

Abstract
The liver is crucial for the maintenance of normal glucose homeostasis — it produces glucose during fasting and stores glucose postprandially. However, these hepatic processes are dysregulated in type 1 and type 2 diabetes mellitus, and this imbalance contributes to hyperglycaemia in the fasted and postprandial states. Net hepatic glucose production is the summation of glucose fluxes from gluconeogenesis, glycogenolysis, glycogen synthesis, glycolysis and other pathways. In this Review, we discuss the in vivo regulation of these hepatic glucose fluxes. In particular, we highlight the importance of indirect (extrahepatic) control of hepatic gluconeogenesis and direct (hepatic) control of hepatic glycogen metabolism. We also propose a mechanism for the progression of subclinical hepatic insulin resistance to overt fasting hyperglycaemia in type 2 diabetes mellitus. Insights into the control of hepatic gluconeogenesis by metformin and insulin and into the role of lipid-induced hepatic insulin resistance in modifying gluconeogenic and net hepatic glycogen synthetic flux are also discussed. Finally, we consider the therapeutic potential of strategies that target hepatosteatosis, hyperglucagonaemia and adipose lipolysis.
Hepatic glucose production (HGP) accounts for ~90% of endogenous glucose production1, and it is crucial for systemic glucose homeostasis2. Net HGP is the summation of fluxes from gluconeogenesis, glycogenolysis, glycogen synthesis, glycolysis and other pathways. In the fasted state, the liver provides glucose to maintain euglycaemia and fuel obligate glucose-consuming cell types, such as neurons, red blood cells and renal medullary cells3. Postprandially, the liver contributes to normal glucose tolerance4. The liver contributes to the disposal of enteral glucose loads by increasing the rate of glycogen synthesis and suppressing hepatic glucose output; these result in a net switch from hepatic glucose output to hepatic glucose uptake2. The suppression of hepatic glucose output involves the suppression of hepatic glycogenolysis and gluconeogenesis. As both glycogenolysis and gluconeogenesis contribute to HGP in humans that have fasted for less than 24 h (REF.5), knowledge of the mechanisms that mediate the postprandial suppression of both processes is relevant to understanding the hyperglycaemia observed in diabetes mellitus. Net hepatic glucose uptake, as measured by splanchnic arteriovenous balance and tracer methods, is estimated to be approximately one-third of a moderate enteral glucose load in humans and dogs2,4,6–9. However, the liver also contributes to the systemic disposal of an enteral glucose load through the suppression of glucose output, thus facilitating the consumption of residual exogenous glucose by extrahepatic tissues, such as skeletal muscle and adipose tissue.
Key regulators of hepatic glucose metabolism act through diverse mechanisms. For example, HGP is regulated by the provision of substrates, such as glucose or glycerol; allosteric control by metabolites, such as acetyl-CoA, glucose and glucose-6-phosphate; the balance of hormones, including insulin, glucagon, catecholamines and corticosteroids; and cellular redox state, which can be modified by treatment with metformin. This list is not comprehensive, which highlights the complexity of the physiological regulation of HGP. In addition, the processes that contribute to net HGP, including glycogen synthesis, glycogenolysis and gluconeogenesis, are regulated by independent mechanisms10–12. As a result of this complexity, hepatic glucose uptake is maximally stimulated by conditions that mimic the postprandial state, such as portal venous hyperglycaemia and hyperinsulinaemia8,13. Of note, the ‘gold-standard’ test of peripheral and hepatic insulin sensitivity — the hyperinsulinaemic–euglycaemic clamp technique14 — fails to recreate the postprandial hepatic sinusoidal milieu. For example, maintaining euglycaemia rather than achieving portal venous hyper glycaemia does not promote net hepatic glycogen synthesis2. Furthermore, the infusion of insulin through a peripheral catheter decreases the normal insulin concentration gradient between the portal vein and systemic vein (normally approximately threefold higher relative to the concentration in the portal vein)15. Consequently, for hyperinsulinaemic–euglycaemic clamps that only increase peripheral insulin concentration by approximately threefold, the insulin level that is detected by the liver will not change. These limitations must be considered in studies of hepatic glucose metabolism using the hyperinsulinaemic–euglycaemic clamp.
This Review aims to detail both the established mechanistic knowledge and newer insights regarding the regulation of HGP in health and in diabetes mellitus. We begin by discussing lipid-induced hepatic insulin resistance, which is one of the primary pathophysiological processes that are involved in the dysregulation of hepatic glucose metabolism in type 2 diabetes mellitus (T2DM). We then consider elements of the regulation of hepatic gluconeogenesis and hepatic glycogen metabolism, and use this knowledge to propose a unified framework for understanding hepatic insulin action. Finally, we highlight some of the therapeutic implications of these advances.
Hepatosteatosis and insulin resistance
Although both insulin and glucose are required for net hepatic glucose uptake, insulin alone suppresses HGP in a dose-dependent manner16,17. In individuals with T2DM, the rate of HGP is increased under basal physiological conditions, and insulin-dependent suppression of HGP is impaired at both physiological and modest supra physiological plasma levels of insulin3,18,19. Although the insulin-dependent suppression of HGP has several physiological components (see below), impaired suppression of HGP is generally considered to represent hepatic insulin resistance. Accordingly, the suppression of HGP is widely used in mechanistic studies as a measure of hepatic insulin action20. Impaired suppression of HGP is reproducibly associated with increased intra hepatic triglyceride (IHTG) content, which is a hallmark of non-alcoholic fatty liver disease (NAFLD). IHTG content is a better predictor of hepatic insulin resistance than visceral adiposity or BMI in individuals without diabetes mellitus and with obesity21, and interventions that decrease IHTG content are associated with the reversal of hepatic insulin resistance in humans with NAFLD and rodent models of this disease20,22–26. Hepatic levels of diacylglycerol — the penultimate intermediate in the triglyceride synthesis pathway — have been proposed to mediate lipid-induced hepatic insulin resistance27. In the four human studies that have measured hepatic diacylglycerol content and hepatic insulin sensitivity, diacylglycerol content was strongly correlated with hepatic insulin resistance in all four, whereas other potential mediators of hepatic insulin resistance (such as ceramides) showed an inconsistent relationship21,28–30. An increase in the level of hepatic diacylglycerol activates protein kinase Cε (PKCε), which impairs the tyrosine kinase activity of the insulin receptor (INSR)31,32 through inhibitory phosphorylation of INSR at Thr1160; mice homozygous for a Thr1150Ala (the homologous residue to human Thr1160) mutation in Insr were protected from lipid-induced hepatic insulin resistance33.
Hepatic insulin resistance is also associated with other abnormalities that might contribute to dysregulated glucose metabolism. For example, the progression of NAFLD to liver fibrosis and non-alcoholic steatohepatitis (NASH) is common, and is associated with increased morbidity and mortality34. The increased deposition of extra cellular matrix (ECM) due to high-fat diet (HFD)-induced hepatic fibrosis results in interactions between the ECM and mediators of insulin signalling, such as AKT, through the scaffolding pseudokinase integrin-linked protein kinase (ILK)35. The liver-specific deletion of Ilk protected mice from HFD-induced hepatic steatosis, and therefore from HFD-induced hepatic insulin resistance, hinting at a bidirectional relationship between fibrosis and hepatic lipid deposition in this model35.
Improvements in our understanding of the regulation of hepatic glucose metabolism and of hepatic insulin resistance might inform potential therapeutic strategies for normalizing hepatic glucose production in T2DM. The physiological and pathophysiological regulation of hepatic gluconeogenesis and glycogen metabolism are examined below.
Control of hepatic gluconeogenesis
Gluconeogenesis contributes approximately half of the total HGP in humans following an overnight fast and is primarily responsible for the increase in fasting HGP in individuals with T2DM5,19,36–39. Major gluconeogenic precursors, including lactate, alanine and glycerol, are subject to diverse regulatory mechanisms. Below, we consider the recent progress in our understanding of gluconeogenic regulation (FIG. 1).
Figure 1. Control of hepatic gluconeogenesis.
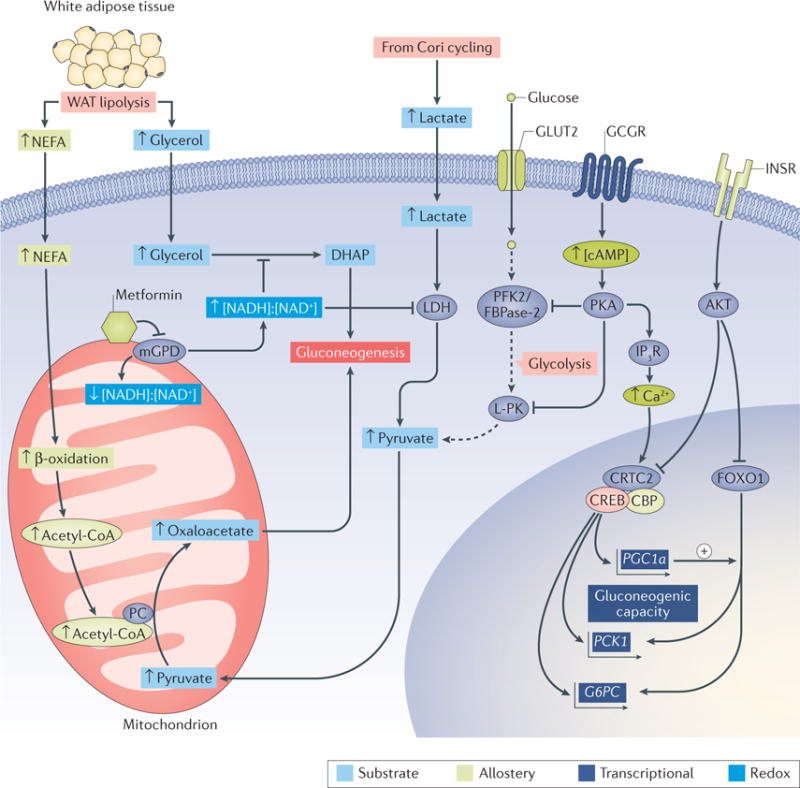
Hepatic gluconeogenesis is regulated by the availability of substrates (light blue boxes), allostery from metabolites (green boxes), transcriptional mechanisms (purple boxes) and cellular redox state (dark blue boxes). Lipolysis in white adipose tissue (WAT) produces nonesterified fatty acids (NEFA) and glycerol, both of which can stimulate gluconeogenesis. The β-oxidation of NEFA yields mitochondrial acetyl-CoA, which promotes gluconeogenesis by allosterically activating pyruvate carboxylase (PC), which, in turn, catalyses the conversion of pyruvate to the gluconeogenic substrate oxaloacetate. Glycerol can be phosphorylated and converted into the gluconeogenic precursor dihydroxyacetone phosphate (DHAP). This process is inhibited by metformin, a non-competitive inhibitor of mitochondrial glycerol-3-phosphate dehydrogenase (mGPD). Inhibition of mGPD impairs the production of DHAP, which results in a decrease in gluconeogenesis from glycerol. Furthermore, the increased cytosolic redox state ([NADH+]:[NAD+]) that results from the inhibition of mGPD inhibits the redox-dependent enzyme lactate dehydrogenase (LDH), thus limiting the production of pyruvate, and thus gluconeogenesis, from lactate. The transcriptional regulation of gluconeogenesis by glucagon and insulin is relatively slow compared with the effects of these hormones on hepatic glycogen metabolism, acting primarily through transcriptional activation and repression, respectively, of the genes that encode the gluconeogenic cytosolic enzymes phosphoenolpyruvate carboxykinase (PCK1) and glucose-6-phosphatase (G6PC). The binding of insulin to the insulin receptor (INSR) leads to the activation of AKT, which phosphorylates and excludes the transcription factor Forkhead box O1 (FOXO1) from the nucleus. In the absence of insulin, FOXO1 promotes gluconeogenic gene transcription with its co-activator peroxisome proliferator-activated receptor-γ co-activator 1α (PGC1α; encoded by PGC1a). Glucagon binding to the glucagon receptor (GCGR) increases intracellular concentrations of cAMP and activates protein kinase A (PKA), which phosphorylates the inositol-1,4,5-trisphosphate receptor (IP3R), increasing cytosolic Ca2+ levels and activating CREB-regulated transcription co-activator 2 (CRTC2). However, GCGR-dependent activation of PKA also acts acutely by inducing inhibitory phosphorylation of glycolytic regulatory enzymes, including 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase (PFK2/FBPase-2) and liver-type pyruvate kinase (L-PK), to decrease glucose oxidation and thereby favour net glucose production. Small up and down arrows represent an increase or decrease, respectively, in protein level or activity. Dotted arrows represent glycolysis. CBP, CREB-binding protein; CREB, cAMP-responsive element-binding protein 1; GLUT2, glucose transporter 2.
Indirect control of hepatic gluconeogenesis by lipolysis
Indirect control (that is, non-hepatocyte-autonomous control) of HGP was first hypothesized to occur more than half a century ago by Levine and Fritz40. It is now clear that multiple indirect mechanisms contribute to the physiological regulation of HGP. For example, insulin strongly regulates the secretion of glucagon from pancreatic α-cells, and the loss of this paracrine mode of regulation contributes to the development of hyperglucagonaemia and increased HGP in diabetes mellitus41,42. HGP is also indirectly regulated by the products of lipolysis — glycerol and nonesterified fatty acids (NEFA). Progress in this field accelerated when investigators began to compare the suppression of HGP during portal venous versus peripheral venous insulin infusions. In an early canine study, these modes of infusion achieved similar peripheral concentrations of insulin but markedly different portal concentrations of insulin; however, both methods of infusion resulted in similar magnitudes of HGP suppression43. In healthy humans, portal and peripheral venous infusions achieved similar portal insulin concentrations and different peripheral insulin concentrations, and revealed that individuals who had the highest peripheral insulin concentrations also displayed the greatest degree of HGP suppression44. Although a specific increase in portal venous insulin levels also caused the suppression of HGP in dogs45 and humans44, a role for the indirect control of HGP by insulin was apparent46. Attention quickly turned to the role of lipolytic products (NEFA and glycerol) in the regulation of hepatic gluconeogenesis. In humans, low-dose infusion of the lipid emulsion Intralipid attenuated fasting-induced decreases in gluconeogenesis, which indicates that NEFA and/or glycerol have a physiological effect on gluconeogenesis47. Furthermore, low-dose infusion of an Intralipid and heparin mixture to prevent insulin-mediated decreases in plasma levels of NEFA abolished the enhanced suppression of HGP observed with peripheral infusion compared with equimolar tolbutamide-stimulated portal insulin secretion in humans48. Insulin-dependent suppression of HGP was also impaired by this ‘fatty acid clamp’ in dogs49,50, which implicates insulin-mediated suppression of lipolysis as a mediator of insulin-dependent suppression of HGP.
Additional studies have advanced the hypothesis that the control of hepatic gluconeogenesis by lipolysis involves the conversion of glycerol to glucose, and, quantitatively, the acetyl-CoA-mediated allosteric activation of pyruvate carboxylase51–53. Conceptually, the effect of lipolysis to increase hepatic gluconeogenesis from glycerol is mechanistically straightforward; it involves a ‘substrate push’ mechanism, in which increased substrate availability drives an increase in the formation of product54. However, the mechanism by which an increased turnover and oxidation of fatty acids might drive gluconeogenesis from pyruvate cannot be explained by a ‘substrate push’ mechanism, as acetyl-CoA and acetate, the products of β-oxidation of fatty acids, are not gluconeogenic substrates; they do not contribute net carbon to gluconeogenesis. Instead, fatty acids can activate hepatic gluconeogenesis by increasing mitochondrial levels of acetyl CoA and, consequently, by allosterically activating pyruvate carboxylase, as was described in the 1960s52,55–57. Indeed, hepatic concentrations of acetyl-CoA and pyruvate carboxylase activity were increased in rat models of insulinopenic diabetic ketoacidosis and suppressed following pharmacological inhibition of lipolysis52,53. These concepts were then extended to the normal physiological suppression of hepatic gluconeogenesis by insulin. Infusion of insulin in fasted rats rapidly and markedly suppressed plasma concentrations of NEFA and glycerol, which was accompanied by decreases in hepatic acetyl-CoA concentration and HGP51. Functionally blocking the insulin-dependent suppression of lipolysis using acetate and glycerol infusions that were calibrated to match basal hepatic levels of acetyl-CoA and rates of whole-body glycerol turnover completely prevented insulin-mediated suppression of HGP51. Importantly, these studies were carried out in fasted rats that had minimal hepatic glycogen stores; therefore, insulin-dependent suppression of HGP solely reflected the suppression of gluconeogenesis.
Further evidence for this indirect mechanism of gluconeogenic suppression was provided by studies in rodents with genetic defects in hepatocellular insulin signalling. Mice in which Akt1, Akt2, and Forkhead box O1 (Foxo1) — genes that encode three crucial effectors of hepatocellular insulin action — were knocked out behaved similarly to wild-type mice, suppressing HGP normally in hyperinsulinaemic–euglycaemic clamp studies but showing increased HGP when acetate and glycerol infusions were superimposed on the clamp51,58. Moreover, rats in which Insr was ablated in liver and white adipose tissue (WAT) by antisense oligonucleotides regained the ability to suppress HGP in an insulin-dependent manner when adipose lipolysis was pharmacologically blocked51. This finding is consistent with other studies in mice that reported that the ablation of hepatic Insr does not prevent the insulin-dependent suppression of HGP59, and that liver-specific rescue of Insr expression does not restore acute insulin-dependent suppression of HGP60. These results demonstrate that hepatic insulin signalling is not essential for the suppression of HGP by insulin in an overnight fasted rodent, which is depleted of hepatic glycogen and is mostly dependent on hepatic gluconeogenesis. Overall, substantial data point to a crucial role for the inhibition of adipose lipolysis, and resultant decreases in the turnover of NEFA and glycerol, in the acute insulin-mediated suppression of hepatic gluconeogenesis.
Whether the impaired lipolytic control of hepatic gluconeogenesis contributes to the increase in HGP that is associated with T2DM is unclear. Humans who have poorly controlled T2DM (fasting plasma glucose >250 mg dl−1) have higher plasma concentrations of NEFA than healthy controls throughout the day, and increased plasma concentrations of NEFA are an independent predictor of incident T2DM61–67. Insulin-dependent suppression of glycerol turnover, which is a readout of lipolytic flux, is impaired in individuals who have insulin resistance, with or without diabetes mellitus68–71. The rates of glycerol turnover and of gluconeogenesis from glycerol are also increased in overnight fasted humans with T2DM72,73. Although these data are largely correlative, they suggest that chronic increases in the rate of lipolysis might promote an increased rate of hepatic gluconeogenesis in T2DM. Of note, the plasma turnover of β-hydroxybutyrate might be a non-invasive surrogate biomarker for hepatic concentrations of acetyl-CoA74. This method might be useful for human studies that examine the lipolytic control of hepatic gluconeogenesis.
Suppression of hepatic gluconeogenesis by metformin
The biguanide compound metformin is used as a first-line therapy for T2DM75, and acts primarily through the suppression of hepatic gluconeogenesis76,77. However, the molecular mechanism that underlies this effect remains a subject of active investigation. Perhaps the best-studied potential mechanism of metformin action is the stimulatory phosphorylation of AMP-activated protein kinase (AMPK) at Thr172 (REF.78). This mechanism was supported by studies in mice with a liver-specific deletion of the gene that encodes serine/threonine liver kinase B1 (LKB1; also known as STK11), which phosphorylates AMPK at Thr172; these mice were refractory to metformin therapy and displayed substantial transcriptional dysregulation as a result of chronic inactivation of AMPK79. The link between metformin, AMPK and gluconeogenesis has been proposed to involve both the AMPK-mediated disassembly of the cAMP-responsive element-binding protein 1 (CREB)–CREB-binding protein (CBP)–CREB-regulated transcription co-activator 2 (CRTC2) transcriptional complex, which positively regulates the expression of the gluconeogenic genes phosphoenolpyruvate carboxykinase 1 (Pck1) and glucose-6-phosphatase (G6pc)80, and improvements in lipid-induced hepatic insulin resistance81. However, metformin continues to inhibit glucose production in Ampk-knockout primary mouse hepatocytes and improves glucose tolerance in Ampk-knockout mice to a similar extent to wild-type control mice82. Furthermore, metformin suppressed glucose production in mouse hepatocytes that overexpressed the gluconeogenic enzymes cytosolic PCK1 and G6PC, which challenges the hypothesis that transcriptional mechanisms are essential for the action of metformin82. In addition, both galegine (a related guanide compound) and metformin suppressed hepatic gluconeogenesis within 20 min of intravenous infusion in rats, a time frame that is inconsistent with that of transcriptional mechanisms83. In hepatocytes, the activation of AMPK in response to guanide and biguanide compounds is well established78,80,82,84–86 and occurs rapidly following the intravenous administration of galegine83. However, interestingly, pharmacological activation of AMPK with A-769662 (which activates AMPK to a similar extent to galegine) did not suppress HGP in awake hepatic glycogen-depleted rats83. Thus, several lines of evidence have cast doubt on the necessity of AMPK activation for the metformin-dependent suppression of hepatic gluconeogenesis, warranting the investigation of alternative mechanisms of action.
An alternative but related hypothesis posits that metformin inhibits the activity of mitochondrial complex I and thus alters the adenine nucleotide energy charge (that is, the cellular [AMP]:[ATP] and [ADP]:[ATP] ratios)87. An increase in the [AMP]:[ATP] ratio would activate AMPK and inhibit the activity of fructose-1,6-bisphosphatase, which is a key gluconeogenic enzyme88. However, some reports suggest that metformin-dependent inhibition of mitochondrial complex I is observed only at suprapharmacological concentrations (millimolar) of metformin, in contrast to the clinically relevant plasma concentration range of 50–100 μM, which is observed in patients who take 2 g of metformin daily83,87. Moreover, many studies disagree on whether pharmacologically relevant concentrations of biguanides can increase intracellular levels of AMP83,84,89. In one study89, long-term treatment of mouse hepatocytes with physiologically relevant biguanide concentrations increased intra cellular levels of AMP, and decreased intracellular levels of cAMP (consistent with the allosteric inhibition of adenylyl cyclase by AMP); this decrease in the concentration of cAMP was hypothesized to reduce HGP by antagonizing the action of glucagon. However, other studies in mouse hepatocytes84 and rats83 have not observed a link between metformin-dependent suppression of HGP and changes in intracellular concentrations of cAMP. Furthermore, metformin did not inhibit glucagon-stimulated HGP in a randomized placebo-controlled double-blind trial in humans with prediabetes90.
Metformin is less potent than other related guanide and biguanide compounds, such as galegine and phenformin91. In particular, phenformin was withdrawn from clinical use owing to adverse effects, including lactic acidosis91. However, the observation that acute galegine treatment decreased hepatic gluconeogenesis and increased plasma concentrations of lactate in rats within 30 min of administration provided an initial clue to a third proposed mechanism for the action of metformin83. In rats, metformin increased the cytoplasmic redox state and decreased the mitochondrial redox state, which suggests that metformin inhibits one of the metabolic shuttles that are involved in equilibrating cytoplasmic and mitochondrial redox states83. Indeed, metformin was shown to non-competitively inhibit mitochondrial glycerol-3-phosphate dehydrogenase (mGPD) in isolated hepatocytes with a clinically relevant Ki of ~50 μM83. An important distinguishing feature of this mechanism, in contrast to all previous proposed mechanisms, is that inhibition of mGPD was predicted to impair gluconeogenesis only from redox-dependent substrates: lactate, as lactate dehydrogenase requires the reduction of NAD+, and glycerol, as mGPD produces the gluconeogenic precursor dihydroxyacetone phosphate (DHAP)83. This prediction was confirmed in cultured hepatocytes that were treated with metformin or small interfering RNA (siRNA) targeting the mGPD transcript83. Consistent with this model, knockdown of hepatic mGPD using antisense oligonucleotides phenocopied metformin treatment in rats, and Gpd2-knockout mice displayed reduced HGP during fasting that was unaltered by treatment with metformin83, which is consistent with a previous report of decreased fasting glycaemia in Gpd2-knockout mice92. As the importance of the glycerol-3-phosphate shuttle in gluconeogenesis in the human liver is unclear93,94, studies in humans are required to determine whether metformin regulates gluconeogenesis by modulating cellular redox state. A unique testable prediction of the redox hypo thesis is that treatment with metformin should decrease gluconeogenesis from lactate and glycerol, but not from pyruvate, alanine or DHAP.
Hormonal control of hepatic gluconeogenesis
Glucagon and catecholamines positively regulate hepatic gluconeogenesis through cAMP-dependent activation of protein kinase A (PKA)95. Glucagon action is essential for hyperglycaemia in rodent models of T2DM; db/db diabetic mice that lack the glucagon receptor (Gcgr) did not develop hyperinsulinaemia or hyperglycaemia96. Glucagon and catecholamines stimulate net hepatic gluconeogenic flux in the acute setting by promoting the phosphorylation of PKA, inhibition of the liver-type isozyme of pyruvate kinase (L-PK; with glucagon only)97 and phosphorylation of the bifunctional enzyme 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 2 (PFK2/FBPase-2)98 at Ser36. Phosphorylation of PFK2/FBPase-2 favours its phosphatase activity, enabling it to decrease the production of fructose-2,6-bisphosphate, an allosteric inhibitor of the gluconeogenic enzyme fructose-2,6-bisphosphatase 1 (FBPase-1)98,99; phosphorylated PFK2/FBPase-2 also facilitates the nuclear translocation and consequent functional inactivation of glucokinase (GCK)100. These mechanisms might also underlie the stimulatory effect of asprosin (an adipose-derived peptide hormone that activates hepatocellular cAMP–PKA signalling) on HGP101. The observation that asprosin stimulated HGP in mice that were fasted for 18 h (REF.10) suggests that asprosin stimulates gluconeogenesis; however, cAMP–PKA-dependent stimulation of hepatic glycogenolysis (see below) would also be expected to contribute to HGP in non-fasted mice.
By contrast, the acute and direct negative control of gluconeogenesis by insulin is not prominent in vivo11,102. High concentrations of insulin can counteract cAMP-mediated effects, such as the phosphorylation of L-PK and PFK2/FBPase-2, within 30 min (REFS97,102,103); however, the insulin-dependent regulation of gluconeogenesis primarily occurs through slow transcriptional mechanisms. The best-characterized pathway for insulin-dependent transcriptional control of gluconeogenic gene expression involves members of the FOXO family of transcription factors (FOXO1, FOXO3a and FOXO4)11. FOXO proteins, in concert with the transcriptional co-activator peroxisome proliferator-activated receptor-γ co-activator 1α (PGC1α; also known as PPARGC1A), positively regulate the expression of PCK1 and G6PC104–107. FOXO proteins are phosphorylated by AKT following stimulation with insulin, which induces their nuclear exclusion and consequent inactivation106. Gain-of-function or loss-of-function perturbations of the FOXO–PGC1α axis have marked effects on G6PC and PCK1 protein levels and glycaemia in rodent studies105,108,109. However, normalization of hyperglycaemia and re-sensitization of HGP to insulin stimulation were observed following the deletion of Foxo1 in mice with liver-specific knockout of Insr, which illustrates both the profound consequences of continuous activation of FOXO and the dispensability of the FOXO–PGC1α axis for acute insulin-mediated suppression of hepatic gluconeogenesis110,111. Overall, the place of the FOXO–PGC1α axis in the hierarchy of gluconeogenic control mechanisms remains ambiguous. For example, it is unclear whether dysregulation of FOXO transcription factors contributes to the increased rate of gluconeogenesis observed in T2DM. Rodents that are fed a HFD have hepatic insulin resistance but unaltered levels of the gluconeogenic enzymes G6PC and PCK1 (REFS24,51). Similarly, the expression levels of G6PC and PCK1 were not altered in humans with T2DM112. Available evidence, although limited, does not currently support a central role for perturbed gluconeogenic gene transcription in the increased gluconeogenesis involved in T2DM.
Glucagon and catecholamines also participate in the slow transcriptional control of gluconeogenesis by stimulating the cAMP–PKA-dependent activation of the CREB–CBP–CRTC2 complex80,113. The key mediator of hormonal control in the CREB–CBP–CRTC2 module seems to be CRTC2. Similarly to FOXO1, CRTC2 is dephosphorylated and localized to the nucleus during fasting113, and is phosphorylated and excluded from the nucleus in response to insulin114. The dephosphorylation of CRTC2 during fasting involves the PKA-dependent inhibition of the serine/threonine kinase SIK2, which normally phosphorylates CRTC2, and the PKA-dependent phosphorylation of inositol-1,4,5-trisphosphate receptors (IP3R) with a resultant increase in intracellular Ca2+ levels, which activates the CRTC2-specific phosphatase calcineurin115. Crosstalk between the FOXO1 and CRTC2 pathways includes the induction of PGC1α expression by CREB113. Consistent with this crosstalk, the CREB–CBP–CRTC2 module exerts stronger control over the expression of G6pc during the first 6 h of fasting in mice, whereas the FOXO1–PGC1α module has a larger role in stimulating G6pc transcription over longer durations (~18 h) of fasting in mice116. As predicted, Crtc2–knockout mice had fasting hypoglycaemia and mice with adeno-associated virus (AAV)-mediated overexpression of constitutively active CRTC2 were hyperglycaemic117,118.
Gluconeogenic capacity of the liver
Oscillations in the FOXO- and CREB-regulated expression of gluconeogenic enzymes have been proposed to dictate the ‘gluconeogenic capacity’ of the liver. These transcriptional oscillations are probably superimposed on normal circadian oscillations119; the circadian clock modulates the activity of CREB (and therefore glucagon and glucocorticoid function)120,121. Liver-specific knockout of the circadian clock component aryl hydrocarbon receptor translocator-like protein 1 (Bmal1, also known as Arntl) in mice was associated with fasting hypoglycaemia; however, this effect was primarily attributable to a marked decrease in the expression of glucose transporter 2 (Glut2; also known as Slc2a2) rather than changes in gluconeogenic gene transcription122. Interesting associations between the circadian control of gluconeogenesis and lipogenesis have been uncovered. Mice with hepatic knockout of the gene that encodes histone deacetylase 3 (Hdac3), which regulates the circadian rhythm of lipogenesis, rerouted gluconeogenic substrates to lipid storage, resulting in modest decreases in fasting glycaemia and improved glucose tolerance123. However, interestingly, these alterations in gluconeogenic flux were not associated with changes in the expression of G6pc or Pck1 (REF.123). Overall, no direct evidence indicates that modest oscillations in the transcription of gluconeogenic genes drive changes in gluconeogenic flux — the pyruvate tolerance test, which is used in many studies of circadian control of gluconeogenesis, is highly responsive to changes in gluconeogenic capacity but might not reflect physiological rates of gluconeogenesis. Indeed, indirect evidence suggests that gluconeogenic capacity might not be a major regulator of gluconeogenic flux in mice. In one study, a >90% decrease in the expression of Pck1 in mice resulted in only a moderate 40% reduction in gluconeogenesis124. Similarly, in G6pc−/− mice, adenoviral-mediated restoration of G6pc expression to only 20% of the activity in wild-type mice alleviated fasting hypoglycaemia125. These remarkable phenotypes suggest that modest oscillations in the expression of Pck1 and G6pc, mediated by insulin, the circadian clock, glucagon and other mechanisms, exert limited control of gluconeogenic flux in the normal liver; however, compensations, such as the activation of alternative metabolic pathways in these mouse models, potentially complicate this simple interpretation.
Neural control of HGP
A role for the central nervous system (CNS) in the regulation of hepatic glucose metabolism has been appreciated since the 1850s126, and this subject has experienced a resurgence in research in the past few years127,128. Leptin, an adipose-derived hormone that controls satiety, acts through several mechanisms to modulate energy balance and hepatic glucose metabolism129. Insulin is transcytosed across the blood– brain barrier130 and can bind to INSR on neurons and glial cells131. Neuron-specific deletion of Insr predisposes mice to diet-induced obesity and concomitant hepatic insulin resistance, probably through increasing appetite132. However, CNS-dependent regulation of hepatic insulin action might occur through mechanisms that are independent of energy balance. For example, insulin action in hypothalamic nuclei can potently suppress HGP127,133. Hepatic vagotomy might also alter hepatic insulin action, although the mechanisms that underlie this are currently unclear134. The intracerebroventricular infusion of insulin suppressed HGP in rodents135; however, a physiological increase in levels of insulin in the brain achieved through intra-arterial infusion did not alter HGP in dogs136,137. Although the physiological mechanisms that link insulin action in the brain and liver are unclear, they involve direct sympathetic and parasympathetic efferents, as well as the activation of the hypothalamic–pituitary–adrenal axis127,128.
Control of hepatic glycogen metabolism
Hepatic glycogen can be synthesized from glucose directly (glucose to glucose-6-phosphate to UDP-glucose to glycogen) or indirectly (glucose to glucose-6-phosphate to pyruvate to glucose-6-phosphate to UDP-glucose to glycogen); these pathways contribute similarly to glycogen synthesis in the liver138. Net hepatic glycogen deposition depends on the coordinated suppression of glycogenolytic flux and the stimulation of glycogen synthetic flux. Both glycogenolysis and glycogen synthesis are subject to complex regulatory mechanisms; however, generally, a useful simplification is to consider glucose as the primary suppressor of hepatic glycogenolysis and insulin as the principal activator of hepatic glycogen synthesis in vivo. This paradigm was illustrated in a study that used 13C magnetic resonance spectroscopy (MRS) measurements of hepatic glycogen in healthy individuals who had fasted overnight and who were subjected to hyperglycaemia (10 mM glucose), hyperinsulinaemia (400 pM insulin), both hyperglycaemia and hyperinsulinaemia, or neither10. Endogenous secretion of insulin and glucagon was suppressed using somatostatin. Control individuals who were not subjected to hyperglycaemia or hyperinsulinaemia exhibited net hepatic glycogenolysis, as expected. Hyperinsulinaemia was required for the stimulation of glycogen synthesis but did not suppress glycogenolysis. By contrast, hyperglycaemia was required for the suppression of glycogenolysis; consequently, the hyperinsulinaemic–hyperglycaemic group achieved maximal hepatic glycogen synthesis10. Of note, the degree of hepatic hyperinsulinaemia that was achieved in this study was modest; higher concentrations of exogenous insulin might suppress glycogenolysis, as observed in cultured human hepatocytes139.
The conclusion that both hyperglycaemia and hyperinsulinaemia are necessary for maximal hepatic glycogen synthesis also has implications for the use of hyperinsulinaemic– euglycaemic clamps. The action of hepatic insulin during the hyperinsulinaemic– euglycaemic clamp is often measured as suppression of HGP; however, suppression of HGP has a large extrahepatic component, owing largely to the lipolytic control of gluconeogenesis. Insulin-dependent stimulation of hepatic glycogen synthesis would be a useful readout of direct hepatic insulin action, but under hyperinsulinaemic–euglycaemic conditions both glycogen synthesis and glycogenolysis are active, which results in glycogen cycling and attenuated net hepatic glycogen synthesis10. Thus, a hyperinsulinaemic–hyperglycaemic clamp is necessary to achieve maximal net hepatic glycogen synthesis and, accordingly, this technique has been used to measure absolute rates of hepatic glycogen synthesis through both direct and indirect glycogen synthesis pathways in rodents33 and dogs140. The prevention of lipid-induced hepatic insulin resistance in InsrT1150A mice (which are insensitive to INSR inhibition by PKCε) was associated with improvements in insulin-stimulated hepatic glycogen synthesis compared with HFD-fed wild-type controls during hyperinsulinaemic–hyperglycaemic clamp studies33. Although they are infrequently used, these in vivo measurements of net hepatic glycogen synthesis probably provide the best direct readout of hepatic insulin action.
Insulin, glucagon and glucose regulate hepatic glycogenolysis and glycogen synthesis through mechanisms that alter the activity of GCK, glycogen synthase and glycogen phosphorylase (FIG. 2).
Figure 2. Control of hepatic glycogen metabolism.
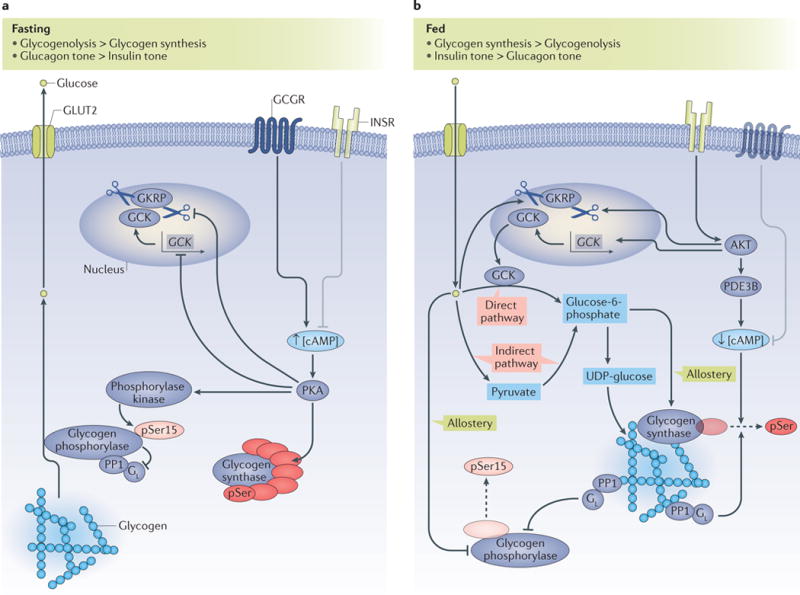
a| Under fasted conditions, glycogenolysis is activated and glycogen synthesis is suppressed. Activation of the glucagon receptor (GCGR) induces increased intracellular concentrations of cyclic AMP (cAMP; indicated by an up arrow), which leads to the activation of protein kinase A (PKA). Activated PKA inhibits the transcription of the glucokinase (GCK) gene; inhibits the dissociation of GCK from glucokinase regulatory protein (GKRP), and thus induces the nuclear sequestration of GCK; phosphorylates, and thus inactivates, glycogen synthase; and phosphorylates and activates phosphorylase kinase, which activates glycogen phosphorylase by phosphorylating Ser15. Phosphorylated, active glycogen phosphorylase also binds to and inhibits the GL subunit of protein phosphatase 1 (PP1), which prevents the PP1-dependent dephosphorylation and inactivation of glycogen synthase. The coordinated activation of glycogen phosphorylase and inhibition of glycogen synthase result in net glycogenolysis. b | Under fed conditions, hormonal and allosteric mechanisms coordinate the stimulation of glycogen synthesis through direct and indirect pathways. Glucose promotes the dissociation of GCK from GKRP, which leads to the cytoplasmic translocation of GCK; glucose also allosterically inhibits glycogen phosphorylase. The ‘direct pathway’ of glycogen synthesis involves the conversion of glucose to glucose-6-phosphate and its subsequent incorporation into glycogen, with all six carbons of the glucosyl unit intact. The ‘indirect pathway’ of glycogen synthesis involves the conversion of glucose to pyruvate, and pyruvate to glucose-6-phosphate, before incorporation into glycogen. Glucose-6-phosphate both allosterically activates glycogen synthase and is a substrate for glycogen synthesis. Insulin activates AKT, which, in turn, induces the transcription of GCK and the cytoplasmic translocation of GCK and activates phosphodiesterase 3B (PDE3B), which decreases intracellular levels of cAMP (indicated by a down arrow) and leads to the inhibition of the PKA-dependent processes described in part a. Active PP1 with its GL targeting subunit dephosphorylates and inactivates glycogen phosphorylase, and dephosphorylates and activates glycogen synthase. The coordinated inhibition of glycogen phosphorylase and activation of glycogen synthase result in net hepatic glycogen synthesis. In parts a, b the insulin receptor (INSR) and glucagon receptor (GCGR) are shown in faded colours for context, and grey inhibitory arrows depict the processes in which they are not dominant. The dashed arrows indicate dephosphorylation. Small up and down arrows indicate an increase or decrease, respectively, in protein level or activity. GLUT2, glucose transporter 2; pSer15, phosphorylated Ser15.
GCK activity and glycogenesis
The transport of glucose into hepatocytes occurs by facilitated diffusion through GLUT2, and, consequently, intrahepatic glucose concentrations parallel those in the plasma. GCK — which catalyses the phosphorylation of glucose to glucose-6-phosphate — is therefore the gatekeeper for glucose metabolism in hepatocytes141,142. GCK strongly regulates hepatic glycogen deposition. Transgenic mice that have an extra copy of the Gck displayed more than threefold greater hepatic glycogen deposition than wild-type mice during hyperglycaemic clamp studies143. Similarly, the glucose intolerance of mice that have liver-specific deletions of the genes that encode AKT (Akt1−/−/Akt2−/−) is normalized by the overexpression of Gck144. Conversely, patients with maturity onset diabetes mellitus of the young type 2 (MODY2) — which is caused by loss-of-function mutations in GCK — have reduced hepatic glycogen synthesis in the postprandial state, which is attributable to both the loss of hepatic GCK activity and a reduction in pancreatic insulin secretion145. GCK activity is largely regulated by its subcellular localization. Glucokinase regulatory protein (GKRP) binds to GCK in the nucleus when cytoplasmic concentrations of glucose are low146 (FIG. 2a). Glucose induces the dissociation of the GCK–GKRP complex, thus enabling the translocation of GCK to the cytosol, where it is active147 (FIG. 2b). Insulin facilitates this glucose-induced dissociation of the GCK–GKRP complex and upregulates the expression of Gck in rat hepatocytes through unclear AKT-dependent mechanisms147. Conversely, glucagon inhibits the dissociation of the GCK–GKRP complex and represses Gck expression in rodents147. GCK exerts a high degree of metabolic control over glycogen synthesis, and only small increases in cytosolic free GCK are required to stimulate glycogen synthesis148.
Glycogen synthase and glycogenesis
Although GCK has a high metabolic control coefficient for glycogen synthesis in the hepatocyte, liver glycogen synthase (GYS2) also shares control147. Indeed, the concomitant overexpression of both Gck and Gys2 in mouse models enhanced hepatocellular glycogen synthesis more than the overexpression of either gene alone141. This shared control of glycogen synthesis is probably partly attributable to the GCK-dependent production of glucose-6-phosphate, which is the key allosteric activator of GYS2 (REFS141,149). Mice than have an Arg582Ala mutation in GYS2, which renders the enzyme insensitive to allosteric activation by glucose-6-phosphate, displayed severely impaired hepatic glycogen synthesis and unstable GYS2, which highlights the essential role of allostery in mediating the activity of GYS2 (REF.150). GYS2 is also regulated by phosphorylation at seven sites; phosphorylated GYS2 has a decreased maximum reaction rate (Vmax)151 (FIG. 2a). This mode of regulation of GYS2 is complex, as it can be phosphorylated by PKA, PKC isoforms, AMPK, glycogen synthase kinase 3 (GSK3) and possibly other kinases151. However, site-directed mutagenesis studies indicate that the most important phosphorylation site for GYS2 activity is Ser7, which is a substrate of AMPK and PKA152,153; mice with adenoviral overexpression of GYS2 with a Ser7Ala mutation displayed increased levels of liver glycogen under both fasting and fed conditions154. These mice had markedly improved glucose tolerance and lower plasma levels of glucose in the fed state but not in the fasted state, which highlights the crucial role of hepatic glycogen synthesis in postprandial glucose disposal154. Insulin, which is essential for hepatic glycogen synthase flux in vivo10, was long thought to act through the AKT-dependent inhibition of GSK3, which would favour the dephosphorylation of GYS2 (REF.155). In fasting–refeeding studies, Akt2−/− mice had impaired hepatic glycogen synthesis, which demonstrates the necessity of AKT for insulin-dependent stimulation of glycogen synthesis; however, Gsk3αSer21Ala/Gsk3βSer9Ala mice displayed normal hepatic glycogen metabolism, suggesting that inhibitory phosphorylation of GSK3 by AKT is unnecessary for glycogen synthesis156. The mechanism for the insulin-dependent activation of GYS2 might involve the activation of phosphodiesterase 3B (PDE3B) by AKT, which antagonizes the cAMP-mediated phosphorylation of GYS2 (REF.157) In addition, this process might involve the dephosphorylation of GYS2 by protein phosphatase 1 (PP1), which involves targeting subunits of PP1, especially GL (REF.158) (FIG. 2b).
Glycogen phosphorylase and glycogenolysis
The stimulation of hepatic glycogenolysis during fasting or adrenergic stimulation occurs largely through well-characterized pathways downstream of cAMP– PKA signalling; both GYS2 and glycogen phosphorylase are phosphorylated by PKA, which results in the coordinated inhibition of GYS2 and activation of glycogen phosphorylase. This elegant reciprocal control of glycogen synthesis and glycogenolysis is mediated by the GL targeting subunit of PP1, which regulates both GYS2 and glycogen phosphorylase through dephosphorylation. The phosphorylated active form of glycogen phosphorylase binds to and inhibits PP1-GL, preventing PP1-dependent dephosphorylation and activation of GYS2 (REF.159); this process prevents futile glycogen cycling (FIG. 2a). Glucose also regulates glycogen phosphorylase activity by allosterically binding to glycogen phosphorylase, which stabilizes a conformation that enables dephosphorylation and inactivation of the enzyme147,160. In this way, glycogen phosphorylase senses plasma levels of glucose to ensure that glycogenolysis is halted when glucose is abundant in plasma (FIG. 2b). Interestingly, a prediction of this model is that hyperglycaemia should lead to the dephosphorylation of glycogen phosphorylase, the release of PP1-GL and the consequent dephosphorylation and activation of GYS2; however, in humans, when the release of insulin and glucagon was prevented by somatostatin infusion, hyperglycaemia could block hepatic glycogenolysis but it could not stimulate hepatic glycogen synthesis10. This finding suggests that the role of insulin in stimulating hepatic glycogen synthesis extends beyond its ability to promote the PDE-mediated attenuation of glucagon signalling. Therefore, further research is required to elucidate the complete mechanistic basis for the regulation of hepatic glycogen synthesis by insulin and glucagon.
Understanding hepatic insulin action
A clinically and therapeutically relevant perspective on hepatic glucose metabolism requires an understanding of how hepatic glucose fluxes fit together in states of health and disease. On the basis of data described in this Review, we attempt to construct a unified framework to describe the control of hepatic glucose metabolism by insulin, both in healthy physiological states and in the state of metabolic dysfunction that causes T2DM (FIG. 3).
Figure 3. Framework for understanding the insulin-dependent regulation of hepatic glucose metabolism.
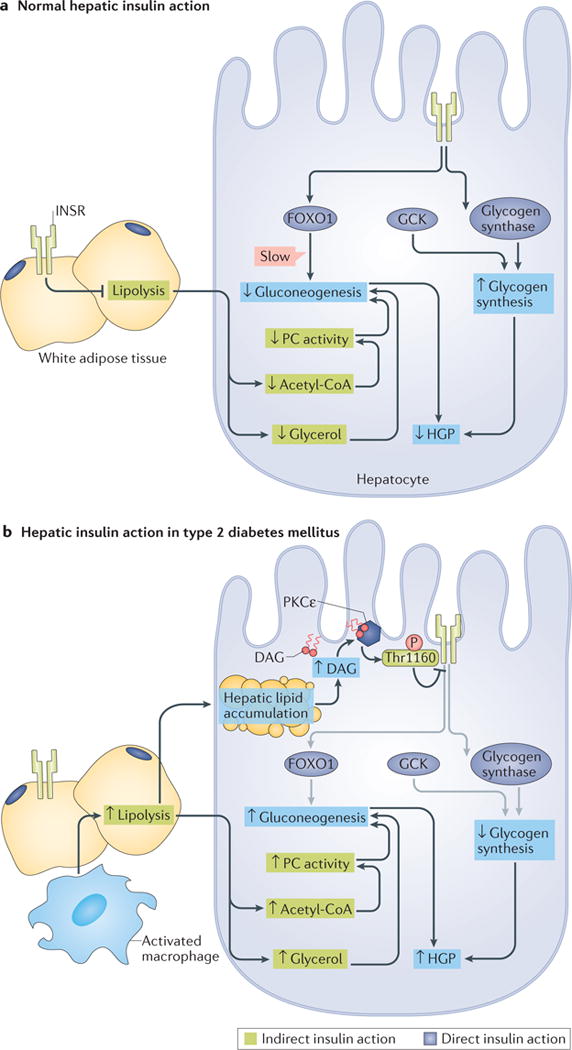
a| Under normal physiological conditions, the direct actions of hepatocellular insulin (dark blue boxes) primarily facilitate net hepatic glycogen synthesis through the activation of glucokinase (GCK) and glycogen synthase. Insulin receptor (INSR)-dependent inactivation of Forkhead box O1 (FOXO1) decreases the transcription of gluconeogenic genes, but this is a relatively slow mechanism that does not mediate the acute suppression of hepatic gluconeogenesis by insulin. Suppression of lipolysis in white adipose tissue (WAT), which is an indirect mechanism of hepatic insulin action (green boxes), acutely suppresses hepatic gluconeogenesis by decreasing the delivery of nonesterified fatty acids (NEFA) and glycerol to the liver, which results in reduced acetyl-CoA-dependent activation of pyruvate carboxylase (PC), and decreased gluconeogenesis from glycerol. The direct stimulation of net hepatic glycogen synthesis and the indirect suppression of hepatic gluconeogenesis collectively suppress hepatic glucose production (HGP). b | In type 2 diabetes mellitus (T2DM), the direct (dark blue boxes) and indirect (green boxes) effects of insulin are impaired through different mechanisms. Lipid-induced hepatic insulin resistance increases hepatic levels of diacylglycerol (DAG), which results in the activation of protein kinase Cε (PKCε) and thereby impairs direct hepatic insulin signalling through PKCε-dependent phosphorylation of INSR at Thr1160. This inhibits the INSR-dependent stimulation of hepatic glycogen synthesis in response to insulin. Impaired INSR signalling is indicated by grey arrows. In WAT, macrophage activation and consequent inflammatory signalling, as well as intrinsic adipocyte dysfunction, increase lipolysis and promote adipocyte insulin resistance, resulting in the continued delivery of NEFA and glycerol to the liver despite high plasma concentrations of insulin. Continued delivery of NEFA and glycerol to the liver promotes hepatic lipid accumulation and PKCε-mediated hepatic insulin resistance, and promotes gluconeogenesis by increasing hepatic acetyl-CoA and gluconeogenesis from glycerol. Impaired net hepatic glycogen synthesis and unrestrained hepatic gluconeogenesis together lead to increases in HGP. Small up and down arrows indicate an increase or decrease, respectively, in protein level or activity. DAG, diacylglycerol.
The most important aspect of this framework is to distinguish the direct (cell intrinsic) effects of insulin on hepatocytes from the indirect (cell non-autonomous) effects: that is, to regard the acute suppression of hepatic gluconeogenesis in response to insulin as primarily an indirect effect that is mediated by the insulin-dependent inhibition of adipocyte lipolysis, and to regard the acute stimulation of net hepatic glycogen metabolism in response to insulin as a direct effect that is mediated by the activation of the INSR in hepatocytes. This is a simplification: for example, insulin also alters the phosphorylation status of several gluconeogenic and glycolytic enzymes (as described above), and suppresses lipolysis in hepatocytes. However, as discussed above, physiological data indicate that these mechanisms contribute relatively little to the insulin-mediated suppression of gluconeogenesis compared with the insulin-mediated inhibition of adipose lipolysis. Insulin-mediated suppression of adipose lipolysis suppresses hepatic gluconeogenesis through two main mechanisms: first, it decreases the delivery of NEFA to the liver, which results in an acute reduction in hepatic mitochondrial acetyl-CoA levels and a consequent decrease in pyruvate carboxylase activity; second, it decreases the turnover of glycerol, which decreases hepatic gluconeogenesis from this substrate. Concomitantly, the direct action of insulin in the liver, which occurs through insulin receptor signalling, stimulates hepatic glycogen synthesis, mostly through the activation of both GCK and glycogen synthase. As noted, maximal net hepatic glycogen synthesis also requires hyperglycaemia to inhibit glycogen phosphorylase in a glucose-dependent manner.
This framework can be adapted to understand the physiological basis of impaired hepatic glucose metabolism in T2DM. Lipid-induced hepatic insulin resistance, which involves diacylglycerol-dependent activation of PKCε and the resulting inhibitory phosphorylation of INSR at Thr1160, would be primarily expected to impair insulin-stimulated hepatic glycogen synthesis33. However, lipid-induced hepatocellular insulin resistance would not be expected to substantially alter the acute suppression of hepatic gluconeogenesis by insulin because that process is mostly controlled indirectly. Rather, impaired insulin-dependent suppression of hepatic gluconeogenesis would be expected to result from adipocyte insulin resistance; that is, an inability of the adipocyte to suppress lipolysis following insulin stimulation. Although the mechanisms of adipose insulin resistance are incompletely understood, and are beyond the scope of this Review, they probably involve macrophage activation and inflammatory signalling, and intrinsic adipocyte defects161,162.
Inflammatory signalling might also drive adipose lipolysis independent of insulin signalling and might therefore have implications for understanding the progression of metabolic disease from subclinical insulin resistance to overt T2DM. The development of NAFLD is an early step in the progression of metabolic disease; most individuals with NAFLD do not have T2DM, although the majority of individuals with both obesity and T2DM have NAFLD163. Lipid-induced hepatic insulin resistance, but not necessarily inflammation-associated adipose insulin resistance, accompanies NAFLD. Accordingly, lipid-induced hepatic insulin resistance might precede impairments in the insulin-mediated suppression of gluconeogenesis. Individuals who have isolated hepatic insulin resistance (that is, without adipose insulin resistance) would be expected to display fasting euglycaemia, and might even show normal suppression of HGP in hyperinsulinaemic–euglycaemic clamp studies, owing to the fact that fasted glycogen-depleted individuals are reliant on gluconeogenesis for the majority of HGP. We hypothesize that progression to the state of fasting hyperglycaemia that defines T2DM involves inflammation-associated lipolysis and associated adipose insulin resistance (in addition to β-cell dysfunction). Importantly, there are likely to be interrelations between the direct and indirect modes of control of hepatic glucose metabolism. For example, the indirect regulation of hepatic NEFA delivery by adipose lipolysis might drive hepatic lipid accumulation and therefore impair direct hepatocellular insulin action.
This framework incorporates observations from human, dog and rodent studies. It is heavily dependent on insights from tracer studies of the integrated physiology of insulin action in vivo, without which the indirect effects of insulin would not have been elucidated. Importantly, this framework is incomplete; for example, it is focused on the acute effects of insulin and does not account for the many relevant transcriptional mechanisms that are discussed above, which probably participate in the chronic control of hepatic glucose metabolism. However, we believe that this framework might prove to be a useful heuristic tool for scientists and clinicians who wish to understand and further probe the complexities of hepatic glucose metabolism in physiological states of health and disease, especially those who seek to interpret hyperinsulinaemic clamp data in rodents. The framework also has therapeutic implications, which we consider below.
Targeting hepatic glucose production
The dysregulated hepatic glucose metabolism in individuals with T2DM is an attractive therapeutic target. Three major pathophysiological mechanisms that can be targeted include the excessive action of glucagon, lipid-induced hepatic insulin resistance and excess adipose lipolysis. Hyperglucagonaemia is a hallmark of both T1DM and T2DM, and antagonizing the action of glucagon has been remarkably effective at decreasing hyperglycaemia in diverse rodent models of diabetes mellitus164. Similarly, the case for targeting lipid-induced hepatic insulin resistance is well supported. Weight loss following lifestyle modification improves, or even resolves, T2DM in humans, in part, by reversing lipid-induced hepatic insulin resistance165. A study that examined the mechanism that underlies these improvements demonstrated that modest weight loss (~8 kg) led to the resolution of NAFLD and the normalization of fasting plasma concentrations of glucose. The improvement in plasma glucose was attributed to decreases in rates of fasting HGP and gluconeogenesis, and increased insulin-mediated suppression of HGP22. The mechanism by which the resolution of NAFLD improves the dysregulated hepatic glucose metabolism that is associated with T2DM might involve the reversal of INSR inhibition through the inactivation of the diacylglycerol–PKCε axis33, and reductions in hepatic acetyl-CoA content that result in decreased activity of pyruvate carboxylase45,52. The American Diabetes Association (ADA) recommends a target of 5% weight loss through lifestyle interventions for individuals with obesity and diabetes mellitus who are ready to lose weight166. However, readiness to lose weight is often elusive; the multitude of physiological, societal and psychosocial barriers to weight loss frequently render even a 5% long-term weight loss target by lifestyle intervention elusive. Therefore, searching for additional therapies that target hepatic steatosis and lipid-induced hepatic insulin resistance is warranted (FIG. 4).
Figure 4. Therapeutic opportunities for dysregulated hepatic glucose metabolism.
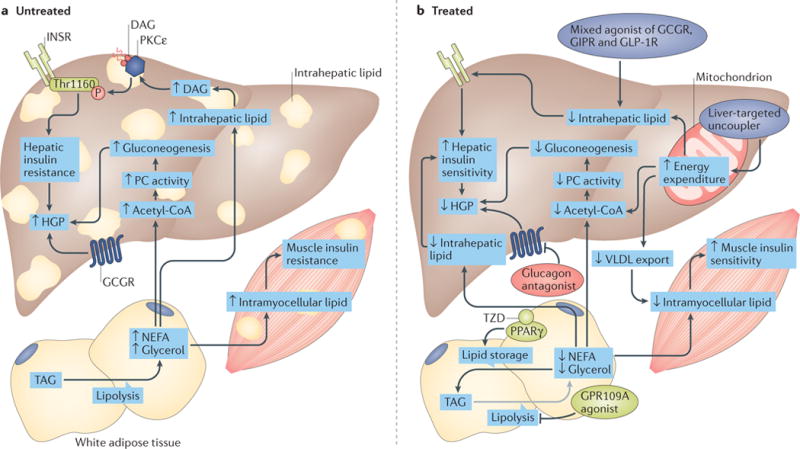
a| In patients with type 2 diabetes mellitus (T2DM), lipid-induced hepatic insulin resistance might result from activation of the diacylglycerol (DAG)–protein kinase Cε (PKCε) axis and the consequent inhibition of insulin receptor (INSR) signalling through inhibitory phosphorylation of INSR at Thr1160. This leads to impaired insulin stimulation of hepatic glycogen synthesis. In parallel, inappropriate increases in adipose lipolysis can drive hepatic gluconeogenesis through increases in hepatic acetyl-CoA and pyruvate carboxylase (PC) activity, and promote ectopic lipid accumulation in liver and muscle. These processes promote the increased hepatic glucose production (HGP) that occurs in T2DM. b | These contributors to increased HGP can be pharmacologically targeted. Mechanisms that target lipid-induced hepatic insulin resistance (purple ovals) include the selective induction of mitochondrial uncoupling in the liver (for example, with 2,4-dinitrophenol (DNP) analogues, such as DNP-methyl ether (DNPME) and other liver-targeted mitochondrial uncoupling agents) to increase hepatic fat oxidation, or mixed agonism of incretin, glucagon and/or thyroid hormone receptors (for example, a mixed agonist of glucagon receptor (GCGR), gastric inhibitory polypeptide receptor (GIPR) and glucagon-like peptide 1 receptor (GLP1R)) to promote fat oxidation. A second therapeutic strategy is to target excessive lipolysis (light green ovals) by promoting the sequestration of lipids in white adipose tissue (WAT). The inhibition of lipolysis or the stimulation of lipogenesis in WAT is predicted to decrease hepatic lipid accumulation and reverse lipid-induced hepatic insulin resistance by decreasing delivery of lipids to the liver. The thiazolidinediones (TZD) are agonists of the lipogenic transcription factor peroxisome proliferator-activated receptor-γ (PPARγ). The inhibition of lipolysis with GPR109A agonists, or with WAT-specific adipose triglyceride lipase (ATGL) inhibitors, would also be predicted to reverse dysregulated glucose metabolism through these mechanisms. Glucagon antagonism (red oval) represents a third potential therapeutic strategy to target the hyperglucagonaemia and excessive hepatic glucose production associated with T2DM if it can be dissociated from on-target adverse effects (for example, hepatic steatosis). Small up and down arrows indicate an increase or decrease, respectively, in protein level or activity. The grey arrow depicts inhibition of lipolysis. ACC, acetyl-CoA carboxylase; TAG, triacylglycerol.
Glucagon antagonism and HGP
The antagonism of circulating glucagon or of GCGR to reduce excess HGP is a long-standing and active area of investigation42,167–170. Plasma levels of glucagon are aberrantly increased in individuals with T2DM171–174. In addition, animal models of reduced or ablated glucagon action show improvements in T2DM96,175,176 (FIG. 4a,b). Although the complete prevention of T2DM observed in some of these rodent models contrasts with the insulin dependence observed in humans with pancreoprivic diabetes mellitus, the marked antidiabetic phenotypes underscore the promise of such anti-glucagon therapeutics. Currently, data in humans is limited to three GCGR antagonists, BAY 27–9955 (REF.177), MK-0893 (REF.178) and LY2409021 (REFS179,180). BAY 27–9955 inhibited glucagon-stimulated glucose production in humans177, but no follow-up studies were published. MK-0893 improved hyperglycaemia in a human clinical trial, but was associated with increases in plasma levels of LDL cholesterol178. In phase I and phase II trials, LY2409021 improved glycaemic control in patients with T2DM, which was complicated only by a modest and reversible increase in the levels of transaminases179,180. One potential caveat of anti-glucagon therapies is that, in rodents, glucagon seems to activate peroxisome proliferator-activated receptor-α (PPARα), which results in an increase in hepatic fatty acid oxidation and a decrease in the export of VLDL181. It is unclear whether this aspect of glucagon action in rodents translates to human physiology; however, hypertriglyceridaemia has not been reported thus far in the human studies of LY2409021 (REFS179,180).
Hepatosteatosis and T2DM
A rational therapeutic approach for the treatment of NAFLD and its sequelae, such as NASH, is to increase hepatic energy expenditure and thereby increase hepatic fat oxidation. Thyroid hormone receptor-β agonists have been tested preclinically for their ability achieve this and, although they can reverse NAFLD, none has been shown to improve the suppression of HGP by insulin (in part due to increased adipose lipolysis)182,183. In addition, a T3–glucagon conjugate improved glycaemia and NAFLD, without causing the rapid development of cardiac hypertrophy and osteopenia observed with T3 alone184; additional studies to evaluate the safety and effects of this conjugate on HGP (which was not evaluated in this study184) will help to assess the promise of this class of compounds.
Liver-targeted mitochondrial uncouplers, which can dissipate the inner mitochondrial membrane proton gradient in the liver, are another promising class of agents for the treatment of NAFLD, NASH and diabetes mellitus. Two modified forms of the mitochondrial protonophore 2,4-dinitrophenol (DNP), DNP-methyl ether (DNPME) and controlled-release mitochondrial protonophore (CRMP), reverse diabetes mellitus, hepatic inflammation and hepatic fibrosis in rodent models of T2DM and NASH24,25. Importantly, the reversal of hyperglycaemia and hepatic insulin resistance by these drugs was attributed to decreases in diacylglycerol-dependent activation of PKCε and decreases in hepatic acetyl-CoA content24,25 (FIG. 4a,b). Both of these liver-targeted mitochondrial uncoupling agents promoted increased energy expenditure exclusively in liver, and, importantly, the reversal of diabetes mellitus and NASH in these rodent models was not associated with detectable changes in body temperature, whole-body energy expenditure or body weight, which minimizes the concerns of toxicity that are inherent to all non-selective mitochondrial uncouplers24,25. These studies provide a proof of concept for the development of novel liver-targeted mitochondrial protonophores to treat NAFLD, NASH and T2DM.
Incretin receptor agonists, and in particular glucagon-like peptide 1 (GLP1) analogues, are used in the treatment of diabetes mellitus and obesity, and might be useful for the treatment of NAFLD. In a phase II clinical trial in patients with NASH, the histology of NASH was resolved more frequently in patients who were treated with the GLP1 analogue liraglutide than in patients who were treated with placebo185. In preclinical studies, a novel compound agonist of the GLP1, gastric inhibitory peptide (GIP) and glucagon receptors improved glycaemic control, decreased food intake, increased energy expenditure and reversed hepatosteatosis in rodents186. This strategy of mixed agonism, also used in the aforementioned glucagon–T3 conjugate, might help to minimize the undesired effects of any one hormone (for example, glucagon-mediated hyperglycaemia and T3-mediated tachycardia), and thus warrants further exploration184.
Targeting the adipocyte
Thiazolidinediones, the best-established pharmacological agents for the treatment for NAFLD and NASH, are PPARγ agonists that have multiple downstream effects. Thiazolidinediones increase adipose function and adipose mass by increasing their capacity for lipid storage and decreasing inappropriate lipolysis187. The treatment of individuals with T2DM with the thiazolidinedione rosiglitazone markedly improved insulin-stimulated glucose metabolism, which was associated with decreased plasma concentrations of fatty acids and lowered hepatic triglyceride content188. These changes were associated with an increase in extra myocellular lipid content and an increase in the sensitivity of peripheral adipocytes to the inhibitory effects of insulin on lipolysis188. These results led to the hypothesis that thiazolidinediones enhance insulin sensitivity in patients with T2DM by promoting increased insulin sensitivity in peripheral adipocytes, resulting in lower plasma concentrations of fatty acids and the redistribution of intracellular lipids from insulin-responsive organs into peripheral adipocytes (FIG. 4a,b). Consistent with these results, in patients with NASH who had either impaired glucose tolerance or overt T2DM, and who were fed a hypocaloric diet, a 6 month treatment regimen with the thiazolidinedione pioglitazone improved hepatic insulin sensitivity and hepatic steatosis compared with placebo189. In patients with NASH, pioglitazone-mediated improvements in hepatic steatosis and histological NASH were correlated with improvements in adipose insulin responsiveness190. However, the clinical use of thiazolidinediones is limited by concerns regarding adverse effects. For pioglitazone, the most widely prescribed thiazolidinedione compound in the United States, adverse effects include weight gain (in an already obese population), peripheral oedema, exacerbation of heart failure and increased frequency of bone fractures191. Thus, although pioglitazone effectively improves adipose function, decreases excess lipolysis and improves insulin sensitivity, ample opportunities exist for development of better-tolerated therapies that target adipose tissue function in T2DM.
Direct inhibitors of lipolytic enzymes, such as adipose triglyceride lipase (ATGL) and hormone-sensitive lipase (HSL), are not used in clinical practice. However, niacin (or nicotinic acid) — a drug that has been used for decades to increase HDL levels and decrease triglyceride levels — also inhibits adipose lipolysis. Niacin binds to, and activates, a small G protein-coupled receptor, GPR109A, which decreases intracellular levels of cAMP, leading to a decrease in the activity of HSL in adipose tissue and reduced adipocyte lipolysis192. However, rather than improving endogenous glucose production, niacin can increase plasma glucose concentrations in some patients193,194. This paradoxical effect could be explained by a ‘rebound increase’ in adipose lipolysis following dissociation of the niacin–GPR109A complex195. Changes in niacin dosing strategy might prevent this rebound increase in plasma levels of NEFA and might improve glycaemic control195. Thus, novel formulations of niacin with different pharmacokinetic profiles, novel GPR109A agonists or other anti-lipolytics (for example, WAT-specific ATGL inhibitors) might indirectly improve hepatic glucose metabolism and therefore merit further investigation.
Conclusions
Well-coordinated hepatic glucose metabolism is essential to health, and dysregulation of hepatic glucose metabolism is central to the pathogenesis and complications of T2DM. The key components of HGP reviewed in this article include gluconeogenesis, which exhibits complex regulation by substrate provision, redox state, gluconeogenic gene transcription and other mechanisms (FIG. 1), and glycogen metabolism, which is remarkably sensitive to both hormonal tone and plasma concentrations of glucose (FIG. 2). We have highlighted the importance of indirect extrahepatic control of gluconeogenesis and of direct hepatic control of glycogen metabolism, and have proposed a paradigm in which dysregulation of both direct and indirect mechanisms contribute to the increased HGP of T2DM (FIG. 3). The multifaceted regulation of HGP through both direct and indirect mechanisms limits the use of in vitro hepatocyte models that are removed from the normal hormonal, anatomical and biochemical context of the in situ liver. Although much remains to be elucidated, current knowledge of the multimodal control of HGP invites rational therapeutic targeting of multiple mechanisms simultaneously. Thus, it is conceivable to imagine a future in which metformin is used to control redox-dependent regulation of gluconeogenesis, a liver-targeted mitochondrial protonophore is used to reverse lipid-induced hepatic insulin resistance, a glucagon antagonist is used to neutralize the excessive HGP that results from hyperglucagonaemia and a thiazolidinedione or antilipolytic agent is used to reduce lipolytic stimulation of hepatic gluconeogenesis (FIG. 4). It is also conceivable that reducing hepatic diacylglycerol and acetyl-CoA content using liver-targeted mitochondrial uncoupling agents alone might be sufficient to reverse both hepatic insulin resistance and increased rates of hepatic gluconeogenesis in T2DM, as shown in preclinical studies24,25. Although many preclinical and clinical studies will be required to test the viability of these novel agents and combinatorial approaches, the global need for effective medical management of dysregulated HGP in T2DM provides an urgent impetus for such work.
Key points.
Hepatic glucose metabolism encompasses several catabolic and anabolic fluxes that have distinct modes of hepatocyte-autonomous (direct) and hepatocyte-non-autonomous (indirect) regulatory mechanisms
Acute regulation of hepatic glucose metabolism is achieved through changes in protein phosphorylation, substrate availability, allostery and redox state
Chronic regulation of hepatic glucose metabolism occurs through transcriptional mechanisms and the development of insulin resistance
Acute suppression of hepatic gluconeogenesis by insulin is largely an indirect effect that is mediated mostly through the suppression of adipose lipolysis, which reduces delivery of nonesterified fatty acids and glycerol to the liver
The major direct effect of insulin on hepatic glucose metabolism is the acute regulation of hepatic glycogen metabolism; however, hyperglycaemia and hyperinsulinaemia are required to maximally stimulate net hepatic glycogenesis
Lipid-induced hepatic insulin resistance, hyperglucagonaemia and excessive adipose lipolysis represent three pathophysiological processes that might be amenable to pharmacological intervention in humans who have impaired hepatic glucose metabolism
Acknowledgments
The authors apologize to those many colleagues whose important contributions could not be discussed owing to word and reference limits. The authors thank A.K. Madiraju for helpful comments. M.C.P. acknowledges grant support from the US National Institutes of Health (NIH; grants F30 DK-104596 and T32 GM-007205). D.F.V. acknowledges grant support from the NIH (grant K23 DK-102874). G.I.S. acknowledges grant support from the NIH (grants R01 DK40936, R01 DK113984 and P30 DK045735).
Glossary
- Hyperinsulinaemic– euglycaemic clamp technique
A technique in which insulin is infused at a constant rate to achieve hyperinsulinaemia and glucose is infused at a variable rate to maintain euglycaemia; once steady-state euglycaemia has been achieved, the glucose infusion rate is proportional to the whole-body insulin sensitivity of the individual
- Pyruvate tolerance test
A test in which a large bolus of the gluconeogenic substrate pyruvate is administered and plasma levels of glucose are measured at defined time intervals; plasma glucose excursion is assumed to be proportional to the rate of pyruvate-stimulated hepatic gluconeogenesis.
- Pancreoprivic diabetes mellitus
Diabetes mellitus caused by medical or surgical loss of pancreatic function, such as after a pancreatectomy or pancreatitis
Footnotes
Author contributions
All authors contributed to all aspects of the preparation of the article. The order of authorship and contribution is M.C.P., D.F.V. and G.I.S.
Competing interests statement
M.C.P. and D.F.V. declare no competing interests. G.I.S. serves on scientific advisory boards for Merck, Novo Nordisk, Celgene, Aegerion and AstraZeneca, receives investigator-initiated support from Gilead Sciences, Inc., and is an inventor on Yale patents for liver-targeted mitochondrial uncoupling agents for the treatment of NAFLD, NASH, type 2 diabetes and related metabolic disorders.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Ekberg K, et al. Contributions by kidney and liver to glucose production in the postabsorptive state and after 60 h of fasting. Diabetes. 1999;48:292–298. doi: 10.2337/diabetes.48.2.292. [DOI] [PubMed] [Google Scholar]
- 2.Moore MC, Coate KC, Winnick JJ, An Z, Cherrington AD. Regulation of hepatic glucose uptake and storage in vivo. Adv Nutr. 2012;3:286–294. doi: 10.3945/an.112.002089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rizza RA. Pathogenesis of fasting and postprandial hyperglycemia in type 2 diabetes: implications for therapy. Diabetes. 2010;59:2697–2707. doi: 10.2337/db10-1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moore MC, et al. Sources of carbon for hepatic glycogen synthesis in the conscious dog. J Clin Invest. 1991;88:578–587. doi: 10.1172/JCI115342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rothman DL, Magnusson I, Katz LD, Shulman RG, Shulman GI. Quantitation of hepatic glycogenolysis and gluconeogenesis in fasting humans with 13C NMR. Science. 1991;254:573–576. doi: 10.1126/science.1948033. [DOI] [PubMed] [Google Scholar]
- 6.Mari A, Wahren J, DeFronzo RA, Ferrannini E. Glucose absorption and production following oral glucose: comparison of compartmental and arteriovenous-difference methods. Metabolism. 1994;43:1419–1425. doi: 10.1016/0026-0495(94)90038-8. [DOI] [PubMed] [Google Scholar]
- 7.Ishida T, et al. Differential effects of oral, peripheral intravenous, and intraportal glucose on hepatic glucose uptake and insulin and glucagon extraction in conscious dogs. J Clin Invest. 1983;72:590–601. doi: 10.1172/JCI111007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pagliassotti MJ, Cherrington AD. Regulation of net hepatic glucose uptake in vivo. Annu Rev Physiol. 1992;54:847–860. doi: 10.1146/annurev.ph.54.030192.004215. [DOI] [PubMed] [Google Scholar]
- 9.Ferrannini E, et al. The disposal of an oral glucose load in healthy subjects A quantitative study. Diabetes. 1985;34:580–588. doi: 10.2337/diab.34.6.580. [DOI] [PubMed] [Google Scholar]
- 10.Petersen KF, Laurent D, Rothman DL, Cline GW, Shulman GI. Mechanism by which glucose and insulin inhibit net hepatic glycogenolysis in humans. J Clin Invest. 1998;101:1203–1209. doi: 10.1172/JCI579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lin HV, Accili D. Hormonal regulation of hepatic glucose production in health and disease. Cell Metab. 2011;14:9–19. doi: 10.1016/j.cmet.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cherrington AD, Edgerton D, Sindelar DK. The direct and indirect effects of insulin on hepatic glucose production in vivo. Diabetologia. 1998;41:987–996. doi: 10.1007/s001250051021. [DOI] [PubMed] [Google Scholar]
- 13.Pagliassotti MJ, Moore MC, Neal DW, Cherrington AD. Insulin is required for the liver to respond to intraportal glucose delivery in the conscious dog. Diabetes. 1992;41:1247–1256. doi: 10.2337/diab.41.10.1247. [DOI] [PubMed] [Google Scholar]
- 14.McGuinness OP, Ayala JE, Laughlin MR, Wasserman DH. NIH experiment in centralized mouse phenotyping: the Vanderbilt experience and recommendations for evaluating glucose homeostasis in the mouse. Am J Physiol Endocrinol Metab. 2009;297:E849–E855. doi: 10.1152/ajpendo.90996.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kowalski GM, Bruce CR. The regulation of glucose metabolism: implications and considerations for the assessment of glucose homeostasis in rodents. Am J Physiol Endocrinol Metab. 2014;307:E859–E871. doi: 10.1152/ajpendo.00165.2014. [DOI] [PubMed] [Google Scholar]
- 16.Steele R, et al. Inhibition by insulin of hepatic glucose production in the normal dog. Am J Physiol. 1965;208:301–306. doi: 10.1152/ajplegacy.1965.208.2.301. [DOI] [PubMed] [Google Scholar]
- 17.Rizza RA, Mandarino LJ, Gerich JE. Dose–response characteristics for effects of insulin on production and utilization of glucose in man. Am J Physiol. 1981;240:E630–E639. doi: 10.1152/ajpendo.1981.240.6.E630. [DOI] [PubMed] [Google Scholar]
- 18.Basu A, Shah P, Nielsen M, Basu R, Rizza RA. Effects of type 2 diabetes on the regulation of hepatic glucose metabolism. J Investig Med. 2004;52:366–374. doi: 10.1136/jim-52-06-30. [DOI] [PubMed] [Google Scholar]
- 19.Magnusson I, Rothman DL, Katz LD, Shulman RG, Shulman GI. Increased rate of gluconeogenesis in type II diabetes mellitus. A 13C nuclear magnetic resonance study. J Clin Invest. 1992;90:1323–1327. doi: 10.1172/JCI115997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Perry RJ, Samuel VT, Petersen KF, Shulman GI. The role of hepatic lipids in hepatic insulin resistance and type 2 diabetes. Nature. 2014;510:84–91. doi: 10.1038/nature13478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kumashiro N, et al. Cellular mechanism of insulin resistance in nonalcoholic fatty liver disease. Proc Natl Acad Sci USA. 2011;108:16381–16385. doi: 10.1073/pnas.1113359108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Petersen KF, et al. Reversal of nonalcoholic hepatic steatosis, hepatic insulin resistance, and hyperglycemia by moderate weight reduction in patients with type 2 diabetes. Diabetes. 2005;54:603–608. doi: 10.2337/diabetes.54.3.603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Petersen KF, et al. Leptin reverses insulin resistance and hepatic steatosis in patients with severe lipodystrophy. J Clin Invest. 2002;109:1345–1350. doi: 10.1172/JCI15001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Perry RJ, Zhang D, Zhang X-M, Boyer JL, Shulman GI. Controlled-release mitochondrial protonophore reverses diabetes and steatohepatitis in rats. Science. 2015;347:1253–1256. doi: 10.1126/science.aaa0672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Perry RJ, et al. Reversal of hypertriglyceridemia, fatty liver disease, and insulin resistance by a liver-targeted mitochondrial uncoupler. Cell Metab. 2013;18:740–748. doi: 10.1016/j.cmet.2013.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim JK, Gavrilova O, Chen Y, Reitman ML, Shulman GI. Mechanism of insulin resistance in A-ZIP/F-1 fatless mice. J Biol Chem. 2000;275:8456–8460. doi: 10.1074/jbc.275.12.8456. [DOI] [PubMed] [Google Scholar]
- 27.Samuel VT, Shulman GI. Mechanisms for insulin resistance: common threads and missing links. Cell. 2012;148:852–871. doi: 10.1016/j.cell.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Magkos F, et al. Intrahepatic diacylglycerol content is associated with hepatic insulin resistance in obese subjects. Gastroenterology. 2012;142:1444–1446 e2. doi: 10.1053/j.gastro.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Luukkonen PK, et al. Hepatic ceramides dissociate steatosis and insulin resistance in patients with nonalcoholic fatty liver disease. J Hepatol. 2016;64:1167–1175. doi: 10.1016/j.jhep.2016.01.002. [DOI] [PubMed] [Google Scholar]
- 30.ter Horst KW, et al. Hepatic diacylglycerol-associated protein kinase Cε translocation links hepatic steatosis to hepatic insulin resistance in humans. Cell Rep. 2017;19:1997–2004. doi: 10.1016/j.celrep.2017.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Samuel VT, et al. Inhibition of protein kinase Cε prevents hepatic insulin resistance in nonalcoholic fatty liver disease. J Clin Invest. 2007;117:739–745. doi: 10.1172/JCI30400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Samuel VT, et al. Mechanism of hepatic insulin resistance in non-alcoholic fatty liver disease. J Biol Chem. 2004;279:32345–32353. doi: 10.1074/jbc.M313478200. [DOI] [PubMed] [Google Scholar]
- 33.Petersen MC, et al. Insulin receptor Thr1160 phosphorylation mediates lipid-induced hepatic insulin resistance. J Clin Invest. 2016;126:4361–4371. doi: 10.1172/JCI86013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Michelotti GA, Machado MV, Diehl AM. NAFLD, NASH and liver cancer. Nat Rev Gastroenterol Hepatol. 2013;10:656–665. doi: 10.1038/nrgastro.2013.183. [DOI] [PubMed] [Google Scholar]
- 35.Williams AS, et al. Integrin-linked kinase is necessary for the development of diet-induced hepatic insulin resistance. Diabetes. 2017;66:325–334. doi: 10.2337/db16-0484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Boden G. Gluconeogenesis and glycogenolysis in health and diabetes. J Investig Med. 2004;52:375–378. doi: 10.1136/jim-52-06-31. [DOI] [PubMed] [Google Scholar]
- 37.Chen X, Iqbal N, Boden G. The effects of free fatty acids on gluconeogenesis and glycogenolysis in normal subjects. J Clin Invest. 1999;103:365–372. doi: 10.1172/JCI5479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Katz J, Tayek JA. Gluconeogenesis and the Cori cycle in 12-, 20-, and 40-h-fasted humans. Am J Physiol. 1998;275:E537–E542. doi: 10.1152/ajpendo.1998.275.3.E537. [DOI] [PubMed] [Google Scholar]
- 39.Landau BR, et al. Contributions of gluconeogenesis to glucose production in the fasted state. J Clin Invest. 1996;98:378–385. doi: 10.1172/JCI118803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Levine R, Fritz IB. The relation of insulin to liver metabolism. Diabetes. 1956;5:209–219. doi: 10.2337/diab.5.3.209. [DOI] [PubMed] [Google Scholar]
- 41.Gaisano H, MacDonald PE, Vranic M. Glucagon secretion and signaling in the development of diabetes. Front Physiol. 2012;3:349. doi: 10.3389/fphys.2012.00349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pearson MJ, Unger RH, Holland WL. Clinical trials, triumphs, and tribulations of glucagon receptor antagonists. Diabetes Care. 2016;39:1075–1077. doi: 10.2337/dci15-0033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ader M, Bergman RN. Peripheral effects of insulin dominate suppression of fasting hepatic glucose production. Am J Physiol. 1990;258:E1020–E1032. doi: 10.1152/ajpendo.1990.258.6.E1020. [DOI] [PubMed] [Google Scholar]
- 44.Lewis GF, Zinman B, Groenewoud Y, Vranic M, Giacca A. Hepatic glucose production is regulated both by direct hepatic and extrahepatic effects of insulin in humans. Diabetes. 1996;45:454–462. doi: 10.2337/diab.45.4.454. [DOI] [PubMed] [Google Scholar]
- 45.Sindelar DK, Balcom JH, Chu CA, Neal DW, Cherrington AD. A comparison of the effects of selective increases in peripheral or portal insulin on hepatic glucose production in the conscious dog. Diabetes. 1996;45:1594–1604. doi: 10.2337/diab.45.11.1594. [DOI] [PubMed] [Google Scholar]
- 46.Prager R, Wallace P, Olefsky JM. Direct and indirect effects of insulin to inhibit hepatic glucose output in obese subjects. Diabetes. 1987;36:607–611. doi: 10.2337/diab.36.5.607. [DOI] [PubMed] [Google Scholar]
- 47.Staehr P, et al. Effects of free fatty acids per se on glucose production, gluconeogenesis, and glycogenolysis. Diabetes. 2003;52:260–267. doi: 10.2337/diabetes.52.2.260. [DOI] [PubMed] [Google Scholar]
- 48.Lewis GF, Vranic M, Harley P, Giacca A. Fatty acids mediate the acute extrahepatic effects of insulin on hepatic glucose production in humans. Diabetes. 1997;46:1111–1119. doi: 10.2337/diab.46.7.1111. [DOI] [PubMed] [Google Scholar]
- 49.Sindelar DK, et al. The role of fatty acids in mediating the effects of peripheral insulin on hepatic glucose production in the conscious dog. Diabetes. 1997;46:187–196. doi: 10.2337/diab.46.2.187. [DOI] [PubMed] [Google Scholar]
- 50.Rebrin K, Steil GM, Mittelman SD, Bergman RN. Causal linkage between insulin suppression of lipolysis and suppression of liver glucose output in dogs. J Clin Invest. 1996;98:741–749. doi: 10.1172/JCI118846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Perry RJ, et al. Hepatic acetyl coa links adipose tissue inflammation to hepatic insulin resistance and type 2 diabetes. Cell. 2015;160:745–758. doi: 10.1016/j.cell.2015.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Perry RJ, et al. Leptin reverses diabetes by suppression of the hypothalamic–pituitary–adrenal axis. Nat Med. 2014;20:759–763. doi: 10.1038/nm.3579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Perry RJ, Peng L, Shulman GI. Mechanism for leptin’s acute insulin-independent effect to reverse diabetic ketoacidosis. J Clin Invest. 2017;127:657–669. doi: 10.1172/JCI88477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Previs SF, Cline GW, Shulman GI. A critical evaluation of mass isotopomer distribution analysis of gluconeogenesis in vivo. Am J Physiol. 1999;277:E154–E160. doi: 10.1152/ajpendo.1999.277.1.E154. [DOI] [PubMed] [Google Scholar]
- 55.Krebs HA, Speake RN, Hems R. Acceleration of renal gluconeogenesis by ketone bodies and fatty acids. Biochem J. 1965;94:712–720. doi: 10.1042/bj0940712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Keech DB, Utter MF. Pyruvate carboxylase. II Properties. J Biol Chem. 1963;238:2609–2614. [PubMed] [Google Scholar]
- 57.Williamson JR, Kreisberg RA, Felts PW. Mechanism for the stimulation of gluconeogenesis by fatty acids in perfused rat liver. Proc Natl Acad Sci USA. 1966;56:247–254. doi: 10.1073/pnas.56.1.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lu M, et al. Insulin regulates liver metabolism in vivo in the absence of hepatic Akt and Foxo1. Nat Med. 2012;18:388–395. doi: 10.1038/nm.2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Buettner C, et al. Severe impairment in liver insulin signaling fails to alter hepatic insulin action in conscious mice. J Clin Invest. 2005;115:1306–1313. doi: 10.1172/JCI23109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Okamoto H, Obici S, Accili D, Rossetti L. Restoration of liver insulin signaling in Insr knockout mice fails to normalize hepatic insulin action. J Clin Invest. 2005;115:1314–1322. doi: 10.1172/JCI23096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chen YD, Golay A, Swislocki AL, Reaven GM. Resistance to insulin suppression of plasma free fatty acid concentrations and insulin stimulation of glucose uptake in noninsulin-dependent diabetes mellitus. J Clin Endocrinol Metab. 1987;64:17–21. doi: 10.1210/jcem-64-1-17. [DOI] [PubMed] [Google Scholar]
- 62.Fraze E, et al. Ambient plasma free fatty acid concentrations in noninsulin-dependent diabetes mellitus: evidence for insulin resistance. J Clin Endocrinol Metab. 1985;61:807–811. doi: 10.1210/jcem-61-5-807. [DOI] [PubMed] [Google Scholar]
- 63.Reaven GM, Hollenbeck C, Jeng CY, Wu MS, Chen YD. Measurement of plasma glucose, free fatty acid, lactate, and insulin for 24 h in patients with NIDDM. Diabetes. 1988;37:1020–1024. doi: 10.2337/diab.37.8.1020. [DOI] [PubMed] [Google Scholar]
- 64.Golay A, Swislocki AL, Chen YD, Reaven GM. Relationships between plasma-free fatty acid concentration, endogenous glucose production, and fasting hyperglycemia in normal and non-insulin-dependent diabetic individuals. Metabolism. 1987;36:692–696. doi: 10.1016/0026-0495(87)90156-9. [DOI] [PubMed] [Google Scholar]
- 65.Swislocki AL, Chen YD, Golay A, Chang MO, Reaven GM. Insulin suppression of plasma-free fatty acid concentration in normal individuals and patients with type 2 (non-insulin-dependent) diabetes. Diabetologia. 1987;30:622–626. doi: 10.1007/BF00277318. [DOI] [PubMed] [Google Scholar]
- 66.Charles MA, et al. The role of non-esterified fatty acids in the deterioration of glucose tolerance in Caucasian subjects: results of the Paris Prospective Study. Diabetologia. 1997;40:1101–1106. doi: 10.1007/s001250050793. [DOI] [PubMed] [Google Scholar]
- 67.Paolisso G, et al. A high concentration of fasting plasma non-esterified fatty acids is a risk factor for the development of NIDDM. Diabetologia. 1995;38:1213–1217. doi: 10.1007/BF00422371. [DOI] [PubMed] [Google Scholar]
- 68.Jocken JWE, et al. Insulin-mediated suppression of lipolysis in adipose tissue and skeletal muscle of obese type 2 diabetic men and men with normal glucose tolerance. Diabetologia. 2013;56:2255–2265. doi: 10.1007/s00125-013-2995-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Heptulla RA, et al. In situ evidence that peripheral insulin resistance in adolescents with poorly controlled type 1 diabetes is associated with impaired suppression of lipolysis: a microdialysis study. Pediatr Res. 2003;53:830–835. doi: 10.1203/01.PDR.0000059552.08913.B7. [DOI] [PubMed] [Google Scholar]
- 70.Gelding SV, Coldham N, Niththyananthan R, Anyaoku V, Johnston DG. Insulin resistance with respect to lipolysis in non-diabetic relatives of European patients with type 2 diabetes. Diabet Med. 1995;12:66–73. doi: 10.1111/j.1464-5491.1995.tb02065.x. [DOI] [PubMed] [Google Scholar]
- 71.Robinson C, et al. Effect of insulin on glycerol production in obese adolescents. Am J Physiol. 1998;274:E737–E743. doi: 10.1152/ajpendo.1998.274.4.E737. [DOI] [PubMed] [Google Scholar]
- 72.Puhakainen I, Koivisto VA, Yki-Järvinen H. Lipolysis and gluconeogenesis from glycerol are increased in patients with noninsulin-dependent diabetes mellitus. J Clin Endocrinol Metab. 1992;75:789–794. doi: 10.1210/jcem.75.3.1517368. [DOI] [PubMed] [Google Scholar]
- 73.Nurjhan N, Consoli A, Gerich J. Increased lipolysis and its consequences on gluconeogenesis in non-insulin-dependent diabetes mellitus. J Clin Invest. 1992;89:169–175. doi: 10.1172/JCI115558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Perry RJ, Peng L, Cline GW, Petersen KF, Shulman GIA. Non-invasive method to assess hepatic acetyl-CoA in vivo. Cell Metab. 2017;25:749–756. doi: 10.1016/j.cmet.2016.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.American Diabetes Association. 7. Approaches to glycemic treatment. Diabetes Care. 2016;39:S52–S59. doi: 10.2337/dc16-S010. [DOI] [PubMed] [Google Scholar]
- 76.Hundal RS, et al. Mechanism by which metformin reduces glucose production in type 2 diabetes. Diabetes. 2000;49:2063–2069. doi: 10.2337/diabetes.49.12.2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Inzucchi SE, et al. Efficacy and metabolic effects of metformin and troglitazone in type II diabetes mellitus. N Engl J Med. 1998;338:867–872. doi: 10.1056/NEJM199803263381303. [DOI] [PubMed] [Google Scholar]
- 78.Zhou G, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108:1167–1174. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Shaw RJ, et al. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science. 2005;310:1642–1646. doi: 10.1126/science.1120781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.He L, et al. Metformin and insulin suppress hepatic gluconeogenesis through phosphorylation of CREB binding protein. Cell. 2009;137:635–646. doi: 10.1016/j.cell.2009.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fullerton MD, et al. Single phosphorylation sites in Acc1 and Acc2 regulate lipid homeostasis and the insulin-sensitizing effects of metformin. Nat Med. 2013;19:1649–1654. doi: 10.1038/nm.3372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Foretz M, et al. Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. J Clin Invest. 2010;120:2355–2369. doi: 10.1172/JCI40671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Madiraju AK, et al. Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase. Nature. 2014;510:542–546. doi: 10.1038/nature13270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Cao J, et al. Low concentrations of metformin suppress glucose production in hepatocytes through AMP-activated protein kinase (AMPK) J Biol Chem. 2014;289:20435–20446. doi: 10.1074/jbc.M114.567271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hawley SA, Gadalla AE, Olsen GS, Hardie DG. The antidiabetic drug metformin activates the AMP-activated protein kinase cascade via an adenine nucleotide-independent mechanism. Diabetes. 2002;51:2420–2425. doi: 10.2337/diabetes.51.8.2420. [DOI] [PubMed] [Google Scholar]
- 86.Howell JJ, et al. Metformin inhibits hepatic mTORC1 signaling via dose-dependent mechanisms involving AMPK and the TSC complex. Cell Metab. 2017;25:463–471. doi: 10.1016/j.cmet.2016.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.El-Mir MY, et al. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J Biol Chem. 2000;275:223–228. doi: 10.1074/jbc.275.1.223. [DOI] [PubMed] [Google Scholar]
- 88.Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. 2012;13:251–262. doi: 10.1038/nrm3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Miller RA, et al. Biguanides suppress hepatic glucagon signalling by decreasing production of cyclic AMP. Nature. 2013;494:256–260. doi: 10.1038/nature11808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Konopka AR, et al. Hyperglucagonemia mitigates the effect of metformin on glucose production in prediabetes. Cell Rep. 2016;15:1394–1400. doi: 10.1016/j.celrep.2016.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Pernicova I, Korbonits M. Metformin — mode of action and clinical implications for diabetes and cancer. Nat Rev Endocrinol. 2014;10:143–156. doi: 10.1038/nrendo.2013.256. [DOI] [PubMed] [Google Scholar]
- 92.Brown LJ, et al. Normal thyroid thermogenesis but reduced viability and adiposity in mice lacking the mitochondrial glycerol phosphate dehydrogenase. J Biol Chem. 2002;277:32892–32898. doi: 10.1074/jbc.M202408200. [DOI] [PubMed] [Google Scholar]
- 93.Saheki T, et al. Citrin/mitochondrial glycerol-3-phosphate dehydrogenase double knock-out mice recapitulate features of human citrin deficiency. J Biol Chem. 2007;282:25041–25052. doi: 10.1074/jbc.M702031200. [DOI] [PubMed] [Google Scholar]
- 94.Baur JA, Birnbaum MJ. Control of gluconeogenesis by metformin: does redox trump energy charge? Cell Metab. 2014;20:197–199. doi: 10.1016/j.cmet.2014.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Exton JH, Park CR. Control of gluconeogenesis in liver. II. Effects of glucagon, catecholamines, and adenosine 3′,5′-monophosphate on gluconeogenesis in the perfused rat liver. J Biol Chem. 1968;243:4189–4196. [PubMed] [Google Scholar]
- 96.Lee Y, et al. Hyperglycemia in rodent models of type 2 diabetes requires insulin-resistant alpha cells. Proc Natl Acad Sci USA. 2014;111:13217–13222. doi: 10.1073/pnas.1409638111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Blair JB, Cimbala MA, Foster JL, Morgan RA. Hepatic pyruvate kinase. Regulation by glucagon, cyclic adenosine 3′-5′-monophosphate, and insulin in the perfused rat liver. J Biol Chem. 1976;251:3756–3762. [PubMed] [Google Scholar]
- 98.Rider MH, et al. 6-Phosphofructo-2-kinase/ fructose-2,6-bisphosphatase: head-to-head with a bifunctional enzyme that controls glycolysis. Biochem J. 2004;381:561–579. doi: 10.1042/BJ20040752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wu C, et al. Perturbation of glucose flux in the liver by decreasing F26P2 levels causes hepatic insulin resistance and hyperglycemia. Am J Physiol Endocrinol Metab. 2006;291:E536–E543. doi: 10.1152/ajpendo.00126.2006. [DOI] [PubMed] [Google Scholar]
- 100.Cullen KS, Al-Oanzi ZH, O’Harte FPM, Agius L, Arden C. Glucagon induces translocation of glucokinase from the cytoplasm to the nucleus of hepatocytes by transfer between 6-phosphofructo 2-kinase/fructose 2,6-bisphosphatase-2 and the glucokinase regulatory protein. Biochim Biophys Acta. 2014;1843:1123–1134. doi: 10.1016/j.bbamcr.2014.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Romere C, et al. Asprosin, a fasting-induced glucogenic protein hormone. Cell. 2016;165:566–579. doi: 10.1016/j.cell.2016.02.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Edgerton DS, et al. Effects of insulin on the metabolic control of hepatic gluconeogenesis. in vivo Diabetes. 2009;58:2766–2775. doi: 10.2337/db09-0328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ramnanan CJ, et al. Molecular characterization of insulin-mediated suppression of hepatic glucose production. in vivo Diabetes. 2010;59:1302–1311. doi: 10.2337/db09-1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Puigserver P, et al. Insulin-regulated hepatic gluconeogenesis through FOXO1–PGC-1α interaction. Nature. 2003;423:550–555. doi: 10.1038/nature01667. [DOI] [PubMed] [Google Scholar]
- 105.Yoon JC, et al. Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1. Nature. 2001;413:131–138. doi: 10.1038/35093050. [DOI] [PubMed] [Google Scholar]
- 106.Nakae J, Kitamura T, Silver DL, Accili D. The forkhead transcription factor Foxo1 (Fkhr) confers insulin sensitivity onto glucose-6-phosphatase expression. J Clin Invest. 2001;108:1359–1367. doi: 10.1172/JCI12876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Schmoll D, et al. Regulation of glucose-6-phosphatase gene expression by protein kinase Bα and the forkhead transcription factor FKHR Evidence for insulin response unit-dependent and -independent effects of insulin on promoter activity. J Biol Chem. 2000;275:36324–36333. doi: 10.1074/jbc.M003616200. [DOI] [PubMed] [Google Scholar]
- 108.Matsumoto M. Dual role of transcription factor FoxO1 in controlling hepatic insulin sensitivity and lipid metabolism. J Clin Invest. 2006;116:2464–2472. doi: 10.1172/JCI27047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Haeusler RA, Kaestner KH, Accili D. FoxOs function synergistically to promote glucose production. J Biol Chem. 2010;285:35245–35248. doi: 10.1074/jbc.C110.175851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.O-Sullivan I, et al. FoxO1 integrates direct and indirect effects of insulin on hepatic glucose production and glucose utilization. Nat Commun. 2015;6:7079. doi: 10.1038/ncomms8079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Titchenell PM, Chu Q, Monks BR, Birnbaum MJ. Hepatic insulin signalling is dispensable for suppression of glucose output by insulin in vivo. Nat Commun. 2015;6:7078. doi: 10.1038/ncomms8078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Samuel VT, et al. Fasting hyperglycemia is not associated with increased expression of PEPCK or G6Pc in patients with type 2 diabetes. Proc Natl Acad Sci USA. 2009;106:12121–12126. doi: 10.1073/pnas.0812547106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Koo S-H, et al. The CREB coactivator TORC2 is a key regulator of fasting glucose metabolism. Nature. 2005;437:1109–1111. doi: 10.1038/nature03967. [DOI] [PubMed] [Google Scholar]
- 114.Dentin R, et al. Insulin modulates gluconeogenesis by inhibition of the coactivator TORC2. Nature. 2007;449:366–369. doi: 10.1038/nature06128. [DOI] [PubMed] [Google Scholar]
- 115.Wang Y, et al. Inositol-1,4,5-trisphosphate receptor regulates hepatic gluconeogenesis in fasting and diabetes. Nature. 2012;485:128–132. doi: 10.1038/nature10988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Liu Y, et al. A fasting inducible switch modulates gluconeogenesis via activator/coactivator exchange. Nature. 2008;456:269–273. doi: 10.1038/nature07349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Wang Y, et al. Targeted disruption of the CREB coactivator Crtc2 increases insulin sensitivity. Proc Natl Acad Sci USA. 2010;107:3087–3092. doi: 10.1073/pnas.0914897107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Hogan MF, et al. Hepatic insulin resistance following chronic activation of the CREB coactivator CRTC2. J Biol Chem. 2015;290:25997–26006. doi: 10.1074/jbc.M115.679266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Bass J, Lazar MA. Circadian time signatures of fitness and disease. Science. 2016;354:994–999. doi: 10.1126/science.aah4965. [DOI] [PubMed] [Google Scholar]
- 120.Zhang EE, et al. Cryptochrome mediates circadian regulation of cAMP signaling and hepatic gluconeogenesis. Nat Med. 2010;16:1152–1156. doi: 10.1038/nm.2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Lamia KA, et al. Cryptochromes mediate rhythmic repression of the glucocorticoid receptor. Nature. 2011;480:552–556. doi: 10.1038/nature10700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Lamia KA, Storch K-F, Weitz CJ. Physiological significance of a peripheral tissue circadian clock. Proc Natl Acad Sci USA. 2008;105:15172–15177. doi: 10.1073/pnas.0806717105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Sun Z, et al. Hepatic Hdac3 promotes gluconeogenesis by repressing lipid synthesis and sequestration. Nat Med. 2012;18:934–942. doi: 10.1038/nm.2744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Burgess SC, et al. Cytosolic phosphoenolpyruvate carboxykinase does not solely control the rate of hepatic gluconeogenesis in the intact mouse liver. Cell Metab. 2007;5:313–320. doi: 10.1016/j.cmet.2007.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Zingone A, et al. Correction of glycogen storage disease type 1a in a mouse model by gene therapy. J Biol Chem. 2000;275:828–832. doi: 10.1074/jbc.275.2.828. [DOI] [PubMed] [Google Scholar]
- 126.Bernard C. Leçons de physiologie expérimentale appliquée a la médecine. J.-B. Baillière; 1855. [Google Scholar]
- 127.Schwartz MW, et al. Cooperation between brain and islet in glucose homeostasis and diabetes. Nature. 2013;503:59–66. doi: 10.1038/nature12709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Myers MG, Olson DP. Central nervous system control of metabolism. Nature. 2012;491:357–363. doi: 10.1038/nature11705. [DOI] [PubMed] [Google Scholar]
- 129.Perry RJ, Petersen KF, Shulman GI. Pleotropic effects of leptin to reverse insulin resistance and diabetic ketoacidosis. Diabetologia. 2016;59:933–937. doi: 10.1007/s00125-016-3909-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Duffy KR, Pardridge WM. Blood–brain barrier transcytosis of insulin in developing rabbits. Brain Res. 1987;420:32–38. doi: 10.1016/0006-8993(87)90236-8. [DOI] [PubMed] [Google Scholar]
- 131.Plum L, Schubert M, Brüning JC. The role of insulin receptor signaling in the brain. Trends Endocrinol Metab. 2005;16:59–65. doi: 10.1016/j.tem.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 132.Brüning JC, et al. Role of brain insulin receptor in control of body weight and reproduction. Science. 2000;289:2122–2125. doi: 10.1126/science.289.5487.2122. [DOI] [PubMed] [Google Scholar]
- 133.Obici S, Zhang BB, Karkanias G, Rossetti L. Hypothalamic insulin signaling is required for inhibition of glucose production. Nat Med. 2002;8:1376–1382. doi: 10.1038/nm1202-798. [DOI] [PubMed] [Google Scholar]
- 134.Pocai A, et al. Hypothalamic KATP channels control hepatic glucose production. Nature. 2005;434:1026–1031. doi: 10.1038/nature03439. [DOI] [PubMed] [Google Scholar]
- 135.Kleinridders A, Ferris HA, Cai W, Kahn CR. Insulin action in brain regulates systemic metabolism and brain function. Diabetes. 2014;63:2232–2243. doi: 10.2337/db14-0568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Edgerton DS, et al. Insulin’s direct effects on the liver dominate the control of hepatic glucose production. J Clin Invest. 2006;116:521–527. doi: 10.1172/JCI27073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Ramnanan CJ, et al. Brain insulin action augments hepatic glycogen synthesis without suppressing glucose production or gluconeogenesis in dogs. J Clin Invest. 2011;121:3713–3723. doi: 10.1172/JCI45472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Shulman GI, Landau BR. Pathways of glycogen repletion. Physiol Rev. 1992;72:1019–1035. doi: 10.1152/physrev.1992.72.4.1019. [DOI] [PubMed] [Google Scholar]
- 139.Syed NA, Khandelwal RL. Reciprocal regulation of glycogen phosphorylase and glycogen synthase by insulin involving phosphatidylinositol-3 kinase and protein phosphatase-1 in HepG2 cells. Mol Cell Biochem. 2000;211:123–136. doi: 10.1023/a:1007159422667. [DOI] [PubMed] [Google Scholar]
- 140.Moore MC, et al. Hepatic glucose disposition during concomitant portal glucose and amino acid infusions in the dog. Am J Physiol. 1998;274:E893–E902. doi: 10.1152/ajpendo.1998.274.5.E893. [DOI] [PubMed] [Google Scholar]
- 141.Gomis RR, Ferrer JC, Guinovart JJ. Shared control of hepatic glycogen synthesis by glycogen synthase and glucokinase. Biochem J. 2000;351:811–816. [PMC free article] [PubMed] [Google Scholar]
- 142.O’Doherty RM, et al. Differential metabolic effects of adenovirus-mediated glucokinase and hexokinase I overexpression in rat primary hepatocytes. J Biol Chem. 1996;271:20524–20530. doi: 10.1074/jbc.271.34.20524. [DOI] [PubMed] [Google Scholar]
- 143.Niswender KD, Shiota M, Postic C, Cherrington AD, Magnuson MA. Effects of increased glucokinase gene copy number on glucose homeostasis and hepatic glucose metabolism. J Biol Chem. 1997;272:22570–22575. doi: 10.1074/jbc.272.36.22570. [DOI] [PubMed] [Google Scholar]
- 144.Titchenell PM, et al. Direct hepatocyte insulin signaling is required for lipogenesis but is dispensable for the suppression of glucose production. Cell Metab. 2016;23:1154–1166. doi: 10.1016/j.cmet.2016.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Velho G, et al. Impaired hepatic glycogen synthesis in glucokinase-deficient (MODY-2) subjects. J Clin Invest. 1996;98:1755–1761. doi: 10.1172/JCI118974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Raimondo A, Rees MG, Gloyn AL. Glucokinase regulatory protein: complexity at the crossroads of triglyceride and glucose metabolism. Curr Opin Lipidol. 2015;26:88–95. doi: 10.1097/MOL.0000000000000155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Agius L. Glucokinase and molecular aspects of liver glycogen metabolism. Biochem J. 2008;414:1–18. doi: 10.1042/BJ20080595. [DOI] [PubMed] [Google Scholar]
- 148.Agius L, Peak M, Newgard CB, Gomez-Foix AM, Guinovart JJ. Evidence for a role of glucose-induced translocation of glucokinase in the control of hepatic glycogen synthesis. J Biol Chem. 1996;271:30479–30486. doi: 10.1074/jbc.271.48.30479. [DOI] [PubMed] [Google Scholar]
- 149.Härndahl L, Schmoll D, Herling AW, Agius L. The role of glucose 6-phosphate in mediating the effects of glucokinase overexpression on hepatic glucose metabolism. FEBS J. 2006;273:336–346. doi: 10.1111/j.1742-4658.2005.05067.x. [DOI] [PubMed] [Google Scholar]
- 150.von Wilamowitz-Moellendorff A, et al. Glucose-6-phosphate-mediated activation of liver glycogen synthase plays a key role in hepatic glycogen synthesis. Diabetes. 2013;62:4070–4082. doi: 10.2337/db13-0880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Bollen M, Keppens S, Stalmans W. Specific features of glycogen metabolism in the liver. Biochem J. 1998;336:19–31. doi: 10.1042/bj3360019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Ros S, García-Rocha M, Domínguez J, Ferrer JC, Guinovart JJ. Control of liver glycogen synthase activity and intracellular distribution by phosphorylation. J Biol Chem. 2009;284:6370–6378. doi: 10.1074/jbc.M808576200. [DOI] [PubMed] [Google Scholar]
- 153.Bultot L, et al. AMP-activated protein kinase phosphorylates and inactivates liver glycogen synthase. Biochem J. 2012;443:193–203. doi: 10.1042/BJ20112026. [DOI] [PubMed] [Google Scholar]
- 154.Ros S, et al. Hepatic overexpression of a constitutively active form of liver glycogen synthase improves glucose homeostasis. J Biol Chem. 2010;285:37170–37177. doi: 10.1074/jbc.M110.157396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Cohen P. The Croonian Lecture 1998. Identification of a protein kinase cascade of major importance in insulin signal transduction. Phil Trans R Soc B. 1999;354:485–495. doi: 10.1098/rstb.1999.0399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Wan M, et al. A noncanonical, GSK3-independent pathway controls postprandial hepatic glycogen deposition. Cell Metab. 2013;18:99–105. doi: 10.1016/j.cmet.2013.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Kitamura T, et al. Insulin-induced phosphorylation and activation of cyclic nucleotide phosphodiesterase 3B by the serine–threonine kinase Akt. Mol Cell Biol. 1999;19:6286–6296. doi: 10.1128/mcb.19.9.6286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Jurczak MJ, Danos AM, Rehrmann VR, Brady MJ. The role of protein translocation in the regulation of glycogen metabolism. J Cell Biochem. 2008;104:435–443. doi: 10.1002/jcb.21634. [DOI] [PubMed] [Google Scholar]
- 159.Alemany S, Cohen P. Phosphorylase a is an allosteric inhibitor of the glycogen and microsomal forms of rat hepatic protein phosphatase-1. FEBS Lett. 1986;198:194–202. doi: 10.1016/0014-5793(86)80404-5. [DOI] [PubMed] [Google Scholar]
- 160.Carabaza A, Ciudad CJ, Baqué S, Guinovart JJ. Glucose has to be phosphorylated to activate glycogen synthase, but not to inactivate glycogen phosphorylase in hepatocytes. FEBS Lett. 1992;296:211–214. doi: 10.1016/0014-5793(92)80381-p. [DOI] [PubMed] [Google Scholar]
- 161.Samuel VT, Shulman GI. The pathogenesis of insulin resistance: integrating signaling pathways and substrate flux. J Clin Invest. 2016;126:12–22. doi: 10.1172/JCI77812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Olefsky JM, Glass CK. Macrophages, inflammation, and insulin resistance. Annu Rev Physiol. 2010;72:219–246. doi: 10.1146/annurev-physiol-021909-135846. [DOI] [PubMed] [Google Scholar]
- 163.Tolman KG, Fonseca V, Dalpiaz A, Tan MH. Spectrum of liver disease in type 2 diabetes and management of patients with diabetes and liver disease. Diabetes Care. 2007;30:734–743. doi: 10.2337/dc06-1539. [DOI] [PubMed] [Google Scholar]
- 164.Lee YH, Wang M-Y, Yu X-X, Unger RH. Glucagon is the key factor in the development of diabetes. Diabetologia. 2016;59:1372–1375. doi: 10.1007/s00125-016-3965-9. [DOI] [PubMed] [Google Scholar]
- 165.Henry RR, Scheaffer L, Olefsky JM. Glycemic effects of intensive caloric restriction and isocaloric refeeding in noninsulin-dependent diabetes mellitus. J Clin Endocrinol Metab. 1985;61:917–925. doi: 10.1210/jcem-61-5-917. [DOI] [PubMed] [Google Scholar]
- 166.American Diabetes Association. 6 Obesity management for the treatment of type 2 diabetes. Diabetes Care. 2016;39:S47–S51. doi: 10.2337/dc16-S009. [DOI] [PubMed] [Google Scholar]
- 167.Lefebvre PJ, Luyckx AS. Glucagon and diabetes: a reappraisal. Diabetologia. 1979;16:347–354. doi: 10.1007/BF01223153. [DOI] [PubMed] [Google Scholar]
- 168.Lotfy M, Kalasz H, Szalai G, Singh J, Adeghate E. Recent progress in the use of glucagon and glucagon receptor antagonists in the treatment of diabetes mellitus. Open Med Chem J. 2014;8:28–35. doi: 10.2174/1874104501408010028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Bagger JI, Knop FK, Holst JJ, Vilsbøll T. Glucagon antagonism as a potential therapeutic target in type 2 diabetes. Diabetes Obes Metab. 2011;13:965–971. doi: 10.1111/j.1463-1326.2011.01427.x. [DOI] [PubMed] [Google Scholar]
- 170.Habegger KM, et al. The metabolic actions of glucagon revisited. Nat Rev Endocrinol. 2010;6:689–697. doi: 10.1038/nrendo.2010.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Reaven GM, Chen YD, Golay A, Swislocki AL, Jaspan JB. Documentation of hyperglucagonemia throughout the day in nonobese and obese patients with noninsulin-dependent diabetes mellitus. J Clin Endocrinol Metab. 1987;64:106–110. doi: 10.1210/jcem-64-1-106. [DOI] [PubMed] [Google Scholar]
- 172.Baron AD, Schaeffer L, Shragg P, Kolterman OG. Role of hyperglucagonemia in maintenance of increased rates of hepatic glucose output in type II diabetics. Diabetes. 1987;36:274–283. doi: 10.2337/diab.36.3.274. [DOI] [PubMed] [Google Scholar]
- 173.Woerle HJ, et al. Mechanisms for abnormal postprandial glucose metabolism in type 2 diabetes. Am J Physiol Endocrinol Metab. 2006;290:E67–E77. doi: 10.1152/ajpendo.00529.2004. [DOI] [PubMed] [Google Scholar]
- 174.Menge BA, et al. Loss of inverse relationship between pulsatile insulin and glucagon secretion in patients with type 2 diabetes. Diabetes. 2011;60:2160–2168. doi: 10.2337/db11-0251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Lee Y, et al. Metabolic manifestations of insulin deficiency do not occur without glucagon action. Proc Natl Acad Sci USA. 2012;109:14972–14976. doi: 10.1073/pnas.1205983109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Neumann UH, et al. Glucagon receptor gene deletion in insulin knockout mice modestly reduces blood glucose and ketones but does not promote survival. Mol Metab. 2016;5:731–736. doi: 10.1016/j.molmet.2016.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Petersen KF, Sullivan JT. Effects of a novel glucagon receptor antagonist (Bay 27–9955) on glucagon-stimulated glucose production in humans. Diabetologia. 2001;44:2018–2024. doi: 10.1007/s001250100006. [DOI] [PubMed] [Google Scholar]
- 178.Guan H-P, et al. Glucagon receptor antagonism induces increased cholesterol absorption. J Lipid Res. 2015;56:2183–2195. doi: 10.1194/jlr.M060897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Kelly RP, et al. Short-term administration of the glucagon receptor antagonist LY2409021 lowers blood glucose in healthy people and in those with type 2 diabetes. Diabetes Obes Metab. 2015;17:414–422. doi: 10.1111/dom.12446. [DOI] [PubMed] [Google Scholar]
- 180.Kazda CM, et al. Evaluation of efficacy and safety of the glucagon receptor antagonist LY2409021 in patients with type 2 diabetes: 12- and 24-week phase 2 studies. Diabetes Care. 2016;39:1241–1249. doi: 10.2337/dc15-1643. [DOI] [PubMed] [Google Scholar]
- 181.Longuet C, et al. The glucagon receptor is required for the adaptive metabolic response to fasting. Cell Metab. 2008;8:359–371. doi: 10.1016/j.cmet.2008.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Vatner DF, et al. Thyroid hormone receptor-β agonists prevent hepatic steatosis in fat-fed rats but impair insulin sensitivity via discrete pathways. Am J Physiol Endocrinol Metab. 2013;305:E89–E100. doi: 10.1152/ajpendo.00573.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Martagón AJ, Lin JZ, Cimini SL, Webb P, Phillips KJ. The amelioration of hepatic steatosis by thyroid hormone receptor agonists is insufficient to restore insulin sensitivity in ob/ob mice. PLoS ONE. 2015;10:e0122987. doi: 10.1371/journal.pone.0122987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Finan B, et al. Chemical hybridization of glucagon and thyroid hormone optimizes therapeutic impact for metabolic disease. Cell. 2016;167:843–857 e14. doi: 10.1016/j.cell.2016.09.014. [DOI] [PubMed] [Google Scholar]
- 185.Armstrong MJ, et al. Liraglutide safety and efficacy in patients with non-alcoholic steatohepatitis (LEAN): a multicentre, double-blind, randomised, placebo-controlled phase 2 study. Lancet. 2016;387:679–690. doi: 10.1016/S0140-6736(15)00803-X. [DOI] [PubMed] [Google Scholar]
- 186.Finan B, et al. A rationally designed monomeric peptide triagonist corrects obesity and diabetes in rodents. Nat Med. 2015;21:27–36. doi: 10.1038/nm.3761. [DOI] [PubMed] [Google Scholar]
- 187.Soccio RE, Chen ER, Lazar MA. Thiazolidinediones and the promise of insulin sensitization in type 2 diabetes. Cell Metab. 2014;20:573–591. doi: 10.1016/j.cmet.2014.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Mayerson AB, et al. The effects of rosiglitazone on insulin sensitivity, lipolysis, and hepatic and skeletal muscle triglyceride content in patients with type 2 diabetes. Diabetes. 2002;51:797–802. doi: 10.2337/diabetes.51.3.797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Belfort R, et al. A placebo-controlled trial of pioglitazone in subjects with nonalcoholic steatohepatitis. N Engl J Med. 2006;355:2297–2307. doi: 10.1056/NEJMoa060326. [DOI] [PubMed] [Google Scholar]
- 190.Gastaldelli A, et al. Importance of changes in adipose tissue insulin resistance to histological response during thiazolidinedione treatment of patients with nonalcoholic steatohepatitis. Hepatology. 2009;50:1087–1093. doi: 10.1002/hep.23116. [DOI] [PubMed] [Google Scholar]
- 191.Rizos CV, Kei A, Elisaf MS. The current role of thiazolidinediones in diabetes management. Arch Toxicol. 2016;90:1861–1881. doi: 10.1007/s00204-016-1737-4. [DOI] [PubMed] [Google Scholar]
- 192.Tunaru S, et al. PUMA-G and HM74 are receptors for nicotinic acid and mediate its anti-lipolytic effect. Nat Med. 2003;9:352–355. doi: 10.1038/nm824. [DOI] [PubMed] [Google Scholar]
- 193.Phan BAP, et al. Effects of niacin on glucose levels, coronary stenosis progression, and clinical events in subjects with normal baseline glucose levels (<100 mg/ dl): a combined analysis of the Familial Atherosclerosis Treatment Study (FATS), HDL-Atherosclerosis Treatment Study (HATS), Armed Forces Regression Study (AFREGS), and Carotid Plaque Composition by MRI during lipid-lowering (CPC) study. Am J Cardiol. 2013;111:352–355. doi: 10.1016/j.amjcard.2012.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Guyton JR. Niacin in cardiovascular prevention: mechanisms, efficacy, and safety. Curr Opin Lipidol. 2007;18:415–420. doi: 10.1097/MOL.0b013e3282364add. [DOI] [PubMed] [Google Scholar]
- 195.Kroon T, Kjellstedt A, Thalén P, Gabrielsson J, Oakes ND. Dosing profile profoundly influences nicotinic acid’s ability to improve metabolic control in rats. J Lipid Res. 2015;56:1679–1690. doi: 10.1194/jlr.M058149. [DOI] [PMC free article] [PubMed] [Google Scholar]