

Summary
Inhibiting protein function selectively is a major goal of modern drug discovery. Here, we report a previously understudied benefit of small molecule Proteolysis Targeting Chimera (PROTACs) that recruit E3 ubiquitin ligases to target proteins for their ubiquitination and subsequent proteasome-mediated degradation. Using promiscuous CRBN- and VHL-recruiting PROTACs that bind >100 kinases, we show that only a subset of bound targets is degraded. The basis of this selectivity relies on protein-protein interactions between the E3 ubiquitin ligase and the target protein, as illustrated by engaged proteins that are not degraded as a result of unstable ternary complexes with PROTAC-recruited E3 ligases. In contrast, weak PROTAC:target protein affinity can be stabilized by high affinity target:PROTAC:ligase trimer interactions, leading to efficient degradation. This study highlights design guidelines for generating potent PROTACs as well as possibilities for degrading undruggable proteins immune to traditional small molecule inhibitors.
Keywords: PROTACs, protein degradation, chemical biology, proteomics, ternary complex, protein-protein interactions, selective degradation, MAPK/p38, VHL, CRBN
eTOC blurb
Bondeson and Smith et al. examined the degradation selectivity of promiscuous PROTACs: heterobifunctional small-molecules based on E3-ligase recruiting molecules conjugated to a non-selective kinase inhibitor. Despite binding to well over 50 kinases, these compounds selectively degrade <15 due to unique protein-protein interactions between the E3 ligase and degraded targets.
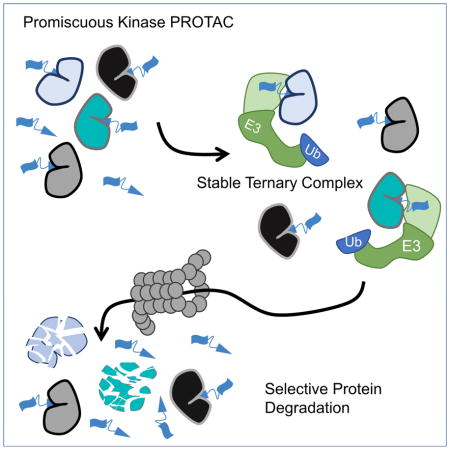
Introduction
Much of modern drug discovery focuses on developing high-affinity ligands that bind to and inhibit enzymatic active sites. This strategy has proven very successful, yet is limited by the number of protein targets currently considered ‘druggable’ (Hopkins and Groom, 2002). Since 2001, our lab and others have worked to develop an alternative targeting strategy that induces selective protein degradation (Sakamoto et al., 2001; Toure and Crews, 2016). Proteolysis Targeting Chimera (PROTACs) are small molecules consisting of a targeting ligand (warhead) for a protein to be degraded, an E3 ubiquitin ligase recruitment ligand, and a chemical linker connecting the two ligands. Upon PROTAC-mediated heterodimerization of the two bound proteins, the target protein is ubiquitinated and degraded by the proteasome (Schneekloth et al., 2004).
Recent years have shown a rapid advancement in the understanding and application of PROTACs due to the discovery of small molecule ligands for E3 ubiquitin ligases. Von Hippel Lindau (VHL) is the substrate adapter for the Cullin 2 E3 ligase complex, inducing the potent and rapid degradation of its substrate HIF1α (Deshaies and Joazeiro, 2009; Min et al., 2002). Through structure-guided design, our lab and others developed high affinity ligands for VHL (Buckley et al., 2012a, 2012b; Galdeano et al., 2014). A second E3 ubiquitin ligase commonly used in PROTAC design is Cereblon (CRBN), which is the substrate adapter for the Cullin 4a ligase complex whose endogenous substrates have not been fully characterized, but likely include MEIS2 and glutamine synthetase (Fischer et al., 2014; Nguyen et al., 2016). The IMiD thalidomide and its analogs have been shown to bind to CRBN and induce degradation of several neosubstrate proteins (Chamberlain et al., 2014; Krönke et al., 2014; Lu et al., 2014).
Many PROTAC molecules have been developed to recruit these E3 ubiquitin ligases to a variety of substrates using high-affinity ligands for the protein of interest. Proteins effectively degraded by PROTACs include RIPK2 and ERRα (Bondeson et al., 2015), BRD4 (Lu et al., 2015; Ohoka et al., 2017; Raina et al., 2016; Winter et al., 2015), BRD9 (Remillard et al., 2017), BCR/Abl and Abl (Lai et al., 2016; Ohoka et al., 2017), and ERα (Ohoka et al., 2017). These proteins represent different structural classes ranging from receptor tyrosine kinases (see accompanying paper Burslem, Smith et al.) to hormone receptors to bromodomains. However, in most cases, these PROTACs have been developed with a single target in mind and have been based on a recruiting element with adequate selectivity towards that target.
Based on these literature examples, one could assume that PROTACs developed from high-affinity ligands will induce efficient target degradation, which raises two important questions related to PROTAC design. What is the correlation, if any, between the target binding affinity and degradation profile of a given PROTAC? And, secondly, if a PROTAC is able to bind to several different target proteins, will they all be degraded? Few literature examples provide initial answers to these questions. Previously, our lab showed that when the targeting warhead of PROTACs was able to efficiently bind both BCR/Abl and c-Abl, the choice of E3 ubiquitin ligase played important roles in the observed degradation (Lai et al., 2016). Only CRBN-recruiting PROTACs were capable of degrading BCR/Abl and c-Abl, whereas PROTACs that recruit VHL only degrade c-Abl. A second set of examples are several CRBN-recruiting PROTACs shown to bind and degrade all BRD proteins to which the targeting ligand binds (Remillard et al., 2017; Winter et al., 2015). In contrast to these studies, in which CRBN seems to promiscuously degrade target proteins, VHL-recruiting PROTACs have been shown to gain specificity towards target proteins with respect to the binding profile of the targeting warhead (Gadd et al., 2017; Zengerle et al., 2015). While not addressing the first question relating to affinity, these examples indicate that PROTACs based on VHL may be able to degrade proteins with enhanced selectivity compared to the targeting warhead.
In this study, we sought to address these questions. We 1) developed CRBN- and VHL-recruiting PROTACs using a promiscuous kinase inhibitor that is potentially capable of degrading more than 100 substrate proteins and 2) asked in a global, unbiased manner how many kinases were degraded by each. Interestingly, we found that both CRBN- and VHL-recruiting PROTACs have drastically increased degradation specificity relative to the parent warhead: the CRBN-recruiting PROTAC degraded fourteen kinases while the VHL-recruiting PROTAC degraded nine. In addition, we found that the biophysical characteristic of a PROTAC:kinase pair that most robustly predicted degradation was not the affinity of the kinase with the PROTAC. Rather, the ability of a PROTAC to induce a stable ternary complex was far more predictive. Indeed, we found a target protein with poor affinity for the PROTAC that was stabilized by protein:protein interactions and led to potent protein degradation.
Results
Development of PROTACs based on the Promiscuous Kinase Inhibitor Foretinib
During our development of receptor tyrosine kinase PROTACs (see accompanying paper Burslem, Smith et al.), we synthesized PROTACs based on the c-Met tyrosine kinase inhibitor foretinib (Vijayan et al., 2015). Given foretinib’s relatively promiscuous inhibition of multiple kinases, we sought to explore other potential targets of our compounds. First, using a high-throughput competitive binding assay (DiscoverX KinomeScan), we found that 10 μM foretinib binds to 133 different kinases with a percent of control (a value with inverse relationship to affinity) value of 35 or less – the recommended cut-off to avoid false-positives for kinase-inhibitor pairs (Figure 1A and Supplemental Table 1) (Fabian et al., 2005; Karaman et al., 2008). Based on the crystal structure of foretinib bound to the c-Met kinase domain (Qian et al., 2009), we reasoned that the disordered and solvent-exposed morpholine of foretinib was dispensable for affinity, and so we used that phenyl ether as a linker attachment point for E3 ubiquitin ligase recruiting molecules. Both a VHL-recruiting PROTAC (compound 1, Figure 1B) and a CRBN-recruiting PROTAC (compound 2) were generated based on the foretinib warhead.
Figure 1. Foretinib PROTACs bind many kinases and potently degrade c-Met.

(A) Foretinib binds to 133 kinases with a Percent of Control less than 35 as measured by a competitive binding assay. (B) Structures of the foretinib-based PROTACs used in this study. Top is the VHL-recruiting PROTAC compound 1. The stereocenter of the hydroxyproline VHL-binding element is R for the active PROTAC while it is S for the inactive control compound 3. Bottom, the Cereblon-recruiting PROTAC compound 2. The nitrogen atom of the glutarimide ring is non-methylated in the active PROTAC, while in the inactive control compound 4 it is methylated. (C) The affinity for most kinases changes upon addition of the linker and E3-ligase recruiting element. Along the X-axis are the 133 kinases that are considered hits for foretinib (i.e. having a percent of control value of 35 or less in KinomeScan data) sorted in order of decreasing affinity for foretinib. The values for foretinib are plotted in grey on both the positive and negative Y-axes. On the positive Y-axis, the values for compound 1 are plotted in black, while the negative X-axis has the values for compound 2. See Supplemental Table 1 and Figure S1 for full KinomeScan data sets. (D) MDA-MB-231 cells were treated with the indicated concentrations of compounds 1–4 for 24 hours, and c-Met protein levels were analyzed by immunoblot.
We first sought to address potential changes in binding affinity that result from the addition of a linker and E3 ubiquitin ligase-recruiting molecule to foretinib. Using a competitive binding assay (DiscoverX, KinomeScan), we tested the selectivity of compounds 1 and 2 at 10 μM across the human kinome (Figure S1A–C). We found that the addition of a linker and E3-recruiting moiety significantly changed the binding profile of the compounds. Compound 1 retained binding to 52 kinases while compound 2 bound 62 kinases (Figure 1C). Interestingly, the binding profiles of compounds 1 and 2 were not identical: even though the aspects of these compounds most proximal to the pocket (i.e. the linker attachment point and linker) are the same, the addition of a different E3-recruiting moiety changed the binding profile of each PROTAC (Figure S1). For example, compound 2 retains binding to p38δ, but the VHL-recruiting PROTAC 1 does not. While conjugation of a linker and an E3-ligase recruiting element raises the selectivity of the PROTACs, both compounds retained binding affinity to a common set of 51 kinases, offering a large set of proteins with which to compare compounds 1 and 2 for their ability to degrade common target proteins.
We first tested the ability of these compounds to degrade c-Met, the clinically relevant target of foretinib, in the triple negative breast-cancer cell line MDA-MB-231. Since triple-negative breast cancer cells have been shown to express ~75% of the kinome (Duncan et al., 2012), they provide an excellent system to study the selectivity of PROTAC-induced degradation. Treatment of MDA-MB-231 cells for 24 hours with compounds 1 and 2 showed a dose-dependent decrease in the levels of c-Met protein as assessed by western blot (Figure 1D). To ensure that the degradation of c-Met was due to the induced ternary complex between the PROTAC, E3 ubiquitin ligase and c-Met, we generated negative control analogs of compounds 1 and 2 (Figure 1B): by inversion of the stereocenter on the hydroxyproline moiety of the VHL ligand or by methylation of the nitrogen on the glutarimide ring of pomalidomide, we synthesized compounds 3 and 4, respectively. Neither compound 3 nor 4 decreased c-Met levels across all concentrations tested.
Global Proteomic Changes upon PROTAC Treatment
Given that these PROTACs possess degradation capacity and retain promiscuous kinase-binding ability, we next sought to assess quantitatively their effect on the entire MDA-MB-231 proteome. We chose eight different treatment groups: a vehicle control, two concentrations of each PROTAC, and a high concentration of foretinib and each of the negative control PROTACs (Figure S1D). We used two concentrations of each PROTAC in an attempt to eliminate false-negatives due to the “hook effect” (Douglass et al., 2013). Negative control compounds were used in this experiment in order to eliminate proteins whose quantitation is decreased due to transcriptional changes or destabilization by the inhibitor alone (Field et al., 2017; Laurence, 2013). After a 24-hour incubation with each treatment condition, the cells were harvested, lysed under denaturing conditions, and proteins tagged for multiplexed isobaric labeling coupled to tandem LC/MS/MS (Mcalister et al., 2014; McAlister et al., 2012). 7,826 unique proteins were identified in parallel with a false discovery rate of less than one percent (Figure 2A and B, and Supplemental Table 2).
Figure 2. Foretinib-based PROTACs degrade only a subset of foretinib-binding kinases.

(A) Multiplexed tandem mass spectrometry was used to assess global changes in the proteome of MDA-MB-231 cells after treatment with VHL PROTAC 1. After 24 hours with either 100 nM (plotted on the X-axis) or 1 μM (Y-axis) of 1, proteins were extracted from cells, labeled with tandem mass tags, and then 7,826 unique proteins were quantified according to the STAR methods section. The values plotted are fractions of the vehicle (DMSO) control. Dotted lines at 0.8 indicate the cut-off for a protein to be degraded. (B) Same as in (A), for CRBN PROTAC 2. (C) Comparison of proteomic changes after treatment with compounds 1 or 2. Fraction of DMSO after treatment with 1 μM VHL PROTAC 1 (X-Axis) or CRBN PROTAC 2 (Y-axis) are compared for each of the 54 foretinib-binding kinases. Dotted lines indicate the 80% cutoff for degradation, and areas of selectivity for 1 or 2 are highlighted. The light blue dots indicate proteins that are degraded in PROTAC treatment and in the control compounds, and so are not classified as bona fide PROTAC targets.
Based on the control compounds (foretinib, compounds 3 and 4), we chose a protein abundance fold-change cut-off value of 0.8 when normalized to DMSO. That is, a protein must have been reduced by 20% or greater in order to be considered a bona fide degradation target. Using this cut-off and compiling the union of both concentrations of each PROTAC, we found that on a proteomic level, compounds 1 and 2 caused the down-regulation of 123 and 204 proteins, respectively (Figure 2A and B). If, however, the proteins which are also down-regulated by the respective negative controls (foretinib and/or the epimer control 3 for VHL PROTAC 1; foretinib and N-methyl control 4 for CRBN PROTAC 2) by more than 10% were filtered out, then only 36 proteins are degraded by compound 1 and 62 proteins are degraded by compound 2. This additional filter is necessary as it controls for proteins down-regulated through binding of foretinib. Such situations include transcriptional effects of foretinib alone or ligand induced destabilization. Thus, the more conservatively filtered set represents downregulation most likely to be caused through a bona fide PROTAC-induced mechanism of degradation. In total, 86 proteins were downregulated by one or the other PROTAC, and only 12 were degraded by both.
Does affinity correlate with degradation potency?
To date, all published PROTAC molecules have been generated using ligands that have high affinity and fairly high selectivity for the protein to be degraded. Since a correlation between affinity and the extent of degradation might be assumed, we focused on those 54 kinases which are both foretinib targets and for which we had quantitative proteomics data (Figure 2C, and Supplemental Table 3). These selected kinases possess a range of affinities for the two PROTACs and provide a large test-set of possibly degraded proteins. Surprisingly, we found that the degradation profile of each PROTAC was far more selective than the binding profile of foretinib. Of these 54 kinases, 9 were degraded by VHL PROTAC 1 and 14 were degraded by CRBN PROTAC 2, with 6 of these kinases in common (Figure 2C).
Interestingly, for the 54 kinases tested, there was no correlation between the affinity of either compound 1 or 2 and the extent of PROTAC-induced degradation (Figure 3A and B). Evident from this analysis are high-affinity pairs (SLK, Axl) that are not degraded by either PROTAC. Additionally, there are several pairs for which the kinase affinity is far above the percent of control cut-off for a bona fide binder, yet the kinase is still efficiently degraded. An excellent example of this is p38α, which has a percent of control of 78, yet is efficiently degraded by PROTAC 1 (Figure 2C, 3A).
Figure 3. Affinity does not correlate with degradation efficiency, and stable ternary complexes more robustly predict degradation.

(A) Comparison of VHL PROTAC 1’s degradation efficiency and binding affinity for 54 kinases. For each foretinib-binding kinase expressed in MDA-MB-231 cells, the fraction remaining by whole cell proteomics after treatment with 1 μM VHL PROTAC 1 is shown on the Y-axis as a fraction of the DMSO-treated samples. PROTAC’s affinity for that target is shown on the X-axis as a percent of control (KinomeScan). A target is considered degraded if it’s percent of DMSO is below 80%, and a target is considered to bind to the PROTAC if it’s percent of control is less than 35%. (B) Same as in (A), except for CRBN PROTAC 2. (C) Degraded kinases form stable ternary complexes with VHL. GST-tagged VHL/Elongin B/Elongin C were immobilized on glutathione beads and incubated with whole cell extract of MDA-MB-231 cells with the indicated concentrations of compounds 1 or 3. The beads were washed and bound proteins eluted with SDS buffer and analyzed by western blot. See Figure S2 for more robust characterization of degradation and affinity, and Figure S3C for further ternary complex blots of degraded targets.
Based on these observations, we conclude that the extent of degradation of a target by a PROTAC does not necessarily correlate with the PROTAC’s affinity for that target. In our dataset, a compound-kinase pair with a higher affinity interaction was no more potently degraded than one with lower affinity. To confirm this relationship, or lack thereof, we more carefully analyzed the affinity and degradation profiles of 12 different kinases, sampling kinases that were degraded as well as kinases that despite high affinity for either PROTAC were not degraded in our quantitative proteomics dataset (Figure S2). In most cases, the western blot data confirmed the proteomics dataset and the extent of degradation for each target agreed well (Figure S2D–E). C-Abl and Arg were decreased by 30% by compound 1 in the proteomics dataset, but showed no degradation by this compound by western blot. Despite this, both proteins were degraded by compound 2 using both techniques, illustrating the importance of secondary confirmation of proteomic datasets. Additionally, for each kinase a dissociation constant (Kd) was determined using multi-point, competitive binding (KinomeScan, Figure S2B–C). The Kd for almost all cases correlated well with that determined from the single point percent of control measurement discussed above (Figure S2F–G), with the only exception being CDK4 which bound to VHL PROTAC 1 with 100% inhibition in the single-point experiment yet had a Kd of 10 μM. The dose responsive changes in protein levels varied for each kinase-PROTAC pair and did not correlate with the affinity of the kinase for the PROTAC. With high affinity for both PROTACs 1 and 2, MerTK and RIPK2 reached more than half-maximal degradation at the lowest concentration tested (30 nM) and demonstrated a ‘hook effect’ at higher concentrations of CRBN PROTAC 2. Strikingly, the VHL- and CRBN-recruiting PROTACs show differential selectivity towards the p38-MAPK family. While p38α and p38δ homologs share 61% identity, they were differentially degraded by the VHL- and CRBN-recruiting PROTACs. p38δ was slightly degraded (~30%) by VHL PROTAC 1, while it was almost completely degraded by the CRBN PROTAC 2 (DC50=27nM, Dmax=91%). Contrary to this, the VHL-recruiting PROTAC 1 degraded p38α potently (DC50=210nM, Dmax=91%), yet compound 2 was unable to degrade p38α.
A stable ternary complex between VHL and a potential substrate is required for degradation
We were surprised to see p38α efficiently degraded by VHL PROTAC 1 with a DC50 value (210 nM, Figure S2A) that is far lower than its binding affinity (Kd = 11 μM). Furthermore, additional experiments support a bona fide PROTAC-based mechanism, despite low binding capacity: 1) proteasome inhibition rescues PROTAC-induced p38α degradation, 2) p38α protein is degraded rapidly within 12 hours of PROTAC treatment, 3) PROTAC 1 rapidly accelerates the basal half-life of p38α from an hour to a minute timescale, and 4) p38α mRNA levels do not change after 24 hours of PROTAC 1 treatment (Figure S3A–D). In contrast to this weak affinity interaction resulting in profound degradation, several kinases with high affinity for PROTAC 1 were not degraded (SLK and Axl). VHL PROTAC 1 binds to Axl with a Kd of 26 nM and potently inhibits Axl signaling at 300 nM, yet does not degrade Axl at concentrations up to 10 μM (Figure S2A–C, S3F).
We next hypothesized that favorable protein:protein interactions between a target protein and a recruited E3 ligase could compensate for a weak target:PROTAC affinity and thus drive target degradation. Conversely, unfavorable protein:protein interactions might impede target protein degradation and might explain why the high-affinity kinase-PROTAC pairs we observed were not degraded. To assess the contribution of ternary complex formation, we used GST-tagged VHL as bait to trap potential ternary complex members in the presence of PROTAC. We elected to use excess VHL as bait, rather than co-immunoprecipitation with endogenous VHL, to avoid false negatives due to competition between potential substrates. When a whole cell lysate from MDA-MB-231 cells was incubated with different concentrations of compounds 1 or 3 and the immobilized VHL bait, interactions for several kinases were detected (Figure 3C and Figure S3F). Encouragingly, we found a strong correlation between those proteins that stably interacted with VHL and those that are degraded. High-affinity kinase:PROTAC pairs that are not degraded, such as SLK (Kd = 450 nM), show no detectable enrichment in the presence of PROTAC, indicating that these kinases are not able to form a ternary complex likely due to steric clashes. In contrast, proteins that are degraded (c-Met, RIPK2, p38α with Kd’s of 310 nM, 1.6 μM, and 11 μM, respectively) are capable of forming a stable complex with VHL only in the presence of the active PROTAC 1 and not in the presence of the inverted stereocenter-containing control compound 3. Intriguingly, VHL-recruiting PROTACs do not induce degradation of either c-Abl or Arg despite a stable interaction in the presence of PROTAC 1. This corresponds to our previous work on PROTACs targeting c-Abl (Lai et al., 2016). In that study, we found that VHL-recruiting PROTACs that incorporated imatinib or bosutinib were unable to degrade c-Abl despite robust target engagement, whereas dasatinib-based PROTACs degraded c-Abl well. The biophysical basis for these results require further study but will likely involve the accessibility of lysine residues that can be efficiently ubiquitinated.
Molecular analysis of PROTAC-mediated protein-protein interactions
Given the paradoxical ability of PROTAC 1 to bind weakly to p38α, yet induce a high affinity interaction between p38α and VHL, we sought a molecular understanding for how p38α forms a high-affinity PROTAC-induced ternary complex with VHL. To do this, we docked previously published structures of p38α and VHL with the PROTAC 1 and used short, 120 nanosecond molecular dynamics simulations to relax the structure into a low energy conformation. This model revealed a vast protein-protein (800 Å2) interaction surface between p38α and VHL.
According to the model, the linker region of VHL PROTAC 1 must adopt a kinked conformation in order to accommodate protein-protein interactions between VHL and p38α. Arginine 69 of VHL provides close, hydrophobic contacts to the linker region while alanine 40 of p38α is poised to accommodate both Arg69 of VHL and the kinked linker of the PROTAC (Figure 4A). To experimentally validate this model, we mutated Ala40 to either lysine or valine, predicting that the former would abrogate any favorable protein-protein interactions while the latter would not. Using purified p38α protein in either a luminescent proximity assay (AlphaScreen) or the VHL pulldown assay, we found that the A40K mutant has no ability to form a stable ternary complex, while the A40V mutant has roughly equivalent ternary complex formation as wildtype underscoring the highly specific interactions that occur within PROTAC-induced ternary complexes (Figure 4B–C). Each of these mutations do not greatly decrease the protein’s stability or kinase activity, nor does the A40K mutation abrogate binding to VHL PROTAC 1 (Figure 4D–E). Finally, when p38α is overexpressed in HeLa cells, the wildtype protein is efficiently degraded by VHL PROTAC 1, whereas the A40K mutant shows no degradation. This indicates that ternary complex formation is necessary for PROTAC-induced degradation.
Figure 4. Favorable protein-protein interactions stabilize the compound 1-induced complex between p38α and VHL.

(A) VHL, PROTAC 1, and p38α were docked and a 120 ns molecular dynamics simulation relaxed the structure. Alanine 40 on p38α interacts with the kinked linker region of PROTAC 1. (B) Proximity-based AlphaLisa detects a ternary complex between GST-VHL and His-p38α. VHL PROTAC 1 and purified p38α and VHL were incubated in the presence of Glutathione Donor and Anti-his acceptor beads, and the extent of ternary complex formation was assessed by excitation at 680 nm and detection of emission at 615 nm. (C) Stable interaction between p38α and VHL are interrupted by the A40K mutation. Immobilized VHL/EloB/EloC was used as a bait to trap purified p38α in the presence of the indicated concentrations of compound 1 or 3. The far right lane represents a 1:25 dilution of the total protein used in each pulldown reaction. (D) Despite inhibition of ternary complex formation, the A40K mutation is inhibited by VHL PROTAC 1 to a similar extent as measured by a Z′Lyte cascade assay in which active p38α phosphorylates inactive MAPKAPK2 which phosphorylates a FRET-pair-containing peptide. Active MAPKAPK2 (without p38α protein) was also included as s control. (E) Thermal Stability of the WT, A40K, and A40 mutants are similar, as assessed by Sypro Orange fluorescence with increasing temperature. (F) Overexpressed wildtype p38α is degraded in cells, but not the A40K mutant. HeLa cells were transfected with a pcDNA5 vector containing FLAG-tagged p38α (wildtype or p38α), and then treated with VHL PROTAC 1 at the indicated concentrations for 24 hours.
Discussion
In this study, we sought to develop principles relating to PROTAC design. At the simplest level, a PROTAC requires both an E3 ubiquitin ligase recruiting ligand and a target protein ligand, each possessing a suitable solvent-exposed position in order to connect the ligands via a linker. However, our group and others have shown that the design of PROTACs is much more nuanced, both in terms of the linker geometry and the substrate-targeting ligand employed (Buckley et al., 2015; Gadd et al., 2017; Lai et al., 2016).
Here, we provide evidence that the factor that best correlates with the degradation potency of a given substrate protein is the ability of that protein to form a stable ternary complex with the PROTAC and recruited E3 ubiquitin ligase. Steric clashes likely explain the lack of degradation of those kinases that bind PROTACs but do not form a stable ternary complex with VHL (e.g. SLK, see Figure 3C). It is possible that longer linker geometries will access these proteins for PROTAC-mediated degradation. Our lab first postulated (Buckley et al., 2015) that protein-protein interactions might be a driver of degradation potency for the model substrate HaloTag7 and an alkyl chloride-conjugated VHL ligand. Specifically, one compound was able to degrade the target protein with negligible hook effect, yet increasing the linker length resulted in a more profound hook effect. Based on these observations, we postulated that the ideal compound oriented VHL and target protein in such a way as to allow favorable protein-protein interactions to occur, thus broadening the effective concentration range of the PROTAC (Buckley et al., 2015; Douglass et al., 2013). In support of this conclusion, we also observed that additional PROTACs with identical linker lengths but decreased VHL affinities could still degrade the target protein comparably, which indicated that protein-protein interactions between VHL and the target could compensate for weakened PROTAC:E3 ligase affinity.
Additional support for this hypothesis comes from a VHL-recruiting PROTAC based on the HER2 kinase inhibitor lapatinib described in the accompanying paper (Burslem and Smith et al., in press). One PROTAC with an optimal linker length between VHL and HER2 degrades HER2 almost completely, whereas a PROTAC with a slightly longer linker (a single ethylene glycol unit) causes nominal degradation. We hypothesize that the shorter linker PROTAC facilitates favorable protein-protein interactions between VHL and HER2, which are lost by linker extension.
A recently published structure of the ternary complex between VHL, BRD4 and a BRD4-degrading PROTAC brings the details of one such E3 ligase:target protein interaction into light (Gadd et al., 2017). These unique PROTAC-mediated protein interfaces between VHL and BRD4 are not possible between VHL and BRD2, explaining the selectivity that this compound shows for BRD4 degradation. Moreover, mutation of these BRD4-specific interfaces to the corresponding residues on BRD2 results in loss of both ternary complex formation and degradation potency in cells.
These results indicate that at least for some PROTAC-substrate pairs, such as p38α (see below), ternary complex formation is the driving force for degradation potency and that a stable ternary complex is highly predictive of the degradation profile of a PROTAC. One can envision that, in contrast to expensive quantitative proteomics, basic protein identification of eluted proteins after E3 ligase pulldown in the presence of PROTAC may provide a quick and inexpensive method to profile that PROTAC’s selectivity. However, our data also reveals a potential exception: degradation of proteins such as c-Abl and Arg was not facilitated by VHL PROTAC 1 despite forming a stable ternary complex with VHL in cell extracts (Figure S3G). In these instances, it is possible that such target proteins have lysine residues that are either suboptimal for ubiquitin transfer or recognition by the proteasome once ubiquitinated (Guharoy et al., 2016; Mattiroli and Sixma, 2014). Different E3 ligases may offer different lysine selectivity profiles and explain why CRBN has had more success in degrading c-Abl than has VHL (Lai et al., 2016). Our lab has recently explored additional E3 ligases and their ability to be hijacked by PROTAC molecules. Five out of the six ligases could degrade non-natural substrates, indicating that expanding the repertoire of E3 ligases for which high affinity small molecule ligands are available will further advance the PROTAC technology (Ottis et al., 2017).
Our study also highlights some differences between the two most commonly used E3 ubiquitin ligases used in PROTACs: VHL and CRBN. Although the chemical linker space explored is limited, compounds 1 and 2 show large differences in both kinase binding ability and total number of degraded substrates, with CRBN shown to be more promiscuous in this current study. CRBN PROTAC 2 does have higher affinity for each target tested (possibly due to hydrophobic collapse of VHL PROTAC 1), but also had more low-affinity targets degraded than VHL. However, several published CRBN-recruiting PROTACs show equivalent selectivity compared to other VHL-recruiting PROTACs, but only when based on a selective warhead (Bondeson et al., 2015; Gadd et al., 2017; Winter et al., 2015). It is, therefore, interesting to speculate on the endogenous role of VHL and CRBN, their ‘natural’ substrates, and how this might inform the degradation profiles we observed in this study.
VHL has a well-characterized substrate whose molecular recognition is exquisitely specific: there is a 1000x fold increase in binding affinity upon hydroxylation of a single proline residue of the VHL protein’s endogenous substrate HIF1α (Hon et al., 2002). The exact details of CRBN recognition of endogenous substrates are still fairly unknown; however, it has been implicated in the degradation of MEIS2 and glutamine synthetase. In addition, a speculative role in binding DNA and interacting with or ubiquitinating core histone complexes has been proposed (Fischer et al., 2014; Hartmann et al., 2014; Nguyen et al., 2016). Furthermore, the binding surface of CRBN is remodeled upon IMiD binding to facilitate the recruitment and ubiquitination of neo-substrates like GSPT1 and ZFP91 (An et al., 2017; Matyskiela et al., 2016; Petzold et al., 2016). Based on these observations, perhaps VHL possess a protein surface near the binding site devoid of ‘hot spots’ of potential protein-protein interfaces, whereas CRBN has a surface highly amenable to such interactions. This should be taken into account when beginning a PROTAC program for a new target. Importantly, both ligases are able to degrade both cytosolic (RIPK2 and c-Abl), membrane-bound (c-Met and other RTKs, see Burslem and Smith et al., in press) and nuclear proteins (ERRα and IKZF1), indicating that within these cellular sub-compartments both ligases have equivalent access to potential PROTAC targets. An additional caveat with CRBN-recruiting PROTACs is the degradation of proteins independent of the targeting warhead through IMiD-dependent remodeling of the surface of CRBN. In our proteomic dataset, we observed degradation of GSPT1 and ZFP91 with the active PROTAC compound 2 but not with the N-methylated negative control 4. This indicates that adding the linker/targeting warhead moiety to lenalidomide does not abrogate recruitment of neo-substrates to CRBN, though this hasn’t been reported in other datasets involving CRBN-recruiting PROTACs(An et al., 2017; Matyskiela et al., 2016; Winter et al., 2015).
Here, p38α presents the most thought-provoking example of a protein-protein interaction mediated by a PROTAC published to date. Though the precise details of the interaction are yet to be elucidated, p38α binds to the VHL PROTAC compound 1 with a dissociation constant greater than 10 μM, yet is degraded and forms a detectable ternary complex 20–50-fold more potently. We ruled out the intriguing hypothesis that p38α might be a bystander protein: one that is degraded without directly binding to the PROTAC, but instead binds a direct PROTAC target and receives collateral or bystander ubiquitination. Using purified protein, we showed that p38α forms a stable ternary complex with VHL only in the presence of the active PROTAC 1. In our molecular dynamics analysis, we identified residues that are necessary for p38α ternary complex formation with VHL. It is worth noting that there are likely multiple ways in which a ternary complex can be stabilized or sterically hindered, even among similar proteins with the canonical kinase fold. The p38α A40K mutation completely hindered the ability of PROTAC 1 to form a complex, but possessing such an alanine or a lysine is not predictive of degradation potency or lack thereof. Of proteins not degraded by compound 1, the corresponding residue ranges from a glutamate (Axl) to a lysine (SLK) while the corresponding residue on proteins that are degraded also range from histidine (c-Met) to a serine (RIPK2). We hypothesize that in each ternary complex, the interacting surfaces of each VHL:compound 1:target protein ternary complex are likely different, making ab initio PROTAC design challenging.
However, the anomaly of p38α highlights a unique opportunity for PROTACs to degrade proteins for which only a low-affinity ligand is available. PROTACs have the potential to expand the ‘drug target space’, i.e., the fraction of the proteome that is considered ‘druggable’ (Lazo and Sharlow, 2016). The vast majority of the proteome lacks enzymatic active sites typically targeted by small-molecule inhibition. Moreover, many disease-causing proteins such as oncogenic drivers function through broad protein-protein interactions that are difficult to inhibit using traditional small molecules (Ostrem and Shokat, 2016). However, if a PROTAC can be designed to induce favorable protein-protein interactions between the target protein and an E3 ligase (a biophysical property amenable to high-throughput screening), then weak affinity ligands or even those that bind to non-inhibitory sites on the protein target could be converted into potent PROTAC molecules. In this situation, high-throughput techniques to screen a larger volume of compounds for ternary complex formation, rather than degradation, could be employed (Milroy et al., 2014).
As the therapeutic significance of the PROTAC platform continues to be realized, the specificity of each PROTAC is crucially important. This study highlights both the PROTAC platform’s added layer of specificity – the requirement for a stable ternary complex – as well as important differences between the use of VHL and CRBN in PROTAC design. Overall, this study illustrates the potential of PROTACs to generate selective chemical probes or therapeutics from poorly selective or weak affinity ligands.
Significance
Proteolysis Targeting Chimera (PROTACs) are an emerging strategy with great therapeutic potential. Here, we demonstrate two intriguing benefits of this technology. Starting with a promiscuous kinase inhibitor which binds >130 kinases, we converted this molecule into a PROTAC which degrades < 10. This indicates that PROTACs may give researchers an additional level of specificity over traditional inhibitors. Additionally, we demonstrate the effective degradation of a therapeutically relevant protein, p38α/MAPK14, despite weak binding affinity. These data support a model by which the PROTAC mediates a protein complex that is stabilized by protein-protein interactions between the E3 ubiquitin ligase and the target protein. This study offers interesting examples to emerging PROTAC design principles and demonstrates the capacity of this technology to expand the scope of what is currently considered ‘druggable.’
STAR Methods
Contact for Reagent and Resource Sharing
Further information and request for resources and reagents should be directed and will be fulfilled by Craig Crews (craig.crews@yale.edu). However, the human p38α (MAPK14) plasmid used in this study was obtained under MTA from Dr. D. Martin Watterson from Northwestern University.
Experimental Model and Subject Details
MDA-MB-231 and HeLa cells were obtained from the American Type Culture Collection (ATCC), cultured in RPMI-1640 (1X) and DMEM (1X) medium, respectively, containing 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin and grown in a humidified incubator at 37°C, 5% CO2.
Method Details
Chemical Syntheses
Details of Chemical Syntheses can be found in the Supporting Information.
Immunoblotting
Lysates from MDA-MB-231 cells were washed once with ice cold PBS (1X), followed by lysis in buffer containing 25 mM Tris [pH 7.5], 0.25% sodium deoxycholate, 1% Triton X-100, supplemented with protease (Roche) and phosphatase inhibitors (10 mM NaF, 1 mM Na3VO4, and 20 mM β-glycerophosphate), unless otherwise noted. Lysates were spun at 15,000 × g for 10 min at 4°C and supernatant was evaluated for protein content using a Pierce BCA Protein Assay (Thermo Fisher Scientific). 25 to 50 μg of protein was loaded onto 8% SDS-PAGE gels or 4–15% Mini-PROTEAN TGX precast gradient gels (Bio-Rad), transferred to nitrocellulose membranes, and probed with the specified antibodies overnight at 4°C in 1X TBS-Tween containing 5% non-fat milk. Immunoblots were visualized using a Bio-Rad ChemiDoc imaging instrument and subsequently processed and quantified using the accompanying Bio-Rad Image Lab software.
Sample preparation for quantitative mass spectrometry analysis
MDA-MB-231 cells were seeded at 2.5 × 106 cells per 100 mm plate, allowed to adhere, and grown in the presence of compound or vehicle at 37°C. After 24 hours, plates were immediately placed on ice, rinsed with ice-cold PBS (1X) three times to remove any remnant serum, and lysed under denaturing conditions (50 mM Tris pH 8.5, 8 M urea, 1% SDS, supplemented with protease (Roche) and phosphatase inhibitors). Lysates were sonicated with a microtip probe and subsequently spun at 14,000 × g for 10 min at 4°C. Supernatant was collected and immediately snap frozen at −80°C before sending samples to the Thermo Fisher Center for Multiplexed Proteomics at Harvard Medical School (TCMP@HMS) for quantitative whole proteome analysis. Sample were prepared as previously described (Weekes et al., 2014) with the following modification. A micro-BCA assay (Pierce) was used to determine the final protein concentration in the cell lysate. Proteins were reduced and alkylated as previously described. Proteins were precipitated using methanol/chloroform. In brief, four volumes of methanol were added to the cell lysate, followed by one volume of chloroform, and finally three volumes of water. The mixture was vortexed and centrifuged to separate the chloroform phase from the aqueous phase. The precipitated protein was washed with one volume of ice cold methanol. The washed precipitated protein was allowed to air dry. Precipitated protein was resuspended in 4 M Urea, 50 mM Tris pH 8.5. Proteins were first digested with LysC (1:50; enzyme:protein) for 12 hours at 25 °C. The LysC digestion is diluted down to 1 M Urea, 50 mM Tris pH8.5 and then digested with trypsin (1:100; enzyme:protein) for another 8 hours at 25 °C. Peptides were desalted using a C18 solid phase extraction cartridges as previously described. Dried peptides were resuspended in 200 mM EPPS, pH 8.0. Peptide quantification was performed using the micro-BCA assay (Pierce). The same amount of peptide from each condition was labeled with tandem mass tag (TMT) reagent (1:4; peptide:TMT label) (Pierce). The 8-plex labeling reactions were performed for 2 hours at 25 °C. Modification of tyrosine residue with TMT was reversed by the addition of 5% hydroxyl amine for 15 minutes at 25 °C. The reaction was quenched with 0.5% TFA and samples were combined at a 1:1:1:1:1:1:1:1 ratio. Combined samples were desalted and offline fractionated into 24 fractions as previously described.
Liquid chromatography-MS3 spectrometry (LC-MS/MS)
12 of the 24 peptide fraction from the basic reverse phase step (every other fraction) were analyzed with an LC-MS3 data collection strategy (Mcalister et al., 2014; McAlister et al., 2012; Ting et al., 2011) on an Orbitrap Lumos mass spectrometer (Thermo Fisher Scientific) equipped with a Proxeon Easy nLC 1000 for online sample handling and peptide separations. Approximately 5 μg of peptide resuspended in 5% formic acid + 5% acetonitrile was loaded onto a 100 μm inner diameter fused-silica micro capillary with a needle tip pulled to an internal diameter less than 5 μm. The column was packed in-house to a length of 35 cm with a C18 reverse phase resin (GP118 resin 1.8 μm, 120 Å, Sepax Technologies). The peptides were separated using a 180 min linear gradient from 3% to 25% buffer B (100% ACN + 0.125% formic acid) equilibrated with buffer A (3% ACN + 0.125% formic acid) at a flow rate of 600 nL/min across the column. The scan sequence for the Fusion Orbitrap began with an MS1 spectrum (Orbitrap analysis, resolution 120,000, 350–1350 m/z scan range with quadrapole isolation, AGC target 1 × 106, maximum injection time 100 ms, dynamic exclusion of 60 seconds). ‘Top N” (the top 10 precursors) was selected for MS2 analysis, which consisted of CID ion trap analysis, AGC 2.5 × 104, NCE 35, maximum injection time 200 ms). Charge state dependent quadrapole isolation was used for MS scans (1.2 Da for m/z =2, 1.0 Da for m/z = 3, and 0.8 Da for m/z 4–6). The top ten fragment ion precursors from each MS2 scan were selected for MS3 analysis (synchronous precursor selection), in which precursors were fragmented by HCD prior to Orbitrap analysis (NCE 55, max AGC 2.2 × 105, maximum injection time 300 ms, MS2 quadrapole isolation was set to 2.0 Da, resolution 60,000.
LC-MS3 data analysis
A suite of in-house software tools were used to for .RAW file processing and controlling peptide and protein level false discovery rates, assembling proteins from peptides, and protein quantification from peptides as previously described. MS/MS spectra were searched against a Uniprot human database (February 2014) with both the forward and reverse sequences. Database search criteria are as follows: tryptic with two missed cleavages, a precursor mass tolerance of 50 ppm, fragment ion mass tolerance of 1.0 Da, static alkylation of cysteine (57.02146 Da), static TMT labeling of lysine residues and N-termini of peptides (229.162932 Da), and variable oxidation of methionine (15.99491 Da). TMT reporter ion intensities were measured using a 0.003 Da window around the theoretical m/z for each reporter ion in the MS3 scan. Peptide spectral matches with poor quality MS3 spectra were excluded from quantitation (<200 summed signal-to-noise across 8 channels and <0.5 precursor isolation specificity).
Gas6 stimulation assays
MDA-MB-231 cells were plated at 3 × 105 cells per well in a 6-well dish, allowed to adhere, and switched to serum-free RPMI-1640 (1X) media the following day, at which point cells were pre-treated with the indicated compounds for 24 hours, followed by a 10-minute pulse of 500 ng mL−1 of Gas6 (R&D systems). After these incubations, cells were immediately placed on ice, rinsed with PBS, lysed, and boiled.
Cycloheximide chase assay
MDA-MB-231 cells were plated at 3 × 105 cells per well in a 6-well dish, allowed to adhere, and switched to serum-free RPMI-1640 media for 16 hr. Cells were then pre-treated with cycloheximide (Sigma) at 100 μg mL−1 for 1 hr prior to adding either HGF (100 ng mL−1), PROTAC (500 nM), or vehicle. At the indicated timepoints, cells were immediately placed on ice, rinsed with PBS, lysed, and boiled.
Transfections
Transfections were carried out using Lipofectamine 2000 (Invitrogen) in HeLa cells seeded at 2.5 × 106 cells per 100 mm plate. 10 μg of of FLAG-containing pcDNA5-p38alphaWT or pcDNA5-p38alphaA40K was used per transfection and Opti-MEM media was changed after 6 hours to DMEM (1X).
Quantitative PCR
MDA-MB-231 cells were plated at 3 × 105 cells per well in a 6-well dish, allowed to adhere, and treated with the indicated compounds for 24 hours, at which point cells were lysed and total RNA was isolated using TRIzol (ThermoFisher). cDNA was synthesized from 2 μg of total RNA per condition according to the manufacturer’s protocol (Applied Biosystems) and real-time PCR was performed with 800 nM primers, diluted with 4 μL SYBR Green Reaction Mix (Applied Biosystems). RT-PCR experiments were performed with the following protocol on a LightCycler 480 Instrument II (Roche): 95°C for 10 min, 40 cycles of 95°C for 15s and 60°C for 45s. qRT-PCR samples were performed and analyzed in triplicate, from two independent experiments. Beta-Tubulin was used for normalization. Primers used in this study were:
Constructs, protein expression and purification
Wild-type human p38alpha-MAPK was received as a gift from Dr. D. Martin Watterson (Northwestern University, under MTA) and is described before (Watterson et al., 2013). Mutants were generated by QuickChange Lightning site-diirected mutagenesis kit (Agilent). The plasmid contains an N-terminal His6 tag and encodes a region spanning amino acids 2-360 of the human p38alpha kinase (NCBI Reference Sequence: NM_139012). BL21-CodonPlus(DE3)-RIPL E. coli cells (Agilent Technologies) were transformed with pMCSG7-His6-p38alpha and were selected in LB medium containing carbenicillin (100 μg/mL), chloramphenicol (15 μg/mL), and spectinomycin (50 μg/mL) at 37°C until OD600=0.6. At this point, cells were induced with 1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) and grown at 25°C for 14 hr. Cell pellets were collected by centrifugation (5,000 rpm, 10 min, 4°C) and homogenized in lysis buffer (10 mM Tris pH 8.3, 500 mM NaCl, 5 mM β-mercaptoethanol, 10 mM imidazole, and 10% glycerol) containing a 1X protease inhibitor tablet (Roche). The homogenized cells were subsequently passed through a microfluidizer three times at 15k PSI and lysate was clarified by centrifugation (16,000 rpm, 45 min, 4°C). The resultant supernatant was then applied to Ni-NTA agarose beads (QIAGEN) with gentle rotation for 1 hr at 4°C, washed once with lysis buffer with 10 mM imidazole (pH 8.3), twice with lysis buffer containing 20 mM imidazole (pH 8.3), and eluted off of the nickel resin in lysis buffer containing 50 mM imidazole (pH 8.3). Eluted protein was assessed for identity and purity via coomassie staining of sample run on an SDS-PAGE gel and pure elutions were pooled, concentrated, and diluted in ion-exchange buffer A (10 mM Tris pH 8.3, 5 mM β-mercaptoethanol) until the salt concentration was 50 mM, before loading onto a Mono Q 5/50 GL column (GE Life Sciences). The protein was subjected to a step-wise wash protocol, followed by a linear gradient from 200–500 mM NaCl using ion-exchange buffer B (10 mM Tris 8.3, 1 M NaCl, 5 mM β-mercaptoethanol). Fractions were then assessed for purity via coomassie, pooled, concentrated, and run on a HiLoad 16/600 Superdex-200 column (GE Healthcare Life Sciences) using size-exclusion buffer (10 mM Tris pH 8.3, 150 mM NaCl, 5 mM β-mercaptoethanol). Pure fractions of p38α were pooled, concentrated to ~10 mg mL−1, aliquoted, and flash-frozen before storing at −80°C.
For the expression of GST-tagged VHL:Elongin B:Elongin C (herein referred to as GST-VHL), wild-type human VHL, Elongin B, and Elongin C were coexpressed in E. coli. BL21(DE3) cells were co-transformed with pBB75-Elongin C and pGEX4T-2-VHL-rbs-Elongin B and selected in LB medium containing carbenicillin (100 μg/mL) and kanamycin (25 μg/mL) at 37°C until OD600=0.8, at which point the culture was chilled to 16°C and induced with 0.4 mM IPTG for 16 hr. Cells were homogenized and lysed, as described above, except the lysis buffer was composed of 30 mM Tris [pH 8.0], 200 mM NaCl, 5% glycerol, 5 mM DTT containing a 1X protease inhibitor tablet (Roche). Clarified cell lysate was applied to Glutathione Sepharose 4B beads (GE Life Science) and gently rotated for 2 hr at 4°C. Beads were washed with four column volumes of lysis buffer, followed by four column volumes of elution buffer (50 mM Tris pH 8.0, 200 mM NaCl, 10 mM Glutathione). Eluted protein was assessed for identity and purity via coomassie staining of sample run on an SDS-PAGE gel and pure elutions were pooled, concentrated, and diluted in ion-exchange buffer A (30 mM Tris pH 8.0, 5% glycerol, 1 mM DTT) until the salt concentration was 50 mM, before loading onto a Mono Q 5/50 GL column (GE Life Sciences). The protein was subjected to a linear gradient of NaCl (0–500 mM NaCl) using ion-exchange buffer B (30 mM Tris 8.0, 1 M NaCl, 5% glycerol, 1 mM DTT). Fractions were then assessed for purity via coomassie, pooled, concentrated, and run on a Superdex-200 column (GE Life Sciences) using size-exclusion buffer (30 mM Tris pH 8.0, 100 mM NaCl, 10% glycerol, 1 mM DTT). Pure fractions of GST-VHL were pooled, concentrated to ~5 mg mL−1, aliquoted, and flash-frozen before storing at −80°C.
Protein target enrichment via PROTAC on immobilized VHL
Glutathione Sepharose 4B was washed twice with 1X TBS-T (Tris buffered saline plus 0.02% Tween 20) and then blocked for one hour at room temperature with 10% BSA in TBST. The beads were then washed again twice with TBS-T and once with wash buffer (50mM HEPES pH 7.5, 150mM NaCl, 1mM DTT, 0.01% NP40, 5 mM MgCl2, 10% Glycerol) and then purified GST-VBC was immobilized for two hours at 4°C at 360 pmole per μL of beads. The beads were then washed thrice with wash buffer, resuspended and p38α protein was added at 500 nM per 50 μL reaction with 5 μL of beads. The bead:p38α mixture was then aliquoted to separate tubes and PROTAC was added at the indicated concentration (PROTACs were intermediately diluted in 10% DMSO and 0.25% CHAPS) and this was incubated at 4°C for two hours. The beads were washed thrice with 10 column volumes of wash buffer and then eluted with SDS loading buffer.
For experiments in which the input substrate is a whole cell lysate, the sample was prepared as follows. Ten 150 mm dishes of confluent MDA-MB-231 cells were washed with TBS and then scraped into a 15 mL falcon tube. The cells were pelleted, resuspended in wash buffer, and then lysed by sonication (Branson sonicator microtip, power = 7, 50% duty cycle for 3 cycles of two minutes on and two minutes rest). The lysate was cleared by centrifugation and then added to the beads as an input substrate, as above.
Molecular Dynamics (MD) Simulations
The starting coordinates for p38α came from the crystal structure downloaded from Protein Data Bank (PDB) entry 1W82. In order to replace the ligand in this structure with foretinib, the PDB entry 5IA4 was used by overlaying its protein backbone with that of 1W82, transferring foretinib to the 1W82 structure and replacing the original ligand. The obtained p38α-foretinib complex was subject to the Protein Preparation Wizard of Maestro 2016-3 program available from Schrodinger Inc. (New York City, NY), with which the hydrogen atoms were added; the missing side chains were built; and the protonation states were assigned assuming a pH of 7.0 for the ionizable groups. An energy minimization of the complex was performed for 500 steps.
The starting coordinates for VHL came from the PDB entry 4W9H. The starting coordinates for the p38α:PROTAC:VHL trimer were prepared as follows. (1) The electrostatic surface was generated for p38α-ligand complex and VHL-ligand complex, respectively. (2) The VHL-ligand complex was set to have different relative dispositions with respect to the p38α-ligand complex in a way that the hydrophobic patch of the VHL-ligand surface opposed different hydrophobic patches and grooves of the p38α-ligand surface, thus producing different starting modes in terms of the relative dispositions between p38α and VHL. (3) For each starting mode, a linker was built to connect foretinib and VHL ligand and form the full PROTAC. And (4) an energy minimization of 500 steps was performed for each starting point of trimer.
OPLS3 force-field was used throughout the calculation steps. The torsional angle parameters were examined with Force Field Builder program and found that the torsional angles between the amide and cyclopropyl group and between the fluorophenyl group and the oxygen ether atom attached to the quinoline group in foretinib needed corrections; and thus the new torsional profiles were generated to match the profiles given by Jaguar quantum mechanical calculations.
Each starting point of the p38α:PROTAC:VHL trimer was subject to molecular dynamics (MD) simulation. The system setup was done using System Builder of Maestro program, in which the periodic boundary condition was used; the box shape was orthorhombic with absolute size of 100×100×100 Å3. The explicit waters were added. The system was neutralized using Na+/Cl− ions and salted into 0.15 M ionic strength. The MD was done using Desmond Multisim version 3.8.5.19 which was an eight-stage process: task; simulation of 100 picosecond with Brownian dynamics NVT with T at 10 K, small time-steps and restraints on solute heavy atoms; simulation of 12 picosecond, NVT ensemble, T at 10 K, small time-steps and restraints on solute heavy atoms; simulation of 12 picosecond, NPT ensemble, T at 10 K and restraints on solute heavy atoms; solvation of potential unfilled pockets; simulation of 12 picosecond up to the target temperature of 310 K, NPT ensemble and restraints on solute heavy atoms; simulation of 24 picosecond, NPT ensemble without restraints at T of 310 K; and finally, production run of 120 nanosecond. During the production run, coordinate frames were saved at every 10 picosecond. The target pressure was set to 1.01325 bar in the related steps.
The post-simulation analysis after each run was done as follows. The last 20 nanosecond of trajectory frames were extracted. A clustering analysis using hierarchical clustering method was performed. The distance between any two members (frames) was the root-mean-square deviation (RMSD) of the solute heavy atoms between the members after overlaying them. The cutoff distance was 2 Å. Every frame was used. The structure closest to the centroid of each cluster was written out as the representative structure of that cluster. The representative structure of the largest cluster for each MD simulation was considered as the representative structure of that simulation run. Such a structure can be considered as the most populated conformation of that run.
The MD simulation was performed using the g2.2×large instances of Amazon Web Service cloud machines. The Desmond GPU-enabled code was used and mainly run using GPU.
AlphaLISA Assay for Ternary Complex Formation
Assays were performed at room temperature and reagents were diluted in buffer containing 50 mM HEPES, 50 mM NaCl, 69 mM BRIJ-35, and 0.1 mg mL-1 BSA. Recombinant GST-VHL-ElonginB-ElonginC (VBC) was mixed with His6-p38α and PROTAC (diluted from a 6x stock) to a final volume of 15 mL per well in a OptiPlate-384 well microplate (PerkinElmer) and incubated for 30 min. VBC and p38α were kept at a constant concentration of 50 nM and 100 nM, respectively. 7.5 mL of Alpha Glutathione Donor beads (PerkinElmer) were added to each well and plates were incubated for 15 min. 7.5 mL of and Anti-6xHis AlphaLISA Acceptor beads (PerkinElmer) were added to each well and plates were incubated for 45 min. Plates were read on a SYNERGY 2 microplate reader (BioTek Instruments) with an excitation wavelength of 680 nm and emission wavelength of 615 nm. Plates were analyzed using the Gen5 microplate reader and imager software (BioTek Instruments).
Differential Scanning Fluorimetry
p38α protein was diluted to 1 mg/mL in Tris-buffered saline and SYPRO Orange (Sigma) was added from a 500x concentrated stock in DMSO. 15 μL of protein was added to each well of a 384-well qPCR plate. 1.5 μL of a vehicle control or a 500 μM solution of foretinib in 50% DMSO was then added to appropriate wells, and the plate was heated to 95 degrees celcius in a Roche Lightcycler 480 II, measuring fluorescence ten times per degree. The protein melt analysis program (Roche) was then used to calculate melting temperatures.
Z′-LYTE Kinase Assay
Z′LYTE kinase assay was performed according to the manufacturer’s protocol (ThermoFisher). Briefly, inactive MAPKAPK2 (13 nM), p38α mutants (between 0.5 and 15 nM, depeding on specific activity), and VHL PROTAC 1 (varying concentrations from 48 nM to 100 μM) were mixed in Kinase Buffer A and the kinase reaction was initiated by addition of ATP (20 μM) and Ser/Thr peptide 4 (1 μM). After a 30 minute reaction, a 1:10,000 dilution of Development Reagent A in Development Reagent B was added at 1/3 the kinase reaction volume. After 30 minutes of development, the trFRET signal between the fluorophores on Ser/Thr peptide 4 was measured using an excitation wavelength of 360 nm and emission wavelengths of 460 and 528. The emission ratio was determined by dividing the emission at 460 nm by the emission at 528, and results were plotted in Graphpad Prism.
Quantification and Statistical Analysis
Western blot data in Figure S3 was quantified by using the band feature in Image Lab, and duplicate values were averaged and analyzed in Graphpad Prism. DC50s and Dmax values were fitted using a three parameter [inhibitor] versus response and reported directly from the Prism output.
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| c-Abl | SantaCruz | 23 |
| Arg | SantaCruz | 81154 |
| Axl | Cell Signaling | 4939 |
| p-AKT | Cell Signaling | 4060 |
| CDK4 | Cell Signaling | 12790 |
| DDR1 | SantaCruz | 532 |
| EphA2 | Cell Signaling | 6997 |
| FLAG M2 | Sigma | F1804 |
| GAPDH | Cell Signaling | 2118 |
| MerTK | Cell Signaling | 4319 |
| p38alpha | Cell Signaling | 9218 |
| p38alpha | Cell Signaling | 9228 |
| p38delta | Cell Signaling | 2308 |
| RIPK2 | Cell Signaling | 4142 |
| c-MET | Cell Signaling | 8198 |
| SLK | Cell Signaling | 41255 |
| Src | Cell Signaling | 2123 |
| CUL2 | Invitrogen | 700179 |
| VHL | Cell Signaling | 68547 |
| Tubulin | Sigma | T9026 |
| HRP linked Mouse IgG | GE Life Sciences | NA931 |
| HRP Linked Rabbit IgG | GE Life Sciences | NA934 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Cycloheximide | Sigma | C104450 |
| Epoxomicin | Crews laboratory | |
| Glutathione Sepharose 4B beads | GE Life Sciences | 17075601 |
| Ni-NTA agarose | QIAGEN | 30250 |
| Alpha Glutathione Donor beads | PerkinElmer | 6765300 |
| Anti-6xHis AlphaLISA Acceptor beads | PerkinElmer | AL128C |
| Recombinant Human Gas6 Protein | R&D Systems | 885-GSB-0503 |
| SYPRO Orange Protein Gel Stain | Sigma | S5692 |
| TRIzol Reagent | ThermoFisher | 15596018 |
| MAPKAPK2 Protein, Inactive | ThermoFisher | PV3316 |
| MAPKAPK2 Protein, Inactive | ThermoFisher | PV3317 |
| Critical Commercial Assays | ||
| Z′-LYTE Kinase Assay Kit – Ser/Thr 4 Peptide | ThermoFisher | PV3177 |
| ThermoFisher | ||
| Experimental Models: Cell Lines | ||
| MDA-MB-231 | ATCC | HTB-26 |
| HeLa | ATCC | CCL-2 |
| Oligonucleotides | ||
| Tub_F: TGGACTCTGTTCGCTCAGGT | Yale School of Medicine | N/A |
| Tub_R: TGCCTCCTTCCGTACCACAT | Yale School of Medicine | N/A |
| p38α_F: TCGCATGAATGATGGACTGAAAT | Yale School of Medicine | N/A |
| p38α_R: CCCGAGCGTTACCAGAACC | Yale School of Medicine | N/A |
| Software and Algorithms | ||
| Image Lab 6.0 | BioRad | N/A |
| Graphpad | Prism | N/A |
| Pymol | Schrodinger LLC | |
| Equipment | ||
| Synergy 2 microplate reader | Biotek | |
Supplementary Material
Supplemental Table 1 – KinomeScan data for foretinib and foretinib-based PROTACs across 428 human kinases, related to Figure 1.
Supplemental Table 2 – Raw peptide quantification of MDA-MB-231 proteomes after 24-hour treatments with foretinib and foretinib-based PROTACs, related to Figure 2.
Supplemental Table 3 – Compilation of KinomeScan data and quantitative proteomics for the 54 kinases bound by foretinib and expressed in MDA-MB-231 cells, related to Figure 3.
Highlights.
Development of VHL- and CRBN-recruiting PROTACs based on a promiscuous kinase inhibitor
Degradation is more selective than parent warhead affinity
Positive protein-protein interactions drive the selectivity and potency
VHL-recruiting PROTACs are more selective than CRBN-PROTACs
Acknowledgments
We thank all members of the Crews laboratory for helpful discussion and A Rice and K Roberts for technical support. DPB is supported by an NCI Predoctoral to Postdoctoral Fellow Transition Award (F99 CA212229-02). GMB is a supported by a Leukemia and Lymphoma Society Career Development Award. JH is supported by an R50 Research Specialists Award from the NCI. C.M.C. gratefully acknowledges support from the Leukemia and Lymphoma Society and the NIH (R35CA197589). CMC is founder, consultant and shareholder in Arvinas, LLC. BH, JW and AI are employees and shareholders of Arvinas, LLC.
Footnotes
Author Contributions
DPB, BES, GMB, ADB, SJF, JW, BH, and AI planned and carried out experiments. DPB wrote the manuscript. All authors reviewed and edited the manuscript. CMC conceived and managed the project, assisted in writing the manuscript, and funded the project.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- An J, Ponthier CM, Sack R, Seebacher J, Stadler MB, Donovan KA, Fischer ES. pSILAC mass spectrometry reveals ZFP91 as IMiD-dependent substrate of the CRL4CRBN ubiquitin ligase. Nat Commun. 2017;8:15398. doi: 10.1038/ncomms15398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondeson DP, Mares A, Smith IED, Ko E, Campos S, Miah AH, Mulholland KE, Routly N, Buckley DL, Gustafson JL, et al. Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat Chem Biol. 2015;11:611–617. doi: 10.1038/nchembio.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley DL, Van Molle I, Gareiss PC, Tae HS, Michel J, Noblin DJ, Jorgensen WL, Ciulli A, Crews CM. Targeting the von Hippel–Lindau E3 Ubiquitin Ligase Using Small Molecules To Disrupt the VHL/HIF-1α Interaction. J Am Chem Soc. 2012a;134:4465–4468. doi: 10.1021/ja209924v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley DL, Gustafson JL, Van Molle I, Roth AG, Tae HS, Gareiss PC, Jorgensen WL, Ciulli A, Crews CM. Small-molecule inhibitors of the interaction between the E3 ligase VHL and HIF1α. Angew Chem Int Ed Engl. 2012b;51:11463–11467. doi: 10.1002/anie.201206231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley DL, Raina K, Darricarrere N, Hines J, Gustafson JL, Smith IE, Miah AH, Harling JD, Crews CM. HaloPROTACS: Use of Small Molecule PROTACs to Induce Degradation of HaloTag Fusion Proteins. ACS Chem Biol. 2015;10:1831–1837. doi: 10.1021/acschembio.5b00442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlain PP, Lopez-Girona A, Miller K, Carmel G, Pagarigan B, Chie-Leon B, Rychak E, Corral LG, Ren YJ, Wang M, et al. Structure of the human Cereblon–DDB1–lenalidomide complex reveals basis for responsiveness to thalidomide analogs. Nat Struct Mol Biol. 2014;21:803–809. doi: 10.1038/nsmb.2874. [DOI] [PubMed] [Google Scholar]
- Deshaies RJ, Joazeiro CaP. RING domain E3 ubiquitin ligases. Annu Rev Biochem. 2009;78:399–434. doi: 10.1146/annurev.biochem.78.101807.093809. [DOI] [PubMed] [Google Scholar]
- Douglass EF, Miller CJ, Sparer G, Shapiro H, Spiegel DA. A comprehensive mathematical model for three-body binding equilibria. J Am Chem Soc. 2013;135:6092–6099. doi: 10.1021/ja311795d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan JS, Whittle MC, Nakamura K, Abell AN, Midland AA, Zawistowski JS, Johnson NL, Granger DA, Jordan NV, Darr DB, et al. Dynamic reprogramming of the kinome in response to targeted MEK inhibition in triple-negative breast cancer. Cell. 2012;149:307–321. doi: 10.1016/j.cell.2012.02.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabian MA, Biggs WH, Treiber DK, Atteridge CE, Azimioara MD, Benedetti MG, Carter TA, Ciceri P, Edeen PT, Floyd M, et al. A small molecule-kinase interaction map for clinical kinase inhibitors. Nat Biotechnol. 2005;23:329–336. doi: 10.1038/nbt1068. [DOI] [PubMed] [Google Scholar]
- Field SD, Arkin J, Li J, Jones LH. Selective Downregulation of JAK2 and JAK3 by an ATP-Competitive pan-JAK Inhibitor. ACS Chem Biol. 2017 doi: 10.1021/acschembio.7b00116. acschembio.7b00116. [DOI] [PubMed] [Google Scholar]
- Fischer ES, Böhm K, Lydeard JR, Yang H, Stadler MB, Cavadini S, Nagel J, Serluca F, Acker V, Lingaraju GM, et al. Structure of the DDB1-CRBN E3 ubiquitin ligase in complex with thalidomide. Nature. 2014;512:49–53. doi: 10.1038/nature13527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadd MS, Testa A, Lucas X, Chan KH, Chen W, Lamont DJ, Zengerle M, Ciulli A. Structural basis of PROTAC cooperative recognition for selective protein degradation. Nat Chem Biol. 2017;13:514–521. doi: 10.1038/nchembio.2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galdeano C, Gadd MS, Soares P, Scaffidi S, Van Molle I, Birced I, Hewitt S, Dias DM, Ciulli A. Structure-Guided Design and Optimization of Small Molecules Targeting the Protein-Protein Interaction between the von Hippel-Lindau (VHL) E3 Ubiquitin Ligase and the Hypoxia Inducible Factor (HIF) Alpha Subunit with in Vitro Nanomolar Affinities. J Med Chem. 2014;57:8657–8663. doi: 10.1021/jm5011258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guharoy M, Bhowmick P, Tompa P. Design principles involving protein disorder facilitate specific substrate selection and degradation by the ubiquitin-proteasome system. J Biol Chem. 2016;291:6723–6731. doi: 10.1074/jbc.R115.692665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann MD, Boichenko I, Coles M, Zanini F, Lupas AN, Hernandez Alvarez B. Thalidomide mimics uridine binding to an aromatic cage in cereblon. J Struct Biol. 2014;188:225–232. doi: 10.1016/j.jsb.2014.10.010. [DOI] [PubMed] [Google Scholar]
- Hon WC, Wilson MI, Harlos K, Claridge TDW, Schofield CJ, Pugh CW, Maxwell PH, Ratcliffe PJ, Stuart DI, Jones EY. Structural basis for the recognition of hydroxyproline in HIF-1 alpha by pVHL. Nature. 2002;417:975–978. doi: 10.1038/nature00767. [DOI] [PubMed] [Google Scholar]
- Hopkins AL, Groom CR. The druggable genome. Nat Rev Drug Discov. 2002;1:727–730. doi: 10.1038/nrd892. [DOI] [PubMed] [Google Scholar]
- Karaman MW, Herrgard S, Treiber DK, Gallant P, Atteridge CE, Campbell BT, Chan KW, Ciceri P, Davis MI, Edeen PT, et al. A quantitative analysis of kinase inhibitor selectivity. Nat Biotechnol. 2008;26:127–132. doi: 10.1038/nbt1358. [DOI] [PubMed] [Google Scholar]
- Krönke J, Udeshi ND, Narla A, Grauman P, Hurst SN, McConkey M, Svinkina T, Heckl D, Comer E, Li X, et al. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science. 2014;343:301–305. doi: 10.1126/science.1244851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai AC, Toure M, Hellerschmied D, Salami J, Jaime-Figueroa S, Ko E, Hines J, Crews CM. Modular PROTAC Design for the Degradation of Oncogenic BCR-ABL. Angew Chemie Int Ed. 2016;55:807–810. doi: 10.1002/anie.201507634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurence H. ATP-competitive inhibitors block protein kinase recruitment to the Hsp90-Cdc37 system. Nat Chem Biol. 2013:1–8. doi: 10.1038/nchembio.1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazo JS, Sharlow ER. Drugging Undruggable Molecular Cancer Targets. Annu Rev Pharmacol Toxicol. 2016;56:23–40. doi: 10.1146/annurev-pharmtox-010715-103440. [DOI] [PubMed] [Google Scholar]
- Lu G, Middleton RE, Sun H, Naniong M, Ott CJ, Mitsiades CS, Wong KK, Bradner JE, Kaelin WG. The myeloma drug lenalidomide promotes the cereblon-dependent destruction of Ikaros proteins. Science. 2014;343:305–309. doi: 10.1126/science.1244917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Qian Y, Altieri M, Dong H, Wang J, Raina K, Hines J, Winkler JD, Crew AP, Coleman K, et al. Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4. Chem Biol. 2015;22:755–763. doi: 10.1016/j.chembiol.2015.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattiroli F, Sixma TK. Lysine-targeting specificity in ubiquitin and ubiquitin-like modification pathways. Nat Struct Mol Biol. 2014;21:308–316. doi: 10.1038/nsmb.2792. [DOI] [PubMed] [Google Scholar]
- Matyskiela ME, Lu G, Ito T, Pagarigan B, Lu CC, Miller K, Fang W, Wang NY, Nguyen D, Houston J, et al. A novel cereblon modulator recruits GSPT1 to the CRL4CRBN ubiquitin ligase. Nature. 2016;535:252–257. doi: 10.1038/nature18611. [DOI] [PubMed] [Google Scholar]
- Mcalister GC, Nusinow DP, Jedrychowski MP, Wühr M, Huttlin L, Erickson BK, Rad R, Haas W, Gygi SP. MultiNotch MS3 Enables Accurate, Sensitive, and Multiplexed Detection of Differential Expression across Cancer Cell Line Proteomes Graeme C. McAlister, 1 David P. Nusinow, 1. Anal Chem. 2014;86:7150–7158. doi: 10.1021/ac502040v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAlister GC, Huttlin EL, Haas W, Ting L, Jedrychowski MP, Rogers JC, Kuhn K, Pike I, Grothe RA, Blethrow JD, et al. Increasing the multiplexing capacity of TMTs using reporter ion isotopologues with isobaric masses. Anal Chem. 2012;84:7469–7478. doi: 10.1021/ac301572t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milroy LG, Grossmann TN, Hennig S, Brunsveld L, Ottmann C. Modulators of protein-protein interactions. Chem Rev. 2014;114:4695–4748. doi: 10.1021/cr400698c. [DOI] [PubMed] [Google Scholar]
- Min JH, Yang H, Ivan M, Gertler F, Kaelin WG, Pavletich NP. Structure of an HIF-1alpha -pVHL complex: hydroxyproline recognition in signaling. Science. 2002;296:1886–1889. doi: 10.1126/science.1073440. [DOI] [PubMed] [Google Scholar]
- Van Nguyen T, Lee JE, Sweredoski MJ, Yang SJ, Jeon SJ, Harrison JS, Yim JH, Lee SG, Handa H, Kuhlman B, et al. Glutamine Triggers Acetylation-Dependent Degradation of Glutamine Synthetase via the Thalidomide Receptor Cereblon. Mol Cell. 2016;61:809–820. doi: 10.1016/j.molcel.2016.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohoka N, Okuhira K, Ito M, Nagai K, Shibata N, Hattori T, Ujikawa O, Shimokawa K, Sano O, Koyama R, et al. In Vivo Knockdown of Pathogenic Proteins via Specific and Nongenetic IAP-dependent Protein Erasers (SNIPERs) J Biol Chem. 2017 doi: 10.1074/jbc.M116.768853. jbc.M116.768853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrem JML, Shokat KM. Direct small-molecule inhibitors of KRAS: from structural insights to mechanism-based design. Nat Rev Drug Discov. 2016;15:771–785. doi: 10.1038/nrd.2016.139. [DOI] [PubMed] [Google Scholar]
- Ottis P, Toure M, Cromm PM, Ko E, Gustafson JL, Crews CM. Assessing Different E3 Ligases for Small Molecule-induced Protein Ubiquitination and Degradation. ACS Chem Biol acschembio. 2017 doi: 10.1021/acschembio.7b00485. 7b00485. [DOI] [PubMed] [Google Scholar]
- Petzold G, Fischer ES, Thomä NH. Structural basis of lenalidomide-induced CK1α degradation by the CRL4CRBN ubiquitin ligase. Nature. 2016;532:127–130. doi: 10.1038/nature16979. [DOI] [PubMed] [Google Scholar]
- Qian F, Engst S, Yamaguchi K, Yu P, Won KA, Mock L, Lou T, Tan J, Li C, Tam D, et al. Inhibition of Tumor Cell Growth, Invasion, and Metastasis by EXEL-2880 (XL880, GSK1363089), a Novel Inhibitor of HGF and VEGF Receptor Tyrosine Kinases. Cancer Res. 2009;69:8009–8016. doi: 10.1158/0008-5472.CAN-08-4889. [DOI] [PubMed] [Google Scholar]
- Raina K, Lu J, Qian Y, Altieri M, Gordon D, Rossi AM, Wang J, Chen X, Dong H, Siu K, et al. PROTAC-induced BET protein degradation as a therapy for castration-resistant prostate cancer. Proc Natl Acad Sci U S A. 2016;113:7124–7129. doi: 10.1073/pnas.1521738113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remillard D, Buckley DL, Paulk J, Brien GL, Sonnett M, Seo HS, Dastjerdi S, Wühr M, Dhe-Paganon S, Armstrong SA, et al. Degradation of the BAF Complex Factor BRD9 by Heterobifunctional Ligands. Angew Chemie - Int Ed. 2017;56:5738–5743. doi: 10.1002/anie.201611281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto KM, Kim KB, Kumagai a, Mercurio F, Crews CM, Deshaies RJ. Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc Natl Acad Sci U S A. 2001;98:8554–8559. doi: 10.1073/pnas.141230798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneekloth JS, Fonseca FN, Koldobskiy M, Mandal A, Deshaies R, Sakamoto K, Crews CM. Chemical Genetic Control of Protein Levels: Selective in Vivo Targeted Degradation. J Am Chem Soc. 2004;126:3748–3754. doi: 10.1021/ja039025z. [DOI] [PubMed] [Google Scholar]
- Ting L, Rad R, Gygi SP, Haas W. MS3 eliminates ratio distortion in isobaric multiplexed quantitative proteomics. Nat Methods. 2011;8:937–940. doi: 10.1038/nmeth.1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toure M, Crews CM. Small-molecule PROTACS: New approaches to protein degradation. Angew Chemie - Int Ed. 2016;55:1966–1973. doi: 10.1002/anie.201507978. [DOI] [PubMed] [Google Scholar]
- Vijayan RSK, He P, Modi V, Duong-Ly KC, Ma H, Peterson JR, Dunbrack RL, Levy RM. Conformational analysis of the DFG-out kinase motif and biochemical profiling of structurally validated type II inhibitors. J Med Chem. 2015;58:466–479. doi: 10.1021/jm501603h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watterson DM, Grum-Tokars VL, Roy SM, Schavocky JP, de Bradaric B, Bachstetter AD, Xing B, Dimayuga E, Saeed F, Zhang H, et al. Development of Novel In Vivo Chemical Probes to Address CNS Protein Kinase Involvement in Synaptic Dysfunction. PLoS One. 2013;8 doi: 10.1371/journal.pone.0066226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weekes MP, Tomasec P, Huttlin EL, Fielding CA, Nusinow D, Stanton RJ, Wang ECY, Aicheler R, Murrell I, Wilkinson GWG, et al. Quantitative temporal viromics: An approach to investigate host-pathogen interaction. Cell. 2014;157:1460–1472. doi: 10.1016/j.cell.2014.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter GE, Buckley DL, Paulk J, Roberts JM, Souza A, Dhe-Paganon S, Bradner JE. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science. 2015;348:1376–1381. doi: 10.1126/science.aab1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zengerle M, Chan KH, Ciulli A. Selective Small Molecule Induced Degradation of the BET Bromodomain Protein BRD4. ACS Chem Biol. 2015;10:1770–1777. doi: 10.1021/acschembio.5b00216. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table 1 – KinomeScan data for foretinib and foretinib-based PROTACs across 428 human kinases, related to Figure 1.
Supplemental Table 2 – Raw peptide quantification of MDA-MB-231 proteomes after 24-hour treatments with foretinib and foretinib-based PROTACs, related to Figure 2.
Supplemental Table 3 – Compilation of KinomeScan data and quantitative proteomics for the 54 kinases bound by foretinib and expressed in MDA-MB-231 cells, related to Figure 3.