: 10.1016/s1074-7613(02)00274-1. [DOI] [PubMed] [Google Scholar]
- 30.Mizoguchi E, Mizoguchi A, Preffer FI, Bhan AK. Regulatory role of mature B cells in a murine model of inflammatory bowel disease. Int Immunol. 2000;12:597–605. doi: 10.1093/intimm/12.5.597. [DOI] [PubMed] [Google Scholar]
- 31.Baumgarth N, Jager GC, Herman OC, Herzenberg LA. CD4+ T cells derived from B cell-deficient mice inhibit the establishment of peripheral B cell pools. Proc Natl Acad Sci USA. 2000;97:4766–4771. doi: 10.1073/pnas.97.9.4766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tedder TF. B10 cells: a functionally defined regulatory B cell subset. J Immunol. 2015;194:1395–1401. doi: 10.4049/jimmunol.1401329. [DOI] [PubMed] [Google Scholar]
- 33.Yanaba K, Bouaziz JD, Haas KM, Poe JC, Fujimoto M, Tedder TF. A regulatory B cell subset with a unique CD1dhiCD5+ phenotype controls T cell-dependent inflammatory responses. Immunity. 2008;28:639–650. doi: 10.1016/j.immuni.2008.03.017. [DOI] [PubMed] [Google Scholar]
- 34.Maynard CL, Harrington LE, Janowski KM, Oliver JR, Zindl CL, Rudensky AY, Weaver CT. Regulatory T cells expressing interleukin 10 develop from Foxp3+ and Foxp3− precursor cells in the absence of interleukin 10. Nat Immunol. 2007;8:931–941. doi: 10.1038/ni1504. [DOI] [PubMed] [Google Scholar]
- 35.Vieira PL, Christensen JR, Minaee S, O’Neill EJ, Barrat FJ, Boonstra A, Barthlott T, Stockinger B, Wraith DC, O’Garra A. IL-10-secreting regulatory T cells do not express Foxp3 but have comparable regulatory function to naturally occurring CD4+CD25+ regulatory T cells. J Immunol. 2004;172:5986–5993. doi: 10.4049/jimmunol.172.10.5986. [DOI] [PubMed] [Google Scholar]
- 36.Smith NH, Hod EA, Spitalnik SL, Zimring JC, Hendrickson JE. Transfusion in the absence of inflammation induces antigen-specific tolerance to murine RBCs. Blood. 2012;119:1566–1569. doi: 10.1182/blood-2011-09-382655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 2001;413:732–738. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- 38.Hendrickson JE, Desmarets M, Deshpande SS, Chadwick TE, Hillyer CD, Roback JD, Zimring JC. Recipient inflammation affects the frequency and magnitude of immunization to transfused red blood cells. Transfusion. 2006;46:1526–1536. doi: 10.1111/j.1537-2995.2006.00946.x. [DOI] [PubMed] [Google Scholar]
- 39.Rawlings DJ, Schwartz MA, Jackson SW, Meyer-Bahlburg A. Integration of B cell responses through Toll-like receptors and antigen receptors. Nat Rev Immunol. 2012;12:282–294. doi: 10.1038/nri3190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vanderlugt CL, Miller SD. Epitope spreading in immune-mediated diseases: implications for immunotherapy. Nat Rev Immunol. 2002;2:85–95. doi: 10.1038/nri724. [DOI] [PubMed] [Google Scholar]
- 41.Mauri C, Bosma A. Immune regulatory function of B cells. Annu Rev Immunol. 2012;30:221–241. doi: 10.1146/annurev-immunol-020711-074934. [DOI] [PubMed] [Google Scholar]
- 42.Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299:1057–1061. doi: 10.1126/science.1079490. [DOI] [PubMed] [Google Scholar]
- 43.Mauri C, Gray D, Mushtaq N, Londei M. Prevention of arthritis by interleukin 10-producing B cells. J Exp Med. 2003;197:489–501. doi: 10.1084/jem.20021293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Toscano MA, Commodaro AG, Ilarregui JM, Bianco GA, Liberman A, Serra HM, Hirabayashi J, Rizzo LV, Rabinovich GA. Galectin-1 suppresses autoimmune retinal disease by promoting concomitant Th2- and T regulatory-mediated anti-inflammatory responses. J Immunol. 2006;176:6323–6332. doi: 10.4049/jimmunol.176.10.6323. [DOI] [PubMed] [Google Scholar]
- 45.Gauld SB, Dal Porto JM, Cambier JC. B cell antigen receptor signaling: roles in cell development and disease. Science. 2002;296:1641–1642. doi: 10.1126/science.1071546. [DOI] [PubMed] [Google Scholar]
- 46.Brinc D, Le-Tien H, Crow AR, Siragam V, Freedman J, Lazarus AH. Transfusion of IgG-opsonized foreign red blood cells mediates reduction of antigen-specific B cell priming in a murine model. J Immunol. 2008;181:948–953. doi: 10.4049/jimmunol.181.2.948. [DOI] [PubMed] [Google Scholar]
- 47.Noizat-Pirenne F, Tournamille C, Bierling P, Roudot-Thoraval F, Le Pennec PY, Rouger P, Ansart-Pirenne H. Relative immunogenicity of Fya and K antigens in a Caucasian population, based on HLA class II restriction analysis. Transfusion. 2006;46:1328–1333. doi: 10.1111/j.1537-2995.2006.00900.x. [DOI] [PubMed] [Google Scholar]
- 48.Reviron D, Dettori I, Ferrera V, Legrand D, Touinssi M, Mercier P, de Micco P, Chiaroni J. HLA-DRB1 alleles and Jk(a) immunization. Transfusion. 2005;45:956–959. doi: 10.1111/j.1537-2995.2005.04366.x. [DOI] [PubMed] [Google Scholar]
- 49.Brantley SG, Ramsey G. Red cell alloimmunization in multi-transfused HLA-typed patients. Transfusion. 1988;28:463–466. doi: 10.1046/j.1537-2995.1988.28588337338.x. [DOI] [PubMed] [Google Scholar]
- 50.Fasano RM, Booth GS, Miles M, Du L, Koyama T, Meier ER, Luban NL. Red blood cell alloimmunization is influenced by recipient inflammatory state at time of transfusion in patients with sickle cell disease. Br J Haematol. 2015;168:291–300. doi: 10.1111/bjh.13123. [DOI] [PubMed] [Google Scholar]
- 51.Bernardo L, Denomme GA, Shah K, Lazarus AH. RhD specific antibodies are not detectable in HLA-DRB1(*)1501 mice challenged with human RhD positive erythrocytes. Adv Hematol. 2014;2014:470242. doi: 10.1155/2014/470242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Scornik JC, Bromberg JS, Norman DJ, Bhanderi M, Gitlin M, Petersen J. An update on the impact of pre-transplant transfusions and allosensitization on time to renal transplant and on allograft survival. BMC Nephrol. 2013;14:217. doi: 10.1186/1471-2369-14-217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Daniels G. Variants of RhD–current testing and clinical consequences. Br J Haematol. 2013;161:461–470. doi: 10.1111/bjh.12275. [DOI] [PubMed] [Google Scholar]
- 54.Flegel WA, Khull SR, Wagner FF. Primary anti-D immunization by weak D type 2 RBCs. Transfusion. 2000;40:428–434. doi: 10.1046/j.1537-2995.2000.40040428.x. [DOI] [PubMed] [Google Scholar]
- 55.Gassner C, Doescher A, Drnovsek TD, Rozman P, Eicher NI, Legler TJ, Lukin S, Garritsen H, Kleinrath T, Egger B, et al. Presence of RHD in serologically D−, C/E+ individuals: a European multicenter study. Transfusion. 2005;45:527–538. doi: 10.1111/j.0041-1132.2004.04211.x. [DOI] [PubMed] [Google Scholar]
- 56.Rosse WF, Gallagher D, Kinney TR, Castro O, Dosik H, Moohr J, Wang W, Levy PS, The Cooperative Study of Sickle Cell Disease Transfusion and alloimmunization in sickle cell disease. Blood. 1990;76:1431–1437. [PubMed] [Google Scholar]
- 57.Seyfried H, Walewska I. Analysis of immune response to red blood cell antigens in multitransfused patients with different diseases. Mater Med Pol. 1990;22:21–25. [PubMed] [Google Scholar]
- 58.Bachmann MF, Rohrer UH, Küundig TM, Büurki K, Hengartner H, Zinkernagel RM. The influence of antigen organization on B cell responsiveness. Science. 1993;262:1448–1451. doi: 10.1126/science.8248784. [DOI] [PubMed] [Google Scholar]
- 59.Batista FD, Neuberger MS. Affinity dependence of the B cell response to antigen: a threshold, a ceiling, and the importance of off-rate. Immunity. 1998;8:751–759. doi: 10.1016/s1074-7613(00)80580-4. [DOI] [PubMed] [Google Scholar]
- 60.Goodnow CC. Balancing immunity and tolerance: deleting and tuning lymphocyte repertoires. Proc Natl Acad Sci USA. 1996;93:2264–2271. doi: 10.1073/pnas.93.6.2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Donjerković D, Scott DW. Activation-induced cell death in B lymphocytes. Cell Res. 2000;10:179–192. doi: 10.1038/sj.cr.7290047. [DOI] [PubMed] [Google Scholar]
- 62.Yarkoni Y, Getahun A, Cambier JC. Molecular underpinning of B-cell anergy. Immunol Rev. 2010;237:249–263. doi: 10.1111/j.1600-065X.2010.00936.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Russell DM, Dembić Z, Morahan G, Miller JF, Büurki K, Nemazee D. Peripheral deletion of self-reactive B cells. Nature. 1991;354:308–311. doi: 10.1038/354308a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ji P, Murata-Hori M, Lodish HF. Formation of mammalian erythrocytes: chromatin condensation and enucleation. Trends Cell Biol. 2011;21:409–415. doi: 10.1016/j.tcb.2011.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Muir AR, Kerr DN. Erythropoiesis: an electron microscopical study. Q J Exp Physiol Cogn Med Sci. 1958;43:106–114. doi: 10.1113/expphysiol.1958.sp001295. [DOI] [PubMed] [Google Scholar]
- 66.Steck TL. The organization of proteins in the human red blood cell membrane. A review. J Cell Biol. 1974;62:1–19. doi: 10.1083/jcb.62.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Moini M, Schilsky ML, Tichy EM. Review on immunosuppression in liver transplantation. World J Hepatol. 2015;7:1355–1368. doi: 10.4254/wjh.v7.i10.1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fishman JA. Infection in solid-organ transplant recipients. N Engl J Med. 2007;357:2601–2614. doi: 10.1056/NEJMra064928. [DOI] [PubMed] [Google Scholar]
- 69.Kunisaki KM, Janoff EN. Influenza in immunosuppressed populations: a review of infection frequency, morbidity, mortality, and vaccine responses. Lancet Infect Dis. 2009;9:493–504. doi: 10.1016/S1473-3099(09)70175-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kang I, Park SH. Infectious complications in SLE after immunosuppressive therapies. Curr Opin Rheumatol. 2003;15:528–534. doi: 10.1097/00002281-200309000-00002. [DOI] [PubMed] [Google Scholar]
- 71.Villanueva C, Colomo A, Bosch A, Concepción M, Hernandez-Gea V, Aracil C, Graupera I, Poca M, Alvarez-Urturi C, Gordillo J, et al. Transfusion strategies for acute upper gastrointestinal bleeding. N Engl J Med. 2013;368:11–21. doi: 10.1056/NEJMoa1211801. [DOI] [PubMed] [Google Scholar]
- 72.Lacroix J, Hébert PC, Fergusson DA, Tinmouth A, Cook DJ, Marshall JC, Clayton L, McIntyre L, Callum J, Turgeon AF, et al. ABLE Investigators; Canadian Critical Care Trials Group Age of transfused blood in critically ill adults. N Engl J Med. 2015;372:1410–1418. doi: 10.1056/NEJMoa1500704. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.