

Abstract
Oxidation of DNA by reactive oxygen species (ROS) yields 8-oxo-7,8-dihydroguanosine (8-oxodG) as primary oxidation product, which can lead to downstream G to T transversion mutations. DNA mutations are nonrandom, and mutations at specific codons are associated with specific cancers, as widely documented for the p53 tumor suppressor gene. Here, we present the first direct LC-MS/MS study (without isotopic labeling or hydrolysis) of primary oxidation sites of p53 exon 7. We oxidized a 32 base pair (bp) double-stranded (ds) oligonucleotide representing exon 7 of the p53 gene. Oxidized oligonucleotides were cut by a restriction endonuclease to provide small strands and enable positions and amounts of 8-oxodG to be determined directly by LC-MS/MS. Oxidation sites on the oligonucleotide generated by two oxidants, catechol/Cu2+/NADPH and Fenton’s reagent, were located and compared. Guanines in codons 243, 244, 245, and 248 were most frequently oxidized by catechol/Cu2+/NADPH with relative oxidation of 5.6, 7.2, 2.6, and 10.7%, respectively. Fenton’s reagent oxidations were more specific for guanines in codons 243 (20.3%) and 248 (10.4%). Modeling of docking of oxidizing species on the ds-oligonucleotide were consistent with the experimental codon oxidation sites. Significantly, codons 244 and 248 are mutational “hotspots” in nonsmall cell and small cell lung cancers, supporting a possible role of oxidation in p53 mutations leading to lung cancer.
Graphical abstract
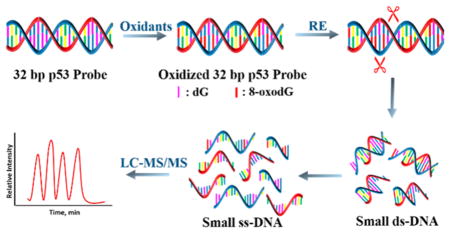
Reactive oxygen species (ROS) such as hydroxyl radical (•OH), singlet oxygen (1O2), peroxynitrite (ONOO–), and superoxide (O2•−) play important roles in living organisms, e.g. as messengers in cell signaling cascades. However, they can also induce cell apoptosis during oxidative stress,1 featuring unregulated accumulation of ROS from endogenous or exogenous processes. Oxidative stress likely play roles in aging, cardiovascular disease, Parkinson’s disease, and cancer.2–6
One source of ROS is oxidation of transition metals by H2O2 to produce •OH, which is usually called Fenton’s reaction, especially when involving ferrous ions.7–9 Another source of ROS is redox cycling of quinones and other metabolites involving Cu2+ and NADPH.10–14 When chemicals or their metabolites lead to DNA damage, they are referred to as genotoxic. Catechol, a model for quinoid metabolites, undergoes redox cycling in the presence of Cu2+ and NADPH that leads to a two-electron oxidation to o-quinone, which subsequently undergoes two one-electron reductions back to catechol by NADPH. ROS, including H2O2, O2•−, and •OH, are formed in these redox cycles (Scheme 1) and can oxidize DNA in vitro.15,16 Also, polycyclic aromatic hydrocarbon (PAH) o-quinones are implicated as causes of mutations in tumor suppressor genes and are known risk factors for lung cancer.17
Scheme 1.

Proposed Redox Cycling Pathway for DNA Oxidation Induced by Catechol, NADPH, and Cu2+.14
Guanine is the main oxidation target in genes due to having the lowest redox potential of the DNA nucleobases (1.3 V vs NHE).18,19 The primary oxidation product of deoxyguanosine is 8-oxo-7,8-dihydrodeoxyguanosine (8-oxodG), which can lead to G to T transversion mutations during DNA replication.20,21 Cancer may result when such mutations occur at tumor suppressor genes or oncogenes.22,23
Tumor suppressor genes provide cancer protection by coding for proteins that inhibit cell proliferation and tumor development.24 TP53 (or p53) was identified as a tumor suppressor gene in the 1980s.25 It encodes p53 protein that regulates the cell cycle and inhibits tumor growth. Mutations in p53 genes are found in >50% of all human cancers.26–28 Extensive databases document mutations found at specific p53 codons from human tumors and cancer cell lines and show that many mutations are well-correlated with specific cancers. Most p53 mutations occur at exons 5–8.29 For example, highly mutated codons 157, 158, 248, and 249 appear in lung cancers; mutated codons 175, 248, and 273 are found in breast cancer; and mutated codons 175, 248, and 282 occur in liver cancer.30,31 Thus, DNA oxidation at specific sites that lead to mutations at the same sites may be possible to correlate with specific cancers.
Early work in p53 gene oxidation employed gel electrophoresis of DNA fragments. The structure of guanine oxidation products was not addressed, and most studies focused on the resulting mutations. Both guanine residues (AGG → ATG, AGG → AGT) in codon 249 mutated to thymine as a result of Fenton oxidation in human fibroblasts.32 Oxidation of human skin fibroblasts by UVB light (280–320 nm) caused G to T mutations at the third position of codon 249 (AGG → AGT).33 PAH-induced oxidation of p53 has been observed at hot spots associated with lung cancer, including codons 245 (GGC → TGC), 249 (AGG → ATG), and 273 (CGT → CTT).17
Mass spectrometry (MS) is a powerful tool to detect structurally damaged DNA.34 LC-MS/MS approaches have been developed to size and sequence DNA oligonucleotides up to 20 bp.35,36 QTOF-MS/MS was employed to detect sequence and agent specificity on the further oxidation products of 8-oxodG in DNA oligonucleotides.37,38 In that study, synthesized 8-oxodG containing oligonucleotides were oxidized to detect the 8-oxodG derived oxidation products, and standard oligonucleotides were not used. Stable isotope labeling of DNA has been widely used with mass spectrometry to examine the reactive sites and cytosine methylation on the formation of 8-oxodG and downstream oxidation products of reactions with nitrosoperoxycarbonate and riboflavin-mediated photolysis.39 This approach detects the amount of 8-oxodG and other products formed, but isotopic labeling syntheses and enzymatic oligonucleotide digestion are required.
To investigate reactive sites, the positional isomeric modified oligonucleotides must be separated. Harsch et al. reported separation of isomeric benzo[c]phenanthrene diol epoxide adducted HRPT gene sequence by using ammonium acetate as ion-pairing reagent.40 Xiong et al. identified nine isomeric (±)-anti-7r,8t-dihydroxy-9t,10-epoxy-7,8,9,10-tetrahydro-benzo[a]pyrene adducted oligonucleotides using ion pair reagent triethylammonium bicarbonate (TEAB).41 Sharma et al. used TEAB to separate oligonucleotides adducted by N-acetylaminofluorene, N-hydroxy-4-aminobiphenyl, and (±)-anti-benzo[a]pyrene diol epoxide to investigate the site selectivity.42,43 These papers provide guidance for HPLC separations of isomeric oligonucleotide products.
We previously described a restriction enzyme-assisted LC-QTOF MS/MS oligonucleotide sequencing methodology to detect covalent adduction on ds-oligonucleotides longer than 20 base pair (bp).45 We used this approach to evaluate reaction products of oligonucleotides with the diol epoxide metabolite of benzopyrene (B[a]P) and examined the effect of cytosine methylation on adduct reaction kinetics of exon 7 of the p53 gene.46 In the present paper, we tailor this approach to directly detect 8-oxodG formation in oxidized ds-DNA strands. We apply the method to the 32 bp exon 7 ds-oligonucleotide fragment of the p53 gene, representing p53 codons 242–253 (Scheme 2). Ion pairing reagent TEAB was used to separate positional isomeric oxidative oligonucleotides. The primary products of exon 7 oligonucleotide from catechol/Cu2+/NADPH and Fenton’s reagent oxidation were compared. Analysis of >20 bp oligonucleotides is facilitated by a restriction enzyme that cuts the oligonucleotide into smaller fragments suitable for MS/MS sequencing. Using this approach, Fenton’s reagent oxidized G at codons 243 and 248; the catechol system oxidized guanines at codons 243, 244, 245, and 248, correlating with lung cancer mutation hot spots at codons 245 and 248. The reacting guanines in codons 245 and 248 are C-phosphate-guanine (CpG) sites, and the guanine in codon 248 is also the most reactive toward SN2 reactions with B[a]P diol epoxide.46 The p53 oxidation sites have been uncovered in a purely chemical reaction, and this is of course much less complex than in vivo in humans. However, we find that the most chemically reactive p53 codons present correlations to mutation sites found in tumors.
Scheme 2. Exon 7 of p53 Gene Showing Reactive Codons in Colora.

aArrows show cut points for restriction enzyme Nla III44 and resulting fragments obtained.
EXPERIMENTAL SECTION
Chemicals and Reagents
Custom oligonucleotides were from Sigma-Aldrich. Sources of chemicals and method details are in the Supporting Information file.
Oligonucleotide Oxidation and Product Preparation
Double stranded 32 bp exon 7 fragment of the p53 gene was oxidized in aqueous solution by catechol/Cu2+/NADPH or Fenton’s reagent (see Supporting Information). Conditions were optimized to limit G oxidations to 8-oxodG without strand breaks and to yield LC-MS/MS results of sufficient intensity for oxidation site location and quantitation. Briefly, 100 μg of oligonucleotide was incubated in 200 μL of Tris buffer with 1 mM catechol,15 50 μM CuCl2, and NADPH regeneration system (10 mM G6P, one unit G6PDH enzyme, 0.8 mM NADP+, 1 mM MgCl2) at 37 °C for 12 h. Alternatively, 100 μg of oligonucleotide was incubated with Fenton’s reagent containing 0.1 mM FeSO4 and 40 mM H2O216 at 37 °C for 4 h with constant stirring. Amicon Ultra-0.5 3K centrifugal filters from MilliporeSigma were used to remove excess catechol, NADPH system, and FeSO4 products. The reaction mixture was put into the vial and centrifuged at 13 000 rpm for 30 min. Ds-oligonucleotide fragments were retained on the filter, which was then reversed, put into a new vial, and centrifuged at 13 000 rpm for 30 min to collect the approximately 50 μL of ds-oligonucleotide. Then, 15 μL (150 units) of restriction enzyme Nla III, 20 μL of 10 × NE buffer (New England Biolabs, 1× buffer contains 50 mM potassium acetate, 20 mM tris-acetate, 10 mM magnesium acetate, 100 ug/mL BSA, pH 7.9) and 115 μL of pure water were added to the reacted oligonucleotide solution, and this solution was incubated 37 °C for 12 h to cut oligonucleotides into smaller fragments.
Removal of Enzymes and Salts
Two hundred microliters of phenol:chloroform:isoamyl alcohol (25:24:1) was added to the mixture from the above process. The mixture was shaken for 15 min and then centrifuged at room temperature for 10 min. Then, the upper aqueous solution was carefully transferred to a fresh tube for subsequent extraction. The solution was extracted with phenol:chloroform:isoamyl alcohol (25:24:1) three times and chloroform:isoamyl alcohol (24:1) twice. The resulting aqueous oligonucleotide solution was about 200 μL.
Ethanol was added to the oligonucleotide solution to precipitate oligonucleotide as a small pellet (full details in Supporting Information). The supernatant was carefully removed, and then 1 mL of 70% cold ethanol in water was used to wash the pellet, which was then dried under N2 and dissolved in 100 μL of HPLC-grade water. This solution was heated at 90 °C for 15 min and cooled rapidly to convert ds- to ss-oligonucleotides and then stored at −20 °C.
UPLC-QTOF MS/MS Analysis
A Dionex Ultimate 3000 UPLC with a Gemini C-18 column (150 × 0.5 mm, 3 μm particle size) was used with mobile phases 25 mM triethylammonium bicarbonate (TEAB, solvent A) and methanol (solvent B).42 TEAB facilitated positional isomer separation of oligonucleotides. A gradient of 15% B for 2 min followed by increasing from 15% B to 20% B over 36 min, then back to 15% B for another 2 min at flow rate 8 μL/min was used. The UHPLC was interfaced to an AB Sciex QSTAR Elite mass spectrometer in negative ion mode with a −4500 V ion spray voltage, −60 V declustering potential at 300 °C. m-Nitrobenzyl alcohol (0.1%) was infused at 4 μL/min to mix with the LC flow postcolumn using a three way connector before entering the QSTAR to enhance oligonucleotide charging and signal intensity.47 The ss-oligonucleotides were analyzed by time-of-flight scan mode for sizing unoxidized and oxidized fragments. Product ion scan mode was used for sequencing oxidized fragments at −40 eV collision energy. In most cases, analysis was restricted to singly oxidized oligonucleotides.
Molecular Modeling
The standard B-DNA form of 32 bp p53 exon 7 ds-oligonucleotide was modeled using web 3DNA48 and solvated using CHIMERA software.49 An Amber solvation model was used with appropriate box size to accommodate water molecules.46 Autodock 4.2.6 was used for docking.50,51 Ligands (Fe2+, Fe3+, H2O2, •OH, catechol, benzoquinone, and Cu(I)OOH) were imported into the model software. A Lamarckian genetic algorithm (LGA) was used in Autodock 4.2.6 to find the binding energy between the oligonucleotides and the ligands.
RESULTS
Method Development
The 32 bp exon 7 p53 fragment was oxidized with catechol or Fenton’s reagent, then restriction enzymes were used to cut the oxidized ds-oligonucleotide to smaller size suitable for LC-MS/MS. Restriction enzyme Nla III cuts both strands of ds-DNA after the sequence CATG. In the p53 exon 7 oligonucleotide, this results in two ds fragments of 13 and 19 bases, leaving unpaired CATG sequences at the ends (Scheme 2). The end 5′-CATG is not cut by Nla III as there is no 3′-complement to be cut. Subsequently, heat was used to convert all ds-oligonucleotides to single strands.
Our focus was to identify the primary oxidation sites on exon 7, so we developed conditions to determine 8-oxodG sites in singly oxidized fragments. Our reaction conditions yielded 8-oxodG as the only oxidation product. If a single dG is oxidized to 8-oxodG, the mass of the reacted ss-oligonucleotide increases by 15.999. The identity of each 8-oxodG-containing oligonucleotide was determined by comparing the measured m/z of the oxidized oligonucleotide to the expected m/z of the corresponding unreacted species calculated using Mongo Oligo Mass Calculator, v2.08.52 m/z values for the four unoxidized and oxidized oligonucleotide fragments are shown in Table S1. For example, m/z 1002.4 was observed for unoxidized fragment 1 (Scheme 2) with z = −4, and the 8-oxodG-containing fragment 1 had m/z = 1006.4 (z = −4). TOF MS spectra of the four oxidized fragments are in Figure S1.
Collision-induced dissociation (CID) provided information on the sequence of oxidized oligonucleotide products by fragmentation of the phosphodiester backbone (Scheme 3), forming an–bn and wn ions.53,54 The position of 8-oxodG was determined by detecting the difference in m/z values of the an–bn and wn ions of corresponding unoxidized and oxidized oligonucleotides.
Scheme 3.

Collision Induced Dissociation (MS/MS) of DNA Fragments Resulting in wn and an–bn ions
Oxidation by Catechol/Cu2+/NADPH
Catechol is an industrial chemical and a major component of tobacco smoke55 and is classified as a group 2B carcinogen.56,57 In the presence of Cu2+ and an enzymatic NADPH regenerating system, redox cycling of catechol, semiquinone radical, and benzoquinone produces ROS, which oxidize dG to 8-oxodG in DNA (Scheme 1).15,16,58
Figure 1A shows an extracted ion chromatogram (XIC) of a selected ion of oxidized exon 7 fragment 1 (Scheme 2) with m/z 1006.4 representing 8-oxodG-containing fragment 1 with z = −4 (the most intense ion was used to present XIC). There are four major peaks in the chromatogram, suggesting that 4 positional isomers for singly oxidized fragment 1 were formed with retention times 13.6, 14.9, 16.2, and 17.9 min. The CID spectrum of fragment ion 1006.4 for peak 1 is shown in Figure 1B and for peak 2 in Figure 1C (total ion chromatogram (TIC) in Figure S2).
Figure 1.

LC-QTOF mass spectrometry of fragment 1 (Scheme 2) from exon 7 oxidized by catechol/Cu2+/NADPH. (A) Extracted ion chromatogram for m/z 1006.4 representing z = −4 of singly oxidized products. (B) CID analysis of m/z 1006.4 for peak 1 eluting at 13.6 min and (C) CID analysis of peak 2 eluting at 14.9 min.
Differences in a–b and w ions between unoxidized and oxidized fragment 1 were used to locate the oxidation sites (i.e., 8-oxodG), as used previously for metabolite adduction sites.35,36,45,59 Table 1 summaries a–b and w ions for unoxidized fragment 1 and singly oxidized fragment 1 for the two peaks in Figures 1B and C. Red numbers indicate the ions increased in mass by 15.999 by oxidation of dG to 8-oxodG in the unoxidized fragment. The MS/MS spectrum for peak 1 of singly oxidized fragment 1 (Figure 1B) shows increase in mass of ions from a6–b6 to a8–b8 compared to unoxidized fragment 1. This indicates that the fifth guanine was oxidized to 8-oxodG, CATGGoxGCGGCATG (Gox = 8-oxodG), which was confirmed by the increase in m/z from w9 among the w ions (Table 1).
Table 1.
Fragment Ions for Unoxidized Fragment 1 (m/z 1002.4) and Singly Oxidized Fragment 1 (1006.4) from LC-MS/MS by Catechol/Cu2+/NADPH
| Fragment ion | m/z | ||
|---|---|---|---|
| Unoxidized | Peak 1 | Peak 2 | |
| a4-b4 | [1003.12]− | [1003.12]− | [1003.20]− |
| a5-b5 | [665.62]−2 | [665.64]−2 | [665.64]−2 |
| a6-b6 | [830.15]−2 | [838.17]−2 | [830.17]−2 |
| a7-b7 | [994.65]−2 | [1002.69]−2 | [994.73]−2 |
| a8-b8 | [759.05]−3 | [764.48]−3 | n/d |
| a9-b9 | [868.78]−3 | [874.20]−3 | [874.17]−3 |
| a10-b10 | [978.47]−3 | [983.78]−3 | [983.68]−3 |
| w4 | [1252.16]− | [1252.31]− | [1252.28]− |
| w5 | [790.13]−2 | [790.14]−2 | [790.16]−2 |
| w6 | [954.60]−2 | [954.70]−2 | [962.68]−2 |
| w7 | [732.42]−3 | [732.43]−3 | [737.79]−3 |
| w8 | [842.12]−3 | [842.17]−3 | [847.46]−3 |
| w9 | [951.77]−3 | [957.19]−3 | [957.14]−3 |
The MS/MS spectrum for peak 2 of singly oxidized fragment 1 (Figure 1C) shows an increase in mass of all ions from a9–b9 compared with that of the unoxidized fragment 1. This shows that the eighth guanine was oxidized to 8-oxodG, CATGGGC-GoxGCATG, and is confirmed by increases in mass of w ions from w6 ion. Similar analysis of the third and the fourth peaks revealed that peak 3 indicates oxidation of the sixth guanine (CATGGGoxCGGCATG) and peak 4 represents oxidation of the fourth guanine in CATGoxGGCGGCATG) (Figure S3, Table S2).
Singly oxidized fragment 2 gave m/z of 971.7 at z = −6 (Figure S1B). The XIC of m/z 971.7 (Figure 2A) showed only one major peak, which indicated that only one base on fragment 2 was oxidized during the reaction. MS/MS spectra for singly oxidized fragment 2, m/z 971.7 (Figure 2B), shows increase in mass of all ions from a6–b6 to a7–b7, suggesting that the fifth guanine was oxidized, AACCGoxGAGGCCCATCCTCA. This is confirmed by increase in mass of w15 and w16 for w ions. All ions below w14 gave the same m/z values as the unoxidized fragment (Table S3).
Figure 2.

LC-QTOF mass spectrometric analysis of 8-oxodG containing fragment 2 oxidized by catechol/Cu2+/NADPH. (A) Extracted ion chromatogram for fragment m/z 971.7, z = −6 of singly oxidized fragment 2. (B) CID analysis of oxidized fragment 2, m/z 971.7 for peak eluting at 24.5 min.
Two unknown peaks were found in oxidized fragments 1 (20.3 min) and 2 (28.7 min) after catechol/Cu2+/NADPH oxidation (Figures 1A and 2A). They are also positional isomers of oxidized fragments 1 and 2 but were not intense enough for reliable identification.
Similar analyses were done on oxidized complementary strands of fragments 3 and 4. Results identified oxidized fragment 3 with m/z 917.1, z = −3 (Figure S1C) and oxidized fragment 4 with m/z 1186.4 (Figure. S1D), z = −6 (Table S1). MS/MS spectra are shown in the Supporting Information (Figures S4 and S5 and Tables S4 and S5). In fragment 3, the third and the last guanines were oxidized, and the fourth guanine in fragment 4 was oxidized. Oxidation sites for all the fragments are summarized in Table 2.
Table 2.
Catechol/Cu2+/NADPH and Fenton’s Reagent Oxidation Results on Fragments of p53 Exon 7 Compared to Mutation Sites from the p53 Database30
| Oxidants | Fragments | Sequence | LC-MS/MS Data | ||
|---|---|---|---|---|---|
|
| |||||
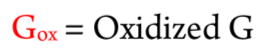
|
Codon (%) | Mutation hot spot | |||
| Catechol/Cu2+/NADPH | Fragment 1 | CATGGGCGGCATG |
|
244 (3.2) | 244a |
|
|
245 (2.6) | 245b,d | |||
|
|
244 (5.0) | 244a | |||
|
|
243 (5.6) | 243c | |||
| Fragment 2 | AACCGGAGGCCCATCCTCA |
|
248 (10.7) | 248a,b,c,d,e | |
| Complementary strands | |||||
| Fragment 3 | CCGCCCATG |
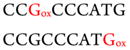
|
|||
| Fragment 4 | TGAGGATGGGCCTCCGGTTCATG |
|
|||
|
| |||||
| Fenton’s Reagent | Fragment 1 | CATGGGCGGCATG |
|
243 (20.3) | 243c |
| Fragment 2 | AACCGGAGGCCCATCCTCA |
|
248 (10.4) | 248a,b,c,d,e | |
| Complementary strands | |||||
| Fragment 3 | CCGCCCATG |
|
|||
| Fragment 4 | TGAGGATGGGCCTCCGGTTCATG |
|
|||
Small cell lung cancer.
Nonsmall cell lung cancer.
Hepatocellular carcinoma (HCC).
Colorectal cancer.
Skin cancer.
We estimated the relative abundance of specific oxidized fragments by comparing areas under extracted ion chromatograms of oxidized fragments to those of unoxidized fragments (Figure S6), assuming that oxidized and unoxidized fragments have similar MS ionization efficiencies (details in Supporting Information).45 The relative amounts of oxidation in the 32 bp fragment were estimated (Table 2). Ratio of relative amounts of oxidation for codons 248/243 was 1.9; codon 248/244 was 1.3, and 248/245 was 4.1.
Oxidation by Fenton’s Reagent
Deoxyguanosine in DNA is easily oxidized to 8-oxodG by Fenton’s reagent. The active oxidant in Fenton’s reagent is thought to be •OH,60,61 and oxidation products of 8-oxodG have also been identified.9,62,63 We found none of these overoxidation products under our conditions.
Oxidation products of p53 exon 7 by Fenton’s reagent revealed several differences from oxidation by catechol/Cu2+/NADPH. Figure 3A shows there is only one major peak in the XIC of singly oxidized fragment 1, indicating only one base was oxidized. The MS/MS spectrum for singly oxidized fragment 1 (Figure 3B) shows an increase in mass of all ions from a5–b5 to a7–b7 compared to the unoxidized fragment. This indicates that the fourth guanine was oxidized to 8-oxodG, CAT-GoxGGCGGCATG, confirmed by increased mass of w ions above w10. All ions from w2 to w9 had the same m/z as that of the unoxidized fragment (Table S6).
Figure 3.

LC-QTOF mass spectrometric analysis of 8-oxodG containing fragment 1 oxidized by Fenton’s reagent. (A) XIC for fragment m/z 1006.4 representing −4 charge of singly oxidized fragment 1. (B) CID analysis of oxidized fragment 1, m/z 1006.4 for peak eluting at 17.9 min.
Oxidized fragments 2 and 4 gave results similar to those of catechol/Cu2+/NADPH oxidation (Figures S7A and C). For fragment 3 (Figure S7B), there is only one major peak for the oxidized fragment 3. Results are summarized in Table 2.
The relative amounts of oxidation by Fenton’s reagent were also estimated (Table 2) by comparing areas under extracted ion chromatograms. Ratio of relative amounts of oxidation for codons 248/243 was 0.5.
Modeling of Oxidant Binding
In DNA oxidation mediated by catechol, NADPH and Cu2+, molecular oxygen is reduced to O2•− and H2O2, and catechol reacts with Cu2+ yielding Cu+ and benzoquinone (Scheme 1). Also, Cu+ reacts with H2O2 to form •OH radical and Cu(I)-hydroperoxyl complex (Cu(I)OOH), which may lead to DNA strand breaks. In Fenton’s reagent oxidation, Fe2+ reacts with H2O2 to form Fe3+ ions and •OH radical. To gain insight into the codon specificity of the oxidations, molecular modeling was done with AutoDock 4.2.6 by docking each possible involved species (Fe2+, Fe3+, H2O2, •OH, catechol, benzoquinone, and Cu(I)-OOH) with the B-DNA form of the exon 7 ds-oligonucleotide. Parameters were set with 25 000 000 evaluations and 50 docked conformations for an individual docking computation. Examples of preferred Fe2+ binding site on exon 7 from this analysis is shown in Figure 4, and preferred catechol sites are shown in Figure 5. The preferred binding positions and binding free energies (ΔGb) were estimated from this analysis. ΔGb values were used to calculate relative apparent binding constants from ln Kb = −ΔGb/RT, where R is the ideal gas constant and T is temperature in Kelvin (Table S7). For Fe2+, 35 out of 50 docked conformations were between the fourth and fifth guanines in fragment 1 (Figure 4), but the binding is quite weak. For catechol, preferred binding was found at multiple positions, including the 4th, 5th, 6th and 8th, and 13th guanines (Figure 5), and the binding is stronger. Results for all possible ROS are listed in the Supporting Information, Table S7.
Figure 4.

Model of Fe2+ docked at preferred site (arrow) near reactive guanine in B form of the 32 bp exon 7 p53 fragment.
Figure 5.

Models of catechol in yellow docking near reactive guanines in the B form of the 32 bp exon 7 p53 fragment showing catechol docked (arrows) (A) with the fourth and fifth guanines, (B) with the sixth guanine, (C) with the eighth guanine, and (D) with the 13th guanine.
The two unknown peaks in oxidized fragment 1 (20.3 min) and fragment 2 (28.7 min) from catechol-mediated oxidation (Figures 1A and 2A) were not intense enough for reliable identification. However, docking results (Figure 5D) are consistent with the possibility that the 13th guanine was oxidized in fragment 1. G in codon 249 was probably oxidized in fragment 2 due to the fact that codon 249 is also a “hotspot” in many cancers such as liver, lung, and hepatocellular carcinoma.30
DISCUSSION
Results above demonstrate successful development of LC-MS/MS methodology to directly locate primary oxidation sites in ds-oligonucleotides of >20 bp without labeling or hydrolysis. This is the first report directly identifying multiple oxidation sites in an intact ds-oligonucleotide. The restriction enzyme assisted methodology is simple, reliable, relatively rapid, and facilitates direct detection and mapping of modified sites. It does not require stable isotope labeling or DNA hydrolysis.
Results reveal that primary oxidation sites involve only guanines on the p53 exon 7 oligonucleotide. This is consistent with guanines as the most easily oxidized of the DNA nucleobases. In general, reactivity of specific guanines within the exon 7 p53 ds-fragment is influenced additionally by neighboring bases and to some extent by secondary structure of the oligonucleotide.46 The most frequently oxidized guanine by catechol/Cu2+/NADPH in the present study was in codon 248 (11%, Table 2), which correlates with codon 248 as a major mutation hot spot in many cancers.30 Guanines within codons 243, 244, and 245 were oxidized to a smaller extent. Thus, there is a quantitative correlation between the high reactive frequency of codon 248 in the p53 exon 7 oligonucleotide with its high mutation frequency in cancers using an oxidation process that mimics oxidations mediated by redox active metabolites (Scheme 1).10–14 However, in Fenton’s reagent oxidation, which may or may not have a directly analogous process in humans, guanine in codon 248 gave a smaller extent of oxidation (Table 2) than codon 243.
According to the p53 database,30 the mutation frequency ratio of codons 248/243 is 24; of codons 248/244 is 7, and of codons 248/245 is 2. Our ratios of relative amounts of oxidation by catechol/Cu2+/NADPH for codons 248/243 was 1.9 and for codons 248/244 was 1.3, which are smaller than the mutation ratios. While we would not expect exact numerical correspondence in codon reactivity and mutation frequency, the lower reactivity ratios involving guanine in codon 243 may be related to it being the first guanine in our exon 7 oligonucleotide and more reactive due to its proximity to the end of a strand.45,64 Also, the guanine in codon 248 (CGG) is a CpG site. In vivo, all cytosines in CpG sites are methylated, which may increase the reactivity and mutation frequency of the neighbor guanine in tumors.39,46,65 For 248/245, the oxidation reactivity ratio was 4.1, which is larger than that for mutations. This could be related to the first guanine in codon 245 (GGC) being a CpG site, making the guanine more reactive in vivo, resulting in a smaller mutation ratio.
The ratio of relative amounts of oxidation for codons 248/243 was 0.5 from Fenton’s reagent oxidation, which is much lower than that in the database. Fenton’s reagent is just an oxidant, not a carcinogen, and may simply oxidize the most accessible guanine with lowest oxidation potential in the sequence. The guanine in codon 243 (ATG) is close to the end of the fragment, adjacent to the AT sequence, and may be more exposed due to a partially unwound duplex with fewer hydrogen bonds.45,64 Thus, the guanine in codon 243 may be more reactive in the fragment than the guanine in codon 248 (CGG) under in vitro conditions.
Oxidations of guanines at codon 245 (GGC), 246 (ATG), 248 (CGG), and 249 (AGG) were observed in isotopic labeled guanines in oligonucleotides.39 Two distinct sites, first guanine in codon 245 (GGC) and first guanine in codon 248 (CGG) had the highest oxidation percentage at 18.7 and 14.2% compared to our values 2.6 and 10.7%, respectively. This may because both guanine sites of highest reactivity had an identical sequence context MeCGG with a methylated cytosine on the 5’ side. In our exon 7, the cytosine is nonmethylated and was less easily oxidized.
Another important observation is the single major peak for oxidized fragment 1 with Fenton’s reagent (Figure 3A), as opposed to four major peaks for oxidized fragment 1 with catechol/Cu2+/NADPH (Figure 1A), suggesting Fenton’s reagent is more specific toward DNA oxidative damage in our system. Several reports indicated that free hydroxyl radical caused DNA damage with no marked site specificity.66,67 But some investigations show iron(II) binding to phosphate groups and a guanine N-7 moiety first, with subsequent oxidative damage of dG, oligomers, and calf-thymus DNA in solution.68–70
Docking studies showed that 35 out of 50 conformations of exon 7 oligonucleotide had Fe2+ docked between the fourth and fifth guanines in fragment 1 (Figure 4, Table S7), somewhat consistent with Fe2+ binding weakly at a specific guanine prior to the actual oxygen transfer, leading to 8-oxodG. However, catechol (Figure 5) was found to bind more strongly at multiple positions, including the 4th, 5th, 6th, 8th, and 13th guanines, which correlates with experimental results showing multiple products (Figure 1, Table 2). Other possible ROS reactants in the catechol reaction also had multiple preferred binding positions on the exon 7 oligonucleotide (Table S7). Thus, the molecular modeling results are consistent with the broader codon specificity found in catechol-induced DNA oxidation.
CONCLUSION
We describe here an LC-MS/MS methodology to directly sequence and quantify the oxidation sites on ds-oligonucleotides of >20 bp. The high oxidation frequency at codon 244 and 248 with catechol/Cu2+/NADPH, a model for quinoid drug metabolites, coincides with high mutation frequency of the p53 gene in lung and other cancers. Fenton’s reagent specificity in oxidation may be consistent with binding of a key oxidizing component on the oligonucleotide near the oxidation site before subsequent oxygen transfer to guanines. On the other hand, multiple binding sites of the model metabolite catechol may explain multiple exon 7 codons oxidized by catechol/Cu2+/NADPH. Our results have implications for understanding the role of oxidation of tumor suppressor genes on carcinogenesis. Future work will focus on investigating tumor suppressor gene oxidation using longer, more representative oligonucleotides and confirming links between oxidation and mutation sites for a broad range of chemicals.
Supplementary Material
Acknowledgments
The authors thank the National Institute of Environmental Health Sciences (NIEHS), NIH, United States, Grant ES03154 for financial support.
Footnotes
Notes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.anal-chem.7b03487.
Detailed experimental section; additional seven figures including negative ion ESI-MS spectra of singly oxidized fragments, TIC for oxidized samples, XIC for oxidized fragments 3 and 4, XIC for unoxidized fragments 1–4, CID analysis of oxidized fragments 1, 3, and 4; six tables describing m/z and fragment ions for singly oxidized fragments 1–4; and one table showing docking results for possible ROS (PDF)
References
- 1.Valko M, Rhodes CJ, Moncol J, Izakovic M, Mazur M. Chem-Biol Interact. 2006;160:1–40. doi: 10.1016/j.cbi.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 2.Cooke MS, Evans MD, Dizdaroglu M, Lunec J. FASEB J. 2003;17:1195–1214. doi: 10.1096/fj.02-0752rev. [DOI] [PubMed] [Google Scholar]
- 3.Hamilton CA, Miller WH, Al-Benna S, Julia Brosnan M, Drummond RD, Mcbride MW, Dominiczak AF. Clin Sci. 2004;106:219–234. doi: 10.1042/CS20030379. [DOI] [PubMed] [Google Scholar]
- 4.Henchcliffe C, Beal MF. Nat Clin Pract Neurol. 2008;4:600–609. doi: 10.1038/ncpneuro0924. [DOI] [PubMed] [Google Scholar]
- 5.Hussain SP, Hofseth LJ, Harris CC. Nat Rev Cancer. 2003;3:276–285. doi: 10.1038/nrc1046. [DOI] [PubMed] [Google Scholar]
- 6.Speina E, Arczewska KD, Gackowski D, Zielinska M, Siomek A, Kowalewski J, Olinski R, Tudek B, Kusmierek JT. J Natl Cancer Inst. 2005;97:384–395. doi: 10.1093/jnci/dji058. [DOI] [PubMed] [Google Scholar]
- 7.Si JC, Lu L, Gao ZF, Zhang Y, Luo HQ, Bing LN. Anal Methods. 2014;6:6536–6540. [Google Scholar]
- 8.Liu G, Huang H, Xie R, Feng Q, Fang R, Shu Y, Zhan Y, Ye X, Zhong C. RSC Adv. 2017;7:71–76. [Google Scholar]
- 9.White B, Smyth MR, Stuart JD, Rusling JF. J Am Chem Soc. 2003;125:6604–6605. doi: 10.1021/ja0343252. [DOI] [PubMed] [Google Scholar]
- 10.Murata M, Tamura A, Tada M, Kawanishi S. Free Radical Biol Med. 2001;30:765–773. doi: 10.1016/s0891-5849(01)00463-4. [DOI] [PubMed] [Google Scholar]
- 11.Ohnishi S, Murata M, Kawanishi S. Jpn J Cancer Res. 2002;93:736–743. doi: 10.1111/j.1349-7006.2002.tb01314.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Naiman K, Martínková M, Schmeiser HH, Frei E, Stiborová M. Mutat Res, Genet Toxicol Environ Mutagen. 2011;726:160–168. doi: 10.1016/j.mrgentox.2011.09.010. [DOI] [PubMed] [Google Scholar]
- 13.Murata M, Yoshiki Y, Tada M, Kawanishi S. Int J Cancer. 2002;102:311–317. doi: 10.1002/ijc.10717. [DOI] [PubMed] [Google Scholar]
- 14.Hepel M, Stobiecka M, Peachey J, Miller J. Mutat Res, Fundam Mol Mech Mutagen. 2012;735:1–11. doi: 10.1016/j.mrfmmm.2012.05.005. [DOI] [PubMed] [Google Scholar]
- 15.Song B, Shen M, Jiang D, Malla S, Mosa IM, Choudhary D, Rusling JF. Analyst. 2016;141:5722–5729. doi: 10.1039/c6an01237j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bist I, Song B, Mosa IM, Keyes TE, Martin A, Forster RJ, Rusling JF. ACS Sensors. 2016;1:272–278. doi: 10.1021/acssensors.5b00189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yu D, Berlin JA, Penning TM, Field J. Chem Res Toxicol. 2002;15:832–842. doi: 10.1021/tx010177m. [DOI] [PubMed] [Google Scholar]
- 18.Steenken S, Jovanovic SV. J Am Chem Soc. 1997;119:617–618. [Google Scholar]
- 19.Neeley WL, Essigmann JM. Chem Res Toxicol. 2006;19:491–505. doi: 10.1021/tx0600043. [DOI] [PubMed] [Google Scholar]
- 20.David SS, O’Shea VL, Kundu S. Nature. 2007;447:941–950. doi: 10.1038/nature05978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shibutani S, Takeshita M, Grollman AP. Nature. 1991;349:431–434. doi: 10.1038/349431a0. [DOI] [PubMed] [Google Scholar]
- 22.Jones RG, Thompson CB. Genes Dev. 2009;23:537–548. doi: 10.1101/gad.1756509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Soussi T. Oncogene. 2007;26:2145–2156. doi: 10.1038/sj.onc.1210280. [DOI] [PubMed] [Google Scholar]
- 24.Cooper GM. The Cell: A Molecular Approach. 2. Sinauer Associates; Sunderland: 2000. [accessed July 2017]. https://www.ncbi.nlm.nih.gov/books/NBK9894/ [Google Scholar]
- 25.May P, May E. Oncogene. 1999;18:7621–7636. doi: 10.1038/sj.onc.1203285. [DOI] [PubMed] [Google Scholar]
- 26.Cheok CF, Verma CS, Baselga J, Lane DP. Nat Rev Clin Oncol. 2011;8:25–37. doi: 10.1038/nrclinonc.2010.174. [DOI] [PubMed] [Google Scholar]
- 27.Soussi T, Wiman KG. Cancer Cell. 2007;12:303–312. doi: 10.1016/j.ccr.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 28.Ozaki T, Nakagawara A. J Biomed Biotechnol. 2011;2011:1–13. doi: 10.1155/2011/603925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Leroy B, Anderson M, Soussi T. Hum Mutat. 2014;35:672–688. doi: 10.1002/humu.22552. [DOI] [PubMed] [Google Scholar]
- 30.Leroy B, Anderson M, Soussi T. [accessed November 17, 2017];The TP53 Mutation database. Available online http://p53.fr/the-database.
- 31.Soussi T. Adv Cancer Res. 2011;110:107–139. doi: 10.1016/B978-0-12-386469-7.00005-0. [DOI] [PubMed] [Google Scholar]
- 32.Hussain SP, Aguilar F, Amstad P, Cerutti P. Oncogene. 1994;9:2277–2281. [PubMed] [Google Scholar]
- 33.Amstad P, Hussain SP, Cerutti P. Mol Carcinog. 1994;10:181–188. doi: 10.1002/mc.2940100402. [DOI] [PubMed] [Google Scholar]
- 34.Tretyakova N, Villalta PW, Kotapati S. Chem Rev. 2013;113:2395–2436. doi: 10.1021/cr300391r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chowdhury G, Guengerich FP. Chem Res Toxicol. 2009;22:1310–1319. doi: 10.1021/tx900115z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liao Q, Shen C, Vouros P. J Mass Spectrom. 2009;44:549–560. doi: 10.1002/jms.1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lim KS, Cui L, Taghizadeh K, Wishnok JS, Chan W, DeMott MS, Babu IR, Tannenbaum SR, Dedon PC. J Am Chem Soc. 2012;134:18053–18064. doi: 10.1021/ja307525h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lim KS, Taghizadeh K, Wishnok JS, Babu IR, Shafirovich V, Geacintov NE, Dedon PC. Chem Res Toxicol. 2012;25:366–373. doi: 10.1021/tx200422g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ming X, Matter B, Song M, Veliath E, Shanley R, Jones R, Tretyakova N. J Am Chem Soc. 2014;136:4223–4235. doi: 10.1021/ja411636j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Harsch A, Sayer JM, Jerina DM, Vouros P. Chem Res Toxicol. 2000;13:1342–1348. doi: 10.1021/tx000140m. [DOI] [PubMed] [Google Scholar]
- 41.Xiong W, Glick J, Lin Y, Vouros P. Anal Chem. 2007;79:5312–5321. doi: 10.1021/ac0701435. [DOI] [PubMed] [Google Scholar]
- 42.Sharma V, Glick J, Vouros P. J Chromatogr A. 2012;1245:65–74. doi: 10.1016/j.chroma.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sharma VK, Xiong W, Glick J, Vouros P. Eur Mass Spectrom. 2014;20:63–72. doi: 10.1255/ejms.1268. [DOI] [PubMed] [Google Scholar]
- 44.Jung V, Pestka SB, Pestka S. Nucleic Acids Res. 1990;18:6156–6156. doi: 10.1093/nar/18.20.6156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Malla S, Kadimisetty K, Fu YJ, Choudhary D, Jansson I, Schenkman JB, Rusling JF. Chem Sci. 2015;6:5554–5563. doi: 10.1039/c5sc01403d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Malla S, Kadimisetty K, Fu Y-J, Choudhary D, Schenkman JB, Rusling JF. Sci Rep. 2017;7:40890. doi: 10.1038/srep40890. Open access: www.nature.com/articles/srep40890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brahim B, Alves S, Cole RB, Tabet J. J Am Soc Mass Spectrom. 2013;24:1988–1996. doi: 10.1007/s13361-013-0732-8. [DOI] [PubMed] [Google Scholar]
- 48.Zheng G, Lu X-J, Olson WK. [accessed November 17, 2017];Nucleic Acids Res. 2009 37:W240–W246. doi: 10.1093/nar/gkp358. (web server issue) Website: http://w3dna.rutgers.edu/index.php/rebuild. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 50. [accessed July 2017];AutoDock 4.2 is distributed free of charge as open source software. whttp://autodock.scripps.edu.
- 51. [accessed May 2017];ADT is distributed free of charge as part of the MGL Tools package. http://mgltools.scripps.edu/downloads.
- 52. [accessed May 2017];Mongo Oligo Mass Calculator v2.08. http://mods.rna.albany.edu/masspec/Mongo-Oligo.
- 53.McLuckey SA, Van Berkel GJ, Glish GL. J Am Soc Mass Spectrom. 1992;3:60–70. doi: 10.1016/1044-0305(92)85019-G. [DOI] [PubMed] [Google Scholar]
- 54.McLuckey SA, Habibi-Goudarzi S. J Am Soc Mass Spectrom. 1994;5:740–747. doi: 10.1016/1044-0305(94)80006-5. [DOI] [PubMed] [Google Scholar]
- 55.Hecht SS. Int J Cancer. 2012;131:2724–2732. doi: 10.1002/ijc.27816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Oikawa S, Hirosawa I, Hirakawa K, Kawanishi S. Carcinogenesis. 2001;22:1239–1245. doi: 10.1093/carcin/22.8.1239. [DOI] [PubMed] [Google Scholar]
- 57.Oikawa S. Environ Health Prev Med. 2005;10:65–71. doi: 10.1007/BF02897995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zuniga MA, Dai J, Wehunt MP, Zhou Q. Chem Res Toxicol. 2006;19:828–836. doi: 10.1021/tx060021s. [DOI] [PubMed] [Google Scholar]
- 59.Chowdhury G, Guengerich FP. Angew Chem, Int Ed. 2008;47:381–384. doi: 10.1002/anie.200703942. [DOI] [PubMed] [Google Scholar]
- 60.Haber F, Weiss Proc R Soc London, Ser A. 1934;147:332–351. [Google Scholar]
- 61.Fenton HJH. J Chem Soc, Trans. 1894;65:899–910. [Google Scholar]
- 62.White B, Tarun MC, Gathergood N, Rusling JF, Smyth MR. Mol BioSyst. 2005;1:373–381. doi: 10.1039/b511756a. [DOI] [PubMed] [Google Scholar]
- 63.Song B, Pan S, Tang C, Li D, Rusling JF. Anal Chem. 2013;85:11061–11067. doi: 10.1021/ac402736q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Calladine CR, Drew HR, Luisi BF, Travers AA. Understanding DNA. 3. Academic Press; Oxford: 2004. pp. 64–93. [Google Scholar]
- 65.Guza R, Kotandeniya D, Murphy K, Dissanayake T, Lin C, Giambasu GM, Lad RR, Wojciechowski F, Amin S, Sturla SJ, Hudson RHE, York DM, Jankowiak R, Jones R, Tretyakova NY. Nucleic Acids Res. 2011;39:3988–4006. doi: 10.1093/nar/gkq1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kawanishi S, Hiraku Y, Oikawa S. Mutat Res, Rev Mutat Res. 2001;488:65–76. doi: 10.1016/s1383-5742(00)00059-4. [DOI] [PubMed] [Google Scholar]
- 67.Margolin Y, Shafirovich V, Geacintov NE, DeMott MS, Dedon PC. J Biol Chem. 2008;283:35569–35578. doi: 10.1074/jbc.M806809200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Colwell BA, Morris DL., Jr J Inorg Biochem. 2003;94:100–105. doi: 10.1016/s0162-0134(02)00626-8. [DOI] [PubMed] [Google Scholar]
- 69.Rai P, Cole TD, Wemmer DE, Linn S. J Mol Biol. 2001;312:1089–1101. doi: 10.1006/jmbi.2001.5010. [DOI] [PubMed] [Google Scholar]
- 70.Ouameur AA, Arakawa H, Ahmad R, Naoui M, Tajmir-Riahi HA. DNA Cell Biol. 2005;24:394–401. doi: 10.1089/dna.2005.24.394. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.