

Summary Statement
Commensal bacteria are believed to play important roles in human health. The mechanisms by which they affect mammalian physiology are poorly understood; however, bacterial metabolites are likely to be key components of host interactions. Here, we use bioinformatics and synthetic biology to mine the human microbiota for N-acyl amides that interact with G-protein-coupled receptors (GPCRs). We found that N-acyl amide synthase genes are enriched in gastrointestinal bacteria and the lipids they encode interact with GPCRs that regulate gastrointestinal tract physiology. Mouse and cell-based models demonstrate that commensal GPR119 agonists regulate metabolic hormones and glucose homeostasis as efficiently as human ligands although future studies are needed to define their potential physiologic role in humans. This work suggests that chemical mimicry of eukaryotic signaling molecules may be common among commensal bacteria and that manipulation of microbiota genes encoding metabolites that elicit host cellular responses represents a new small molecule therapeutic modality (microbiome-biosynthetic-gene-therapy).
Keywords: GPCR, microbiome, metagenome, signaling, N-acyl amide
Introduction
Although the human microbiome is believed to play an important role in human physiology the mechanisms by which bacteria affect mammalian physiology remain poorly defined.1 Bacteria rely heavily on small molecules to interact with their environment.2 While it is likely that the human microbiota similarly relies on small molecules to interact with its human host, the identity and functions of microbiota-encoded effector molecules are largely unknown. The study of small molecules produced by the human microbiota and the identification of the host receptors they interact with should help to define the relationship between bacteria and human physiology and provide a resource for the discovery of small molecule therapeutics.
We recently reported on the discovery of commendamide, a human microbiota encoded, G protein-coupled receptor (GPCR) active, long-chain N-acyl amide that suggests a structural convergence between human signaling molecules and microbiota encoded metabolites.3 N-acyl amides, like the endocannabinoids, are able to regulate diverse cellular functions due, in part, to their ability to interact with GPCRs. GPCRs are the largest family of membrane receptors in eukaryotes and are likely to be key mediators of host-microbial interactions in the human microbiome. The importance of GPCRs to human physiology is reflected by the fact that they are the most common targets of therapeutically approved small molecule drugs. The GPCRs with which human N-acyl amides interact are implicated in diseases including diabetes, obesity, cancer, and inflammatory bowel disease among others.4,5 With numerous possible combinations of amine head groups and acyl tails, long-chain N-acyl amides represent a potentially large and functionally diverse class of microbiota-encoded GPCR-active signaling molecules.
Here, we combined bioinformatic analysis of human microbiome sequencing data with targeted gene synthesis, heterologous expression and high-throughput GPCR activity screening to identify GPCR-active N-acyl amides encoded by the human microbiota. The bacterial effectors we identified provide mechanistic insights into potential functions of the human microbiome and suggest these GPCR-active small molecules and their associated microbial biosynthetic genes have the potential to regulate human physiology.
Isolation of commensal N-acyl amides
To identify N-acyl synthase (NAS) genes within human microbial genomes, the Human Microbiome Project (HMP) sequence data was searched with BLASTN using 689 NAS genes associated with the N-acyl synthase protein family PFAM13444.3 The 143 unique human microbial N-acyl synthase genes (hm-NASs) we identified fall into four major clades (clades A–D, Fig. 1a) that are divided into a number of distinct sub-clades (Fig. 1a). Forty-four phylogenetically diverse hm-NAS genes were selected for synthesis and heterologous expression. This set included all hm-NAS genes from clades sparsely populated with hm-NAS sequences and representative examples from clades heavily populated with hm-NAS sequences (Fig. 1a).
Figure 1. hm-NAS genes in gastrointestinal microbiota.

a, Phylogenetic tree of N-acyl genes from PFAM13444. hm-NAS genes have a circle at the branch tip. Black dots were not synthesized, red dots were synthesized but no molecule was detected and large grey dots mark genes that produced N-acyl amides. Branches are colored by bacterial phylogeny. b, The major heterologously produced metabolite from each N-acyl family (1–6) is shown. c, hm-NAS gene distribution and abundance [Reads per Kilobase of Gene Per Million Reads (RPKM)] based on molecule family (1–6). Body site and molecule designations in a are based on the analysis shown in c and b. Box plots are median from 1st to 3rd quartile. HMP patient samples analyzed include N = 133 tongue, 127 plaque, 148 stool, 122 buccal.
Liquid chromatography-mass spectrometry (LCMS) analysis of ethyl acetate extracts derived from E. coli cultures transformed with each construct revealed clone specific peaks in 31 cultures. hm-NAS gene functions could be clustered into 6 groups based on the retention time and mass of the heterologously produced metabolites (Extended Data Fig. 1 and Supplementary Table 1). Molecule isolation and structural elucidation studies were carried out on one representative culture from each group (Supplementary Information). This analysis identified six N-acyl amide families that differ by amine head group and fatty acid tail (Fig. 1b, families 1–6): 1) N-acyl glycine, 2) N-acyloxyacyl lysine, 3) N-acyloxyacyl glutamine, 4) N-acyl lysine/ornithine, 5) N-acyl alanine, 6) N-acyl serinol. Each family was isolated as a collection of metabolites with different acyl substituents. The most common analog within each family is shown in Figure 1b. Long-chain N-acyl ornithines, lysines and glutamines have been reported as natural products produced by soil bacteria and some human pathogens.6,7,8
Functional differences in NAS enzymes follow the pattern of the NAS phylogenetic tree, with hm-NAS genes from the same clade or sub-clade largely encoding the same metabolite family (Fig. 1a). With the exception of one NAS that is predicted to use lysine and ornithine as substrates, hm-NASs appear to be selective for a single amine-containing substrate. The most common acyl chains incorporated by hm-NASs are from 14–18 carbons in length. These can be modified by β-hydroxylation or a single unsaturation. Three hm-NAS enzymes contain two domains. The second domain is either an aminotransferase that is predicted to catalyze the formation of serinol from glycerol (Fig. 1b, family 6, Extended Data Fig. 2) or an additional acyltransferase that is predicted to catalyze the transfer of a second acyl group (Fig. 1b, families 2, 3). To explore NAS gene synteny we looked for gene occurrence patterns around NAS genes in the human microbiome. The only repeating pattern that we saw was that some NAS genes appear adjacent to genes predicted to encode acyltransferases. This is reminiscent of the two domain NASs that we found produce di-acyl lipids (families 2 and 3). There were rare instances where NASs potentially occur in gene clusters, but none of these were used in this study.
To look for native N-acyl amide production by commensal bacteria, organic extracts from cultures of species containing the hm-NAS genes we examined were screened by LCMS. Based on retention time and mass we detected the production of the expected N-acyl amides by commensal species predicted to produce N-acyl glycines, N-acyloxyacyl lysines, N-acyl lysine/ornithines and N-acyl serinols. The only case where we did not detect the expected N-acyl amide was for N-acyloxyacyl glutamines (Extended Data Fig. 1).
hm-NAS genes are enriched in GI bacteria
A BLASTN search of NAS genes against human microbial reference genomes and metagenomic sequence data from the HMP revealed that NAS genes are enriched in gastrointestinal (GI) bacteria relative to bacteria found at other body sites (Fischer’s exact test p < 0.05, gastrointestinal versus non gastrointestinal sites, Supplementary Table 2, Figure 1). Within gastrointestinal sites that were frequently sampled in the context of the HMP (e.g., stool, buccal mucosa, supragingival plaque, tongue) hm-NAS gene families show distinct distribution patterns (Fig. 1c, two way ANOVA p < 2e-16). Despite tremendous person-to-person variation in microbiota species composition, most N-acyl amide synthase gene families we studied can be found in over 90% of patient samples. N-acyoxyacyl glutamine (12%) and N-acyl alanine (not detected) synthase genes are the only exceptions. Taken together, these data suggest that NAS genes are highly prevalent in the human microbiome and unique sites within the gastrointestinal tract are likely exposed to different sets of N-acyl amide structures.
When we searched existing metatranscriptome sequence data from stool and supragingival plaque microbiomes to look for evidence of hm-NAS gene expression in the gastrointestinal tract we observed site-specific hm-NAS gene expression that matches the predicted body site localization patterns for hm-NAS genes in metagenomic data. Across patient samples hm-NAS genes are transcribed to varying degrees relative to the average level of transcription for each gene in the bacterial genome (Fig. 2a). In the stool metatranscriptome dataset both RNA and DNA sequencing datasets were available allowing for a more direct sample-to-sample comparison of hm-NAS gene expression levels. When metatranscriptome data were normalized using the number of hm-NAS gene specific DNA sequence reads detected in each sample, we observed what appears to be differential expression of hm-NAS genes in different patient samples (Fig. 2b). Datasets whereby bacterial genes, transcripts and metabolites can be tracked in a single sample will be necessary to explore how hm-NAS gene transcription variation impacts metabolite production.
Figure 2. N-acyl synthase gene expression in vivo.

a, Gene expression analysis for an N-acyl glycine hm-NAS gene in a stool metatranscriptome dataset and an N-acyloxyacyl lysine hm-NAS gene in a supragingival plaque metatranscriptome dataset. Gene expression is normalized to the expression of all genes from a bacterial genome containing the hm-NAS gene that was heterologously expressed – Bacteroides dorei in stool, Capnocytophaga ochracea in plaque (1 highly expressed, 0 not expressed). b, Comparison of hm-NAS gene abundance based on RNA or DNA derived reads obtained from individual patient stool samples. Abundance is measured in RPKM. N = 24 patient stool samples analyzed – 8 patients with 3 samples per patient. N = 38 patient plaque samples analyzed.
hm-N-acyl-amides interact with GI GPCRs
The major N-acyl amide isolated from each family was assayed for agonist and antagonist activity against 240 human GPCRs (Fig. 3 and Extended Data Fig. 3). The strongest agonist interactions were: activation of GPR119 by N-palmitoyl serinol (EC50 9 µM), activation of sphingosine-1-phosphate receptor 4 (S1PR4) by N-3-hydroxypalmitoyl ornithine (EC50 32 µM) and activation of G2A by N-myristoyl alanine (EC50 3 µM). Interactions between bacterial N-acyl amides and GPCRs were also specific (Fig. 3a and b). In each survey experiment, no other GPCRs reproducibly showed greater than 35% activation relative to the endogenous ligands. The strongest antagonist activities were observed for N-acyloxyacyl glutamine against two prostaglandin receptors, PTGIR and PTGER4 (Fig. 3c, PTGIR IC50 15 µM, PTGER4 IC50 43 µM). PTGIR was specifically antagonized by N-acyloxyacyl glutamine, while PTGER4 was antagonized by N-acyloxyacyl glutamine as well as other N-acyl amides [Fig. 3c(i) and 3c(ii)]. Alternative GPCR screening methods could identify interactions in addition to those uncovered here.
Figure 3. hm-N-acyl GPCR activity screen.

Screen of N-acyl amides (color coded) for agonist activity against 168 GPCRs with known ligands (a) as well as 72 orphan GPCRs (b). Dot plots display data for all N-acyl amides assayed against all GPCRs. Screen performed in singlicate. Bar graphs show the strongest N-acyl GPCR agonist interactions compared to all GPCRs. Insets show dose response curves and EC50 data (each dose performed in duplicate). c, Screen of N-acyl amides as antagonists in the presence of endogenous ligands. Screen performed in singlicate. i, PTGIR is specifically inhibited by N-acyloxyacyl glutamine. ii, PTGER4 is inhibited by structurally diverse hm-N-acyl amides. Error bars are mean +/− SEM.
Based on data from the Human Protein Atlas (HPA) GPCRs targeted by human microbial N-acyl amides are localized to the gastrointestinal tract and its associated immune cells. In mouse models, this collection of gastrointestinal tract localized GPCRs have been reported to affect diverse mucosal functions including metabolism (GPR119), immune cell differentiation (S1PR4, PTGIR, PTGER4), immune cell trafficking (S1PR4, G2A) and tissue repair (PTGIR).9–14 It is not possible at this time to look for co-localization of GPCR and hm-NAS gene expression in specific gastrointestinal niches, as neither the HMP nor the HPA are sufficiently comprehensive in their survey of human body sites. Nonetheless, 16S and metagenomic deep sequencing studies link bacteria containing hm-NAS genes or hm-NAS genes themselves to specific locations in the gastrointestinal tract where GPCRs of interest are expressed (Extended Data Fig. 4).
Bacterial and human ligands share structure and function
Human microbiota-encoded N-acyl amides bear structural similarity to endogenous GPCR-active ligands (Fig. 4). The clearest overlap in structure and function between bacterial and human GPCR-active ligands is for the endocannabinoid receptor GPR119 (Fig. 4 and 5). Endogenous GPR119 ligands include oleoylethanolamide (OEA) and the dietary lipid derivative 2-oleoyl glycerol (2-OG).15,16 In our heterologous expression experiment we isolated both the palmitoyl and oleoyl analogs of N-acyl serinol. The latter only differs from 2-OG by the presence of an amide instead of an ester and from OEA by the presence of an additional ethanol substituent. N-oleoyl serinol is a similarly potent GPR119 agonist compared to the endogenous ligand OEA (EC50 12 µM vs. 7 µM) but elicits almost a 2-fold greater maximum GPR119 activation (Fig. 5a). N-palmitoyl derivatives of all 20 natural amino acids were synthesized and none activated GPR119 by more than 37% relative to OEA (Fig. 5b). The generation of a potent and specific long-chain N-acyl-based GPR119 ligand therefore necessitates a more complex biosynthesis than the simple N-acylation of an amino acid as is commonly seen for characterized NAS enzymes. In this case, the biosynthesis of N-acyl serinols is achieved through the coupling of an NAS domain with an aminotransferase that is predicted to generate serinol from glycerol (Extended Data Fig. 2).
Figure 4. Structural mimicry of GPCR ligands.

Comparison of microbiota encoded and human GPCR ligands suggests a structural and functional complementarity.
Figure 5. N-acyl serinols affect GLP-1 secretion in vitro and glucose homeostasis in vivo.

a, β-arrestin GPR119 activation assay using microbiota (green) and human (blue) ligands (each dose performed in duplicate). b, β-arrestin assay comparing microbiota ligands and 20 synthesized N-palmitoyl amino acids (screen performed in singlicate). c, Release of GLP-1 by GLUTag cells (ANOVA, p < 0.05, data combined from 2 independent experiments, N = 4 for DMSO and 2-oleoyl glycerol, N = 6 for OEA and N-oleoyl serinol). d, Oral glucose tolerance test (OGTT) in gnotobiotic mice. Treatment mice (n = 6 mice, data combined from 2 independent experiments) were colonized with E. coli producing N-acyl serinols and control mice (n = 8 mice, data combined from 2 independent experiments) were colonized with E. coli containing an empty vector (two way ANOVA, bonferroni post-hoc) e, OGTT after withholding IPTG to stop N-acyl gene expression (no difference, two way ANOVA, N is the same as in d). f, OGTT in an antibiotic treated mouse cohort (n = 9 mice in both groups, data combined from 2 independent experiments, two way ANOVA, bonferroni post-hoc). g, Insulin (n = 6 mice in both groups, one experiment, technical triplicates) and h, GLP-1 (n = 9 control mice, n = 10 treatment mice, data combined from 2 independent experiments, technical replicates) measured at 15 min after glucose gavage in the antibiotic treated cohort (unpaired T test, two tailed). Key Bacterial (green) and human (blue) GPR119 ligands reference figure colors. Error bars (mean +/− SEM) * p < 0.05, ** p < 0.01 *** p < 0.001.
The endogenous agonist for S1PR4, sphingosine-1-phosphate (S1P) and the N-3-hydroxypalmitoyl ornithine/lysine family of bacterial agonists share similar head group charges. S1P is a significantly more potent agonist (EC50 0.09 µM vs. EC50 32 µM); however, the bacterial agonists are more specific for S1PR4. The bacterial N-3-hydroxypalmitoyl ornithine did not activate S1PR1, 2, or 3 in our GPCR screen, whereas S1P activates all four members of the S1P receptor family tested.
No direct comparison could be made between the microbiota-derived and endogenous ligands for PTGIR or PTGER4, as there are no known endogenous antagonists for these receptors. Many human GPCRs remain orphan receptors lacking known endogenous ligands. Ligands for at least some of these receptors will undoubtedly be found among the small molecules produced by the human microbiota. G2A is an orphan receptor and therefore does not have a well-defined endogenous agonist, although it has been reported to respond to lysophosphatidylcholine.17,18 We found that the bacterial metabolites N-3-hydroxypalmitoyl glycine (commendamide) and N-palmitoyl alanine, both activate G2A. Mammals produce N-palmitoyl glycine, which differs from commendamide by the absence of the β-hydroxyl and, based on our synthetic N-acyl studies, activates G2A.19
GPR119 is the most extensively studied of the GPCRs activated by bacterial ligands we identified (Fig 4). Mechanisms that link endogenous GPR119 agonists (OEA, 2-OG) to changes in host phenotype are well defined as a result of the exploration of GPR119 as a therapeutic target for diabetes and obesity.20–23 GPR119 agonists are thought to primarily affect glucose homeostasis but also gastric emptying and appetite through both GPR119-dependent hormone release from enteroendocrine cells (GLP-1, GIP, PYY) and pancreatic β-cells (insulin) as well as GPR119-independent mechanisms including PPARα modulation.9,16,24–30 Murine enteroendocrine GLUTag cells have been used as a model system for measuring the ability of potential GPR119 agonists to induce GLP-1 release. When administered to GLUTag cells at equimolar concentrations, microbiota-encoded N-oleoyl serinol or the endogenous ligands OEA and 2-OG induced GLP-1 secretion to the same magnitude (Fig. 5c). To provide an orthogonal measurement of GPR119 activation by N-acyl serinols, HEK293 cells were stably transfected with a GPR119 expression construct. Both OEA and N-oleoyl serinol increased cellular cAMP concentrations in a GPR119 dependent fashion (Extended Data Fig 5).
hm-NAS expression alters blood glucose in mice
The functional overlap between endogenous and bacterial metabolites suggested that bacteria expressing microbiota-encoded GPR119 ligands might elicit host phenotypes that mimic those induced by eukaryotic ligands. Endogenous and synthetic GPR119 ligands have been associated with changes in glucose homeostasis that are relevant to the etiology and treatment of diabetes and obesity including a study where mice were orally administered bacteria engineered to produce a eukaryotic enzyme that increases endogenous GPR119 ligand (OEA) precursors.9,16,24–27,31 The metabolic effect of the endogenous GPR119 ligands is believed to occur at the intestinal mucosa as the delivery of OEA intravenously fails to lower blood glucose in mice during an oral glucose tolerance test (OGTT).26 Consequently, we sought to determine whether mice colonized with bacteria engineered to produce human microbiota N-acyl serinols would exhibit predictable host phenotypes. notobiotic mice were colonized with E. coli engineered to express the N-acyl serinol synthase gene in an IPTG dependent manner. Control mice were colonized with E. coli containing an empty vector. Based on the number of colony forming units detected in fecal pellets both cohorts of mice were colonized to the same extent (Extended Data Fig. 7). After one week of exposure to IPTG both cohorts were fasted overnight and subjected to an OGTT. At 30 minutes post challenge we observed a statistically significant decrease in blood glucose levels for the group colonized with E. coli expressing the N-acyl serinol synthase gene (Fig. 5d). MS analysis of metabolites present in cecal samples revealed the presence of N-acyl serinols in the treatment cohort but not in the control cohort (Extended Data Fig. 6). After two weeks of withdrawing IPTG from the drinking water we no longer observed a difference in blood glucose between the two cohorts in an OGTT (Fig. 5e).32
To further explore the metabolic phenotype induced by N-acyl serinols we repeated the OGTT experiment in an antibiotic treated mouse model. In this study we compared mice colonized with E. coli expressing an active N-acyl serinol synthase to mice colonized with E. coli expressing an NAS point mutant (Extended Data Fig. 8, p.Glu91Ala) that no longer produced N-acyl serinols. In this model the glucose lowering effect of colonization with N-acyl serinol producing E. coli remained significant (Fig. 5f). In the antibiotic treated mice we measured GLP-1 and insulin concentrations after glucose gavage. Both hormones were significantly increased in the treatment group compared to the control group (Fig. 5 g, h). In all mouse models the observed correlation between hm-NAS gene induction and increased glucose tolerance is similar in magnitude to several studies with small molecule GPR119 agonists including glyburide, an FDA approved therapeutic for diabetes.24,25
Discussion
Our characterization of human microbial N-acyl amides, together with other investigations of the human microbiota, suggests that host-microbial interactions may rely heavily on simple metabolites built from the same common lipids, sugars, and peptides that define many human signaling systems (e.g., neurotransmitters, bioactive lipids, glycans). This is not surprising, as the genomes of the bacterial taxa common to the human gastrointestinal tract (e.g., Bacteroidetes, Firmicutes and Proteobacteria) are often lacking in gene clusters that encode for the production of complex secondary metabolites (e.g., polyketides, nonribosomal peptides, terpenes). It appears that biosynthesis of endogenous mammalian signaling molecules as well as those produced by the human microbiota may rely on the modest manipulation of primary metabolites. As a result, the structural conservation between metabolites used in host-microbial interactions and endogenous mammalian signaling metabolites may be a common phenomenon in the human microbiome. Evolutionarily, the convergence of bacterial and human signaling systems through structurally related GPCR ligands is not unreasonable as GPCRs are thought to have developed in eukaryotes to allow for structurally simple signaling molecules to regulate increasingly complex cellular interactions.33–35 The structural similarities between microbiota-encoded N-acyl amides and endogenous GPCR-active lipids may be indicative of a broader structural and functional overlap among bacterial and human bioactive lipids including other GPCR-active N-acyl amides, eiconasoids (prostaglandins, leukotrienes) and sphingolipids. Sphingolipid based signaling molecules may also be common in the human microbiome as prevalent bacterial species are known to synthesize membrane sphingolipids.36
The GPCRs with which bacterial N-acyl amides were found to interact are all part of the same “lipid-like” GPCR gene family. The potential importance of this GPCR family to the regulation of host-microbial interactions is suggested by their localization to areas of gastrointestinal track enriched in bacteria that are predicted to synthesize GPCR ligands (Extended Data Fig. 4). Lipid-like GPCRs have been shown to play roles in disease models that are correlated with changes in microbial ecology including colitis (S1PR4, PTGIR, PTGER4), obesity (GPR119), diabetes (GPR119), autoimmunity (G2A) and atherosclerosis (G2A, PTGIR).9,10,13,14 The fact that the expression of an NAS gene in a gastrointestinal colonizing bacterium is sufficient to alter host physiology suggests that the interaction between lipid-like GPCRs and their N-acyl amide ligands could be relevant to human physiology and warrants further study. By LCMS analysis we observed most of the microbiota encoded N-acyl amides reported here in human stool samples (Extended Data Fig. 9). Further studies will be needed to better define the distribution and concentration of these metabolites throughout the gastrointestinal tract especially at the mucosa where the physiologic activity of these metabolites likely occurs. Interestingly, Gemella spp. predicted to encode N-acyl serinols are tightly associated with the small intestinal mucosa supporting this site as a potentially important location for N-acyl amide mediated interactions.37 As the mouse model system used here relies on induced expression of NAS genes it will also be important to understand how these genes are natively regulated.
Current strategies for treating diseases associated with the microbiome such as inflammatory bowel disease or diabetes are not believed to address the dysfunction of the host-microbial interactions that are likely part of the disease pathogenesis. Bacteria engineered to deliver bioactive small molecules produced by the human microbiota have the potential to help address diseases of the microbiome by modulating the native distribution and abundance of these metabolites. Regulation of GPCRs by microbiota-derived N-acyl amides is a particularly attractive therapeutic strategy for the treatment of human diseases as GPCRs have been extensively validated as therapeutic targets. As our mechanistic understanding of how human microbiota-encoded small molecules effect changes in host physiology grows, the potential for using “microbiome-biosynthetic-gene-therapy” to treat human disease by complementing small molecule deficiencies in native host-microbial interactions with microbiota derived biosynthetic genes should increase accordingly. The use of functional metagenomics to identify microbiota encoded effectors combined with bioinformatics and synthetic biology to expand effector molecule families provides a generalizable platform to help define the role microbiota-encoded small molecules play in host-microbial interactions.
Methods
Bioinformatics analysis of human N-acyl synthase genes
Protein sequences for members of the PFAM family 13444 Acetyltransferase (GNAT) domain (http://pfam.xfam.org/family/PF13444) (n=689) were downloaded and corresponding gene sequences identified based on European Bioinformatics Institute (EBI) number. A multiple sequence alignment was performed using Clustal Omega (http://www.ebi.ac.uk/Tools/msa/clustalo/), generating a phylogenetic tree in Newick format with the “--guidetree-out” option. The 689 PFAM sequences were queried against the Human Microbiome Project (HMP) clustered gene index datasets and reference genome datasets with BLASTN (http://hmpdacc.org/HMGC/). The PFAM13444 sequences that aligned to a HMP gene [expectation (E) value < e−40 and > 70% identity] were identified and comprise the human N-acyl synthase (hm-NAS) gene dataset (143 hm-NAS genes). Reference genomes for 111/143 hm-NAS genes were identified (Supplementary Table 2).
To determine the abundance of hm-NAS genes within microbiomes at specific human body sites, hm-NAS genes were queried against HMP whole metagenome shotgun sequencing data on a per body site basis (http://hmpdacc.org/HMASM/). Each hm-NAS gene was BLASTN searched against the non-redundant gene sets from the following body sites: buccal mucosa, anterior nares, mid vagina, posterior fornix, vaginal introitus, retroauricular crease (combined left and right), stool, supragingival plaque and tongue dorsum. These body sites were chosen because they contained sequence data from the largest number of unique patients.38 hm-NAS genes and highly similar genes in the HMP non redundant gene set (E-value < e−40) were aligned to shotgun sequencing reads from each patient sample taken from different sites in the human microbiome. Aligned reads were normalized to hm-NAS gene length and sequencing depth of each dataset. The normalized count of the reads aligned to each hm-NAS gene or its highly similar gene from the HMP non redundant gene set were scaled [0–1] and color coded per body site, and added as concentric rings around the phylogenetic tree (Fig. 1A). To determine the variability and distribution of hm-NAS genes that correspond to specific N-acyl amide families 1–6 (Fig. 1) in the human microbiome normalized read counts for hm-NAS gene from each N-acyl amide family were plotted separately per body site as Reads per Kilobase of Gene Per Million Reads (RPKM) (Fig. 1c). The tree in Figure 1 was plotted using graphlan (https://huttenhower.sph.harvard.edu/graphlan).
Analysis of metatranscriptome datasets
Two RNAseq datasets were identified with multiple patient samples taken from separate sites in the human microbiome.39,40 One RNAseq dataset was part of the HMP (http://hmpdacc.org/RSEQ/) and generated from supragingival samples taken from twin pairs with and without dental caries. The second RNAseq dataset was generated from stool samples and compared different RNA extraction methods. We used only samples labeled “whole” which functioned as controls for the original study.39 Alignment of all hm-NAS genes to each dataset only identified hm-NAS genes from N-acyl amide family 1 and 2 in each of the RNAseq datasets (1 in stool, 2 in supragingival plaque). To explore whether hm-NAS gene expression might vary in patient samples we performed two different analyses. In the first analysis we identified reference genomes containing hm-NAS genes identical to those we used in heterologous expression experiments for molecule families 1 and 2 (Bacteroides dorei for compound 1, Capnocytophaga ochracea for compound 2). RNAseq reads were aligned to all of the genes from each reference genome. For each genome the average per gene read density normalized for gene length was compared to the read density seen for the hm-NAS gene. The percentile of the normalized expression of each hm-NAS gene was then plotted (0 for not expressed, 1 for the most expressed) and compared between patient samples for each RNAseq dataset (Fig. 2a). In the second analysis the direct correlation between DNA and RNA abundance was determined for the stool metatranscriptome dataset for which DNA reads were also available.39 RNAseq and shotgun-sequenced DNA reads were aligned to the 15 hm-NAS genes from N-acyl amide family 1 that encoded for N-acyl glycines (Supplementary Table 1). The reads were normalized (RPKM) and each hm-NAS gene from each patient sample was plotted as a single point with DNA and RNA read counts on the X and Y axis (Fig. 2b).
Heterologous expression of PFAM13444 genes in Escherichia coli
The 44 hm-NAS genes we examined by heterologous expression were codon optimized, appended with NcoI and NdeI sites at the N and C terminus respectively and synthesized by Gen9. Genes obtained from Gen9 were digested with NdeI and NcoI and ligated into the corresponding restriction sites in pET28c (Novagen). For heterologous expression purposes the resulting constructs were transformed into E. coli EC100 containing the T7 polymerase gene integrated into its genome (E. coli EC100:DE3). E. coli EC100:DE3 hm-NAS containing strains were inoculated into 10 ml of Luria-Bertani (LB) broth supplemented with kanamycin (50 µg/ml) and grown overnight (37 °C with shaking 200 rpm). One ml of overnight culture was used to inoculate 50 ml of LB supplemented with kanamycin (50 µg/ml) and isopropyl β-D-1-thiogalactopyranoside (IPTG) (25 µM). Cultures were incubated at 30 °C for 4 days with shaking (200 rpm). Each culture broth was extracted with an equal volume of ethyl acetate and the resulting crude extracts were dried in vacuo. Crude extracts were resuspended in 50 µL of methanol and analyzed by reversed phase HPLC-MS (XBridgeTm C18 4.6 mm × 150 mm) using a binary solvent system (A/B solvent of water/acetonitrile with 0.1% formic acid: 10% B isocratic for 5 minutes, gradient 10% to 100% B over 25 minutes). Clone specific metabolites encoded by each hm-NAS gene were identified by comparing experimental extracts with extracts prepared from cultures of E. coli EC100:DE3 transformed with an empty pET28c vector.
N-acyl amide isolation and structure determination
For each group of clones that, based on LCMS analysis, were predicted to produce a different N-acyl amide family we chose one representative clone for use in molecule isolation studies. Each representative clone was grown in 1.5 L of LB in a 2.7 L Fernach flask (30 °C, 200 RPM). After 4 days, cultures were extracted 2 times with an equal volume of ethyl acetate. Dried ethyl acetate extracts were partitioned by reversed phase flash chromatography (Teledyne Isco, C18 RediSep RF GoldTm 15 g) using the following mobile phase conditions: water:acetonitrile with 0.1% formic acid, 10% acetonitrile isocratic for 5 minutes, gradient to 100% acetonitrile over 20 minutes (30 mL/minute). Fractions containing clone specific metabolites were pooled and semi preparative reversed phase HPLC was used to separate individual N-acyl amide molecules (Supplementary Information). The structures of compounds 2–6 were determined using a combination of HRMS, 1H, 13C, and 2D NMR data (Supplementary Information). Compound 1 was described in our previous study.3
hm-NAS gene containing bacterial species culture broth analysis
Capnocytophaga ochracea F0287 (compound 2), Klebsiella pneumoniae WGLW1–5 (compound 3), Neisseria flavescens SK114 (compounds 4a and 4b), and Gemella haemolysans M341 (compound 6) were obtained from the Biodefense and Emerging Infections Research Resources Repository (BEI Resources) HMP catalogue. Compound 1 was previously identified in culture broth extracts from cultures of Bacteroides vulgatus.3 Each chosen bacteria contains an hm-NAS gene related to that which was heterologously expressed to produce compound 2, 3, 4a, 4b or 6. Strains were inoculated under sterile conditions into 2 L of LYBHI medium [brain–heart infusion medium supplemented with 0.5% yeast extract (Difco), 5 mg/L hemin (Sigma), 1 mg/ml cellobiose (Sigma), 1 mg/ml maltose (Sigma), 0.5 mg/ml cysteine (Sigma)] and grown anaerobically (C. ochracea) or aerobically (N. flavescens, G. haemolysans, K. pneumoniae) for 7 days. Culture broths were extracted with an equal volume of ethyl acetate. To look for the presence of N-acyl amides these extracts were examined by HPLC-MS as was done in the original heterologous expression experiments. With the exception of family 3, the N-acyl metabolite that was heterologously expressed could be identified in the culture broth extracts from the bacteria that harbored that hm-NAS gene (Extended Data Fig. 1).
GPCR screen of N-acyl amide small molecules
For each of the 6 N-acyl amide families (1–6) the analog produced at the highest level in our heterologous expression experiments was assayed for GCPR activity. In the case of family 4 the major lysine analog (N-3-hydroxyoleoyl lysine) was also screened. For family 1 the major glycine analog (N-3-hydroxypalmitoyl glycine) was previously screened.3 Using β-arrestin cell-based assays at 10 µM ligand concentration, agonist and antagonist activity was assessed by DiscoveRx Corporation against 168 GPCRs with known ligands as well as 72 orphan GPCRs. The most potent interactions between N-acyl amides and GPCRs were validated by repeating the assay in duplicate and generating dose response curves. Synthetic N-acyl amides were assayed in the same fashion. HEK293 cells expressing GPR119 were exposed to equimolar concentrations of N-palmitoyl serinol or OEA. cAMP was used as an orthogonal assay to measure GPR119 activation. cAMP was measured in HEK293 cells engineered to express a cAMP sensitive ion channel that permits cAMP measurement in live cells by monitoring calcium eflux (ACTOne cells, Codex Biosolutions).41 Calcium eflux was measured using the ACTone membrane potential kit (Codex Biosolutions CB-80500-201). For our analysis ACTOne HEK293 cells transfected with GPR119 (ACTOne-GCPR 119) were compared to cells not transfected with GPR119. Each cell line was exposed to equimolar concentrations of OEA, N-oleoyl serinol or [5–(N-Ethylcarboxamido)adenosine] which stimulates GPCR ADORA2B in the parental HEK293 cell line. For a reference to the quality control of each GPCR reporter cell line from DiscoveRx please refer to https://www.discoverx.com/targets/cell-based-assay-list?class=gpcr&pcat=stable%20cell%20lines&readout=arrestin or Codex Biosolutions http://codexbiosolutions.com/actone_cell_lines.php.
Synthesis of proteinogenic amino acid containing N-acyl-palmitoyl analogs
Wang resins with preloaded amino acids were purchased from Matrix Innovation. Coupling reagents (PyBOP and Cl-HOBt) were purchased from P3 BioSystems. Palmitoyl chloride and all other reagents were purchased from Sigma-Aldrich. Dimethylformamide (DMF) was added to preloaded Wang resins (~80 mg) and incubated for 30 minutes. Removal of N-Fmoc from swollen resins was accomplished by two rounds of piperidine treatment [20% solution in DMF (v/v), 3 ml] for 3 and 10 minutes, followed by several washes with DMF. Palmitoyl chloride (1 equivalent) in DMF was then added and the resin suspension was shaken for 2 hours at room temperature. The N-acylated amino acid product was cleaved from the resins by treatment with trifluoroacetic acid (TFA) supplemented with 2.5% (v/v) water and 2.5% (v/v) triisopropylsilane (TIPS). After evaporation of TFA the crude product was purified by automated reversed phase flash chromatography (Teledyne Isco, C18 RediSep RF GoldTm 15 g), binary solvent system: water and acetonitrile supplemented with 0.1% acetic acid. All final products were verified by MS (Supplementary Table 3).
In-vitro study of GLP-1 release from GLUTag cells
Oleoylethanolamide (OEA) and 2-oleoyl glycerol (2-OG) were purchased from Cayman Chemical Company and resuspended in DMSO to a concentration of 10 mM. N-oleoyl serinol was isolated and purified in the same manner as N-palmitoyl serinol described above and its identity was confirmed by 1H NMR and HRMS. N-oleoyl serinol was resuspended at 10 mM concentration in DMSO. GLUTag cells were obtained from the Mangelsdorf Lab (University of Texas Southwestern) with permission from Daniel Drucker (Mount Sinai Hospital Toronto). GLUTag cells were grown in DMEM, low glucose, GlutaMAX (ThermoFisher) supplemented with 10% FBS and 1% Penicillin/Streptomycin. Once cells grew to 80% confluence they were harvested and plated 1:1 into 24 well culture plates in fresh culture media at 50,000 cells per well. After overnight growth in culture plates, cells were washed twice with Krebs buffer supplemented with 20 µL per ml of DPP4 inhibitor (Millipore). GLUTag cells were incubated for 30 minutes in supplemented Krebs buffer and compounds were added at 1 µM and 100 µM. Cells were incubated with compounds for 2 hours. Media was then collected, centrifuged at 500×g (4 °C) for 5 minutes and cell free supernatant was analyzed for GLP-1 level using the Active GLP-1 V2 kit (Mesoscale Discovery). Data from 2 independent experiments were analyzed together for Figure 5c (N = 6 wells for OEA, N-oleoyl serinol and N = 4 wells for 2-OG and DMSO).
Colonization of germ-free and antibiotic treated mice with N-acyl serinol producing E. coli
All experimental procedures were approved by the Animal Care and Use Committee of The Rockefeller University. Germ free C57BL/6 mice were maintained in sterile isolators with autoclaved food and water in the Gnotobiotic Facility of the Mucida Laboratory at The Rockefeller University. Wild type C57BL/6 mice were purchased from Jackson Labs. 8-week-old mice were used for all experiments. For colonization studies 5 ml of an overnight culture (LB with 50 µg/ml kanamycin) of E. coli transformed with pET28c:hm-NAS N-acyl serinol synthase (treatment group) or E. coli transformed with the empty pET28c vector (control group) was centrifuged at 500 × g for 2 minutes, the supernatant was decanted and the cells were resuspended in 2 ml of sterile PBS. Germ free mice were gavaged with 100 µL of bacterial culture immediately upon removal from sterile isolators. Antibiotic treated mice were given water supplemented with 500 µg/ml ampicillin for 1 week followed by gavage with E. coli clones. Suppression of endogenous microbiota was confirmed prior to gavage by culturing stool on non-selective LB agar and demonstrating no colony formation. After colonization mice were housed in specific-pathogen-free conditions and fed with autoclaved water and food. Water was supplemented with 35 µg/ml kanamycin and 25 mM IPTG.32 Fecal pellets from mice were analyzed each week for 3 weeks to confirm colonization by the appropriate bacteria and to check for contamination by plating on LB agar with and without kanamycin 50 µg/ml (Extended Data Fig 7). A single fresh mouse fecal pellet was weighed and resuspended in PBS at a fixed concentration (400 µL per 40 mg). 1 µL of this suspension was diluted 1:100 in PBS and plated in duplicate, the average number of colonies per plate (2 plates) was recorded. Plasmids were isolated and restriction mapped from these colonies to confirm the presence of the correct hm-NAS gene insert or lack thereof. Ethyl acetate extracts from broth cultures were also examined as previously discussed to confirm the production of N-acyl serinols by bacteria in the treatment group. In the first experiment of germ free, 7 mice were studied - 3 (1M, 2F) in the treatment group and 4 (2M, 2F) in the control group. The independent replicate experiment of germ free mice also consisted of 7 mice (all female, 3 in the treatment group and 4 in the control group). Mice were all individually caged and at the end of each week food consumption and weight were measured. In the antibiotic treated mouse cohort 18 mice were studied [two independent experiments of 9M in the treatment group and 9M in the control group]. Mice were of the same size and gender distribution in each replicate. In the antibiotic treated study the control mouse group was E. coli transformed with pET28c:hm-NAS, which contains an N-acyl serinol synthase gene containing a single point mutation that rendered the enzyme non-functional (E94A) (see method section below). The animal experiments were not randomized and the investigators were not blinded to the allocation during experiments and outcome assessment. No statistical methods were used to predetermine sample size. All mice which completed the experiments were analyzed.
Generation of active site pET28c:hm-NAS N-acyl serinol synthase mutant
Conserved active site residues in bacterial NASs were identified in previous biochemical and X-ray crystalography studies.42 To create a catalytically inactive N-acyl serinol synthase we changed a key glutamic acid residue (Glu91) to alanine. The point mutation was created by PCR using pET28c:hm-NAS N-acyl serinol synthase vector as template and the following primers F – GTTCTGTGCGATACGTCTCC and R – GCCTTTCACAGGCAGATATTC. The position of point mutation is underlined in the F primer. The resulting PCR reaction was digested with DpnI to remove any remaining methylated vector. The PCR product was then phosphorylated, column purified and blunt end ligated (End-It, Epicentre). The vector was transformed into EC100:DE3 cells and the point mutation was confirmed by Sanger sequencing. When transformed into E. coli, the resultant E94A mutant construct did not confer the production of any detectable N-acyl serinols. Cultures were grown under the same conditions as the original N-acyl serinol synthase producing clone (Extended Data Fig 8).
Oral glucose tolerance test
One week post colonization mice were fasted overnight (16 hours) and then administered a 2 g/kg OGTT (40% glucose solution). Blood glucose was measured by tail bleed (Breeze 2 Bayer) at time 0 prior to the glucose gavage, then 15, 30, 60, 90 and 120 minutes post gavage. After one week the IPTG was removed from the mouse drinking water and mice were allowed to equilibrate for an additional 2 weeks.32 Three weeks post colonization the oral glucose tolerance test was repeated. Blood glucose levels at each time point during the OGTT tests were compared between groups using a Students T-test with significance threshold of p < 0.05.
Insulin and GLP-1 measurement
Mice were given an OGTT as previously described. At 15 minutes blood was collected by submandibular bleed and immediately mixed with 10 µL of 0.5M EDTA and 5 µL of DPPIV inhibitor (Millipore, DPP4-010) per 500 µL of blood. Treated blood was spun at 2,000 × g for 15 minutes at 4 °C. Plasma was collected and immediately placed at −80 °C. Insulin was measured using the Crystal Chem Ultra Sensitive Mouse ELISA kit and active GLP-1 was measured using the Mesoscale Discovery Active GLP-1 V2 kit. Samples were analyzed for insulin in triplicate and GLP-1 in duplicate. Insulin was measured from mice in one experiment (N = 6 mice in each group). GLP-1 was measured from mice in two independent experiments (N = 9 mice in control, N = 10 mice in treatment). All mice were male and of the same size distribution.
N-acyl serinol metabolite measurement in mouse cecal samples and human stool
After withholding IPTG for two weeks, mice from the first experimental set were re-exposed to IPTG in the drinking water for 1 week to induce hm-NAS gene expression and N-acyl serinol production. Mice were sacrificed and cecal samples taken. Fresh cecal stool from two control mice and two treated mice was resuspended in 5 ml of sterile PBS and extracted 1:1 with ethyl acetate. Crude extracts were dried in vacuo and resuspended in methanol normalized by crude extract weight. Each extract was then analyzed by reversed phase liquid chromatography coupled to a 6550 Q-TOF mass spectrometer (Agilent Technologies). Peak identities were confirmed by accurate mass, and also by comparison of chromatographic retention time and MS/MS spectra to those of the purified N-palmitoyl serinol standard. In both mice in the treatment group N-palmitoyl serinol could be detected in the cecal samples whereas N-palmitoyl serinol was detected in neither of the mice in the control group (Extended Data Fig. 6). Stool samples were collected from human subjects prior to bone marrow transplant as part of a previous clinical trial conducted at Memorial Sloan Kettering in collaboration with the author J.C.. Fresh stool samples were processed in the same manner as the mouse cecal samples described above.
Data Availability Statement
All figure data is available in the source data sheet. Gene accession numbers for all cloned genes are provided in Supplementary Table 1. Publically available DNA and RNA datasets analyzed in this study are referenced accordingly and references contain links to datasets available for download.
Extended Data
Extended Data Figure 1. Analysis of hm-NAS clone families.
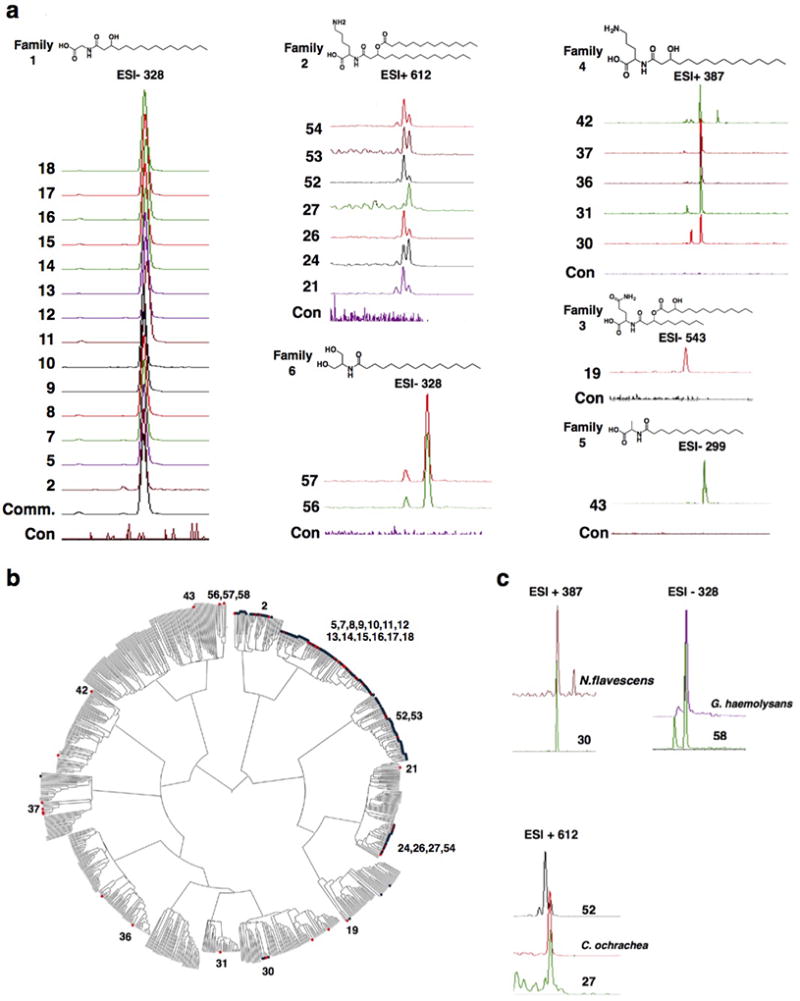
a, LCMS analysis of crude extracts prepared from E. coli transformed with each hm-NAS gene expression construct (number 1–43, see Supplementary Table 1 for details about each clone number) compared to negative control extracts derived from E. coli containing an empty vector (con). Based on metabolite retention time and observed mass hm-NAS genes could be grouped into 6 N-acyl amide families (1–6). The mass of the major metabolite (pictured) from each N-acyl amide family is shown in either the ESI(+) or ESI(−) MS detection mode for each hm-NAS extract including the control extract. Functional differences in NAS enzymes follow the pattern of the NAS phylogenetic tree, with hm-NAS genes from the same clade or sub-clade largely encoding the same metabolite family. Commendamide was previously isolated and is part of family 1.3 b, Phylogenetic tree of PFAM13444 showing the location of each hm-NAS gene that we synthesized and examined by heterologous expression. c, Crude ethyl acetate extracts were prepared from cultures of bacterial species that harbor the same or highly related (>80% nucleotide identity) hm-NAS gene that was expressed by heterologous expression. The only exception was for N-acyl alanines for which a representative cultured commensal bacterial species was not available. N-acyl glycines were previously analyzed in the same manner.3 The extracted ion for the hm-NAS gene family is shown for the E. coli clone compared to the crude extract from the commensal species. For family 6 hm-NAS clone 58 is pictured in c and not a due to formatting constraints.
Extended Data Figure 2. Proposed biosynthesis of N-acyl serinol.
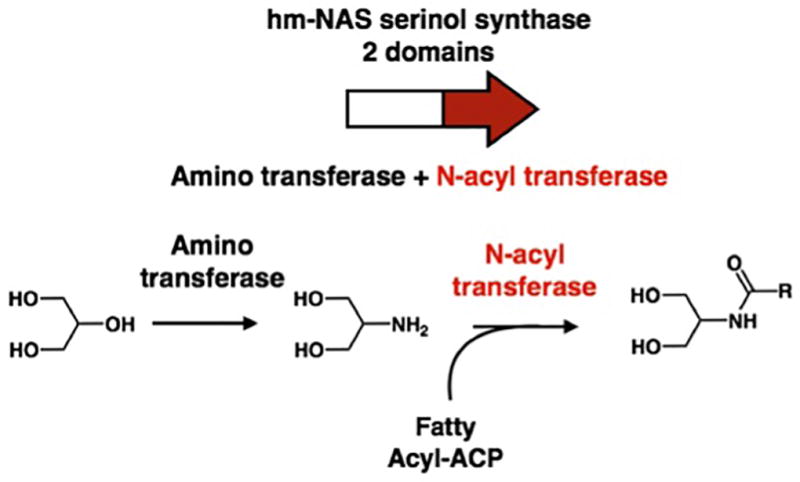
Proposed two-step biosynthesis of N-acyl serinol using the two domains found in the enzyme predicted to be encoded by the hm-NAS N-acyl serinol synthase gene. Simple N-palmitoyl derivatives of all 20 natural amino acids did not activated GPR119 by more than 37% relative to OEA.
Extended Data Figure 3. Validation of hits from the high throughput GPCR screen.
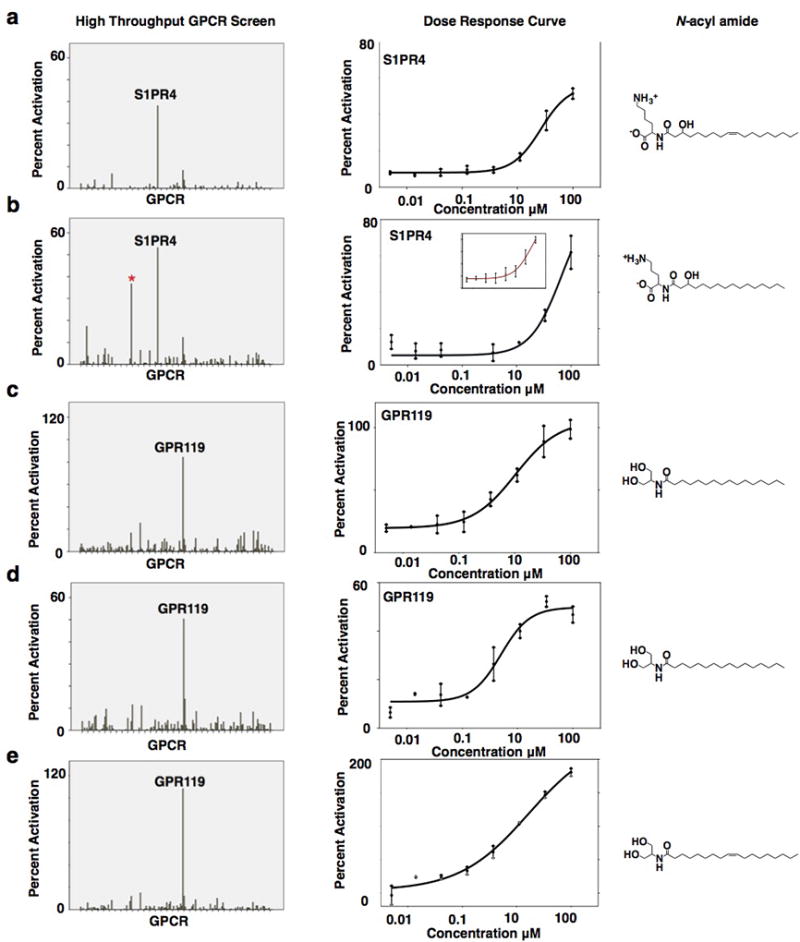
When structural analogs were independently screened in the GPCR panel (e.g., N-oleoyl and palmitoyl serinol or N-3-hydroxypalmitoyl lysine and ornithine) they yielded the same GPCR profile and when N-acyl serinol was re-assayed across all GPCRs in the panel, it also yielded the same GPCR activity profile. a, b, N-3-hydroxypalmitoyl lysine and ornithine both interact with S1PR4. * Did not repeat. b, inset technical repeat of N-3-hydroxypalmitoyl ornithine dose response curve. c, d technical repeats of N-palmitoyl serinol. c, d, e N-palmitoyl and oleoyl serinol both interact with GPR119. Screening data performed in singlicate, dose response curves performed in duplicate. Error bars are mean +/− SEM.
Extended Data Figure 4. Combined analysis of protein and transcript expression of GPCR in the gastrointestinal tract.
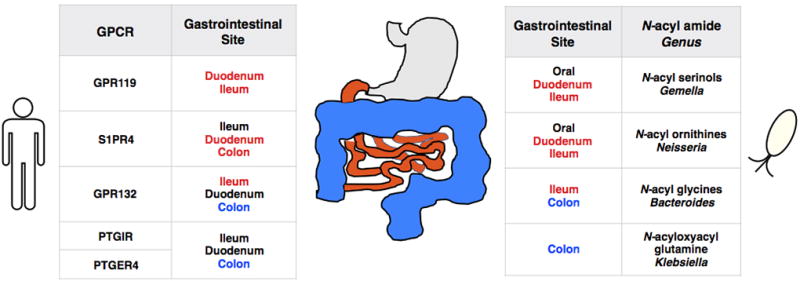
Table links GPCR, N-acyl amide, bacterial genus and the site where these co-occur in the gastrointestinal tract (colored). Based on protein expression data (Human Protein Atlas) GPR119 is most highly expressed in the pancreas and duodenum, S1PR4 in the spleen and lymph node, G2A in the lymph node and appendix, PTGIR in the lung and appendix and PTGER4 in the bone marrow and small intestine. From gene expression data in the colon (GTEx dataset, N = 88 patient samples from small intestine, 345 patient samples from colon) GPR132, PTGER4, and PTGIR are all expressed alongside the N-acyl synthase genes known to encode metabolites that target these GPCR (Figure 1). In the gastrointestinal tract GPR119 and S1PR4 are most highly expressed in the small intestine where 16S studies have identified bacteria from the genera Gemella and Neisseria. All known reference genomes (NCBI) from these genera contain N-acyl synthase genes that are highly similar (blastN, e value 2e-132) to those we found to encode GPR119 or S1PR4 ligands.37,43,44
Extended Data Figure 5. Secondary assay of GPR119.
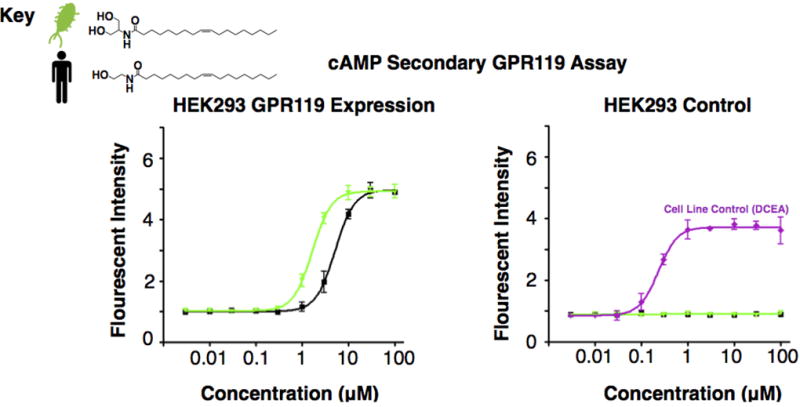
ACTOne HEK293 cells (control) and ACTOne HEK293 cells transfected with GPR119 were exposed to equimolar concentrations of the endogenous GPR119 ligand oleoylethanolamide or the bacterial ligand N-oleoyl serinol. Relative fluorescent intensity was recorded for each ligand concentration compared to background signal. All data points were performed in quadruplicate and error bars represent SD around the mean. An increase in cAMP concentration was observed in HEK293 cells expressing GPR119 but not in native HEK293 cells. The DCEA [5–(N-Ethylcarboxamido)adenosine] control is presented to confirm cAMP response of the parental cell line. The EC50 for N-oleoyl serinol (bacterial) was 1.6 µM and for oleoylethanolamide was 5.1 µM, which are consistent with data from the β-arrestin assay (Figure 5a).
Extended Data Figure 6. Identification of N-acyl serinol biosynthesis in vivo.
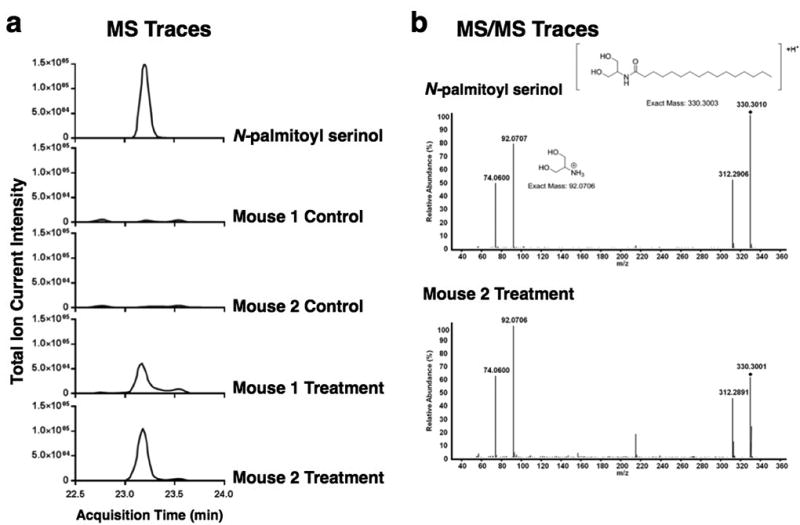
a, LC-MS analysis of crude cecal extracts. Extracted-ion chromatograms for palmitoyl serinol ([M+H]+ m/z: 330.3003) are shown. A peak with the same exact mass and chromatographic retention time as the N-palmitoyl serinol standard was present in treatment mice but not control mice. Treatment mice were colonized with E. coli containing the N-acyl serinol synthase gene. Control mice were colonized with E. coli containing the empty pET28c vector. b, Identification of N-palmitoyl serinol by MS/MS fragmentation of the m/z 330.3003 ion. In the MS2 spectrum the diamond indicates N-palmitoyl serinol parent ion and the product ion at m/z: 92.0706 shows presence of the serinol head group.
Extended Data Figure 7. Bacterial colonization of mouse model systems.
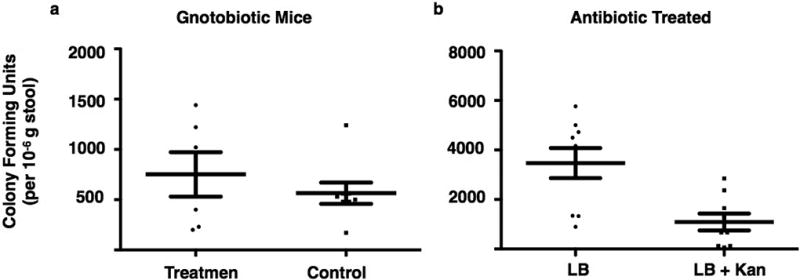
One week after inoculation with E. coli a single fecal pellet from a colonized mouse was collected, resuspended in 400 µL PBS and plated at a 1/100 dilution onto LB agar plates with or without kanamycin 50 µg/mL. a) The number of colony forming units per 10−6 g of feces observed on LB agar plates with kanamycin was similar for the treatment group (E. coli with hm-NAS gene, N = 6 mouse stool samples) and the control group (E. coli with empty vector, N = 8 mouse stool samples). b) In the antibiotic treated mouse cohort there are other colonizing bacteria present. Stool samples produced threefold more colony forming units on unselected LB agar plates compared to LB agar plates with kanamycin. Error bars in both a, b are mean +/− SEM. In both cases when random colonies were picked from the LB/kanamycin plates they were all found to contain the cloning vector indicating these were in fact E. coli colonizing bacteria.
Extended Data Fig 8. N-acyl serinol synthase point mutant.
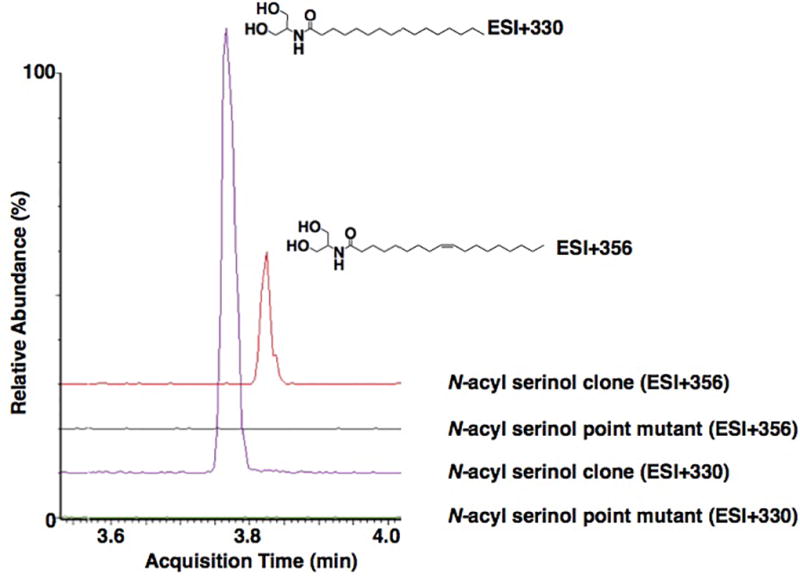
LC-MS analysis of crude extracts prepared from cultures of E. coli expressing either the N-acyl serinol synthase gene or the N-acyl serinol synthase gene with an active site point mutation (E94A). N-acyl serinol metabolites (e.g., N-palmitoyl serinol and N-oleoyl serinol) are absent from the point mutant culture broth (ESI(+) mode). This mutant was created to address the possibility that the observed mouse phenotype might be due to over-production of any protein by E. coli and not specifically from N-acyl serinol production.
Extended Data Fig 9. Detection of N-acyl amides in human fecal samples.
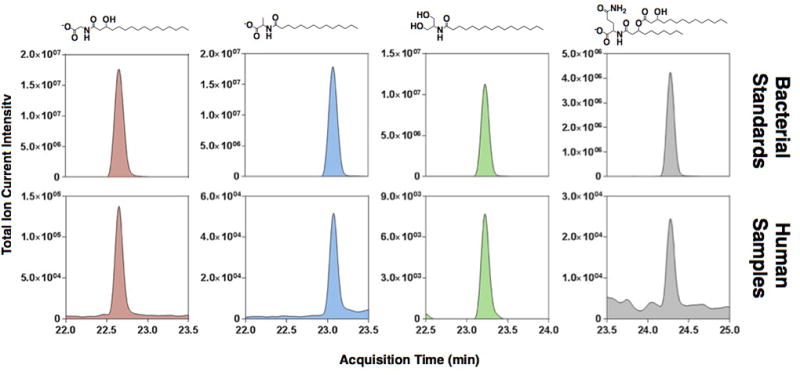
High-resolution reversed-phase LC-MS analysis of human fecal extract pooled from 128 samples representing 21 individuals. Extracted ion chromatograms for individual N-acyl amides are shown within a 2 ppm tolerance of the exact mass (M+H). Compounds observed to be present in the human fecal extract were confirmed by alignment to authentic standards (top panel), and by spiked addition of the pure compound (data not shown). No zwitterionic N-acyl amides (N-acyl or N-acyloxyacyl ornithine/lysines) were detected.
Supplementary Material
Acknowledgments
The authors would like to acknowledge the High-Throughput and Spectroscopy Resource Center, Center for Clinical and Translational Science, and the Comparative Bioscience Center at Rockefeller University for the use of their facilities; the Mangelsdorf Laboratory at UT Southwestern and Dr. Daniel Drucker at Mt Sinai Hospital, Toronto for the use of the GLUTag cell line; Alex Milshteyn, Andreia Estrela and Jeffrey Craig for their critical review of the manuscript. This work was supported in part by a grant from the Robertson Foundation, the Center for Basic and Translational Research on Disorders of the Digestive System Through the generosity of the Leona M. and Harry B. Helmsley Charitable Trust, Rainin Foundation, U01 GM110714-1A1 (S.F.B.), GM122559-01 (S.F.B.), the Crohn’s and Colitis Foundation Career Development Award (L.J.C.) and NIDDK K08 DK109287-01 (L.J.C.).
Footnotes
Author Contributions:
L.J.C. and S.F.B. designed research; L.J.C. assisted with all experiments; S-H.K. assisted with molecule characterization; E.A.G., P.Y.C., J.K.B., and R.R.A. assisted with gene cloning; D.E., A.B.E., S.M.H., C.H. and A.R. assisted with mouse experiments; J.C., X.V-F., J.K. assisted with molecule synthesis; A.J.P. and J.R.C. assisted with metabolite analysis in human/mouse samples; L.J.C. and C.L. analyzed data; L.J.C. and S.F.B. wrote the paper
Extended data figures and tables are provided to accompany the main text and methods. Supplementary information contains the methods, figures and tables related to the structural determination of compounds.
Competing Financial Interest Statement
The authors of this study have no competing financial interests to declare.
References
- 1.Koppel N, Balskus EP. Exploring and Understanding the Biochemical Diversity of the Human Microbiota. Cell Chem Biol. 2016;23:18–30. doi: 10.1016/j.chembiol.2015.12.008. [DOI] [PubMed] [Google Scholar]
- 2.Meinwald J, Eisner T. Chemical ecology in retrospect and prospect. Proc Natl Acad Sci U S A. 2008;105:4539–4540. doi: 10.1073/pnas.0800649105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cohen LJ, et al. Functional metagenomic discovery of bacterial effectors in the human microbiome and isolation of commendamide, a GPCR G2A/132 agonist. Proceedings of the National Academy of Sciences of the United States of America. 2015 doi: 10.1073/pnas.1508737112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cani PD, et al. Endocannabinoids - at the crossroads between the gut microbiota and host metabolism. Nature reviews. Endocrinology. 2015 doi: 10.1038/nrendo.2015.211. [DOI] [PubMed] [Google Scholar]
- 5.Pacher P, Kunos G. Modulating the endocannabinoid system in human health and disease--successes and failures. FEBS J. 2013;280:1918–1943. doi: 10.1111/febs.12260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moore EK, et al. Lysine and novel hydroxylysine lipids in soil bacteria: amino acid membrane lipid response to temperature and pH in Pseudopedobacter saltans. Front Microbiol. 2015;6:637. doi: 10.3389/fmicb.2015.00637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Geiger O, Gonzalez-Silva N, Lopez-Lara IM, Sohlenkamp C. Amino acid-containing membrane lipids in bacteria. Prog Lipid Res. 2010;49:46–60. doi: 10.1016/j.plipres.2009.08.002. [DOI] [PubMed] [Google Scholar]
- 8.Zhang X, Ferguson-Miller SM, Reid GE. Characterization of ornithine and glutamine lipids extracted from cell membranes of Rhodobacter sphaeroides. J Am Soc Mass Spectrom. 2009;20:198–212. doi: 10.1016/j.jasms.2008.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Flock G, Holland D, Seino Y, Drucker DJ. GPR119 regulates murine glucose homeostasis through incretin receptor-dependent and independent mechanisms. Endocrinology. 2011;152:374–383. doi: 10.1210/en.2010-1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schulze T, et al. Sphingosine-1-phospate receptor 4 (S1P(4)) deficiency profoundly affects dendritic cell function and TH17-cell differentiation in a murine model. FASEB J. 2011;25:4024–4036. doi: 10.1096/fj.10-179028. [DOI] [PubMed] [Google Scholar]
- 11.Le LQ, et al. Mice lacking the orphan G protein-coupled receptor G2A develop a late-onset autoimmune syndrome. Immunity. 2001;14:561–571. doi: 10.1016/s1074-7613(01)00145-5. [DOI] [PubMed] [Google Scholar]
- 12.Konya V, Marsche G, Schuligoi R, Heinemann A. E-type prostanoid receptor 4 (EP4) in disease and therapy. Pharmacol Ther. 2013;138:485–502. doi: 10.1016/j.pharmthera.2013.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kabashima K, et al. The prostaglandin receptor EP4 suppresses colitis, mucosal damage and CD4 cell activation in the gut. The Journal of clinical investigation. 2002;109:883–893. doi: 10.1172/JCI14459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Manieri NA, et al. Mucosally transplanted mesenchymal stem cells stimulate intestinal healing by promoting angiogenesis. The Journal of clinical investigation. 2015;125:3606–3618. doi: 10.1172/JCI81423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hansen KB, et al. 2-Oleoyl glycerol is a GPR119 agonist and signals GLP-1 release in humans. J Clin Endocrinol Metab. 2011;96:E1409–1417. doi: 10.1210/jc.2011-0647. [DOI] [PubMed] [Google Scholar]
- 16.Overton HA, et al. Deorphanization of a G protein-coupled receptor for oleoylethanolamide and its use in the discovery of small-molecule hypophagic agents. Cell metabolism. 2006;3:167–175. doi: 10.1016/j.cmet.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 17.Khan SY, et al. Lysophosphatidylcholines activate G2A inducing G(αi)−1-/G(αq/)11- Ca2(+) flux, G(βγ)-Hck activation and clathrin/β-arrestin-1/GRK6 recruitment in PMNs. The Biochemical journal. 2010;432:35–45. doi: 10.1042/BJ20091087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kabarowski JH. G2A and LPC: regulatory functions in immunity. Prostaglandins & other lipid mediators. 2009;89:73–81. doi: 10.1016/j.prostaglandins.2009.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rimmerman N, et al. N-palmitoyl glycine, a novel endogenous lipid that acts as a modulator of calcium influx and nitric oxide production in sensory neurons. Molecular pharmacology. 2008;74:213–224. doi: 10.1124/mol.108.045997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ritter K, Buning C, Halland N, Poverlein C, Schwink L. G Protein-Coupled Receptor 119 (GPR119) Agonists for the Treatment of Diabetes: Recent Progress and Prevailing Challenges. J Med Chem. 2016;59:3579–3592. doi: 10.1021/acs.jmedchem.5b01198. [DOI] [PubMed] [Google Scholar]
- 21.Nunez DJ, et al. Gut hormone pharmacology of a novel GPR119 agonist (GSK1292263), metformin, and sitagliptin in type 2 diabetes mellitus: results from two randomized studies. PloS one. 2014;9:e92494. doi: 10.1371/journal.pone.0092494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ha TY, et al. Novel GPR119 agonist HD0471042 attenuated type 2 diabetes mellitus. Arch Pharm Res. 2014;37:671–678. doi: 10.1007/s12272-013-0209-0. [DOI] [PubMed] [Google Scholar]
- 23.Katz LB, et al. Effects of JNJ-38431055, a novel GPR119 receptor agonist, in randomized, double-blind, placebo-controlled studies in subjects with type 2 diabetes. Diabetes Obes Metab. 2012;14:709–716. doi: 10.1111/j.1463-1326.2012.01587.x. [DOI] [PubMed] [Google Scholar]
- 24.Chu ZL, et al. A role for beta-cell-expressed G protein-coupled receptor 119 in glycemic control by enhancing glucose-dependent insulin release. Endocrinology. 2007;148:2601–2609. doi: 10.1210/en.2006-1608. [DOI] [PubMed] [Google Scholar]
- 25.Chu ZL, et al. A role for intestinal endocrine cell-expressed g protein-coupled receptor 119 in glycemic control by enhancing glucagon-like Peptide-1 and glucose-dependent insulinotropic Peptide release. Endocrinology. 2008;149:2038–2047. doi: 10.1210/en.2007-0966. [DOI] [PubMed] [Google Scholar]
- 26.Lauffer LM, Iakoubov R, Brubaker PL. GPR119 is essential for oleoylethanolamide-induced glucagon-like peptide-1 secretion from the intestinal enteroendocrine L-cell. Diabetes. 2009;58:1058–1066. doi: 10.2337/db08-1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Serrano A, et al. Oleoylethanolamide: effects on hypothalamic transmitters and gut peptides regulating food intake. Neuropharmacology. 2011;60:593–601. doi: 10.1016/j.neuropharm.2010.12.007. [DOI] [PubMed] [Google Scholar]
- 28.Fu J, et al. Oleylethanolamide regulates feeding and body weight through activation of the nuclear receptor PPAR-a. Nature. 2003;425:90–93. doi: 10.1038/nature01921. [DOI] [PubMed] [Google Scholar]
- 29.Lan H, et al. GPR119 is required for physiological regulation of glucagon-like peptide-1 secretion but not for metabolic homeostasis. J Endocrinol. 2009;201:219–230. doi: 10.1677/JOE-08-0453. [DOI] [PubMed] [Google Scholar]
- 30.Lauffer L, Iakoubov R, Brubaker PL. GPR119: "double-dipping" for better glycemic control. Endocrinology. 2008;149:2035–2037. doi: 10.1210/en.2008-0182. [DOI] [PubMed] [Google Scholar]
- 31.Chen Z, et al. Incorporation of therapeutically modified bacteria into gut microbiota inhibits obesity. The Journal of clinical investigation. 2014;124:3391–3406. doi: 10.1172/JCI72517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mimee M, Tucker, Alex C, Voigt, Christopher A, Lu, Timothy K. Programming a Human Commensal Bacterium, Bacteroides thetaiotaomicron, to Sense and Respond to Stimuli in the Murine Gut Microbiota. Cell Systems. 2015 doi: 10.1016/j.cels.2015.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vaudry H. Molecular evolution of GPCRs: What we know and what the future holds. J Mol Endocrinol. 2014;52:E1–2. doi: 10.1530/JME-14-0103. [DOI] [PubMed] [Google Scholar]
- 34.Lovejoy DA, Chang BS, Lovejoy NR, del Castillo J. Molecular evolution of GPCRs: CRH/CRH receptors. J Mol Endocrinol. 2014;52:T43–60. doi: 10.1530/JME-13-0238. [DOI] [PubMed] [Google Scholar]
- 35.Hla T. Genomic insights into mediator lipidomics. Prostaglandins Other Lipid Mediat. 2005;77:197–209. doi: 10.1016/j.prostaglandins.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 36.Wieland Brown LC, et al. Production of alpha-Galactosylceramide by a Prominent Member of the Human Gut Microbiota. PLoS biology. 2013;11:e1001610. doi: 10.1371/journal.pbio.1001610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ou G, et al. Proximal small intestinal microbiota and identification of rod-shaped bacteria associated with childhood celiac disease. The American journal of gastroenterology. 2009;104:3058–3067. doi: 10.1038/ajg.2009.524. [DOI] [PubMed] [Google Scholar]
- 38.Structure, function and diversity of the healthy human microbiome. Nature. 2012;486:207–214. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Franzosa EA, et al. Relating the metatranscriptome and metagenome of the human gut. Proceedings of the National Academy of Sciences of the United States of America. 2014 doi: 10.1073/pnas.1319284111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Peterson SN, et al. Functional expression of dental plaque microbiota. Front Cell Infect Microbiol. 2014;4:108. doi: 10.3389/fcimb.2014.00108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tang Y, Li X, Han X, Lu J, Diwu Z. Functional analysis of endogenous beta-adrenergic receptor through fluorimetric monitoring of cyclic nucleotide-gated ion channel. Analytical biochemistry. 2007;360:303–305. doi: 10.1016/j.ab.2006.04.051. [DOI] [PubMed] [Google Scholar]
- 42.Van Wagoner RM, Clardy J. FeeM, an N-acyl amino acid synthase from an uncultured soil microbe: structure, mechanism, and acyl carrier protein binding. Structure. 2006;14:1425–1435. doi: 10.1016/j.str.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 43.Chen Y, et al. Dysbiosis of small intestinal microbiota in liver cirrhosis and its association with etiology. Scientific reports. 2016;6:34055. doi: 10.1038/srep34055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cheng J, et al. Duodenal microbiota composition and mucosal homeostasis in pediatric celiac disease. BMC Gastroenterol. 2013;13:113. doi: 10.1186/1471-230X-13-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All figure data is available in the source data sheet. Gene accession numbers for all cloned genes are provided in Supplementary Table 1. Publically available DNA and RNA datasets analyzed in this study are referenced accordingly and references contain links to datasets available for download.