

Summary
Although the cAMP-dependent protein kinase (PKA) is ubiquitously expressed, it is sequestered at specific subcellular locations throughout the cell, thereby resulting in compartmentalized cellular signaling that triggers site-specific behavioral phenotypes. We developed a three step engineering strategy to construct an optogenetic PKA (optoPKA) and demonstrated that, upon illumination, optoPKA migrates to specified intracellular sites. Furthermore, we designed intracellular spatially segregated reporters of PKA activity and confirmed that optoPKA phosphorylates these reporters in a light-dependent fashion. Finally, proteomics experiments reveal that light activation of optoPKA results phosphorylation of known endogenous PKA substrates as well as potential novel substrates.
Keywords: Optogenetics, Protein engineering, Protein kinases, Synthetic biology, Proteomics
eTOC Blurb
O’Banion et al. describe the engineering, validation, and phosphoproteomics assessment of light-activated, organelle-directed, cAMP-dependent protein kinase
Introduction
Optogenetics has emerged as a profoundly impactful tool used to correlate biochemical action with dynamic cellular and organismal behavior. Genetically encoded, light-responsive proteins provide the means to control when, where, and how much biochemical activity is triggered. In the late 20th century, light-activatable proteins were prepared by isolating the enzyme of interest, chemically modifying the active site with a light-cleavable moiety, and subsequently reintroducing the engineered protein into cells (Lawrence, 2005). Beginning in the first decade of the 21st century, genes coding for light-responsive ion channels from lower organisms were commandeered and introduced into higher organisms to primarily study the effect that neuronal activation in specific regions of the brain has on behavior (Deisseroth, 2015). More recently, mammalian proteins have been fused to light-sensing domains and genetically re-engineered to serve as light-activatable species. However, a major challenge remains eliminating the “dark activity” of these constructs, namely the inherent natural activity of the protein in the absence of light (Pathak et al., 2013; Weitzman and Hahn, 2014; Yin and Wu, 2013). We’ve recently described a strategy that addresses this engineering challenge and applied it to proteins that drive cell motility and trigger cell death (Hughes et al., 2015; Hughes and Lawrence, 2014). However, these previous studies focused on non-enzymatic proteins. In this report, we describe our efforts in constructing an optogenetic analog of the cAMP-dependent protein kinase (PKA), an enzyme whose structure is the template for the protein kinase family (Taylor et al., 2013).
PKA regulates a variety of cellular processes, including apoptosis, motility, glycolysis, and gene expression to name but a few. However, these distinct behaviors emanate from PKA activity at specific intracellular sites (Dessauer, 2009; Zaccolo et al., 2006). Indeed, intracellular PKA activity serves as the quintessential model of compartmentalized signaling (Lefkimmiatis and Zaccolo, 2014). This enzyme is sequestered to various subcellular locations by A-Kinase Anchoring Proteins, which create localized signaling nodes that respond to both global and local environmental changes in cAMP levels (Scott et al., 2013). For example, PKA activity at the outer mitochondrial membrane (OMM) is thought to regulate mitochondrial dynamics and metabolism (Dickey and Strack, 2011), whereas PKA signaling at the plasma membrane (PM) is believed to control migration and membrane dynamics (Newell-Litwa and Horwitz, 2011; Tkachenko et al., 2011). There are a number of optogenetic and chemogenetic tools available to control PKA’s upstream activator, cAMP (Raffelberg et al., 2013; Ryu et al., 2010; Sample et al., 2012; Stierl et al., 2011). However, cAMP interacts with a number of downstream effectors, including EPAC (Gancedo, 2013), cyclic nucleotide-gated ion channels (Matulef and Zagotta, 2003), and POPDC proteins (Amunjela and Tucker, 2016). Consequently, light- or chemical-triggered cAMP production does not provide the means to specifically activate compartmentalized PKA. In this regard, an optogenetic PKA (optoPKA), which is decoupled from cAMP production and activated at distinct intracellular locations, could prove invaluable for examining the roles of subcellular pools of PKA.
RESULTS AND DISCUSSION
Construction of a cAMP-independent, low activity, PKA
We addressed the “dark activity” challenge associated with engineered optogenetic proteins via a three-step strategy. First, to ensure that the engineered protein functions independently of the biochemistry of the cell, PKA was mutated to a constitutively active cAMP-independent enzyme. cAMP binds to the regulatory (R) subunits of PKA, a binding event that activates the corresponding catalytic (C) subunits. A W196R mutation (Gibson and Taylor, 1997), as well as a L206R mutation (Beuschlein et al; 2014, Goh et al; 2014), in the C subunit compromises the repressive action of the R subunit, thereby rendering PKA cAMP independent. We constructed, expressed, and purified the W196R mutant and found cAMP independent activity as well (Table S1). Second, we reduced the activity of PKA by generating C subunit mutants (Moore et al., 2003; Yang et al., 2009) known to deleteriously impact kcat/Km (E203A, Y204A, and F327A). The E203 and Y204 residues are present in the “activation loop”, a structural motif found in other protein kinases (Steichen et al., 2012). These residues contribute to binding of the protein/peptide substrate (Moore et al., 2003), whereas the F327 residue plays a key role in binding the second substrate, ATP (Yang et al., 2009). The three C subunit double mutants were separately expressed in E. coli and purified via affinity chromatography. As expected, the catalytic activities of these mutants are compromised relative to that of the wild type enzyme (Table S2): CW196R/E203A (43 ± 6%), CW196R/Y204A (5.6 ± 1.3%), and CW196R/F327A (1.2 ± 0.1%). Third, we employed the cryptochrome 2 (Cry2)/Cib photodimerizing protein pair (Kennedy et al., 2010) as a means to recruit, and therefore concentrate [~15-fold; (Hughes et al., 2015; Hughes and Lawrence, 2014)], the constitutively active, but moderately phosphotransferase impaired, C subunit mutants to specific intracellular sites (vide infra).
We examined the phosphotransferase selectivity of the CW96R/Y204A subunit double mutant relative to that of its wild type counterpart using the vasodilator-stimulated phosphoprotein (VASP) as a substrate. VASP is phosphorylated by both PKA and the cGMP-dependent protein kinase (PKG). However, although these two enzymes display very similar substrate specificity profiles, they act on VASP in a dramatically different fashion. Although both enzymes phosphorylate S157 in IERRVS157NAGGP, only PKG phosphorylates S239 in KLRKVS239KQEEA (Priestman et al., 2011). We confirmed that the double PKA mutant, CW96R/Y204A, retains the selectivity of its wild type counterpart by only phosphorylating S157 (Figure S1).
Construction of Cry2-mCh-[C subunit] (optoPKA)
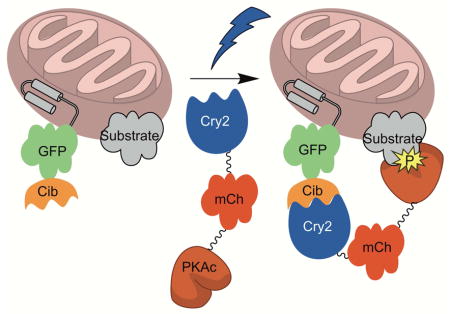
We fused each C subunit double mutant, at either the C- or N-terminus, to Cry2-mCherry (Cry2-mCh) via a 16 amino acid flexible linker (see STAR methods). PKA is known to undergo N-terminal myristoylation (Gaffarogullari et al., 2011). Although the C-terminal fusions of PKA to Cry2-mCh (Cry2-mCh-[C-subunit]) will not by myristoylated due to an absolute requirement of an N-terminal Gly residue for myristoylation (Towler et al., 1987), the N-terminal fusions of PKA to Cry2-mCh ([C-subunit]-Cry2-mCh) may be myristoylated. To determine whether or not the N-terminal residues of PKA affected light-induced localization, we generated N-terminal fusions to Cry2-mCh of both full length PKA and a mutant lacking 10 N-terminal residues that cannot be myristoylated. These fused constructs, heretofore referred to as optoPKA, were separately co-expressed with the Cib protein localized at the OMM, PM, or the actin cytoskeleton in MVD7 cells. optoPKAs, in the dark, are designed to be cytoplasmically diffuse. However, upon illumination, Cry2 undergoes a light-induced conformational change that promotes transient binding to Cib (Figure 1). We found that the [C-subunit]-Cry2-mCh constructs poorly accumulate at their designated locations upon illumination regardless of whether or not the 10 N-terminal residues are present and were therefore not used in further studies (data not shown). By contrast, all Cry2-mCh-[C-subunit] constructs robustly migrate to the desired subcellular locations in a light-triggered fashion (Figure 2, Movies S1 – S3). We quantified the light driven association and dissociation dynamics of the optoPKAs via fluorescent microscopy (Figure 2, Table S3). A single 100 ms 488 nm pulse is sufficient to drive maximal subcellular localization in 1 – 2 min. Subsequent dissociation to the cytoplasmically diffuse dark state transpires with a t1/2 of 6 – 7 min. Although a single 100 ms light pulse is sufficient for subcellular recruitment under the microscope due to the large photon flux experienced by a few cells, analogous “bench-top” studies with large cell numbers (e.g. Western blot, immunofluorescence and proteomic studies) were performed with longer illumination times (vide infra) to achieve the maximal response.
Figure 1. Schematic of the light-triggered transfer of optoPKA [Cry2-mCh-(C-subunit)] from the cytoplasm to the outer mitochondrial membrane.

The C subunit of PKA is a reduced activity mutant that is cAMP-independent. Tables S1 and S2 provide PKA mutant activities.
Figure 2. Light-triggered optoPKA Association with and Subsequent Dissociation from the OMM, Cytoskeleton, and PM.

All optoPKA constructs contain the mCherry (mCh) fluorescent protein: Cry2-mCh-CW196R/E203A (A – D, F), Cry2-mCh (E, control), Cry2-mCh-CW196R (E), Cry2-mCh-CW196R/Y204A (F, G, H), Cry2-mCh-CW196R/F327A (F, G, H). The following Cib constructs were employed to recruit optoPKA to specific intracellular sites: Tom20-Cib-GFP (OMM-Cib in A - E, F) at the OMM; LifeAct-GFP-Cib (LifeAct-Cib in G) at the actin cytoskeleton; Cib-GFP-CAAX (PM-Cib in H) at the plasma membrane. Visualization of the mCh label in optoPKAW196R/E203A (A) before and (B) 1 min after Cry2 stimulation with a 100 ms 488 nm light pulse. (C) Visualization of the GFP label in OMM-Cib (Tom20-Cib-GFP), where (D) is an overlay of (B) and (C). (E – H) Association and subsequent dissociation of optoPKA with and from the PM, OMM, and the cytoskeleton were monitored via mCh fluorescence. A single 100 ms pulse (FITC cube) was employed to initiate recruitment of the optoPKA constructs to their designated sites. Experiments were performed on either a widefield (OMM-Cib, LifeAct-Cib) or confocal (PM-Cib) microscope. N = 3 cells per group. Scale bar = 50 μm. Table S3 provides association times for optoPKA to achieve maximal OMM localization and t1/2 dissociation times from the OMM. Data expressed as mean +/− SEM.
An Organelle-Localized PKA Activity Reporter [(organelle targeting sequence)-mTq2-(VASP)]
We subsequently assessed whether the light-triggered recruitment of optoPKAs to specific intracellular sites correlates with spatially focused phosphoryltransferase activity. Unfortunately, available live cell PKA activity imaging tools, such as the AKARs and related biosensors (Mo et al., 2017), contain two fluorescent proteins that spectrally overlap with Cry2 activation (impeding our ability to perform “dark” experiments, Figure S1). Red shifted variants of AKARs are available (Ni et al., 2011), but these tools still utilize GFP as a FRET donor. Cry2 has an FAD chromophore that absorbs light at wavelengths up to 520 nm (Banerjee et al., 2007) and therefore any light exposure at blue wavelengths activates Cry2 due to the spectral overlap illustrated in Figure S2. An alternative approach for assessing PKA activity is the detection of endogenous phosphorylated substrates via immunostaining. However, this is hampered by, with few exceptions, the general lack of antibodies against the phosphorylated forms of spatially localized endogenous substrates. Consequently, we developed a PKA-Reporter that is sequentially comprised of an N-terminal subcellular targeting sequence, the fluorescent protein mTurquoise2 (mTq2), and a C-terminal PKA substrate peptide composed of amino acids 148 – 164 of the protein VASP (Figure 3). VASP is a well-characterized PKA substrate that is phosphorylated at Ser157 (Lambrechts et al., 2000), but distributed throughout the cell at distinct locations. The N-terminal targeting sequence is used to control the specific intracellular location of the PKA-Reporter. In particular, we employed the Tom20 (Kanaji et al., 2000) or Lyn (Depry et al., 2011) kinase N-terminal domains to target the PKA-Reporter to the OMM or the PM (Figure 3), respectively. We employed mTq2 fluorescence, after completion of the Cry2 photoactivation experiment, to confirm that the PKA-Reporter is spatially localized in the desired fashion.
Figure 3. Validation of Intracellular PKA-Reporter Localization.

(A) Schematic of PKA-Reporter architecture, where the targeting sequence (green) positions the PKA-Reporter at specific intracellular sites, mTq2 (blue) offers a visual confirmation of intracellular placement, VASP148–164 (red) is phosphorylated by PKA, and phospho-VASP148–164 (yellow) is recognized by a commercially available antibody (and a secondary antibody containing Alexa 647). (B, C) Endogenous PKA was activated via treatment of MVD7 cells with forskolin (activates adenylate cyclase, which produces cAMP) or Br-cAMP (a cell permeable analog of cAMP). Cells were lysed after treatment and the extent of PKA-Reporter phosphorylation quantified via Western blot using a phospho-VASP antibody. (B) Quantification of PKA-Reporter phosphorylation at the OMM (black) or the PM (grey) in response to forskolin or Br-cAMP. Changes in PKA-Reporter phosphorylation levels are defined as relative to untreated cells. (C) Western blot of phosphorylated PKA-Reporter targeted to either the OMM or the PM as a function of various treatment protocols: (top) The dynamic range of the PKA-Reporter at the OMM (7-fold) is greater than that of the PKA-Reporter at the PM (2-fold). The reduced dynamic range of the latter is due to higher levels of PM phosphorylated PKA-Reporter in untreated cells. This may be a consequence of more robust endogenous PM PKA activity and/or reduced PM protein phosphatase activity relative to that at the OMM. (bottom) Staining of the HA tag on the PKA-Reporter was used to assess total PKA-Reporter levels (Note: only the region of the Western blot that contains the bands of interest is shown. The rest of the blot has been cropped for clarity). (D, E) Validation of the PKA-Reporter, with representative images for experiments conducted at the OMM and PM in MVD7 cells. Left panels display the OMM or PM-localized PKA-reporter (mTq2) and middle panels display the presence/absence of phosphorylated-reporter (using a pVASP antibody). There is no PKA-Reporter phosphorylation (D, E) in the absence of PKA activation, but extensive PKA-Reporter phosphorylation (D, E) upon treatment with 100 μM forskolin. Finally, (D, E) PKA-Reporter phosphorylation is not observed in cells exposed to both forskolin and a PKA inhibitor (H89). Scale bar = 50 μm.
As noted above, there is a dearth of available antibodies that recognize phosphorylated PKA substrates at specific subcellular locations. phosphoVASPS157 antibodies are an exception to this general trend. We employed anti-phosphoVASPS157 to detect the phosphorylated PKA-Reporter via cell immunofluorescence (using an Alexa 647-conjugated) and Western blot analyses. Since MVD7 cells lack all VASP family member proteins (Bear et al., 2000), the phosphoVASPS157 antibody enables quantification of PKA-Reporter phosphorylation via immunofluorescence without interference from endogenous VASP. In addition, the molecular weights of the PKA-Reporters are 30.9 and 37.6 kDa for the PM and OMM-targeted species, respectively, whereas the molecular weight of endogenous VASP is 46 kDa. Therefore, Western blot analysis for PKA-Reporter phosphorylation can be used in the presence of natural VASP and thus appropriate for any cell line.
Both the OMM- and PM-localized PKA-Reporters were validated in MVD7 cells. Upon exposure of these cells to the adenylate cyclase activator forskolin, cAMP is generated, which activates endogenous PKA. Immunostaining with the phosphoVASPS157 antibody revealed PKA-Reporter phosphorylation at the expected intracellular sites (Figure 3, D, E). Furthermore, forskolin exposure in the presence of a PKA inhibitor (H89) blocks PKA-Reporter phosphorylation indicating a PKA associated phosphorylation event (Figure 3, D, E). Analogous Western blot experiments confirmed these results (Figure 3, B, C).
Evaluation of Dark and Light optoPKA Activity
We examined the relative activities of the optoPKA variants by co-expression with OMM-Cib and the OMM-PKA-Reporter in MVD7 cells (Figures 4 and S3). The cells were illuminated for 1 min in the presence or absence of the PKA inhibitor H89 and subsequently analyzed by immunofluorescence. OptoPKAW196R, which is the fully active cAMP-independent enzyme construct, displays undesired “dark activity”, emblematic of the difficulties that can be encountered when appending a wild-type active enzyme to a photoresponsive protein. Furthermore, optoPKAW196R/E203A, which is 43% as active as the wild type enzyme, likewise displays undesired phosphorylation of the PKA-Reporter in the dark. By contrast, the less enzymatically active optoPKAW196R/Y204A and optoPKAW196R/F327A constructs are silent in the dark. As anticipated, illumination (1 min) of cells expressing optoPKAW196R/Y204A furnishes the desired phosphorylation of the PKA-Reporter. optoPKAW196R/F327A, which is 5-fold less active than optoPKAW196R/Y204A, requires longer illumination times to generate equivalent levels of PKA-Reporter phosphorylation (Figure 5). Finally, pre-treatment with H89 blocks the light-stimulated phosphorylation event, which is in keeping with the notion that phosphorylation is PKA-dependent. These results are consistent with the predicted design element: light-triggered recruitment of partially impaired enzymes to specific intracellular sites furnishes localized phosphotransferase activity with reduced dark activity.
Figure 4.
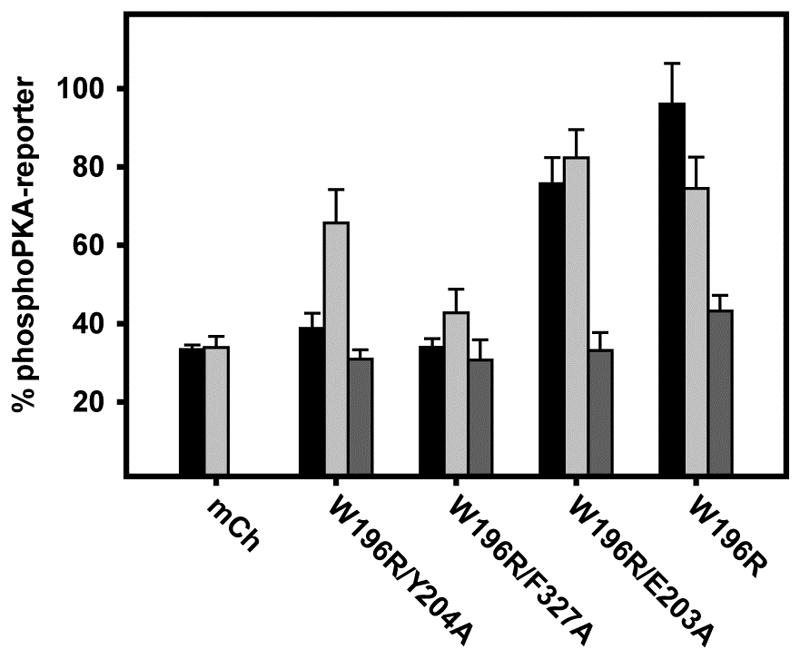
Relative phosphorylation of OMM PKA-Reporter in response to 1 min light stimulation with different optoPKA constructs, where mCherry (Cry2-mCh) serves as a non-kinase control. optoPKAW196R/Y204A elicits robust phosphorylation of the PKA-Reporter upon illumination (light grey bar) compared to dark (medium grey bar) or (light + H89) (black bar) conditions. All data presented as mean +/- SEM, N ≥ 10 cells per bar. Representative images are found in Figure S3.
Figure 5. Light titration of PKA-Reporter phosphorylation by optoPKAs.

(A) Both optoPKAW196R/Y204A (black) and optoPKAW196R/F327A (grey) display an illumination time-dependent increase in PKA-Reporter phosphorylation when both the optoPKAs and the PKA-Reporter are positioned at the OMM. (B) Both optoPKAW196R/Y204A and optoPKAW196R/F327A display an illumination time-dependent increase in PKA-Reporter phosphorylation when both optoPKAs and the PKA-Reporter are situated at the PM. Changes in PKA-Reporter phosphorylation levels are defined as relative to non-illuminated cells. optoPKAW196R/Y204A displays more robust activity (more PKA-reporter phosphorylation at shorter illumination times) than its optoPKAW196R/F327A counterpart. This may be due to the 4-fold higher catalytic activity displayed by optoPKAW196R/Y204A. Data reported as mean +/− SEM n = 10 – 20 cells per group.
Time-Dependence and Site-Selectivity of optoPKA Activity
As noted in Figure 4, optoPKAW196R/Y204A displays more robust phosphorylation of the PKA-Reporter than optoPKAW196R/F327A following 1 minute of illumination. We subsequently examined the relationship between light exposure time (0 – 300 s) and phosphorylated PKA-Reporter levels. Short illumination times (≤120 s) reveal that optoPKAW196R/Y204A generates a higher level of phosphorylated PKA-Reporter than its less active counterpart optoPKAW196R/F327A (Figure 5). However, at 300 s of illumination, both optoPKA constructs phosphorylate the PKA-Reporter to the same extent. We also assessed the spatial selectivity of the optoPKAs by targeting optoPKAW196R/Y204A or optoPKAW196R/F327A to the PM, OMM, or actin cytoskeleton while varying the location of the PKA-Reporter. A time and location dependent increase in PKA-Reporter phosphorylation is only observed when the opto-PKAs and the PKA-Reporter are localized to the same subcellular compartments (Figures 6 and S4).
Figure 6. Phosphorylation of OMM-localized PKA-Reporter by optoPKAW196R/Y204A as a function of location and illumination time.

(A) OMM PKA-Reporter phosphorylation occurs when optoPKAW196R/Y204A is recruited to the OMM, but not to the PM or the cytoskeleton. In an analogous fashion (B) PM PKA-Reporter phosphorylation only transpires when optoPKAW196R/Y204A is recruited to the PM. In addition, the magnitude of PKA-Reporter phosphorylation correlates with total light exposure. Data is presented as mean +/− SEM, N ≥ 10 cells per experimental group. Data for optoPKAW196R/F327A can be found in Figure S4.
We also investigated the reversibility of optoPKAW196R/Y204A phosphotransferase action at the OMM. MVD7 cells expressing optoPKAW196R/Y204A, Tom20-Cib, and OMM-PKA-Reporter were exposed to 5 min of blue light and fixed at indicated time points. The reporter is maximally phosphorylated after 5 min light exposure and the signal decays nearly to baseline over 45 min (Figure S5). Linear regression analysis reveals a t1/2 for reporter dephosphorylation of 20.25 ± 0.38 min. It is interesting to note that the rate of dephosphorylation of the OMM-PKA-Reporter is nearly three times slower than the off rate of optoPKA, which implies that phosphatase action is rate limiting. A comparison of the t1/2 for dephosphorylation of the OMM-PKA-Reporter to the AKAR reporters show similar if not faster time domains for dephosphorylation (Zhang et al., 2001). These results indicate that consequences of optoPKA activation are reversible.
Phosphoproteomics Reveals that optoPKA Activation Results in the Phosphorylation of Endogenous Proteins in an Organelle-Targeted Fashion
We employed phosphoproteomics to examine the consequences of optoPKA recruitment to the OMM and the PM (Table S4). We searched the acquired phosphoproteomics data sets for validated PKA substrates and phosphorylation sites using Qiagen’s Ingenuity® Pathway Analysis (IPA®) tool and the PhosphoSitePlus Resource (Hornbeck et al., 2015). The results are summarized in a number of different contexts:
optoPKA-activation induces phosphorylation of endogenous PKA substrates
optoPKA phosphorylates an array of known PKA substrates at validated phosphorylation sites (Table S5). For example, optoPKAW196R/Y204A recruitment to the OMM results in the phosphorylation of BAD at Ser118 (Polzien et al., 2009; Virdee et al., 2000), a well-characterized PKA substrate. By contrast optoPKAW196R/Y204A light-mediated migration to the PM, generates a β-catenin that is phosphorylated at Ser552 and Ser675 (Taurin et al., 2006). A number of protein kinases that are phosphorylated by PKA are likewise phosphorylated in response to optoPKAW196R/Y204A activation. For example, upon OMM recruitment, Ser21 of glycogen synthase kinase-3α (Fang et al., 2000), Ser259 of Raf1 (Dhillon et al., 2002), and Ser12 of the cyclin-dependent kinase 16 (Graeser et al., 2002) are all phosphorylated. Furthermore, photo-transfer of optoPKA to the PM, generates phospho-Ser447 of B-Raf, phospho-Ser205 of protein kinase D (Gerarduzzi et al., 2014) and phospho-Ser21 of Fyn (Yeo et al., 2011). In addition to validated PKA substrates, we identified a number of other proteins that are phosphorylated at potential PKA consensus sites in response to optoPKA photo-activation (Table S6). As with the other studies that have identified putative PKA substrates via phosphoproteomics (de Graaf et al., 2014; Giansanti et al., 2013; Hamaguchi et al., 2015; Imamura et al., 2017), these proteins remain to be validated as authentic substrates of the cAMP-dependent protein kinase. These results validate the ability of optoPKAW196R/Y204A to serve as a trigger for the phosphorylation of known PKA substrates at confirmed phosphorylation sites, while raising the prospect that additional proteins are phosphorylated in a subcellular compartment fashion as a direct or indirect consequence of PKA activity.
optoPKA recruitment to the OMM and PM elicits the phosphorylation of organelle-associated proteins
Analysis of our dataset using IPA and the PhosphoSitePlus databases revealed that 15 out of the 17 proteins identified to reside at the OMM display enhanced phosphorylation in a light-induced manner relative to the dark control in response to optoPKAW196R/Y204A OMM recruitment (Figure 7a–b). Some of these proteins are known PKA substrates (BAD, Raf1), whereas others are phosphorylated at sequences that are not canonical PKA consensus sites [voltage dependent anion channel 1 (VDAC1), caveolin-1 (CAV1)], implying that the latter are phosphorylated by other protein kinases as a consequence of PKA activity. In an analogous vein, 12 out of the 15 proteins, classified as PM-localized, likewise exhibit an increase in phosphorylation levels following PM recruitment of optoPKAW196R/Y204A. We do note that the phosphorylation status of some “OMM” proteins is impacted by the light-induced migration of optoPKAW196R/Y204A to the PM and, analogously, some “PM” proteins are phosphorylated upon transfer of optoPKAW196R/Y204A to the OMM. However, these results are not necessarily inconsistent with the PKA-Reporter validated compartmental activity of the optoPKAs. For example, more than 50% of intracellular proteins are located in multiple compartments (e.g. RAF1, AKAP12, AKAP13) (Thul et al., 2017) or are cytoplasmically diffuse (e.g. PKD) and thus the targeted action of the optogenetic construct at distinct sites could result in the phosphorylation of substrates common to multiple locations.
Figure 7. optoPKAW196R/Y204A recruitment triggers changes in the phosphorylation status of known PM and OMM associated PKA substrates as well as a general enrichment of the canonical phosphoPKA consensus site.

Phosphoproteomic analysis of light-triggered optoPKAW196R/Y204A recruitment to the (A) PM and (B) OMM relative to the corresponding samples in the dark. Validated PKA substrates from the phosphoproteomics data were identified using the IPA and PhosphoSitePlus databases and subsequently grouped according to PM- or OMM-localization using the IPA program. Node colors indicate hyper- (red) or hypo-phosphorylation (green) of the identified phospho-site relative to the non-illuminated sample. Proteins not observed in the indicated dataset are not colored. Proteins with multiple phosphorylation sites are denoted with an asterisk (*), and the site with the maximum light/dark fold change is represented. Although the proteins represented here are validated PKA substrates, the specific phosphorylation sites are not necessarily known PKA sites. Proteins containing pSer motifs identified (p-value <0.05 and light/dark fold change >1.5) upon light-triggered recruitment of optoPKAW196R/Y204A to the (A) PM and (B) OMM samples using Motif-X (possibility threshold, p < 10−6). The enriched consensus sites identified at the (C) PM and (D) OMM. Tables S4 – S7 provide related phosphoproteomics information.
optoProtein constructs induce changes in the phosphorylation status of the generic PKA substrate motif
We employed the software tool motif-x to perform an unbiased extraction of overrepresented phospho-sequences induced by activation of the optogenetic constructs. The canonical PKA consensus site (Arg-Arg-Xaa-pSer-Xaa, where Xaa is any residue) is overrepresented (3-fold) upon recruitment of optoPKAW196R/Y204A to the OMM or the PM (Figure 7c–d). By contrast, light-triggered recruitment of the Cry2-mCh control does not elicit a change in the relative phosphorylation status of the canonical PKA phosphorylation consensus site.
optoPKA and forskolin induce differential, but overlapping, substrate phosphorylation
As previously noted, forskolin is an activator of adenylate cyclase and, in this capacity, induces cell-wide generation of cAMP. We compared phosphoprotein formation induced by forskolin with that of optoPKA targeted to the OMM. Furthermore, we only analyzed the mitochondrial fraction from cell lysates. Of the 1153 proteins identified (see STAR methods), 266 (23.1%) display an increase in phosphorylation due to optoPKA alone, 348 (30.2%) due to forskolin alone, and 218 (18.9%) due to both (Table S7). These results imply a shared, yet distinct, impact on the phosphoproteome by optoPKA and forskolin. We utilized NetPhorest to predict the upstream protein kinases directly responsible for the phosphorylated proteins revealed by the OMM phosphoproteomics data. NetPhorest is a database that contains the linear motifs recognized by individual members of the protein kinase family (Miller et al., 2008). The percentage of phosphorylated proteins only generated in response to optoPKA activation can be traced to the cyclin dependent kinases (29%), CLK kinases (15%), and MAPKs (13%). The analogous subset of phosphorylated proteins that is only activated by forskolin is attributed to the cyclin dependent kinases (24%), casein kinase 2 (20%), and the MAPKs (10%). Finally, the protein kinases responsible for the phosphorylation of proteins observed with optoPKA or forskolin include casein kinase 2 (24%), the cyclin dependent kinases (15%), and PKA (14%). These results confirm that optoPKA and forskolin elicit the phosphorylation of proteins common to both triggers. However, these common proteins represent a fraction of the proteins phosphorylated in response to forskolin or optoPKA alone, implying that these triggers activate distinct pathways as well. In this regard, it is interesting that these distinct pathways appear to ultimately converge on common protein kinase(s) as predicted by NetPhorest. These results corroborate the recognized complexity of signaling pathways and confirm the need for biochemical tools capable of teasing apart the mechanisms that enable cells to employ common biochemical pathways for distinct purposes.
Implications and Opportunities for optoProteinKinases
We’ve developed an optogenetic engineering strategy that employs constitutively active, yet enzymologically reduced, mutants of PKA. PKA itself serves as the structural prototype for the entire protein kinase family (Smith et al., 1999), raising the possibility that this strategy could prove applicable to other protein kinases present in the human proteome (~500 protein kinases that are segregated into 7 major subgroups). The mutations introduced to generate the dark-silent/light-active optoPKAs display reduced activities (1 – 5%) relative to wild type PKA. These residues reside in regions that are conserved amongst the protein kinase family (the activation loop or ATP binding site). Indeed, equivalent mutations produce analogous reductions in phosphotransferase activities in other protein kinases (Edwards and Gill, 1999; Jacobs et al., 2006; Manser et al., 1997; Ohashi et al., 2000). Consequently, studies are in progress to examine the applicability of this engineering strategy to other members of the protein kinase family. We do note that, as with any tool that probes or manipulates intracellular biochemistry, judicious care be taken with its application and subsequent interpretation of results. The introduction of mutations, no matter how seemingly innocuous, can impact signaling behavior. Our proteomics data confirms that the activation of optoPKAs induces the phosphorylation of known PKA substrates at previously validated residues. In addition, these proteomics studies have identified heretofore-unreported phospho sites in proteins that are either directly phosphorylated by optoPKA or indirectly by an optoPKA-activated protein kinase. Indeed, other phosphoproteomic studies have likewise described newly identified substrates phosphorylated in response to activation of the PKA pathway under a variety of conditions (de Graaf et al., 2014; Giansanti et al., 2013; Hamaguchi et al., 2015; Imamura et al., 2017). These studies employed small molecule stimulators of the PKA pathway (e.g. prostaglandin E2, forskolin), which trigger adenylate cyclase activity and cAMP production. However, cAMP is known to act through multiple effectors (Gancedo, 2013), raising the prospect that some of the observed phospho-products are generated in a PKA-independent fashion. By contrast, an optoPKA in particular, and optoProteinKinases in general, offer control over specific signaling molecules with high temporal resolution. Unlike small molecule PKA signaling activators, optoPKA is readily targeted to individual cellular compartments, enabling correlation of spatially focused signaling [e.g. the mitochondrial cAMP “signalsome”; (Monterisi and Zaccolo, 2017)] with downstream biochemical action and cellular phenotype (apoptosis, epithelial-mesenchymal transition, aging, pathological conditions, and other PKA-implicated processes).
STAR Methods
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, David S. Lawrence (lawrencd@email.unc.edu)
Experimental Model and Subject Details
HEK 293T I/17 cells are the I/17 clone of female human embryonic kidney cells and were purchased from the UNC Tissue Culture Facility. Cells grown in DMEM + 10% FBS, 1X GlutaMAX, and 1X PenStrep at 37 °C in 5% CO 2. Cells were tested for mycoplasma by PCR by the UNC Tissue Culture Facility prior to purchase.
MVD7 cells were a gift from Dr. James E. Bear and are a double knockout VASP and Mena embryonic mouse fibroblast clonal line with no detectable expression of the EVL protein. Cells express a temperature sensitive large T antigen and are grown in DMEM + 10% FBS, 1X GlutaMAX, 1X antimycotic/antibiotic, and 50 IU mouse recombinant interferon-γ at 32 °C in 5% CO2. Sex of these cells is not available (Bear et al., 2000) and these cells have not been tested for mycoplasma.
Method Details
Cloning
Cloning and Mutagenesis of C Subunit for Bacterial Expression
The C subunit was amplified by PCR with primers that appended nucleotides necessary for ligation independent cloning into pMCSG11 (DNASU). Briefly, the PCR-amplicon was treated with T4 DNA Polymerase with dCTP for 30 min at 22 °C, followed by heat inactivation for 20 min at 75 °C and reaction clean up by spin column. Separately, the vector pMCSG11 was digested with SspI, gel isolated, and then treated with T4 DNA Polymerase with dGTP for 30 min at 22 °C, followed by heat inactivation and spin column clean up. The resulting PCR fragment (4 μL) and vector (1 μL) were then annealed by incubation for 20 min at room temperature, followed by the addition of 2 μL of 100 mM EDTA. The resulting solution was heated to 75 °C for 2 min and subsequently allowed to slowly cool to room temperature. An aliquot of the resulting mixture (2 μL) was then transformed into chemically competent DH5α E. coli cells, and resulting clones analyzed for inserts via HindIII restriction digest and sequence verified. Following successful isolation of WT C subunit in pMCSG11, mutations (W196R, E203A, Y204A, F327A) were introduced via site-directed mutagenesis. All cloning materials were purchased from New England Biolabs (see Key Resources and Methods S1).
Construction of Cry2-mCh-[C subunit] (optoPKA)
The C subunit was amplified by PCR with primers that appended both BsrgI and NotI restriction sites as well as a sixteen-amino acid N-terminal tether (SAGGSAGGSAGGSAGG). After isolation and restriction digest of the PCR product, it was ligated into a plasmid containing Cry2-mCh that was also cut with BsrgI and NotI enzymes. The resulting construct encodes the fusion protein Cry2-mCh-[C subunit] (see Key Resources and Methods S1).
Cloning of PM-Cib
A construct encoding Cib-GFP-CAAX (Addgene 26867) was modified via PCR-mediated site-directed mutagenesis to insert a poly-lysine sequence-CAAX box [GGSGKKKKKKSKTK-CVIM] and stop codon between Cib and GFP. The resulting construct, Cib-CAAX (PM-Cib), lacks GFP, which proved necessary when used in conjunction with the PKA-Reporter (which contains mTq2). We found that GFP fluorescence bleeds into the mTq2 window.
Site-Directed Mutagenesis
Primers for site-directed mutagenesis were designed to have a minimum Tm of 72 °C. 125 ng of each primer were combined with 50 ng of template DNA, 4 μL of 2.5 mM dNTPs, 5 μL of 10X PfuUltra buffer, 1 μL of PfuUltra Hotstart DNA Polymerase, and ddH2O added to 50 μL. The resultant PCR products were then treated with DpnI for 1 h at 37 °C and 1 μL of this solution transformed into 50 μL of Invitrogen Max Efficiency DH5α E. coli cells. Individual colonies were then isolated and sequence verified (see Key Resources and Methods S1).
PKA-Reporter Cloning and Characterization
The PKA-Reporter was constructed by appending nucleotides encoding the PKA consensus site of the VASP protein (residues 148–164) to the 3′ end of mTq2 via primer elongation. The OMM targeted construct was cloned by generating a PCR fragment appending an HA-tag preceded by an in-frame KpnI site onto the 5′ end and the VASP 148–164 sequence followed by a BamHI site onto the 3′ end of mTq2 (a gift from Dr. Theodorus Gadella, University of Amsterdam, Netherlands). The PCR product was digested with KpnI and BamHI and ligated into a pEF1alpha-Tom20 vector (modified from pEF1alpha-MCS-IRES-DsRedExpress2 from Clonetech) digested with KpnI and BamHI such that the insert was in-frame with the Tom20 mitochondrial localization sequence. The PM targeted construct was generated via PCR using a 5′ primer containing an EcoRI site followed by the Lyn kinase N-terminal myristoylation sequence and a gene specific region against the HA-tag. The 3′ primer was the same primer used above to append the VASP peptide to mTq2. The PCR product was digested with EcoRI/BamHI and ligated into a pEF1alpha vector digested with EcoRI/BamHI. Positive clones were identified via restriction digest with EcoRI and BamHI and confirmed via sequencing using a set of sequencing primers (see Key Resources and Methods S1) generated for the specific pEF1alpha vector used. The plasmids were then transfected into cells and characterized for 1) subcellular localization via fluorescent microscopy, 2) molecular weight via Western blot, 3) ability to be specifically phosphorylated by PKA via Western blot and immunofluorescence. We detected peptide phosphorylation using a mouse anti-pVASP S157 antibody from abcam (ab58555) at 1:100 or 1:1000 for immunofluorescence and Western blotting respectively.
In Vitro Assessment of C Subunit Activity
Over-expression and Purification of C Subunit Mutants
BL21DE3 E. coli cells (New England Biolabs) were transformed with the pMSCG11-C subunit plasmids and grown in LB media supplemented with chloramphenicol (Sigma). Cells were incubated in a shaker at 37 °C to an optical density of 0.5 and then induced with 500 μM Isopropyl β-D-1-thiogalactopyranoside (Sigma). Immediately after induction the temperature was dropped to 18 °C and the cells were incubated for 20 h.
During purification steps a mammalian cell protease cocktail (Sigma) was used to prevent proteolytic cleavage of the C subunit mutants. Purification of C subunit mutants was carried out at 4 °C via an AKTA FPLC. Cell lysates in PBS pH 7.4 were loaded onto a 10 mL Ni-NTA agarose (Thermo Scientific) column and unbound protein was washed off with PBS followed by stepwise elution at 10, 25, 75 and 250 mM Imidazole in PBS pH 7.4. Fractions were analyzed by SDS-PAGE, determined to be > 90 % pure, concentrated and stored at −80 °C. Protein concentrations were determined by using the Bradford reagent from Pierce.
Assessment of PKA activity
The in vitro activities of C subunit enzymes were determined using a coupled enzyme assay with kemptide (LRRASLG) as the substrate. Briefly, phosphorylation of the peptide was coupled to pyruvate kinase and lactate dehydrogenase resulting in the oxidation of NADH. Formation of the latter was monitored at 340 nm. Pyruvate kinase and lactate dehydrogenase were maintained as non-rate limiting enzymes. The activities of the C subunit enzymes (10 – 500 nM) were determined in 50 mM Tris pH 7.5, 100 mM NaCl, 10 mM MgCl2, 10% glycerol, 1 mM kemptide, 1 mM ATP, 1mM DTT, 1 mM phosphoenol pyruvate and 0.2 mM NADH. To determine the effect of the regulatory subunit, assays were run with or without 50 nM regulatory subunit (5 times the catalytic concentration) and with or without 100 μM cAMP. All coupled assay reagents were purchased from Sigma.
Assessment of CW196R/Y204A Substrate Specificity
VASP (80 ng) was incubated with or without 500 ng PKG/PKA in buffer (50 mM Tris + 100 NaCl + 10 mM MgCl2 + 1 mM DTT + 1 mM EDTA + 1 mM ATP) with a final volume of 100 μL at room temperature for 30 min. Western blots of all samples, 10 μL per well, were performed on 4–20% SDS Tris-HCl gels (Biorad) and transferred to nitrocellulose membranes. Membranes were blocked with 5% BSA in PBS + 0.1% tween 20 for 2 h before incubation with 1:100 of the primary antibody in the blocking solution at 4 °C overnight. Membranes were washed with PBS + 0.1% tween 20, incubated with 1:10,000 secondary HRP conjugated antibody for 2 h and washed with PBS. Visualization was performed according to Pierce recommended protocols for their ECL Pico kit. Images were acquired using an Alpha Innotech Fluorchem FC2 using the chemiluminescent settings.
Cell-Based PKA Activity Assessment
Cell culture
MVD7 cells (a gift from Dr. James Bear at UNC Chapel Hill) were grown on MatTek 12-well # 1.5 glass bottom plates or 35 mm # 1.5 glass bottom dishes. The day of the microscopy experiments the media was replaced with Leibovitz’s L-15 medium without phenol red containing 10% FBS, 2 mM Glutamax, and 50 U/mL interferon γ.
Imaging of the Light-triggered Association of optoPKA Constructs with Subcellular Sites
MVD7 cells were transfected with Cry2-mCh, optoPKAW196R/Y204A, optoPKAW196R/F327A, optoPKAW196R/E203A, or optoPKAW196R and OMM-Cib, PM-Cib, or LifeAct-Cib at a ratio of 1:1 with a total of 2 μg DNA per well. 1:2 ratio of DNA:JetPrime (mass:vol) transfection cocktails were incubated at 10 min at RT before adding to dishes. Cells were incubated with DNA for 3 h at 32 °C before media was exchanged. On the day of imaging, media was exchanged for L-15/10 % FBS/1X GlutaMAX/50 UI interferon γ. Dishes were allowed to equilibrate in heated incubators (32 °C, 45% humidity) for at least 30 min prior to imaging. Lifeact-Cib and OMM-Cib imaging was performed on an inverted Olympus IX81 microscope equipped with a Hamamatsu C8484 camera, 60X oil immersion Plan S-Apo objective and FITC and TxRed filter cubes from Semrock. Images were acquired every 30 s over 15 min. Light stimulation was provided via a 100 ms pulse from the FITC cube in the first frame and translocation of optoPKA was monitored via mCh fluorescence by a 500 ms pulse from the TxRed cube. Image analysis was performed by defining regions of interest (ROIs) at the mitochondria and the actin cytoskeleton. At least three ROIs were analyzed per cell and three total cells were analyzed. Background fluorescence measurements were subtracted from each ROI and a normalized change in fluorescence was calculated. PM-Cib imaging was performed on an Olympus FV1000 scanning confocal microscope with an IX81 base using the 488 nm and 559 nm laser lines. Two frames of the 559 nm mCh channel were recorded before a single stimulus of the 488 nm laser at 10 μs per pixel and 2% power followed by imaging with the 559 nm laser every 30 s. Image analysis was performed as described above.
Initial cellular activity characterization and immunofluorescence
Nontransfected MVD7 cells were treated with 100 μM forskolin (Fisher) for 30 min at 32 °C before fixation using the protocol outlined under Fluorescent Microscopy Imaging Analysis. Cells were transfected with optoPKA and OMM-Cib and allowed to grow overnight. For these studies, we employed an OMM-Cib that does not contain GFP as spectral bleed through between the CFP and GFP channels on our microscope impacted quantification of data. The cells were then either 1) kept in the dark, 2) exposed to light for 1 min using a 470 nM LED flood light (PAR38; Westinghouse 0315100; 1.7 mW power) or 3) exposed to light for 1 min in the presence of 10 μM H89 (PKA inhibitor, Sigma). Immediately after removal from light, cells were fixed by adding a 2X solution of fixing reagent so the final concentration was 4% paraformaldehyde (Electron Microscopy Sciences) in PBS. Cells were incubated for 10 min at room temperature before being washed twice with PBS. Cells were blocked with 5% BSA + 0.1% triton X-100 in PBS for 1 h at room temperature. Fixed cells were incubated with a 1:100 dilution of mouse anti-phosphoVASP157 (abcam ab58555) in 1% BSA + 0.1% triton X-100 in PBS overnight at 4 °C. Cells were then washed 3 times with PBS and incubated with 1:1000 anti-mouse Alexa 647 antibody (Life Technologies) at room temperature for 2 h. Cells were washed 3 times with PBS and placed in PBS for imaging. Image analysis was performed as outlined under Fluorescent Microscopy Imaging Analysis. Images for figures were subjected to 100 pixel radius rolling ball background subtraction and intensities for the phosphorylated PKA-Reporter were normalized to positive controls.
Light Titrations and Reversibility
Control cells were treated with 100 μM forskolin + 100 μM isobutylmethylxanthine (IBMX, Sigma) for 30 min at 32 °C before being fixed using the same protocol outlined above. Light titrations were conducted by exposing the cultured cells to 0, 30, 60, 120, or 300 s of light using a 470 nM LED flood light (PAR38; Westinghouse 0315100; 1.7 mW power). Cells were fixed and stained as outlined above. Reversibility experiments were performed as outlined above. In brief, cells were exposed to 5 min blue light and fixed at indicated time points. Fixation, staining, imaging, and analysis were performed as outlined above.
Fluorescent Microscopy Imaging Analysis
All fluorescent microscopy imaging experiments were performed on an inverted Olympus IX81 microscope equipped with a Hamamatsu C8484 camera, 60X oil immersion Plan S-Apo objective and CFP, TxRed, and Cy5.5 filter cubes from Semrock. Images were taken at 250 ms exposure for CFP fluorescence, 500 ms exposure for mCh fluorescence, and 1000 ms exposure for Alexa 647 fluorescence. An ROI surrounding the entire cell was defined for cells that contain both the PKA-Reporter and Cry2-mCh. The minimum background fluorescence for each image was subtracted from each cell and then the ratio of the phosphoPKA-Reporter fluorescence/PKA-Reporter fluorescence (Alexa 647/CFP) was calculated. Values were normalized to the ratio from cells that were never exposed to light. All imaging analysis was done using Image J software and reported as the mean ± standard error of 10 – 25 cells per experiment.
Phosphoproteomics Studies
Mitochondrial optoPKA Lysate for Phosphoproteomics Analysis
HEK293T cells were plated in 10 cm plates in galactose (4.5 g/L) containing media [DMEM, 10% FBS, 1% GlutaMAX, 1% Penicillin/Streptomycin, 4.5 g/L D-galactose (Sigma)] and grown to 80 – 90% confluence. Cells were transfected in media without antibiotics with 20 μg (16 μg optoPKAW196R/Y204A and 4 μg OMM-Cib) DNA/plate using lipofectamine 2000:DNA at 3:1 volume:mass for 3 h. Cells were collected via manual dissociation (pipetting) and spun down in 2 mL eppendorf tubes. Cells were pelleted and treated with 10 μM forskolin or light (100% 470 nm on colbri LED) for 30 min at room temperature. Cells were then lysed and fractionated using the Pierce mitochondrial isolation kit for cultured cells (ThermoFisher Scientific, 89874) according to manufacturer’s protocol. H89 and HALT were included in all post-experimental steps in order to preserve phosphorylation status at the time of lysis. The lysates were acetone precipitated, reconstituted in 7 M urea, reduced and alkylated, and then digested with trypsin overnight. The peptide samples were desalted using SepPak C18 columns (Waters) and then enriched for phosphopeptides with TiO2 tips (GL Sciences). TiO2 eluates were dried via vacuum centrifugation and then stored at −80 °C until analysis.
Plasma Membrane optoPKA Lysate for Phosphoproteomics Analysis
HEK293T cells were plated on 10 cm tissue-culture dishes at 80% confluency and transfected with 10 μg of total DNA per plate (5 μg PM-Cib; 5 μg optoPKAW196R/F327A or Cry2-mCh) using Lipofectamine 2000 transfection reagent (32 μL Lipofectamine reagent:1 mL Opti-MEM) in Opti-MEM with 10% FBS. 4 h post-transfection, cell media was changed to DMEM + 10% FBS, and the cells were stored overnight, protected from light, in a humidified tissue culture incubator (37 °C; 5% CO2). The next day, cells were serum starved in PBS for 15 min in a humidified tissue culture incubator (37 °C; 5% CO2), and exposed to light (4 min; 470 nm LED flood lamp; continuous illumination) or kept in the dark (4 min). Cells were then lysed using 2 mL of M-PER + 1x HALT protease and phosphatase inhibitor per plate. Lysates were cleared by centrifugation (5 min; 1000 x g; 4 °C), and concentrations determi ned via the Bradford assay. The lysates were acetone precipitated, reconstituted in 7 M urea, reduced and alkylated, and then digested with trypsin overnight. The peptide samples were desalted using SepPak C18 columns (Waters) and then enriched for phosphopeptides with TiO2 tips (GL Sciences). TiO2 eluates were dried via vacuum centrifugation and then stored at −80 °C until analysis.
LC/MS/MS Phosphoproteomics Data Acquisition
Each sample was analyzed by LC/MS/MS using an Easy nLC 1000 coupled to a QExactive HF (Thermo Scientific). Samples were injected onto an Easy Spray PepMap C18 column (75 μm id × 25 cm, 2 μm particle size) (Thermo Scientific) and separated over a 60 min method. The gradient for separation consisted of 5 – 32% mobile phase B at a 250 nL/min flow rate, where mobile phase A was 0.1% formic acid in water and mobile phase B consisted of 0.1% formic acid in acetonitrile. The QExactive HF was operated in data-dependent mode where the 15 most intense precursors were selected for subsequent HCD fragmentation. Resolution for the precursor scan (m/z 350 – 1600) was set to 120,000, while MS/MS scan resolution was set to 15,000. The normalized collision energy was set to 27% for HCD. Peptide match was set to preferred, and precursors with unknown charge or a charge state of 1 and ≥ 7 were excluded.
Phosphoproteomics Data Analysis
A label-free quantitative phosphoproteomics analysis was employed for the whole cell lysate and mitochondrial samples. Briefly, raw data files were processed using MaxQuant (Cox and Mann, 2008) (version 1.5.3.17). Data were searched against a human UniProt database (containing 20,159 sequences) using the integrated Andromeda search engine. The following parameters were used to identify tryptic peptides for protein identification: up to two missed trypsin cleavage sites; carbamidomethylation (C) was set as a fixed modification; and oxidation (M) and phosphorylation (STY) were set as variable modifications. A false discovery rate (FDR) of 1% was used to filter all results, and match between runs was enabled. The results were imported into Perseus (version 1.5.3.0) for statistical analysis (student’s t-test). To identify over-represented pSer motifs among the statistically significant (p-value ≤0.05; light/dark fold change ≥1.5) phosphopeptides, a motif analysis was conducted using Motif-X (Chou and Schwartz, 2011) with default settings (possibility threshold, p < 10−6). The individual datasets were further analyzed using IPA phosphorylation analysis (Qiagen). Phosphoproteomics datasets for the OMM and PM were filtered using KNIME (https://www.knime.org/). First, the OMM or PM datasets were filtered for PKA substrates using the PhosphositePlus database. Then, the resulting identified PKA substrates from the OMM and PM datasets were filtered through the PhosphoSitePlus kinase-substrate database to search for PKA substrates in our datasets that are protein kinases. Finally, the phosphopeptides identified as both PKA substrates and protein kinases were hand curated for the specific pSer sequences known to be phosphorylated by PKA using the PhosphoSitePlus database.
Kinase Prediction Analysis
Raw datasets from the OMM phosphoproteomics study were used. The datasets included phosphoproteomics results for forskolin, not treated, and light and dark treated cells. Forskolin and not treated data were run a single time. Therefore, a stringent filter was placed on all data. Only data points that contained full data for all treatment groups were used. We applied a cutoff of 1.3 fold upregulation in phosphorylation as another filter and categorized phosphopeptides as upregulated by forskolin, optoPKA, or both. Of the 4910 phosphopeptides identified across all groups, 1153 were represented with full datasets and used. We used the resulting lists to predict the kinases that were activated in response to forskolin or optoPKA activation. Lists were run on the NetPhorest kinase prediction tool (http://netphorest.info/) (Horn et al., 2014; Miller et al., 2008) and subsequent analysis was performed in Excel.
Quantification and Statistical Analysis
All image analysis was performed in Fiji (imageJ). Image quantification methods are described in Fluorescent Microscopy Imaging Analysis. All data is presented as mean ± SEM. N values are indicated in figure legends. For all cellular studies, n is a measure of the number of individual cells counted. For PKA kinetic studies, n represents the number of experimental replicates. All graphs and regression analysis for in vitro studies were performed in SigmaPlot 12. Proteomics quantification performed in MaxQuant (version 1.5.3.17) and Perseus (version 1.5.3.0). Analysis of the phosphoproteomics data is described in Phosphoproteomics Data Analysis.
Data Availability
All datasets generated from phosphoproteomics studies are available in Tables S4 – S7.
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-phospho VASP Ser 157 | Abcam | Ab58555 |
| Rabbit anti-HA tag | Cell Signaling Technologies | 3724 |
| Rabbit anti-phosphoPKA substrate | Cell Signaling Technologies | 9624 |
| Rabbit anti-GAPDH | Cell Signaling Technologies | 5174 |
| Rabbit anti-mCherry | Abcam | Ab167453 |
| anti-mouse Alexa 647 | LifeTechnologies | A28181 |
| anti-mouse IRDYE™ 800CW | Licor | 926-32212 |
| anti-rabbit IRDYE™ 680RD | Licor | 926-68071 |
| Anti-rabbit IgG HRP | Cell Signaling Technologies | 7074P |
| Rabbit anti-VASP Ser239 | Cell Signaling Technologies | 3114S |
| Rabbit anti-VASP | Cell Signaling Technologies | 3132S |
| Bacterial and Virus Strains | ||
| E. coli DH5α Max Efficiency | Life Technologies | 18258012 |
| E. coli BL21-DE3 | New England Biotechnologies | C2527I |
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| Forskolin | Fisher | BP252010 |
| H-89 dihydrochloride | Sigma Aldrich | B1427-5MG |
| 8 Bromo-cAMP | Sigma Aldrich | B5386 |
| Pyruvate Kinase/Lactate Dehydrogenase | Sigma Aldrich | P0294 |
| Kemptide | Sigma Aldrich | K1127 |
| 3′5′-cyclic adenosine monophosphate | Sigma Aldrich | A9501 |
| Bovine PKA regulatory subunit | Sigma Aldrich | P2645 |
| Jet Prime Poly Plus Transfection Reagent | PolyPlus (via VWR) | 114–15 |
| Lipofectamine 2000 | ThermoFisher Scientific | 11668019 |
| Isobutylmethylxanthine | Sigma Aldrich | I5879 |
| Mouse Interferon-gamma | ThermoFisher Scientific | PMC4033 |
| Ni-NTA resin | ThermoFisher Scientific | R90101 |
| Protein A Sepharose 4B | ThermoFisher Scientific | 101041 |
| Mitochondrial Isolation kit for cultured cells | ThermoFisher Scientific | 89874 |
| DMEM | ThermoFisher Scientific | 11995065 |
| Fetal Bovine Serum | Hyclone | SH30071.03 |
| GlutaMAX-I | ThermoFisher Scientific | 35050-061 |
| Penicillin/Streptomycin | ThermoFisher Scientific | 15140122 |
| D-Galactose | Sigma | G5388 |
| BsrGI Restriction Enzyme | New England Biotech | R0575S |
| NotI Restriction Enzyme | New England Biotech | R0189L |
| BamHI Restriction Enzyme | New England Biotech | R0136L |
| KpnI Restriction Enzyme | New England Biotech | R0142L |
| EcoRI Restriction Enzyme | New England Biotech | R0101L |
| IPTG | Sigma | I5502 |
| LB/LB Agar | Difco | 244610/244510 |
| Mammalian cell protease inhibitor cocktail | Sigma | P8340 |
| 660 nm protein assay | ThermoFisher Scientific | 22660 |
| Bradford protein assay | ThermoFisher Scientific | 23238 |
| NADH | Acros Ogranics | 271100010 |
| Leibovitz L-15 media, no phenol red | ThermoFisher Scientific | 21083027 |
| BSA | Fisher | BP1600-100 |
| Triton-X 100 | Fisher | BP151-500 |
| Paraformaldehyde | Electron Microscopy Sciences | 15710 |
| M-PER lysis buffer | ThermoFisher Scientific | 78501 |
| Halt Protease and Phosphatase Inhibitor Cocktail | ThermoFisher Scientific | 78445 |
| 4–15% Tris-Glycine Gels | BioRad | 4561084 |
| 4–12% Bis-Tris Gels | ThermoFisher Scientific | NP0336BOX |
| 20X MOPS running buffer | ThermoFisher Scientific | NP0001 |
| 0.2 μm PVDF membrane | BioRad | 1620239 |
| Formic Acid | Fisher | BP1215-500 |
| Acetonitrile | Fisher | A955-1 |
| Opti-MEM | Gibco | 31985062 |
| BioRad Clarity Western ECL Substrate | BioRad | 1705061 |
| RESTORE stripping buffer | ThermoFisher Scientific | 21059 |
| Adenosine Triphosphate | Sigma | A6417 |
| Dithiothreitol | Sigma | D9779 |
| Antibiotic-Antimycotic | ThermoFisher Scientific | 15240062 |
| VASP protein | abnova | |
| Recombinant PKG I63S | Priestman et al, ACS Chem. Biol., 2011, 6 (4), pp 377–384 | |
| Recombinant PKA C-subunit (and mutants) | This Paper | |
| Phosphoenol Pyruvate | Sigma Aldrich | 10108294001 |
| Critical Commercial Assays | ||
| QuikChange II Site Directed Mutagenesis Kit | Agilent | 200523 |
| Deposited Data | ||
| Experimental Models: Cell Lines | ||
| Mouse: MVD7 cells: Ena/Mena/VASP KO mouse embryonic fibroblasts | Dr. James E. Bear | N/A |
| Human: HEK293T I17 cells | UNC Tissue Culture Facility | |
| Experimental Models: Organisms/Strains | ||
| Oligonucleotides | ||
| LIC-C subunit-F TACTTCCAATCCAATGCCATGGGCAACGCCGCCGCCGCCAAGAA |
This Paper/IDTDNA | N/A |
| LIC-C subunit-R TTATCCACTTCCAATGTTACTAAAACTCAGTAAACTCCTTGCC |
This Paper/IDTDNA | N/A |
| C subunit site directed mutagenesis primer: W196R-F GTTTTGCCAAGCGTGTGAAAGGCCGTACTCGGACCTTGTGTGGGACC |
This Paper/IDTDNA | N/A |
| C subunit site directed mutagenesis primer: W196R-R GGCCAAGTACTCAGGGGTCCCACACAAGGTCCGAGTACGGCCTTTCAC |
This Paper/IDTDNA | N/A |
| C subunit site directed mutagenesis primer: F327A-F CCTGGGGACACGAGTAACGCCGACGACTATGAGGAGGAAGAG |
This Paper/IDTDNA | N/A |
| C subunit site directed mutagenesis primer: F327A-R CTCTTCCTCCTCATAGTCGTCGGCGTTACTCGTGTCCCCAGG |
This Paper/IDTDNA | N/A |
| C subunit site directed mutagenesis primer: Y204A-F GTGTGGGACCCCTGAGGCCTTGGCCCCCGAGATTATC |
This Paper/IDTDNA | N/A |
| C subunit site directed mutagenesis primer: Y204A-R GATAATCTCGGGGGCCAAGGCCTCAGGGGTCCCACAC |
This Paper/IDTDNA | N/A |
| C subunit site directed mutagenesis primer: E203A-F GACCTTGTGTGGGACCCCTGCCTACTTGGCCCCCGAGATTATC |
This Paper/IDTDNA | N/A |
| C subunit site directed mutagenesis primer: E203A-R GATAATCTCGGGGGCCAAGTAGGCAGGGGTCCCACACAAGGTC |
This Paper/IDTDNA | N/A |
| C subunit lacking 10 N terminal residues: 302-PKA-DeltaN10-F ATGGGCCTCGAGATGGAGCAGGAGAGCGTGAAAGAG |
This Paper/IDTDNA | N/A |
| Insertion of CAAX-stop into Cib-GFP: CIB-CAAX-F GGAGAAAACGGATTATATTCATGTAGGCGGCAGCGGTAAAAAGAAGAAAAAGAAGTCAAAGACAAAGTGTGTAATTATGTAGACCGGTCGCCACCATGGTGAGC |
This Paper/IDTDNA | N/A |
| Insertion of CAAX-stop into Cib-GFP: CIB-CAAX-R GCTCACCATGGTGGCGACCGGTCTACATAATTACACACTTTGTCTTTGACTTCTTTTTCTTCTTTTTACCGCTGCCGCCTACATGAATATAATCCGTTTTCTCC |
This Paper/IDTDNA | N/A |
| Primer for amplification of core mTurquoise2 for reporter: KpnI-HA-mTurquoise2 Forward ATGACTAGTGGTACCTACCCATACGATGTTCCAGATTACGCTATGGTGAGCAAGGGCGAGGAG |
This Paper/IDTDNA | N/A |
| Primer for addition of N-terminal PM targeting sequence to reporter: EcoRI-LynPMSS-HA Forward GAATTCGCCACCATGGGCTGCATCAAGAGCAAGGGCAAGGACAGCGCCTACCCATACGATGTTCCAGATTACGC |
This Paper/IDTDNA | N/A |
| Primer for addition of VASP peptide: mTurquoise1-VASP148–164 Reverse CATCACGGATCCTCAGGCGGGGGGGCCGCCGGCGTTGCTCACCCGCCGCTCGATGTGCTCGCTCTTGTACAGCTCGTCCATGCCGAGAGTGATCCCG |
This Paper/IDTDNA | N/A |
| Forward Sequencing Primer for pEF1alpha: AGATCTCGAGCTCAAGCTTC |
This Paper/IDTDNA | N/A |
| Reverse Sequencing Primer for pEF1alpha: GTAACCATTATAAGCTGCAA |
This Paper/IDTDNA | N/A |
| Recombinant DNA | ||
| pMSCG11 vector | DNASU repository | pMSCG11 |
| pEF1alpha vector (modified from Clontech # 631980) | Clontech | 631980 |
| PKA-prSET | Dr. Susan S. Taylor | |
| pmCherryN1(Cry2-mCh) | Addgene | 26866 |
| CibN(ΔNLS)-GFP-CAAX | Addgene | 268267 |
| Tom20-Cib-GFP | Hughes et al. 2015 | |
| Lifeact-Cib-GFP | Hughes et al. 2014 | |
| Tom20-Cib | This Paper | N/A |
| Cib-CAAX | This Paper | N/A |
| pmCherryN1-Cry2-mCh-PKA (and associated mutants) | This Paper | N/A |
| pMSCG11-PKA C subunit (and associated mutants) | This Paper | N/A |
| pEF1alpha-Tom20-HA-mTurquoise2-VASP 148–164 (OMM Reporter) | This Paper | N/A |
| pEF1alpha-LynPMSS-HA-mTurquoise2-VASP 148–164 (PM Reporter) | This Paper | N/A |
| mTurquoise2 plasmid | Dr. Theodorus Gadella, University of Amsterdam, Netherlands | |
| Software and Algorithms | ||
| Fiji/imageJ | Schindelin, K et al. 2012 | https://fiji.sc/ |
| QuikChange II Site Directed Mutagenesis Primer Design Tool | Agilent | http://www.genomics.agilent.com/primerDesignProgram.jsp |
| Sequest with Proteome Discoverer v1.4 | ThermoFisher Scientific | https://www.thermofisher.com/order/catalog/product/IQLAAEGABSFAKJMAUH |
| MaxQuant v1.5.3.17 | Cox and Mann 2008 | http://www.coxdocs.org/doku.php?id=maxquant:start |
| NetPhorest 2.1 | Kinome Explorer | http://netphorest.info/ |
| University of Arizona Spectral Database | http://www.spectra.arizona.edu/ | |
| KNIME Analytics Platform | KNIME | https://www.knime.org/ |
| Other | ||
| 470 nm flood lamp PAR38 1.7 mW | Westinghouse | 0315100 |
| Colibri LED light source | Colibri | |
| MatTek glass bottom dishes (#1.5) | MatTek | https://www.mattek.com/product-category/cultureware/ |
| Easy nLC 1000-QExactive HF LC/MS/MS | Thermo Scientific | LC120/IQLAAEGAAPFALGMBFZ |
| TiO2 Tips for phosphopeptide enrichment | GL Sciences | http://www.glsciencesinc.com/ProductInfo/Proteomics/Phosphopeptides.aspx |
Supplementary Material
Time-lapse microscopy of optoPKA association with and disassociation from the cytoskeleton.
Time-lapse microscopy of optoPKA association with and disassociation from the cytoskeleton.
Time-lapse microscopy of optoPKA association with and disassociation from the plasma membrane.
Table S4. Related to Figure 7. Summary of OMM and PM phosphoproteomics datasets. See appended Excel file.
Table S7. Related to Figure 7. Data used for NetPhorest prediction.
SIGNIFICANCE.
The design and acquisition of optogenetic analogs of endogenous mammalian proteins has received considerable attention. However, a major engineering impediment is the “dark activity” observed with these constructs. We’ve developed a structure-based strategy to design reduced activity protein kinase mutants that reside in the cytoplasm in an enzymologically impotent state. This strategy targets a structural motif near the kinase active site that is common to many protein kinases and thus offers the potential application to other members of this large enzyme family. Illumination of the cytoplasmically diffuse, enzymatically reduced kinase triggers a concentration jump to the intracellular site of interest and consequent phosphotransferase activity. In addition, a PKA-Reporter has been constructed that resides at designated intracellular locations and thus serves as a barometer of subcellular protein kinase activity. Furthermore, we demonstrated that optoPKA recruitment to the mitochondria or plasma membrane results in the phosphorylation of validated endogenous substrates of PKA, displays the anticipated organelle-selective phosphorylation of substrates, and generates a proteome-wide enriched phosphorylated generic PKA sequence. The ability to trigger signaling activity in a spatially-focused, temporally-resolved fashion, offers a means to correlate the relationship between intracellular biochemical activity at a given time and place with the attendant downstream signaling (via proteomics) and genomic responses, and subsequent alterations in cellular (and organismal) behavior.
Highlights.
An engineered organelle-targeted optogenetic protein kinase
Design of organelle-specific reporters of protein kinase activity
Proteomics of light induced kinase activity at the mitochondria and plasma membrane
Acknowledgments
We thank the National Institutes of Health for financial support (1R21NS093617 and 1U01CA207160).
Footnotes
Supplemental Information includes three figures, six tables, three movies, and sequence information for five constructs.
AUTHOR CONTRIBUTIONS
D.S.L, C.P.O, R.M.H, M.A.P, and L.E.H. designed the experiments. C.P.O. and D.S.L. wrote the manuscript. R.M.H cloned bacterial PKA and optoPKA constructs. M.A.P. performed in vitro kinetics experiments. C.P.O. developed the PKA reporter, performed association and disassociation, and 1 min light experiments. M.A.P. performed the experiments described in Figure 5. R.M.H, C.P.O, S.J.C., and L.E.H. performed phosphoproteomics experiments.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Amunjela JN, Tucker SJ. POPDC proteins as potential novel therapeutic targets in cancer. Drug Discov Today. 2016;21:1920–1927. doi: 10.1016/j.drudis.2016.07.011. [DOI] [PubMed] [Google Scholar]
- Amunjela JN, Tucker SJ. POPDC proteins as potential novel therapeutic targets in cancer. Drug Discov Today. 2016;21:1920–1927. doi: 10.1016/j.drudis.2016.07.011. [DOI] [PubMed] [Google Scholar]
- Banerjee R, Schleicher E, Meier S, Viana RM, Pokorny R, Ahmad M, Bittl R, Batschauer A. The signaling state of Arabidopsis cryptochrome 2 contains flavin semiquinone. J Biol Chem. 2007;282:14916–14922. doi: 10.1074/jbc.M700616200. [DOI] [PubMed] [Google Scholar]
- Bear JE, Loureiro JJ, Libova I, Fässler R, Wehland J, Gertler FB. Negative Regulation of Fibroblast Motility by Ena/VASP Proteins. Cell. 2000;101:717–728. doi: 10.1016/s0092-8674(00)80884-3. [DOI] [PubMed] [Google Scholar]
- Chou MF, Schwartz D. Biological sequence motif discovery using motif-x. Curr Protoc Bioinformatics. 2011;Chapter 13(Unit 13):15–24. doi: 10.1002/0471250953.bi1315s35. [DOI] [PubMed] [Google Scholar]
- Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol. 2008;26:1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- de Graaf EL, Giansanti P, Altelaar AF, Heck AJ. Single-step enrichment by Ti4+-IMAC and label-free quantitation enables in-depth monitoring of phosphorylation dynamics with high reproducibility and temporal resolution. Mol Cell Proteomics. 2014;13:2426–2434. doi: 10.1074/mcp.O113.036608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deisseroth K. Optogenetics: 10 years of microbial opsins in neuroscience. Nat Neurosci. 2015;18:1213–1225. doi: 10.1038/nn.4091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depry C, Allen MD, Zhang J. Visualization of PKA activity in plasma membrane microdomains. Mol Biosyst. 2011;7:52–58. doi: 10.1039/c0mb00079e. [DOI] [PubMed] [Google Scholar]
- Dessauer CW. Adenylyl cyclase--A-kinase anchoring protein complexes: the next dimension in cAMP signaling. Mol Pharmacol. 2009;76:935–941. doi: 10.1124/mol.109.059345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhillon AS, Pollock C, Steen H, Shaw PE, Mischak H, Kolch W. Cyclic AMP-dependent kinase regulates Raf-1 kinase mainly by phosphorylation of serine 259. Mol Cell Biol. 2002;22:3237–3246. doi: 10.1128/MCB.22.10.3237-3246.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickey AS, Strack S. PKA/AKAP1 and PP2A/Bbeta2 regulate neuronal morphogenesis via Drp1 phosphorylation and mitochondrial bioenergetics. J Neurosci. 2011;31:15716–15726. doi: 10.1523/JNEUROSCI.3159-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards DC, Gill GN. Structural features of LIM kinase that control effects on the actin cytoskeleton. J Biol Chem. 1999;274:11352–11361. doi: 10.1074/jbc.274.16.11352. [DOI] [PubMed] [Google Scholar]
- Fang X, Yu SX, Lu Y, Bast RC, Jr, Woodgett JR, Mills GB. Phosphorylation and inactivation of glycogen synthase kinase 3 by protein kinase A. Proc Natl Acad Sci U S A. 2000;97:11960–11965. doi: 10.1073/pnas.220413597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaffarogullari EC, Masterson LR, Metcalfe EE, Traaseth NJ, Balatri E, Musa MM, Mullen D, Distefano MD, Veglia G. A myristoyl/phosphoserine switch controls cAMP-dependent protein kinase association to membranes. J Mol Biol. 2011;411:823–836. doi: 10.1016/j.jmb.2011.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gancedo JM. Biological roles of cAMP: variations on a theme in the different kingdoms of life. Biol Rev Camb Philos Soc. 2013;88:645–668. doi: 10.1111/brv.12020. [DOI] [PubMed] [Google Scholar]
- Gerarduzzi C, He Q, Antoniou J, Di Battista JA. Quantitative phosphoproteomic analysis of signaling downstream of the prostaglandin e2/g-protein coupled receptor in human synovial fibroblasts: potential antifibrotic networks. J Proteome Res. 2014;13:5262–5280. doi: 10.1021/pr500495s. [DOI] [PubMed] [Google Scholar]
- Giansanti P, Stokes MP, Silva JC, Scholten A, Heck AJ. Interrogating cAMP-dependent kinase signaling in Jurkat T cells via a protein kinase A targeted immune-precipitation phosphoproteomics approach. Mol Cell Proteomics. 2013;12:3350–3359. doi: 10.1074/mcp.O113.028456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson RM, Taylor SS. Dissecting the cooperative reassociation of the regulatory and catalytic subunits of cAMP-dependent protein kinase. Role of Trp-196 in the catalytic subunit. J Biol Chem. 1997;272:31998–32005. doi: 10.1074/jbc.272.51.31998. [DOI] [PubMed] [Google Scholar]
- Graeser R, Gannon J, Poon RY, Dubois T, Aitken A, Hunt T. Regulation of the CDK-related protein kinase PCTAIRE-1 and its possible role in neurite outgrowth in Neuro-2A cells. J Cell Sci. 2002;115:3479–3490. doi: 10.1242/jcs.115.17.3479. [DOI] [PubMed] [Google Scholar]
- Hamaguchi T, Nakamuta S, Funahashi Y, Takano T, Nishioka T, Shohag MH, Yura Y, Kaibuchi K, Amano M. In vivo screening for substrates of protein kinase A using a combination of proteomic approaches and pharmacological modulation of kinase activity. Cell Struct Funct. 2015;40:1–12. doi: 10.1247/csf.14014. [DOI] [PubMed] [Google Scholar]
- Horn H, Schoof EM, Kim J, Robin X, Miller ML, Diella F, Palma A, Cesareni G, Jensen LJ, Linding R. KinomeXplorer: an integrated platform for kinome biology studies. Nat Methods. 2014;11:603–604. doi: 10.1038/nmeth.2968. [DOI] [PubMed] [Google Scholar]
- Hornbeck PV, Zhang B, Murray B, Kornhauser JM, Latham V, Skrzypek E. PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res. 2015;43:D512–520. doi: 10.1093/nar/gku1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes RM, Freeman DJ, Lamb KN, Pollet RM, Smith WJ, Lawrence DS. Optogenetic apoptosis: light-triggered cell death. Angew Chem Int Ed Engl. 2015;54:12064–12068. doi: 10.1002/anie.201506346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes RM, Lawrence DS. Optogenetic engineering: light-directed cell motility. Angew Chem Int Ed Engl. 2014;53:10904–10907. doi: 10.1002/anie.201404198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imamura H, Wagih O, Niinae T, Sugiyama N, Beltrao P, Ishihama Y. Identifications of Putative PKA Substrates with Quantitative Phosphoproteomics and Primary-Sequence-Based Scoring. J Proteome Res. 2017;16:1825–1830. doi: 10.1021/acs.jproteome.7b00087. [DOI] [PubMed] [Google Scholar]
- Jacobs M, Hayakawa K, Swenson L, Bellon S, Fleming M, Taslimi P, Doran J. The structure of dimeric ROCK I reveals the mechanism for ligand selectivity. J Biol Chem. 2006;281:260–268. doi: 10.1074/jbc.M508847200. [DOI] [PubMed] [Google Scholar]
- Kanaji S, Iwahashi J, Kida Y, Sakaguchi M, Mihara K. Characterization of the signal that directs Tom20 to the mitochondrial outer membrane. J Cell Biol. 2000;151:277–288. doi: 10.1083/jcb.151.2.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy MJ, Hughes RM, Peteya LA, Schwartz JW, Ehlers MD, Tucker CL. Rapid blue-light-mediated induction of protein interactions in living cells. Nat Methods. 2010;7:973–975. doi: 10.1038/nmeth.1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambrechts A, Kwiatkowski AV, Lanier LM, Bear JE, Vandekerckhove J, Ampe C, Gertler FB. cAMP-dependent protein kinase phosphorylation of EVL, a Mena/VASP relative, regulates its interaction with actin and SH3 domains. J Biol Chem. 2000;275:36143–36151. doi: 10.1074/jbc.M006274200. [DOI] [PubMed] [Google Scholar]
- Lawrence DS. The preparation and in vivo applications of caged peptides and proteins. Curr Opin Chem Biol. 2005;9:570–575. doi: 10.1016/j.cbpa.2005.09.002. [DOI] [PubMed] [Google Scholar]
- Lefkimmiatis K, Zaccolo M. cAMP signaling in subcellular compartments. Pharmacol Ther. 2014;143:295–304. doi: 10.1016/j.pharmthera.2014.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manser E, Huang HY, Loo TH, Chen XQ, Dong JM, Leung T, Lim L. Expression of constitutively active alpha-PAK reveals effects of the kinase on actin and focal complexes. Mol Cell Biol. 1997;17:1129–1143. doi: 10.1128/mcb.17.3.1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matulef K, Zagotta WN. Cyclic nucleotide-gated ion channels. Annu Rev Cell Dev Biol. 2003;19:23–44. doi: 10.1146/annurev.cellbio.19.110701.154854. [DOI] [PubMed] [Google Scholar]
- Miller ML, Jensen LJ, Diella F, Jorgensen C, Tinti M, Li L, Hsiung M, Parker SA, Bordeaux J, Sicheritz-Ponten T, et al. Linear motif atlas for phosphorylation-dependent signaling. Sci Signal. 2008;1:ra2. doi: 10.1126/scisignal.1159433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo GC, Ross B, Hertel F, Manna P, Yang X, Greenwald E, Booth C, Plummer AM, Tenner B, Chen Z, et al. Genetically encoded biosensors for visualizing live-cell biochemical activity at super-resolution. Nat Methods. 2017;14:427–434. doi: 10.1038/nmeth.4221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monterisi S, Zaccolo M. Components of the mitochondrial cAMP signalosome. Biochem Soc Trans. 2017;45:269–274. doi: 10.1042/BST20160394. [DOI] [PubMed] [Google Scholar]
- Moore MJ, Adams JA, Taylor SS. Structural basis for peptide binding in protein kinase A. Role of glutamic acid 203 and tyrosine 204 in the peptide-positioning loop. J Biol Chem. 2003;278:10613–10618. doi: 10.1074/jbc.M210807200. [DOI] [PubMed] [Google Scholar]
- Newell-Litwa KA, Horwitz AR. Cell migration: PKA and RhoA set the pace. Curr Biol. 2011;21:R596–598. doi: 10.1016/j.cub.2011.06.032. [DOI] [PubMed] [Google Scholar]
- Ni Q, Ganesan A, Aye-Han NN, Gao X, Allen MD, Levchenko A, Zhang J. Signaling diversity of PKA achieved via a Ca2+-cAMP-PKA oscillatory circuit. Nat Chem Biol. 2011;7:34–40. doi: 10.1038/nchembio.478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohashi K, Nagata K, Maekawa M, Ishizaki T, Narumiya S, Mizuno K. Rho-associated kinase ROCK activates LIM-kinase 1 by phosphorylation at threonine 508 within the activation loop. J Biol Chem. 2000;275:3577–3582. doi: 10.1074/jbc.275.5.3577. [DOI] [PubMed] [Google Scholar]
- Pathak GP, Vrana JD, Tucker CL. Optogenetic control of cell function using engineered photoreceptors. Biol Cell. 2013;105:59–72. doi: 10.1111/boc.201200056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polzien L, Baljuls A, Rennefahrt UE, Fischer A, Schmitz W, Zahedi RP, Sickmann A, Metz R, Albert S, Benz R, et al. Identification of novel in vivo phosphorylation sites of the human proapoptotic protein BAD: pore-forming activity of BAD is regulated by phosphorylation. J Biol Chem. 2009;284:28004–28020. doi: 10.1074/jbc.M109.010702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priestman MA, Sun L, Lawrence DS. Dual wavelength photoactivation of cAMP- and cGMP-dependent protein kinase signaling pathways. ACS Chem Biol. 2011;6:377–384. doi: 10.1021/cb100398e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raffelberg S, Wang L, Gao S, Losi A, Gartner W, Nagel G. A LOV-domain-mediated blue-light-activated adenylate (adenylyl) cyclase from the cyanobacterium Microcoleus chthonoplastes PCC 7420. Biochem J. 2013;455:359–365. doi: 10.1042/BJ20130637. [DOI] [PubMed] [Google Scholar]
- Ryu MH, Moskvin OV, Siltberg-Liberles J, Gomelsky M. Natural and engineered photoactivated nucleotidyl cyclases for optogenetic applications. J Biol Chem. 2010;285:41501–41508. doi: 10.1074/jbc.M110.177600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sample V, DiPilato LM, Yang JH, Ni Q, Saucerman JJ, Zhang J. Regulation of nuclear PKA revealed by spatiotemporal manipulation of cyclic AMP. Nat Chem Biol. 2012;8:375–382. doi: 10.1038/nchembio.799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott JD, Dessauer CW, Tasken K. Creating order from chaos: cellular regulation by kinase anchoring. Annu Rev Pharmacol Toxicol. 2013;53:187–210. doi: 10.1146/annurev-pharmtox-011112-140204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CM, Radzio-Andzelm E, Madhusudan Akamine P, Taylor SS. The catalytic subunit of cAMP-dependent protein kinase: prototype for an extended network of communication. Prog Biophys Mol Biol. 1999;71:313–341. doi: 10.1016/s0079-6107(98)00059-5. [DOI] [PubMed] [Google Scholar]
- Steichen JM, Kuchinskas M, Keshwani MM, Yang J, Adams JA, Taylor SS. Structural basis for the regulation of protein kinase A by activation loop phosphorylation. J Biol Chem. 2012;287:14672–14680. doi: 10.1074/jbc.M111.335091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stierl M, Stumpf P, Udwari D, Gueta R, Hagedorn R, Losi A, Gartner W, Petereit L, Efetova M, Schwarzel M, et al. Light modulation of cellular cAMP by a small bacterial photoactivated adenylyl cyclase, bPAC, of the soil bacterium Beggiatoa. J Biol Chem. 2011;286:1181–1188. doi: 10.1074/jbc.M110.185496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taurin S, Sandbo N, Qin Y, Browning D, Dulin NO. Phosphorylation of beta-catenin by cyclic AMP-dependent protein kinase. J Biol Chem. 2006;281:9971–9976. doi: 10.1074/jbc.M508778200. [DOI] [PubMed] [Google Scholar]
- Taylor SS, Zhang P, Steichen JM, Keshwani MM, Kornev AP. PKA: lessons learned after twenty years. Biochim Biophys Acta. 2013;1834:1271–1278. doi: 10.1016/j.bbapap.2013.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thul PJ, Akesson L, Wiking M, Mahdessian D, Geladaki A, Ait Blal H, Alm T, Asplund A, Bjork L, Breckels LM, et al. A subcellular map of the human proteome. Science. 2017;356 doi: 10.1126/science.aal3321. [DOI] [PubMed] [Google Scholar]
- Tkachenko E, Sabouri-Ghomi M, Pertz O, Kim C, Gutierrez E, Machacek M, Groisman A, Danuser G, Ginsberg MH. Protein kinase A governs a RhoA-RhoGDI protrusion-retraction pacemaker in migrating cells. Nat Cell Biol. 2011;13:660–667. doi: 10.1038/ncb2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Towler DA, Eubanks SR, Towery DS, Adams SP, Glaser L. Amino-terminal processing of proteins by N-myristoylation. Substrate specificity of N-myristoyl transferase. J Biol Chem. 1987;262:1030–1036. [PubMed] [Google Scholar]
- Virdee K, Parone PA, Tolkovsky AM. Phosphorylation of the pro-apoptotic protein BAD on serine 155, a novel site, contributes to cell survival. Curr Biol. 2000;10:1151–1154. doi: 10.1016/s0960-9822(00)00702-8. [DOI] [PubMed] [Google Scholar]
- Weitzman M, Hahn KM. Optogenetic approaches to cell migration and beyond. Curr Opin Cell Biol. 2014;30:112–120. doi: 10.1016/j.ceb.2014.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Kennedy EJ, Wu J, Deal MS, Pennypacker J, Ghosh G, Taylor SS. Contribution of non-catalytic core residues to activity and regulation in protein kinase A. J Biol Chem. 2009;284:6241–6248. [Google Scholar]
- Yeo MG, Oh HJ, Cho HS, Chun JS, Marcantonio EE, Song WK. Phosphorylation of Ser 21 in Fyn regulates its kinase activity, focal adhesion targeting, and is required for cell migration. J Cell Physiol. 2011;226:236–247. doi: 10.1002/jcp.22335. [DOI] [PubMed] [Google Scholar]
- Yin T, Wu YI. Guiding lights: recent developments in optogenetic control of biochemical signals. Pflugers Archiv: European journal of physiology. 2013;465:397–408. doi: 10.1007/s00424-013-1244-x. [DOI] [PubMed] [Google Scholar]
- Zaccolo M, Di Benedetto G, Lissandron V, Mancuso L, Terrin A, Zamparo I. Restricted diffusion of a freely diffusible second messenger: mechanisms underlying compartmentalized cAMP signalling. Biochem Soc Trans. 2006;34:495–497. doi: 10.1042/BST0340495. [DOI] [PubMed] [Google Scholar]
- Zhang J, Ma Y, Taylor SS, Tsien RY. Genetically encoded reporters of protein kinase A activity reveal impact of substrate tethering. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:14997–15002. doi: 10.1073/pnas.211566798. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Time-lapse microscopy of optoPKA association with and disassociation from the cytoskeleton.
Time-lapse microscopy of optoPKA association with and disassociation from the cytoskeleton.
Time-lapse microscopy of optoPKA association with and disassociation from the plasma membrane.
Table S4. Related to Figure 7. Summary of OMM and PM phosphoproteomics datasets. See appended Excel file.
Table S7. Related to Figure 7. Data used for NetPhorest prediction.
Data Availability Statement
All datasets generated from phosphoproteomics studies are available in Tables S4 – S7.