

Abstract
Chloramphenicol (Cam) is a broad-spectrum antibiotic used to combat bacterial infections in humans and animals. Cam export from bacterial cells is one of the mechanisms by which pathogens resist Cam's antibacterial effects, and several different proteins are known to facilitate this process. However, to date no report exists on any specific transport protein that facilitates Cam uptake. The proton-coupled oligopeptide transporter (POT) YdgR from Escherichia coli is a prototypical member of the POT family, functioning in proton-coupled uptake of di- and tripeptides. By following bacterial growth and conducting LC-MS–based assays we show here that YdgR facilitates Cam uptake. Some YdgR variants displaying reduced peptide uptake also exhibited reduced Cam uptake, indicating that peptides and Cam bind YdgR at similar regions. Homology modeling of YdgR, Cam docking, and mutational studies suggested a binding mode that resembles that of Cam binding to the multidrug resistance transporter MdfA. To our knowledge, this is the first report of Cam uptake into bacterial cells mediated by a specific transporter protein. Our findings suggest a specific bacterial transporter for drug uptake that might be targeted to promote greater antibiotic influx to increase cytoplasmic antibiotic concentration for enhanced cytotoxicity.
Keywords: antibiotics, membrane transport, molecular docking, site-directed mutagenesis, transporter
Introduction
Amphenicols are phenolpropanoid-based antibiotics that interact with the P-site of bacterial ribosomes to prevent protein synthesis, thereby leading to bacteriostasis (1). The amphenicols include chloramphenicol (Cam),2 thiamphenicol, florfenicol, and azidamfenicol. Amphenicols are broad-spectrum antibiotics, which can kill a number of Gram-positive and Gram-negative bacteria (2). Cam was the first amphenicol to be identified during studies of the bacterium Streptomyces venezuelae (3), and is still being used for treatment of infections in both humans and animals, albeit as a last resort due to possible serious side effects, such as aplastic anemia and genotoxicity (4).
Protein-mediated cross-membrane transport of Cam in bacteria has been described upon two instances: first, when Cam enters Gram-negative bacteria through the outer membrane channels; for example, by OmpC and OmpF from Escherichia coli (5), Omp P1 and P2 from Haemophilus influenza (6, 7), and OmpC, OmpF, and OmpD from Salmonella typhi (8) and then upon efflux from the cytoplasm through multidrug resistance transporters, such as MdfA from E. coli (9), CmlA from Psuedomonas aeruginosa (10) both from the major facilitator superfamily (11), and MexEF and OprN (P. aeruginosa) (12) and AcrB (E. coli) from the resistance nodulation division (RND) family (13). Entry of Cam into the cytoplasm is believed to occur not only through passive diffusion due to its relatively lipophilic character but also through secondary active transporters (14, 15), and to our knowledge, no specific transporters of the cytosolic membrane have been shown to transport Cam into the cells.
During over-expression studies of the proton-coupled oligopeptide transporter (POT) YdgR from E. coli in BL21(DE3)pLysS (pLysS carries a gene encoding T7 lysozyme and Cam acetyltransferase (CAT), respectively) cells in the presence of Cam, we noticed bacteriostasis upon addition of isopropyl β-d-thiogalactoside (IPTG) to YdgR expression cultures, despite the presence of CAT in the host cells. Similar observations were also reported previously by others (16). This led us to hypothesize that the YdgR transporter is facilitating the entry of Cam the cells. Here we follow up on our hypothesis and show directly and indirectly that YdgR is able to facilitate uptake of Cam. To our knowledge, this is one of the first reports of Cam uptake, mediated by a specific transporter.
Results
During over-expression of YdgR with Cam in the growth medium using Cam-resistant BL21(DE3)pLysS cells, we observed a tendency of bacteriostasis upon induction with IPTG. As this could suggest high levels of intracellular Cam, we next tested whether Cam would inhibit YdgR-mediated uptake of a peptide substrate. For this we utilized the well known YdgR substrate β-Ala-Lys(AMCA) (17) and YdgR expressed in BL21(DE3)pLysS cells. In the presence of Cam (0.5 mm) we did not observe any inhibition of β-Ala-Lys(AMCA) uptake (Fig. 1B), which indicates that Cam is a poor competitor of peptide uptake. This prompted us to perform a series of investigations based on growth curves, minimum inhibitory concentration (MIC), and LC-MS–based assays to determine whether Cam is a substrate of YdgR.
Figure 1.
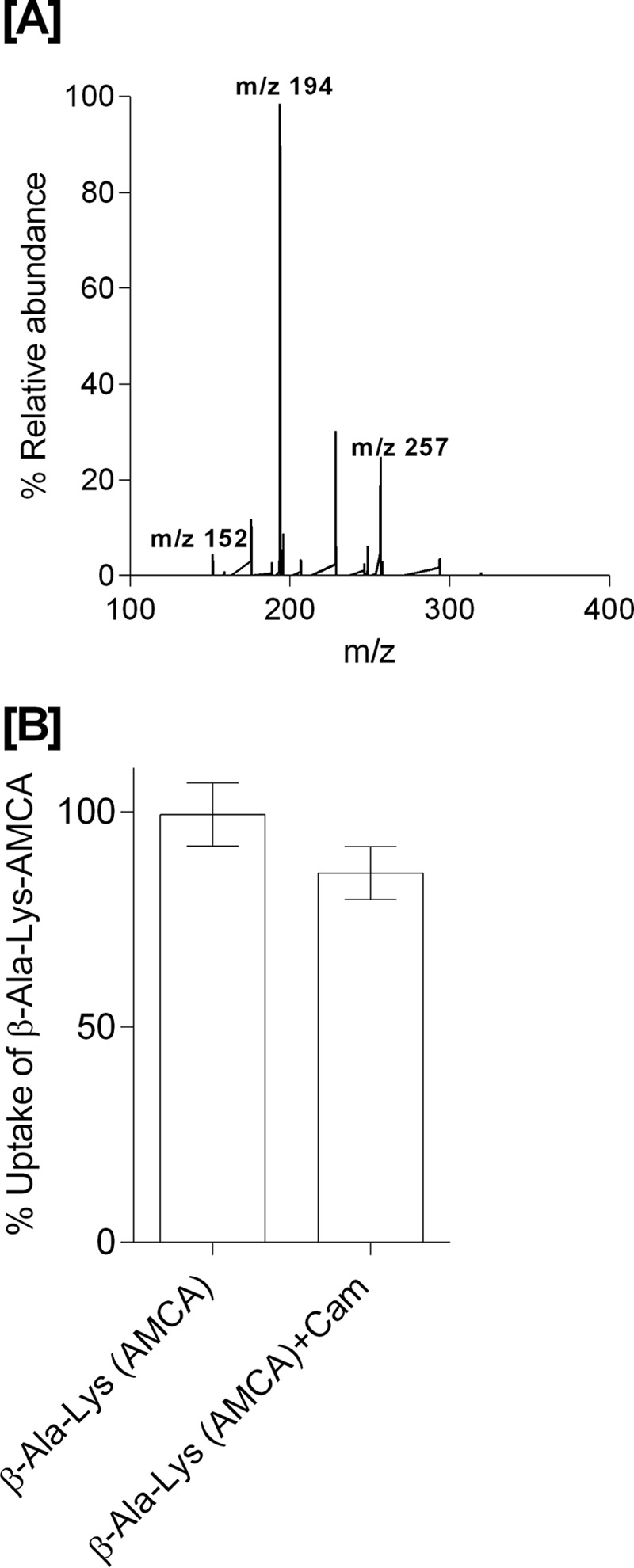
A, LC-MS electrospray ion-trap spectrum of chloramphenicol standard (15 μg/ml) dissolved in cell lysates. The parent ion at m/z 321 is observed but of low intensity. The most prominent product ion at m/z 194 was monitored in the uptake assays. B, β-Ala-Lys(AMCA) (0.5 mm) uptake and inhibition in the presence of Cam (0.5 mm) as a competitor (p value < 0.05, n = 3).
Bacterial growth in the presence and absence of Cam
Growth curves of BL21(DE3)pLysS cells transformed with pTTQ18–ydgR, pTTQ18 plasmid, and the pTTQ18—ydgR–E33Q plasmid, respectively, were followed in the presence of Cam (34 μg/ml) and ampicillin (100 μg/ml). YdgR–E33Q is a variant that has a very low transport activity even when being over-expressed to WT–YdgR levels in BL21(DE3)pLysS cells (18). In YdgR-expressing cells, the growth became stagnant after addition of IPTG (Fig. 2A). Cells transformed with the pTTQ18 plasmid grew continuously irrespective of induction (Fig. 2B) and a similar trend was observed for YdgR–E33Q (Fig. 2C). These results suggest that growth inhibition is related to YdgR being expressed and active. Next, we tested whether a halt in growth would also be observed in BL21(DE3) cells harboring the pTTQ18–ydgR plasmid, i.e. the active transporter, grown in media containing 100 μg/ml of ampicillin only for selection and induced at the appropriate time; these cells grew uninhibited (Fig. 2D). Expression of WT–YdgR in BL21(DE3)pLysS cells has been established previously (18) and Western blot analysis confirmed that WT–YdgR was also expressed in BL21(DE3) cells (Fig. 2D).
Figure 2.

Growth curves of YdgR, pTTQ18 vector, and YdgR–E33Q mutant in BL21(DE3)pLysS cells (in the presence of 100 μg/ml of ampicillin and 34 μg/ml of chloramphenicol). Arrow marks indicates time of induction with IPTG. Closed symbols for the cells induced with IPTG and open symbols for cells without IPTG. A, YdgR; B, pTTQ18 vector; and C, YdgR–E33Q (the insert of Western blot bands are spliced from the same gel). Open squares indicate the growth of BL21(DE3) cells in the presence of 100 μg/ml of ampicillin and absence of 34 μg/ml of chloramphenicol. D, YdgR; n = 3.
Taken together these results suggest that in the absence of Cam neither YdgR over-expression, nor activity have any effect on growth. Both the presence of Cam in the medium and the expression of functional YdgR are needed for growth inhibition of these cells.
To investigate the effect of the extracellular Cam concentration on the growth of YdgR expressing Bl21(DE3)pLysS cells, we followed their growth on agar plates in the presence or absence of IPTG. The Cam concentrations were 32, 48, 64, 96, and 128 μg/ml (Fig. 3). In the absence of IPTG no growth inhibition was observed at any of the given Cam concentrations, however, in the presence of IPTG, growth was apparently reduced already at a Cam concentration of 48 μg/ml (Fig. 3A) and completely inhibited at 64 μg/ml (Fig. 3B) for cells harboring pTTQ18–ydgR but not for pTTQ18 cells. These results support that over-expression of ydgR in combination with relatively high extracellular levels of Cam results in the BL21 (DE3)pLysS growth inhibition. Experiments using Bl21(DE3) cells, i.e. absence of pLysS, were also performed, however, we failed to reach reproducibility in mic values. We believe that this is due to a very narrow Cam concentration difference between colony growth and inhibition of cells harboring pTTQ18.
Figure 3.

MIC determination of YdgR and pTTQ18 (pTTQ18 vector) expressing BL21(DE3)pLysS cells and BL21DE3. A, plates supplemented with 0.1 mm IPTG. B, plates without IPTG induction. n = 3.
Effects of YdgR expression on intracellular Cam levels
To investigate Cam concentrations in YdgR over-expressing cells we used a previously established LC-MS–based transport assay (19). For these assays, we used Bl21(DE3) cells to avoid enzymatic conversion of Cam by CAT. The Cam product ion of m/z 194 (Fig. 1A) was found to be the major Cam product ion in lysates and this ion was used to detect Cam in cell lysates of cells carrying pTTQ18–ydgR, YdgR variants, and pTTQ18 plasmid. Cells harboring the pTTQ18–ydgR plasmid showed a 5-fold higher peak height for the product ion m/z 194 compared with cells harboring the pTTQ18 vector (Fig. 4A). The peak for the pTTQ18—ydgR-transformed cells was quenched 5-fold upon co-incubation with the dipeptide Ala–Ala in 10-fold excess of the Cam concentration (Fig. 4A). So there clearly are increased Cam levels in cells over-expressing YdgR, and this effect is quenchable by Ala–Ala supporting that Cam is likely transported by YdgR. Cam is also very weakly detectable in cells harboring the pTTQ18 vector. This could be due to the transport by endogenous YdgR or by other endogenous E. coli peptide transporters (17, 20, 22, 23).
Figure 4.

A, mass chromatograms of chloramphenicol detected in the BL21(DE3) cell lysates of YdgR (thin line), pTTQ18 vector (small dots), YdgR in competition with Ala–Ala (broken lines). B, time-dependent uptake of chloramphenicol. The optimal uptake of YdgR is at pH 6.5 (100%) and C and E are shown relative to D. C, uptake of chloramphenicol at pH 5.5. D, uptake of chloramphenicol at pH 6.5. E, uptake of chloramphenicol at pH 7.5, n = 3. F, ethidium bromide efflux in the presence (closed circles) and absence (pTTQ18, circles) of YdgR. G, mass chromatogram of chloramphenicol in an E. coli parent strain (thin line) and YdgR deletion strain (broken lines). n = 3.
To characterize whether the proton ionophore CCCP (carbonyl cyanide m-chlorophenylhydrazone), a compound that dissipates the proton electrochemical gradient, had any effects on the intracellular Cam levels, we performed the assays in the presence of CCCP (20) and observed that Cam levels were similar to those harboring pTTQ18-transfected cells alone (Fig. 4A).
To investigate whether the increased levels of Cam are due to YdgR-mediated uptake or due to a more indirect effect on the cells' Cam efflux mechanisms, we performed an ethidium bromide efflux assay in the presence and absence of over-expressed YdgR. Transporters that export ethidium bromide are known to also export Cam (14). The efflux assay showed that there was a 10% increase in the efflux of ethidium bromide in cells over-expressing YdgR (Fig. 4F). Thus, Cam efflux has not been compromised by YdgR over-expression, if anything, it appears to be slightly higher.
Taken together, these findings suggest that Cam is an YdgR substrate. The led us to characterize the substrate's properties. We tested the uptake of Cam as a function of time and observed fast kinetics (Fig. 4B). Based on the cell volume (24) and Cam uptake, the internal concentration in cells bearing the pTTQ18 vector was calculated to be 800 μg/ml. Uptake of Cam was also found to be dependent upon external pH (Fig. 4, C–E); apparently pH optimum was found to be at 6.5 (Fig. 4D) and at a minimum at a pH of 5.5 (Fig. 4C). To further validate our hypothesis that Cam uptake is facilitated by YdgR, we performed uptake assays on E. coli K12 cells and its derivative ydgR deletion strain (Keio collection, GE Life Sciences). The uptake of Cam in YdgR deletion cells was reduced by 40% compared with the parent strain (Fig. 4G). This strengthens our hypothesis that YdgR facilitates Cam uptake.
Cam uptake in YdgR variants compromised in peptide uptake
Having established that YdgR facilitates Cam transport, we next investigated whether transport of Cam would be effected in YdgR variants with altered the ability to transport β-Ala-Lys(AMCA). It is known that E33Q and K130Q are unable to transport β-Ala-Lys(AMCA), Y38F and Y71F show an intermediate level of transport, and Y292F is hyperactive compared with WT–YdgR (18). All of these residues are either in close proximity or directly interacting with the peptide as observed from a POT:peptide crystal structure (25). Using Western blots, we verified that all variants were expressed and their uptake was subsequently normalized compared with WT–YdgR (Fig. 5A). As observed for β-Ala-Lys(AMCA), -E33Q was unable to transport Cam (Fig. 5B), whereas Y292F did not show any hyperactivity as it transported to 60% activity (Fig. 5C). Y38F and Y71F exhibited transport activity that was <35% (Fig. 5, D and E) thus comparable with β-Ala-Lys(AMCA) uptake. Uptake by K130Q was 22% (Fig. 5F) in contrary with the transport β-Ala-Lys(AMCA) (18). Overall, these results indicate that the YdgR Cam-binding site is similar to that of β-Ala-Lys(AMCA) but not strictly identical.
Figure 5.

A, Western blots showing expression of YdgR–mutants in BL21(DE3) cells. B, mass chromatograms showing expression normalized uptake of chloramphenicol in the BL21(DE3) cell in expressing YdgR (thin line), pTTQ18 vector (small dots), YdgR–E33Q (broken lines). C, YdgR (thin line), pTTQ18 vector (small dots), YdgR–Y292F (broken lines). D, YdgR (thin line), pTTQ18 vector (small dots), YdgR–Y38F (broken lines). E, YdgR (thin line), pTTQ18 vector (small dots), YdgR–Y71F (broken lines). F, YdgR (thin line), pTTQ18 vector (small dots), YdgR–K130Q (broken lines). n = 3.
Investigating the YdgR Cam-binding site
To rationalize our functional results and gain more insight on the YdgR Cam-binding site, we turned to structural analyses using computational approaches. Docking of Cam into a homology model of YdgR came up with various conformations (Fig. 6A) of which a single, the highest scoring conformation, was selected and used for further analyses. Interestingly, in this conformation (Fig. 6B) Cam is bound such that a hydrophobic region consisting of in particular Phe-288 but also Phe-289, interacts with the nitro-benzyl moiety, and a polar region consisting primarily of Asn-325 and Glu-396; the latter in particular is in proximity of the Cam hydroxyl groups. Furthermore, a hydrogen bond between Tyr-38 and the Cam peptide backbone is observed. Based on these predicted interactions we prepared alanine variants of residues Tyr-38, Phe-288, Phe-289, and Glu-396 and, additionally, Glu-396 was mutated to glutamine. First, expression of all variants was verified by Western blotting (Fig. 7A) and characterized for their activity by measuring the uptake of β-Ala-Lys(AMCA) in BL21(DE3)pLysS cells. F288A was found to be hyperactive with an uptake of about 300% relative to YdgR. All other mutants had an uptake between 5 and 10% (Fig. 7B). The pronounced effects observed suggest that the predicted Cam-binding site is at least overlapping with the YdgR peptide-binding site. Second, we tested Cam uptake by BL21(DE3) cells expressing all Phe-288, Phe-289, and Glu-396 variants (Fig. 7, C–E). Although F289A and E396Q showed reduced uptake, most interestingly, F288A (Fig. 7E), which was hyperactive with regards to peptide uptake, was also reduced to approximately 10% activity. Thus, the effects of these mutations are in support of the calculated Cam-binding site.
Figure 6.

A, various conformations of Cam (gray color; one in yellow is the most preferred confirmation) out by docking it into the binding pocket YdgR. B, green, blue, and magenta colors indicate protonation, hydrophilic, and hydrophobic regions in the active site. Interactions between Cam (yellow color) and YdgR (white color) from the docking pose.
Figure 7.

Expression and functional characterization of mutants prepared based on predicted docking of Cam at the active site. A, Western blots of mutants expressed in BL21(DE3) and BL21(DE3)pLysS cells. B, β-Ala-Lys(AMCA) (0.5 mm) uptake profiles of ydgR/ydgR–mutants. Mass chromatograms showing expression normalized uptake of chloramphenicol in the BL21(DE3) cell lysates. Broken lines represent mutants and thin solid line represent wild type YdgR. C, YdgR (thin line), YdgR–E396Q (broken lines). D, YdgR (thin line), YdgR–F288A (broken lines). E, YdgR (thin line), YdgR–F289A (broken lines). n = 3.
Discussion
Substrate promiscuity has been observed for a number of transporters and in a number of different transporter families. P-glycoprotein is known to transport colchicine, quinine, vinblastine, and digoxin (26). MdfA is an efflux transporter and is capable of transporting chloramphenicol, ciprofloxacin, kanamycin, and tetracycline (27). Likewise, the human POT, hPepT1, is a highly promiscuous transporter, as it is able to transport virtually all di- and tripeptide and a number of drugs such as β-lactams, ACE inhibitors, sulpiride, and valacyclovir (28). YdgR is a well characterized prototypical POT from E. coli with several similarities compared with hPepT1.
Our growth experiments strongly support that YdgR facilitates the uptake of Cam into E. coli cells. Ethidium bromide efflux assay shows that there is minimal effect (10% increase in efflux) of YdgR over-expression on the efflux systems of cells and the LC-MS/MS results on YdgR together with YdgR variants show elevated levels of Cam inside the cells upon over-expression of YdgR. Finally, a YdgR deletion strain accumulates significantly less than its parents strain, further substantiating YdgR-mediated Cam transport. YdgR variants based on docking also exhibited compromised uptake of Cam and thereby validate and support our proposed Cam-binding site. Furthermore, Cam uptake has similarities to peptide uptake by YdgR as it is found to be dependent on extracellular pH and on the proton electrochemical gradient. Also Cam being a poor competitor of β-Ala-Lys(AMCA) was still translocated by YdgR, and a similar observation was also reported earlier where ampicillin was reported to be a poor competitor of β-Ala-Lys (AMCA) although it was a substrate of YdgR in the absence of β-Ala-Lys(AMCA) (19). Previous studies on uptake of Cam in E. coli reported an inside Cam concentration of 690 μg/ml, which was almost a 136-fold accumulation compared with the outside concentration (15), and the author suggested energized uptake of Cam. The intracellular concentration attained in our studies was 800 μg/ml in the cells carrying pTTQ18 vector, and thus similar to the previously reported value; in the presence of over-expressed YdgR, however, uptake was increased by 5 times. It is also intriguing that apparently Cam uptake facilitated by YdgR over-expressed in BL21(DE3)pLysS leads to bacteriostatis despite a functional CAT harbored by the pLysS plasmid. We speculate that this may be due to saturation of CAT by the YdgR activity.
Recently, structural information on a transporter–Cam complex has become available; the crystal structure of MdfA in complex with Cam (29) shows that Cam is bound in a region between two 6-helix domains of MdfA, a common binding site of major facilitator superfamily transporters including the POTs (Fig. 8A) (29). In this structure only three polar interactions are observed between Cam and MdfA (Fig. 8A), whereas a number of non-polar interactions are observed. As such, the MdfA Cam-binding site may be divided into a hydrophilic part and a hydrophobic part (Fig. 8A). Accumulated structural and functional analyses on POTs (30) have established the importance of both peptide termini and a peptide bond for binding to the POT active site through highly conserved residues (Fig. 8B). The N terminus of peptide binds to a glutamate residue (Glu-396 in YdgR), whereas the C terminus interacts with a lysine (Lys-130 in YdgR); the peptide bond hydrogen bonds with a tyrosine side chain (Tyr-38 in YdgR) (18, 31). The side chains are accommodated in the rather large cavity of the POTs. Cam has neither of the peptide termini, however, it does have a peptide bond, and overall it resembles a dipeptide in size and shape. According to our docking and mutational studies Cam is able to utilize the YdgR-binding site in a similar manner as observed for MdfA, and is able to do so by interacting with residues that are highly conserved residues Tyr-38, and Glu-396 and lesser conserved phenylalanine residues 288 and 289 (Fig. 6B). Furthermore, an obvious exception is that no apparent interactions are observed with Lys-130. Interestingly, previously the K130Q–YdgR variant showed no measurable peptide uptake (18), however, here we observe a significant Cam uptake. This indicates a less important role in Cam uptake, which could be explained by the lack of C terminus in Cam. Lys-130 is part of an additional hydrophilic cluster in YdgR, which also included the so-called EXXER motif that along with Lys-130 has been suggested to couple proton and peptide translocation (32) (Fig. 6B).
Figure 8.

The red, blue, and green color patches represent the hydrophobic, hydrophilic, and protonation sites, respectively. A, interactions between Cam (yellow color) and MdfA (wheat color) in a crystal structure (PDB code 4ZOW). B, interactions between the Ala–Phe peptide (yellow color) and PepTSt (gray color) in the crystal structure (PDB code 5D59).
In summary, we have shown that the POT YdgR facilitates Cam uptake in E. coli cells. An interesting additional observation in the BL12(DE3)pLysS cell experiments was that the Cam transport activity of YdgR could be related to the growth of these cells, i.e. the inactive YdgR variant E33Q grew uninhibited after addition of IPTG compared with WT–YdgR. It can be assumed that the basis of growth inhibition is due to saturation of the inherent resistance mechanism Cam-acetyl transferase, encoded by the pLysS plasmid. A number of studies have described the inverse of what we observe, i.e. a transporter facilitating drug resistance by drug export. Furthermore, the inhibition of such efflux transporters has been suggested as an approach to counteract pathogenic resistant bacteria (33–37). Our study suggests that perhaps uptake transporters present in bacteria could be targeted with new derivatives of already known antibiotics, designed to have optimal influx. This could increase the cytoplasmic concentration of the antibiotic and hereby enhance cytotoxicity.
Materials and methods
The expression construct for WT–ydgR used in this study have been described previously (18, 23). YdgR mutants E396Q, E396A, F289A, and F288A were generated using the QuikChange Lightning Multi Site-directed Mutagenesis Kit (Agilent Technologies). Primers were designed using the web-based QuikChange Primer Design Program (Agilent Technologies) and the following primers were used: 5′-gaccagagatcatcagctgcccgatgctctgcagg-3′ (E396Q), 5′-ccagagatcatcagtgccccgatgctctgca-3′ (E396A), 5′-gctcgaagccattatcttcgccgtgctgtacagccagatg-3′ (F289A) and 5′-gatgctcgaagccattatcgccttcgtgctgtacagcc-3′ (F288A).
Expression
Over-expression of YdgR was performed as described previously (38). Briefly, a single colony of E. coli BL21(DE3)pLysS cells containing the plasmids pTTQ18–ydgR/ydgR–mutant or pTTQ18 (pTTQ18 vector) were inoculated in 3 ml of LB media containing 100 μg/ml of ampicillin and 34 μg/ml of chloramphenicol and allowed to grow overnight. Overnight cultures were transferred to 10 ml of LB media with the same amount of antibiotics using a dilution of 1:50. The cells were allowed to grow until A600 of 0.6–0.8 before induction with 1 mm IPTG. The cells were harvested 3 h after induction with IPTG. Over-expression of ydgR/ydgR–mutants in BL21(DE3) cells was performed in a media containing 100 μg/ml of ampicillin and without any chloramphenicol. The rest of the procedures for this cell line were the same as for the BL21(DE3)pLysS cells (38).
Western blot
Western blots were performed in the same way as reported earlier (22, 23). Briefly, 500 μl of cells at A600 = 10 were resuspended in lysis buffer and one protease inhibitor tablet (Roche) was added (22). Samples were incubated on ice for 30 min, sonicated for 30 s, and finally centrifuged at 13,000 × g for 30 min at 4 °C. The solubilized membranes were separated using SDS-PAGE and immediately blotted on to PVDF membrane using X cell module (Invitrogen). Immunodetection was performed using mouse anti-His6 and HRP-conjugated rabbit anti-mouse antibodies (IBA, 1:1000 dilutions) using the Supersignal West Pico chemiluminescent substrate (Pierce). Signals were detected with a MicroChemi imaging system. We have prepared blots by normalizing all the blots with Coomassie-stained membranes.
Uptake assays and efflux assays
The uptake assays were performed on E. coli cells transformed with the pTTQ18–ydgR plasmid and on cells transformed with plasmid only to assess background transport. Harvested cells were resuspended in modified Kreb's buffer (18). An amount of 50 μl of cells (A600 = 10) was added to Eppendorf tubes with 100 μg/ml of chloramphenicol yielding a total assay volume of 100 μl. After 10 min of incubation the assay was terminated by addition of 500 μl of ice-cold modified Kreb's buffer. The cells were then centrifuged at 16,000 × g for 1 min and the pellet was washed three times with 500 μl of ice-cold modified Kreb's buffer. The pellet was resuspended in 100 μl of ice-cold MQ water and vortexed for 30 s (22, 32). The cell suspension was mixed with −22 °C methanol and stored in a freezer overnight. The cells were centrifuged at 14,000 rpm for 15 min and the supernatant was used for further analysis. The method to calculate accumulation has been described previously (32). Efflux assays were done essentially as done previously (42). Briefly, the cells were incubated with ethidium bromide at 37 °C for 10 min and the cells were then centrifuged at 16,000 × g for 1 min and the pellet was washed three times with 500 μl of ice-cold modified Kreb's buffer. The cell suspension was measured for fluorescence by Safire2 (Tecan) at 526 and 605 nm as excitation and emission wavelengths.
Determination of MICs
Cam transport capability of the YdgR transporter in a whole cell-based assay was determined by a MIC assay as described by Wiegand and co-workers (43) with slight modification.
In this assay, E. coli BL21(DE3)pLysS cells were transformed with pTTQ18 and pTTQ18/YdgR plasmids. Cam susceptibility was determined on LB agar containing Cam at concentrations of 32, 48, 64, 96, and 128 μg/ml as well as ampicillin at 50 μg/ml to sustain pTTQ18 and pTTQ18/YdgR plasmids inside the E. coli BL21(DE3)pLysS cells. The protein expression was induced by adding 0.1 mm IPTG to the agar plate. A cell inoculum of 104 cfu/ml was used in the assay and each spot was placed in triplicate. The plates were incubated at 37 °C for 20–24 h, and then growth was evaluated by visual inspection (33, 43).
Liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis
BL21(DE3) cells harboring the pTTQ18–ydgr plasmid or the pTTQ18 vector were cultured in parallel, induced with IPTG, incubated with Cam for 10 min, followed by wash and lysis. The cleared cell lysates were then subjected to LC-MS/MS analysis, whereas monitoring the peak with m/z 194 originating from Cam. Here an amount of 100 μl of the lysed cell supernatant was transferred to Chromacol LC-MS glass vials. 5 μl of sample was injected into the LC Agilent 1100 series connected to a Esquire 3000 plus mass spectrometer (Bruker Daltonics). The analyte was separated on a Agilent poroshell C-18 column (5 μm, 2.1 × 75 mm, temperature 40 °C). The temperature in the autosampler was maintained at 10 °C. A linear gradient of 0–90% acetonitrile in water was used at a flow rate of 0.2 ml/min. The total run time was 10 min. The injected analyte was sprayed into Bruker Daltonics Esquire 3000 after passing through the column. ESI with a capillary voltage of 2750 V under negative ionization was used, a maximum of 50,000 ions filling the trap at a speed of 13,000 m/z per s was used. The gas temperature was set at 240 °C at a flow rate of 10 liters/min, and nebulizer gas pressure at 50 p.s.i. Single ion monitoring was used for chloramphenicol (m/z 321) utilizing the prominent product ion with m/z 194 see (Fig. 1A) (41). The data analysis was done using Bruker Daltonics 5.3 software. All the mass spectra were exported into .cdf files, which were processed in MATLAB and imported to GraphPad Prism 6.0.
Homology modeling and docking
All available bacterial POT sequences (EBI; https://www.ebi.ac.uk)3 were aligned using MAFFT (39) Online version 7 (https://mafft.cbrc.jp/alignment/software/)3 to determine the sequence similarities. A suitable template for YdgR was PeptSo2; with a highest sequence similarity of 36%. One hundred YdgR models were generated using Modeler (40) as implemented in Chimera 1.10.1 (21). The models were validated using Ramachandran plot.
The Schrödinger Software Release 2014-1 was used to prepare the ligand and protein; and to perform docking. The LigPrep procedure was used to generate the 3D molecular model of chloramphenicol in the lowest energy conformation. The homology model of YdgR with the highest Z-score was chosen for the docking preparation using the Protein Preparation Wizard. The assignment of hydrogen bonds (donors and acceptors) was done using default settings. Restrained minimization was performed until the average root mean square deviation of heavy atoms reached a value of 0.3 Å. A Docking grid of about 20 Å was generated using the receptor grid generation panel. This grid included all the residues of the YdgR-binding pocket; as judged by the aligned dipeptide co-crystallized PeptSo2 structure (PDB code 4TPH). Chloramphenicol was docked into the binding pocket of YdgR using the Glide module in standard precision mode (SP). Following this, 5 conformational poses were generated where the optimal conformation was selected based on the docking score, glide energy, and glide emodel values.
Author contributions
P. B. K. and O. M. conceived, designed, and the wrote paper. P. B. K., A. N. G., M. I. G., A. S., P. R. H., N. S., M. I., M. R., A. S., and C. B. performed experiments.
This work was supported by the Faculty of Health and Medical Sciences, University of Copenhagen (to A. N. G. and P. B. K.). The authors declare that they have no conflicts of interest with the contents of this article.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- Cam
- chloramphenicol
- POT
- proton-coupled oligopeptide transporter
- IPTG
- isopropyl β-d-thiogalactoside
- MIC
- minimum inhibitory concentration
- CCCP
- carbonyl cyanide m-chlorophenylhydrazone
- PDB
- Protein Data Bank.
References
- 1. Sutcliffe J. A. (2005) Improving on nature: antibiotics that target the ribosome. Curr. Opin. Microbiol. 8, 534–542 10.1016/j.mib.2005.08.004 [DOI] [PubMed] [Google Scholar]
- 2. Franje C. A., Chang S. K., Shyu C. L., Davis J. L., Lee Y. W., Lee R. J., Chang C. C., and Chou C. C. (2010) Differential heat stability of amphenicols characterized by structural degradation, mass spectrometry and antimicrobial activity. J. Pharm. Biomed. Anal. 53, 869–877 [DOI] [PubMed] [Google Scholar]
- 3. Gruhzit O. M., et al. , (1949) Chloramphenicol (chloromycetin), an antibiotic. pharmacological and pathological studies in animals. J. Clin. Invest. 28, 943–952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hanekamp J. C., and Bast A. (2015) Antibiotics exposure and health risks: chloramphenicol. Environ. Toxicol. Pharmacol. 39, 213–220 10.1016/j.etap.2014.11.016 [DOI] [PubMed] [Google Scholar]
- 5. Mortimer P. G., and Piddock L. J. (1993) The accumulation of five antibacterial agents in porin-deficient mutants of Escherichia coli. J. Antimicrob. Chemother. 32, 195–213 10.1093/jac/32.suppl_B.195, [DOI] [PubMed] [Google Scholar]
- 6. Burns J. L., and Smith A. L. (1987) A major outer-membrane protein functions as a porin in Haemophilus influenzae. J. Gen. Microbiol. 133, 1273–1277 [DOI] [PubMed] [Google Scholar]
- 7. Srikumar R., Dahan D., Arhin F. F., Tawa P., Diederichs K., and Coulton J. W. (1997) Porins of Haemophilus influenzae type b mutated in loop 3 and in loop 4. J. Biol. Chem. 272, 13614–13621 [DOI] [PubMed] [Google Scholar]
- 8. Toro C. S., Lobos S. R., Calderón I., Rodríguez M., and Mora G. C. (1990) Clinical isolate of a porin less Salmonella typhi resistant to high levels of chloramphenicol. Antimicrob. Agents Chemother. 34, 1715–1719 10.1128/AAC.34.9.1715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kumar S., and Varela M. F. (2012) Biochemistry of bacterial multidrug efflux pumps. Int. J. Mol. Sci. 13, 4484–4495 10.3390/ijms13044484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bissonnette L., Champetier S., Buisson J. P., and Roy P. H. (1991) Characterization of the nonenzymatic chloramphenicol resistance (cmlA) gene of the In4 integron of Tn1696: similarity of the product to transmembrane transport proteins. J. Bacteriol. 173, 4493–4502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lewinson O., Adler J., Sigal N., and Bibi E. (2006) Promiscuity in multidrug recognition and transport: the bacterial MFS Mdr transporters. Mol. Microbiol. 61, 277–284 [DOI] [PubMed] [Google Scholar]
- 12. Poole K. (2000) Efflux-mediated resistance to fluoroquinolones in Gram-negative bacteria. Antimicrob. Agents Chemother. 44, 2233–2241 10.1128/AAC.44.9.2233-2241.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wong K., Ma J., Rothnie A., Biggin P. C., and Kerr I. D. (2014) Towards understanding promiscuity in multidrug efflux pumps. Trends Biochem. Sci. 39, 8–16 [DOI] [PubMed] [Google Scholar]
- 14. Lewinson O., et al. (2003) The Escherichia coli multidrug transporter MdfA catalyzes both electrogenic and electroneutral transport reactions. Proc. Natl. Acad. Sci. U.S.A. 100, 1667–1672 10.1073/pnas.0435544100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Abdel-Sayed S. (1987) Transport of chloramphenicol into sensitive strains of Escherichia coli and Pseudomonas aeruginosa. J. Antimicrob. Chemother. 19, 7–20 10.1093/jac/19.1.7 [DOI] [PubMed] [Google Scholar]
- 16. Harder D. (2008) Characterization of the PTR peptide transporter family of Escherichia coli, in Fakultät Wissenschaftszentrum Weihenstephan für Ernährung, p. 121, Technische Universität, München [Google Scholar]
- 17. Weitz D., Harder D., Casagrande F., Fotiadis D., Orbdlik P., Kelety B., and Daniel H. (2007) Functional and structural characterization of a prokaryotic peptide transporter with features similar to mammalian PEPT1. J. Biol. Chem. 282, 2832–2839 10.1074/jbc.M604866200 [DOI] [PubMed] [Google Scholar]
- 18. Jensen J. M., Ismat F., Szakonyi G., Rahman M., and Mirza O, (2012) Probing the putative active site of YjdL: an unusual proton-coupled oligopeptide transporter from E. coli. PLoS ONE 7, e47780 10.1371/journal.pone.0047780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Prabhala B. K., Aduri N. G., Iqbal M., Rahman M., Gajhede M., Hansen P. R., and Mirza O. (2017) Several hPepT1-transported drugs are substrates of the Escherichia coli proton-coupled oligopeptide transporter YdgR. Res. Microbiol. 168, 443–449 10.1016/j.resmic.2017.01.005 [DOI] [PubMed] [Google Scholar]
- 20. Harder D., Stolz J., Casagrande F.,Obrdlik P., Weitz D., Fotiadis D., and Daniel H. (2008) DtpB (YhiP) and DtpA (TppB, YdgR) are prototypical proton-dependent peptide transporters of Escherichia coli. FEBS J. 275, 3290–3298 [DOI] [PubMed] [Google Scholar]
- 21. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., and Ferrin T. E. (2004) UCSF Chimera: a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 [DOI] [PubMed] [Google Scholar]
- 22. Ernst H. A.,Pham A. Hald H., Kastrup J. S., Rahman M., and Mirza O. (2009) Ligand binding analyses of the putative peptide transporter YjdL from E. coli display a significant selectivity towards dipeptides. Biochem. Biophys. Res. Commun. 389, 112–116 10.1016/j.bbrc.2009.08.098 [DOI] [PubMed] [Google Scholar]
- 23. Prabhala B. K., Aduri N. G., Jensen J. M., Ernst H. A., Iram N., Rahman M., and Mirza O. (2014) New insights into the substrate specificities of proton-coupled oligopeptide transporters from E. coli by a pH sensitive assay. FEBS Lett. 588, 560–565 10.1016/j.febslet.2014.01.004 [DOI] [PubMed] [Google Scholar]
- 24. Volkmer B., and Heinemann M. (2011) Condition-dependent cell volume and concentration of Escherichia coli to facilitate data conversion for systems biology modeling. PLoS ONE 6, e23126 10.1371/journal.pone.0023126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Guettou F., Quistgaard E. M., Raba M., Moberg P., Löw C., and Nordlund P. (2014) Selectivity mechanism of a bacterial homolog of the human drug-peptide transporters PepT1 and PepT2. Nat. Struct. Mol. Biol. 21, 728–731 [DOI] [PubMed] [Google Scholar]
- 26. Chang C., Bahadduri P. M., Polli J. E., Swaan P. W., and Ekins S. (2006) Rapid identification of P-glycoprotein substrates and inhibitors. Drug Metab. Dispos. 34, 1976–1984 [DOI] [PubMed] [Google Scholar]
- 27. Edgar R., and Bibi E. (1997) MdfA, an Escherichia coli multidrug resistance protein with an extraordinarily broad spectrum of drug recognition. J. Bacteriol. 179, 2274–2280 10.1128/jb.179.7.2274-2280.1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ford D., Howard A., and Hirst B. H. (2003) Expression of the peptide transporter hPepT1 in human colon: a potential route for colonic protein nitrogen and drug absorption. Histochem. Cell Biol. 119, 37–43 [DOI] [PubMed] [Google Scholar]
- 29. Heng J., Zhao Y., Liu M., Liu Y., Fan J., Wang X., Zhao Y., and Zhang X. (2015) Substrate-bound structure of the E. coli multidrug resistance transporter MdfA. Cell Res. 25, 1060–1073 10.1038/cr.2015.94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Newstead S. (2015) Molecular insights into proton coupled peptide transport in the PTR family of oligopeptide transporters. Biochim. Biophys. Acta 1850, 488–499 10.1016/j.bbagen.2014.05.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jensen J. M., Aduri N. G., Prabhala B. K., Jahnsen R., Franzyk H., and Mirza O. (2014) Critical role of a conserved transmembrane lysine in substrate recognition by the proton-coupled oligopeptide transporter YjdL. Int. J. Biochem. Cell Biol. 55, 311–317 [DOI] [PubMed] [Google Scholar]
- 32. Aduri N. G., Prabhala B. K., Ernst H. A., Jørgensen F. S., Olsen L., and Miza O. (2015) Salt bridge swapping in the EXXERFXYY motif of proton-coupled oligopeptide transporters. J. Biol. Chem. 290, 29931–29940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shaheen A., Ismat F., Iqbal M., Haque A., De Zorzi R., Mirza O., Walz T., and Rahman M. (2015) Characterization of putative multidrug resistance transporters of the major facilitator-superfamily expressed in Salmonella typhi. J. Infect. Chemother. 21, 357–362 [DOI] [PubMed] [Google Scholar]
- 34. Schindler B. D., Jacinto P., and Kaatz G. W. (2013) Inhibition of drug efflux pumps in Staphylococcus aureus: current status of potentiating existing antibiotics. Future Microbiol. 8, 491–507 10.2217/fmb.13.16 [DOI] [PubMed] [Google Scholar]
- 35. Aygul A. (2015) The importance of efflux systems in antibiotic resistance and efflux pump inhibitors in the management of resistance. Mikrobiyol. Bul. 49, 278–291 10.5578/mb.8964 [DOI] [PubMed] [Google Scholar]
- 36. Opperman T. J., and Nguyen S. T. (2015) Recent advances toward a molecular mechanism of efflux pump inhibition. Front. Microbiol. 6, 421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Xu Z., and Yan A. (2015) Multidrug efflux systems in microaerobic and anaerobic bacteria. Antibiotics 4, 379–396 10.3390/antibiotics4030379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Prabhala B. K., et al. (2015) Investigation of the substrate specificity of the proton coupled peptide transporter PepTSo from Shewanella oneidensis. Int. J. Peptide Res. Therap. 21, 1–6 [Google Scholar]
- 39. Katoh K., and Standley D. M. (2013) MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 10.1093/molbev/mst010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sali A., and Blundell T. L. (1993) Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779–815 10.1006/jmbi.1993.1626 [DOI] [PubMed] [Google Scholar]
- 41. Lee S. H., and Choi D. W. (2013) Comparison between source-induced dissociation and collision-induced dissociation of ampicillin, chloramphenicol, ciprofloxacin, and oxytetracycline via mass spectrometry. Toxicol. Res. 29, 107–114 10.5487/TR.2013.29.2.107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Paixão L., Rodrigues L, Couto I., Martins M., Fernandes P., de Carvalho C. C., Monteiro G. A., Sansonetty F., Amaral L., and Viveiros M. (2009) Fluorometric determination of ethidium bromide efflux kinetics in Escherichia coli. J. Biol. Eng. 3, 18–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wiegand I., Hilpert K., and Hancock R. E. (2008) Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances. Nature Protocols 3, 163–175 10.1038/nprot.2007.521 [DOI] [PubMed] [Google Scholar]