

Abstract
dUTPases are essential enzymes for maintaining genome integrity and have recently been shown to play moonlighting roles when containing extra sequences. Interestingly, the trimeric dUTPase of white spot syndrome virus (wDUT) harbors a sequence insert at the position preceding the C-terminal catalytic motif V (pre-V insert), rarely seen in other dUTPases. However, whether this extra sequence endows wDUT with additional properties is unknown. Herein, we present the crystal structures of wDUT in both ligand-free and ligand-bound forms. We observed that the pre-V insert in wDUT forms an unusual β-hairpin structure in the domain-swapping region and thereby facilitates a unique orientation of the adjacent C-terminal segment, positioning the catalytic motif V onto the active site of its own subunit instead of a third subunit. Consequently, wDUT employs two-subunit active sites, unlike the widely accepted paradigm that the active site of trimeric dUTPase is contributed by all three subunits. According to results from local structural comparisons, the active-site configuration of wDUT is similar to that of known dUTPases. However, we also found that residues in the second-shell region of the active site are reconfigured in wDUT as an adaption to its unique C-terminal orientation. We also show that deletion of the pre-V insert significantly reduces wDUT's enzymatic activity and thermal stability. We hypothesize that this rare structural arrangement confers additional functionality to wDUT. In conclusion, our study expands the structural diversity in the conserved dUTPase family and illustrates how sequence insertion and amino acid substitution drive protein evolution cooperatively.
Keywords: DNA viruses, enzyme structure, nucleic acid enzymology, protein evolution, protein folding, beta-hairpin, WSSV, amino acid substitution, dUTPase, indel
Introduction
dUTPase (EC 3.6.1.23) catalyzes the hydrolysis of dUTP to dUMP and PPi, playing essential roles in cellular nucleotide metabolism (1). First, the enzyme supplies dUMP to synthesize dTTP, a DNA building block. Second, the enzyme lowers the dUTP/dTTP ratio in the cell, thus preventing dUTP misincorporation into genome DNA; deficiency of the enzyme would result in too high a uracil content in DNA and could finally lead to genome instability and thymineless cell death due to overdosed DNA repair activity (2, 3). Because of their essential roles, dUTPases have been proposed as potential drug targets for the therapy of cancer (4, 5) and infectious diseases (6).
dUTPases have been intensively studied structurally and enzymatically (1). According to their structure and oligomerization state, dUTPases are classified into three families: homotrimeric, homodimeric, and monomeric. The homotrimeric and monomeric dUTPases consist mainly of β-pleated sheets and are evolutionarily related (7), whereas the homodimeric dUTPases are all-α proteins and evolutionarily distinct from the other two families (8). Among the three, the homotrimeric family comprises the majority of dUTPases and has been the best studied. So far, a number of trimeric dUTPase structures are available, with those of Escherichia coli (eDUT)2 (9) and humans (hDUT) (10) as prototypes. The trimeric dUTPase has a 3-fold symmetry with the C terminus of each subunit swapped. Seven β-strands from one subunit plus one β-strand from the swapped C-terminal arm of the neighboring subunit form a β-barrel with a jellyroll fold, as a repeating structural unit to assemble the trimer. A striking feature of trimeric dUTPase is that each of its three active sites is formed by five conserved motifs from all three subunits (10), which is rare among enzymes. In such a three-subunit active site, motifs I, II, and IV from one subunit and motif III from another are preformed, whereas the C-terminal P-loop–like motif V from the third subunit is rather flexible in the absence of substrate and undergoes a disorder-to-order transition upon ligand binding, hence capping the active site (11–14). Regarding the catalytic mechanism, previous studies have revealed several interesting features of the enzyme. For example, motif V is not required for substrate binding, but it is essential for organizing the active site into a catalytically competent conformation (11), by contributing several key residues, such as a “Phe-lid” (10) and an arginine finger (15). Interestingly, besides the key individual residues, a proper amino acid interacting network within the active site is also important for catalysis. Mutation of a single residue that does not directly interact with the substrate could disrupt the interacting network and thus significantly reduce enzyme activity (16).
Recently, the finding that in addition to their involvement in nucleotide metabolism, dUTPases can have moonlighting functions in various cellular processes, such as immune system modulation (17, 18), pathogenicity island transferring (19–22), and apoptosis (23), has raised a lot of interest. These “moonlighting” dUTPases often bear an extra sequence insert, called motif VI (24). For example, in the dUTPase of Mycobacterium smegmatis, next to the motif V is a 5-residue insert, deletion of which did not affect the enzyme's activity but was lethal for bacterial viability by an unknown mechanism (25). In staphylococcal phages, motif VI at the central domain is not required for enzyme activity but critical for transferring the pathogenicity island (22). The rat dUTPase contains an N-terminal motif VI, important for interacting with the peroxisome proliferator–activated nuclear receptor α (26). These examples show that active evolution occurs in the putatively conserved dUTPase family, where sequence insertion is a major driving force.
White spot syndrome virus (WSSV) is a deadly pathogen for crustacean animals (27), causing severe annual economic loss in the aquaculture industry. It is a large DNA virus and the sole member of the genus Whispovirus and the family Nimaviridae. So far, its replication mechanism is not understood, and no effective treatment is available (28). Genome sequence analysis uncovered that the ORF wsv112 encodes a dUTPase, although the host can provide dUTPase for viral DNA synthesis (29). The viral dUTPase contains 461 amino acid residues; the N-terminal 171 residues comprise a typical homotrimeric dUTPase domain that shows competitive enzyme activity on its own (30), whereas the C-terminal 290 residues form a region of unknown function. Interestingly, sequence alignment reveals that the N-terminal dUTPase domain harbors an extra 20-residue insert with reference to hDUT, which makes its sequence length between motif IV and V significantly longer than in any other known trimeric dUTPase (Fig. 1); this insert is defined as pre-V insert (sequence insert preceding the catalytic motif V). We propose that the unusual pre-V insert and the long C-terminal tail might endow WSSV dUTPase with additional functions. Otherwise, a short version of the enzyme would be more economical for the virus, if the role of the viral dUTPase is only to complement the host dUTPase for more enzymatic activity. To provide more clues for its potential new functions and also facilitate antiviral drug design targeting this enzyme, we subjected WSSV dUTPase to a structural study.
Figure 1.

Sequence alignment of trimeric dUTPases that have structure solved. FIV, feline immunodeficiency virus; EIAV, equine infectious anemia virus; PBCV, Paramecium bursaria chlorella virus; VACV, vaccinia virus; BSUB, B. subtilis; MTUB, M. tuberculosis; ECOL, E. coli; SCER, Saccharomyces cerevisiae; ATHA, Arabidopsis thaliana; DMEL, Drosophila melanogaster; HSAP, human. Strictly and relatively conserved residues are highlighted in red and yellow boxes, respectively. The secondary structure elements of wDUT and hDUT are shown above and below the alignment, respectively. Positions of motifs I–V and the pre-V insert are indicated. The position of the conserved arginine/lysine on motif IV of classic dUTPases is indicated by a blue triangle, whereas the position of its function-subrogate residue in wDUT (Arg24) is indicated by a red triangle.
Here we present the crystal structures of the N-terminal dUTPase domain of WSV112 (residues 1–171, hereafter referred to as WSSV dUTPase (wDUT), unless otherwise specified), in both its free and ligand-bound forms. Surprisingly, wDUT employs two-subunit active sites, challenging the well-accepted concept that active sites of trimeric dUTPases are contributed by all three subunits. We show how this novel structural form is realized as a combined result of sequence insert and amino acid substitution.
Results
wDUT contains an unusual β-hairpin structural motif in the swapping region
The wDUT crystal grew in space group P212121 with six molecules in one asymmetric unit, forming two trimers. Although our protein construct comprises 171 residues of wDUT, we were only able to model residues 3–150 for four monomers and 3–149 for the other two monomers. The remaining C-terminal residues of all monomers lacked defined electron density, supposedly due to disorder. Most modeled residues showed definite electron density as well as a good quality of geometry, whereas several residues in loop regions and at surfaces exhibited relatively weaker electron density for side chains. Ramachandran plot analysis marked residue Gly85 in all monomers as an outlier, slightly out of the allowed region. However, electron density clearly showed that this residue was correctly modeled. Such an “outlier” conformation is supposed to be necessary for function, as a Ramachandran outlier was also observed for a residue (Ala75) in the same region in hDUT (10).
The wDUT homotrimer has 3-fold symmetry, with a narrow central channel (Fig. 2A). PISA analysis showed that the accessible surface area buried in the interface is 9060 Å2, corresponding to about half of the accessible surface area of all subunits (19,300 Å2), indicating a stable trimer in solution, which is in agreement with the estimation from the gel filtration experiment (data not shown). All of the monomers in the asymmetric unit share essentially identical structures, with a root mean square deviation (RMSD) of 0.2–0.8 Å for all Cα positions when superposed in a pairwise manner (Fig. S1). Compared with residues located in the main body of the protein, residues Tyr134–Glu141 that protrude from the main body exhibit larger RMSD in Cα positions, reflecting their conformational flexibility. The monomer of wDUT adopts the typical dUTPase fold (Fig. 2A). The N-terminal region (residues Ser3–Arg123) forms a β-barrel core; β1, β3, β6, β5b, β4, β7, and β2 form a distorted β-barrel, with β2b, β6b, β5a, and α1 capping the barrel. From residue Tyr124 on, the polypeptide traces outward from the body and contributes a β-strand to arm the adjacent subunit. Up to residue Val133, the structure of wDUT resembles those of known dUTPases. Then follows the extra pre-V insert (residues Tyr134–Asn153), which unexpectedly forms a β-hairpin structure instead of a loop (Fig. 2 (A and B) and Fig. S2). In detail, residues Tyr134–Asn136 continue the β8 strand, making this strand longer than that of any known trimeric dUTPase. Residues Asn136–Gly140 form a tight 5-residue turn, with the backbone oxygen of Asn136 accepting one hydrogen bond from the backbone nitrogen of Gly140, matching the criteria of an α-turn. Residues Gly140–Lys150 form β9, a long β-strand that folds onto β8 in an antiparallel manner, with extensive interstrand hydrogen bonding. Within β9, a single-residue bulge occurs at the position of Ile145. In summary, whereas “classic” dUTPase structures contain one β-strand in the swapping region, wDUT contains a β-hairpin structural motif (including β8, the α-turn, and β9). As a notable result, the C-terminal orientation of wDUT is reversed in comparison with that of classic dUTPases.
Figure 2.

Overall structure of ligand-free wDUT. A, top view (left) and side view (right) of the trimer. Secondary structure elements are indicated in one monomer. The pre-V insert is highlighted in red, and the β-hairpin in the swapping region is indicated with a rectangle. The first residue of the swapping C terminus (Tyr124), the first residue of the pre-V insert (Tyr134), and the last modeled residue (Lys150) are indicated. B, stereo view of the β-hairpin in the swapping region.
The C terminus of classic dUTPase carrying the catalytic motif V would reach the third subunit to cap the active site, after a disorder-to-order transition upon substrate binding (11–14). Because of its direction reversal caused by the β-hairpin, the C-terminal region of wDUT would have to fold back again beyond Lys150 for the C terminus to reach the third subunit in the classic manner. Such a “detour” path would require at least about 25 residues in the C-terminal region following the β-hairpin, even in an extended conformation (Fig. S3). However, only 20 residues are present in the remaining C-terminal region; therefore, there must be an “out-of-reach” difficulty in this scenario. It is very likely that the C-terminal segment of wDUT (which is not defined by electron density) continues in the current direction to reach the active site of its own subunit (Fig. S3). To test this hypothesis, a crystal structure with ordered C-terminal region would be desirable.
The C-terminal region of wDUT takes a novel path to reach the active site of its own subunit in the complex structure
Previous studies have shown that ligand binding can induce ordering of the C-terminal region in dUTPases. Co-crystallization of wDUT with dUTP induced the C-terminal ordering of the enzyme in the crystal structure. (However, the bound ligand is not dUTP, as discussed below). Crystal of the enzyme-ligand complex grew in space group P21, different from the P212121 space group of the free wDUT, with six monomers forming two trimers in one asymmetric unit. The structure of the first 150 residues, including the β-hairpin in the swapping region, is similar to that of the ligand-free form (Fig. 3). The C-terminal residues beyond Lys150, which are invisible in the ligand-free structure, appeared with clear electron density (Fig. S4). However, the degree of order of the C-terminal region is not the same for all monomers. The C-terminal regions in four monomers show a closed conformation to cap the active site of their own monomer; among these, three exhibit excellent and the fourth one exhibits weaker but discernible electron density. The C-terminal region of the fifth monomer is partially ordered (up to Arg158); however, it folds onto the neighboring trimer, which seems physiologically irrelevant. The last monomer lacks any electron density at all for the C-terminal region. This asymmetric ordering is commonly seen in dUTPase structures, reflecting the mobile nature of the C-terminal region. Nevertheless, superposing all monomers in the asymmetric unit shows that the C-terminal regions with closed conformation overlaid well (Fig. S5). Considering that these monomers are located in different crystallographic environments, the observed conformation is unlikely to be a packing artifact but rather reflects a physiologically relevant state in solution.
Figure 3.
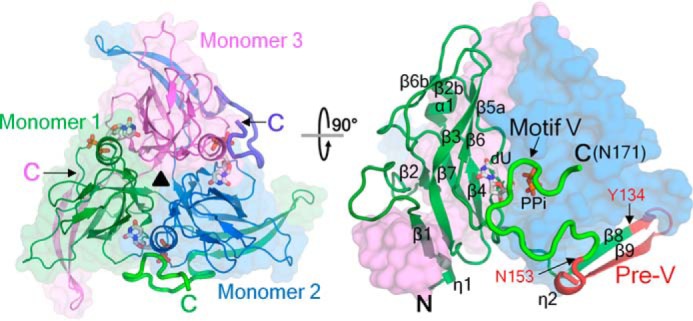
Overall structure of the wDUT-dU-PPi-Mg2+ complex in a top (left) and side view (right). Secondary structure elements are indicated in one monomer. The C-terminal regions that become ordered upon ligand binding are in boldface type. The pre-V insert is highlighted in red. dU and PPi are shown as sticks and indicated. The first and last residues of the pre-V insert (Tyr134 and Asn153, respectively), motif V, and the C-terminal residue (Asn171) are indicated.
In the closed conformation, the C-terminal tail of wDUT continues to reach the active site of its own subunit (Fig. 3) instead of reaching the third subunit as seen in classic dUTPases, supporting our previous hypothesis. This novel orientation is attributed to the chain reversal effect of the β-hairpin in the swapping region. The C-terminal tail covers a rather flat surface of the trimer body and exhibits a meandering conformation (Fig. 4A) and finally brings the catalytic motif V onto the active site. Most interactions between the tail and the trimer body take place in the active-site region, where the ligands interact extensively with both motif V in the C-terminal region and other motifs on the trimer body to stick the two parts together. The details of ligand-protein interactions will be discussed below. Serving as an anchoring point, Lys150 forms hydrogen bonds with residues Pro28 and Asp126 from the neighboring subunit and somehow restricts the bending direction of the following C-terminal region. Mutation of Lys150 to alanine or glutamate caused slight reduction of both the catalytic rate and the substrate-binding affinity (Table 1 and Fig. S6). In addition, the backbone oxygen of Asn155 forms a hydrogen bond with the guanidinium group of Arg24 (neighboring subunit); the backbone nitrogen of Ala157 forms a hydrogen bond with the backbone oxygen of Ala26 (neighboring subunit). Meanwhile, there are several water molecules located in a small pocket between the tail and the body (covered by Lys150–Gln156), contributing indirect hydrogen bonds to fix the tail as well. On the whole, the interaction between the C-terminal tail and the main body is rather weak, explaining the mobile nature of the C terminus.
Figure 4.
The C-terminal tail takes a novel path to reach the active site in the wDUT-dU-PPi-Mg2+ structure. A, the conformation of the C-terminal tail (schematic) and its interactions with the trimer body (transparent surface). Amino acid residues and ligands are displayed as sticks, and their carbons are in subunit-based color and white, respectively. Water molecules are displayed as red balls. Hydrogen bonds are displayed as red dotted lines. The interactions mediated by the water molecules that are covered by Lys150–Gln156 are extensive but are not shown here for clarity. B, superposing wDUT and hDUT monomers shows divergence of their C-terminal regions. Residues at divergent points and motifs V are indicated. C, superposing wDUT and hDUT trimers reveals convergence of the latter parts of their C-terminal regions. For clarity, only the wDUT trimer is shown as a surface, and its bound ligands are shown as sticks to indicate the active-site position. Residues at the convergent point are indicated.
Table 1.
Enzyme kinetics data
Every data point was repeated three times, and S.E. was calculated by the fitting software. Relative efficiency is defined as enzyme efficiency (kcat/Km) divided by that of WT. ND, not determined.
| Enzyme | kcat | Km | kcat/Km | Relative efficiency |
|---|---|---|---|---|
| s−1 | μm | m−1 s−1 | % | |
| wDUTWT | 7.4 ± 0.2 | 1.7 ± 0.2 | 4.4 × 106 | 100 |
| wDUTR24T | 0.45 ± 0.02 | 2.9 ± 0.3 | 1.6 × 105 | 3.6 |
| wDUTD34N | 0.92 ± 0.06 | 13 ± 2 | 7.1 × 104 | 1.6 |
| wDUTR71E | ND | ND | ND | ND |
| wDUTD88N | ND | ND | ND | ND |
| wDUTS115R | 2.6 ± 0.1 | 4.5 ± 0.5 | 5.8 × 105 | 13 |
| wDUTK150A | 5.9 ± 0.1 | 2.5 ± 0.2 | 2.4 × 106 | 54 |
| wDUTK150E | 5.5 ± 0.1 | 2.6 ± 0.2 | 2.1 × 106 | 49 |
| wDUTR158A | 3.2 ± 0.1 | 2.6 ± 0.2 | 1.2 × 106 | 28 |
| wDUTR158E | 2.6 ± 0.1 | 2.6 ± 0.1 | 1.0 × 106 | 23 |
| wDUTR158W | 3.1 ± 0.1 | 3.2 ± 0.2 | 9.7 × 105 | 22 |
| wDUTV160D | 7.4 ± 0.2 | 2.0 ± 0.2 | 3.7 × 106 | 85 |
| wDUTR161K | 0.053 ± 0.002 | 5.4 ± 0.7 | 9.8 × 103 | 0.23 |
| wDUTF166Y | 1.2 ± 0.1 | 5.0 ± 0.2 | 2.4 × 105 | 5.5 |
| wDUTD170G | 6.2 ± 0.2 | 2.1 ± 0.3 | 3.0 × 106 | 68 |
| wDUTD170L | 0.53 ± 0.01 | 7.1 ± 0.4 | 7.5 × 104 | 1.7 |
| wDUTR24T/S115R | 0.51 ± 0.01 | 6.7 ± 0.6 | 7.6 × 104 | 1.8 |
| wDUTR24T/S115R/D170G | 3.1 ± 0.1 | 4.2 ± 0.4 | 7.4 × 105 | 17 |
| wDUTΔ134−153 | 0.67 ± 0.01 | 21 ± 1 | 3.2 × 104 | 0.73 |
| wDUTΔ161−170 | ND | ND | ND | ND |
| wDUTΔ170−171 | 1.7 ± 0.1 | 2.8 ± 0.2 | 6.1 × 105 | 14 |
| eDUTWT | 9.3 ± 0.2 | 0.93 ± 0.11 | 1.0 × 107 | 100 |
| eDUTR116S | 2.7 ± 0.1 | 5.2 ± 0.7 | 5.2 × 105 | 5.2 |
| eDUTT22R/R116S | 3.3 ± 0.2 | 13 ± 2 | 2.5 × 105 | 2.5 |
On the other hand, several hydrogen bonds within the tail itself are important in maintaining the C-terminal conformation (Fig. 4A). Particularly interesting interactions are from Arg158, which is located just before catalytic motif V. It forms hydrogen bonds with Val160, Arg161, and Ser168, introducing a bending conformation to correctly position motif V onto the active site (Fig. 4A). Mutation of Arg158 to alanine, glutamate, or tryptophan removed these interactions and interfered with motif V positioning, reflected by an about 2.5-fold reduction of kcat, whereas dUTP binding affinity in these mutants was slightly decreased (Table 1 and Fig. S6). As a negative control, mutation of Val160, whose side chain points toward the solvent region, to an aspartate showed little effect on enzyme activity (Table 1 and Fig. S6). Based on these mutagenesis results, we conclude that the C-terminal conformation observed in the crystal structure is most likely physiologically relevant, and perturbation of the conformation by site mutation (outside the motif V region) reduces the enzyme activity to a certain extent.
Superposing monomers of wDUT and hDUT (with an RMSD of 1.14 Å for 123 aligned Cα atoms) clearly shows that their C-terminal tails divert into different orientations at a certain point on the swapped β-strand, which is residue Tyr134 in wDUT or Gln146 in hDUT (Fig. 4B). Whereas the C terminus of hDUT proceeds forward to wrap the third subunit, the C terminus of wDUT takes a go-and-return mode and reaches the subunit to which it belongs. Interestingly, superposing wDUT and hDUT trimers demonstrates that the latter portions of their C-terminal tails converge again after residue wDUT Val160 or hDUT Glu152 (Fig. 4C). As a result, motif V at the C terminus caps the active site in essentially the same manner in both enzymes, independently of whether the C-terminal tail reaches the active site of its own subunit (in wDUT) or the third subunit (in hDUT).
wDUT employs noncanonical two-subunit active sites
The ordering of motif V in the complex structure enables us to see a complete active site, which is situated at the intersubunit cleft. Distinct from other dUTPases, the C-terminal region of wDUT is reversed by the β-hairpin in the swapping region and thus orients toward its own subunit. As a remarkable result, the active site of wDUT is actually formed by two subunits only: the catalytic motifs I, II, and IV from one subunit and motifs III and V from the other (Fig. 5A and Fig. S7). This is at variance with the well-accepted concept that the active site of trimeric dUTPase is formed by five conserved motifs from all of its three subunits (Fig. S7). Thus, this complex structure provides definite evidence for the presence of two-subunit active sites in trimeric dUTPases.
Figure 5.

The active site of wDUT shares structural similarity with that of other dUTPases. A, the active site of wDUT is composed of residues from two subunits. Amino acid residues are colored in a subunit-based manner, with the conserved motifs indicated in parentheses. The Mg2+ ion is shown as a green ball, and the water molecules are shown as red balls. Hydrogen bonds are indicated as red dotted lines. B, superimposition of the active sites of wDUT (green) and hDUT (wine). C, superimposition of the active sites of wDUT (green) and bDUT (magenta).
At the active site, we can see clear electron density for ligands. After careful inspection, we modeled this electron density as dU, PPi, and Mg2+ (Fig. S4). The appearance of dU seemed confusing, because we co-crystallized the protein with dUTP. We found that wDUT cannot further hydrolyze the dUMP product to yield dU under standard assay conditions; however, a small amount of dU can be spontaneously formed from dUMP after a certain period of time, which may result from the instability of dUMP itself (Fig. S8). Thus, dU in the crystal seems to be a degradation product formed during the crystallization process. In the complex structure, the binding of ligands is not equivalent in all active sites. dU is found in all six active sites, PPi in five active sites, and Mg2+ in four active sites that also contain dU and PPi. In the following, we present one complete active site that contains all of the above-mentioned ligands to elucidate details of protein-ligand interactions.
dU is deeply buried in the active-site pocket (Fig. 5A and Fig. S9). The uracil ring is sandwiched between Ile87 of motif III and the “Phe-lid” Phe166 of motif V. It forms hydrogen bonds with the main-chain oxygen and nitrogen of Lys96 and with a conserved structural water molecule, thereby mimicking Watson–Crick pairing. The deoxyribose moiety is stabilized in position by stacking with Tyr91, which is at the bottom of the pocket and can discriminate against ribose. The deoxyribose moiety also forms hydrogen bonds with the main-chain nitrogen of Asp88, the side-chain hydroxyl of Ser72, and a water molecule.
The PPi molecule roughly occupies the position where the β- and γ-phosphate groups of the dUTP substrate are located and extensively interacts with the residues at the active site (Fig. 5A and Fig. S9). Its first phosphate group forms hydrogen bonds with Ser72 of motif II and Gly73. The second phosphate group forms extensive hydrogen bonds with the residues of motif V, including Gly167, Ser168, and Thr169, as well as Arg161, which can act as an arginine finger to coordinate the nucleotide substrate (15). Both phosphates form hydrogen bonds with Arg71, which is supposed to play a key role in neutralizing the charge developed in the catalytic process. Moreover, two oxygens of PPi and four water molecules coordinate a Mg2+ in an octahedral configuration. Via water-mediated hydrogen bonding, PPi also interacts with several other residues at the active site, such as the catalytic residue Asp88 of motif III and Asp34, a residue important for maintaining the right interaction network at the active site (16).
Independently of the distinct manner of assembly, the active sites of wDUT and hDUT actually share high structural similarity (Fig. 5B). Superposing all five catalytic motifs gives an RMSD of 0.42 Å for all atoms, with small conformational alterations for individual residues. With respect to ligands, dU in wDUT is imposed well onto the uracil moiety of dUPNPP, whereas the PPi occupies the corresponding position of the β- and γ-phosphate groups of dUPNPP. However, the Mg2+ positions are obviously different; in wDUT, it is coordinated by the PPi group, whereas in hDUT, it is coordinated by all of the α-, β-, and γ-phosphates of dUPNPP. Accordingly, the water positions differ a lot. In fact, the binding of dU, PPi, and Mg2+ was previously observed in the Bacillus subtilis dUTPase (bDUT; PDB code 4B0H), where these ligands together with AlF3 were intentionally added to the crystallization buffer to mimic the transition state (14). When the active site of wDUT is superposed onto that of bDUT, the geometries of the dU, PPi, Mg2+, and water molecules around them are almost identical (Fig. 5C), indicating that the conformation we see in the active site of wDUT is probably a mimic of the pseudo transition state, as described in the bDUT study (14).
To test the roles of individual active-site residues in catalysis, we carried out a mutagenesis study of wDUT (Table 1 and Fig. S6). Deletion of motif V (wDUTΔ161–170) abolished the catalytic power, demonstrating its crucial role that has been well characterized in other dUTPases (31, 32). Mutating the putative catalytic residue Asp88 to an asparagine, thereby removing its role as a general base in catalysis, also eliminated the enzyme's activity. Mutating Arg71, which is supposed to neutralize the negative charge developed in the catalytic process, to a glutamate diminished the enzymatic activity to an undetectable level. Replacing Arg161, which may function as an arginine finger to organize the active-site conformation, with a lysine caused an about 140-fold reduction of the catalytic rate. Change of the “Phe-lid” Phe166 to tyrosine reduced the catalytic efficiency by 20-fold. Mutation of Asp34 to asparagine deteriorated both the catalytic rate and dUTP binding to result in an about 50-fold reduction of catalytic efficiency, supporting the critical role of Asp34 in maintaining a proper interaction network at the active site. Taken together, the mutagenesis results and the structural analysis support the critical roles of these residues in catalysis, as shown in previous studies, suggesting that wDUT shares a similar catalytic mechanism with the known trimeric dUTPases, despite using a different way to assemble the active site.
Residues in the second-shell region of the active site are reconfigured in wDUT as an adaption to its unique C-terminal orientation
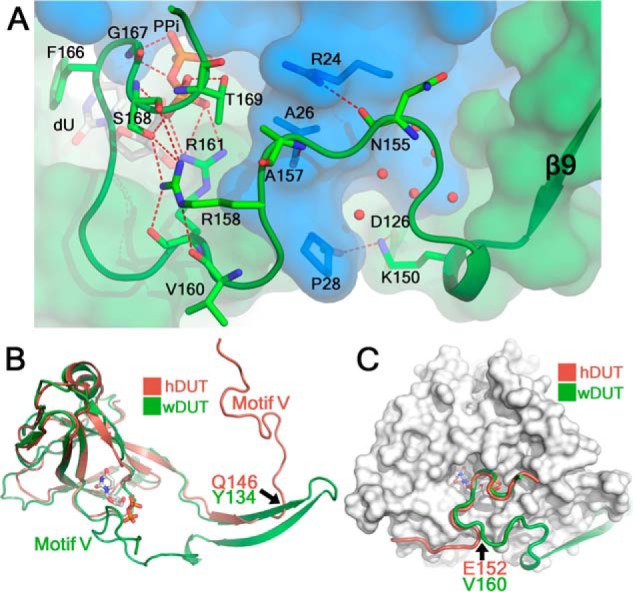
In addition to the pre-V insert, sequence alignment revealed that a conserved arginine/lysine residue of motif IV (corresponding to Arg128 in hDUT) in the known trimeric dUTPases is replaced by a serine in wDUT (residue Ser115) (Fig. 1). The role of this conserved arginine/lysine in dUTPase catalysis has not been well investigated. In the hDUT-dUPNPP structure (PDB code 2HQU), Arg128 does not contact the substrate; instead, it forms two hydrogen bonds with Asp49, a residue playing a key role in maintaining ordered interactions at the active site (16). It also forms a hydrogen bond with Thr161, contributing to the ordering of the C terminus (Fig. 6A). To test its role, we carried out a mutagenesis study (Table 1 and Fig. S6). Because hDUT was difficult to produce, it was only used for structural analysis, whereas eDUT was used for mutagenesis and enzyme kinetics. Mutation of this conserved arginine in eDUT (Arg116) to a serine reduces both the catalytic rate and binding affinity to cause a 20-fold reduction of catalytic efficiency, indicating that the hydrogen-bonding interactions provided by this arginine play a significant role in catalysis. This result also shows that not only a proper interaction network within the active site (16), but also that within the second shell of the active site, is important for dUTPase activity.
Figure 6.
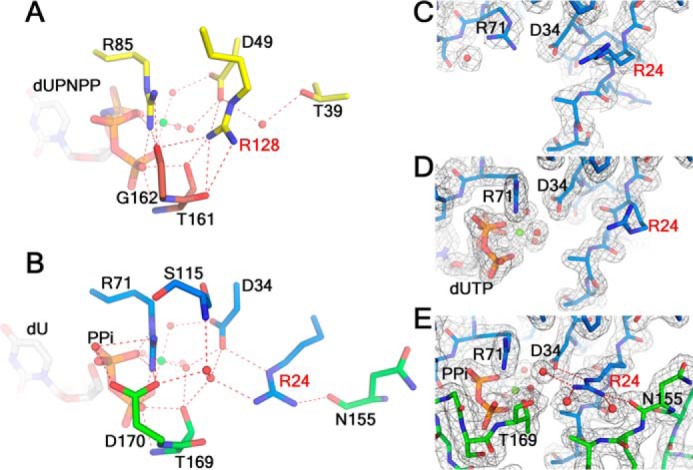
Residues in the second-shell region of the active site are reconfigured in wDUT. A, the second-shell region around the conserved Arg128 in hDUT. B, the second-shell region around Arg24 in wDUT, in an aligned view of A. C–E, the conformations of Arg24 in the free wDUT (C), wDUTD88N/R158E-dUTP-Mg2+ (D), and wDUT-dU-PPi-Mg2+ (E), shown in aligned views. The σA-weighted 2Fo − Fc electron density maps (contoured at 1σ level) are shown as gray mesh. For all panels, hydrogen bonds are shown as red dotted lines. Residues are in subunit-based colors. Ligands are shown as sticks.
Puzzlingly, whereas the eDUTR116S mutant suffers from substantial activity loss, wDUT exhibits activity comparable with that of the wild-type eDUT (Table 1 and Fig. S6), even if the conserved arginine/lysine is also substituted by a serine. To address the discrepancy, we inspected the region around Ser115 in the wDUT complex structure and identified another arginine, residue Arg24, which is located at a position in front of motif I and not conserved in dUTPases (its counterpart residue is Thr39 in hDUT or Thr22 in eDUT) (Figs. 1 and 6B). Its guanidinium moiety sits at the corresponding position of the guanidinium moiety of hDUT Arg128 and forms hydrogen bonds with Asp34, whose orientation is supposed to be critical for enzyme activity (16). Arg24 also interacts with the C-terminal Thr169 through a water-mediated hydrogen bond. Moreover, Arg24 forms a hydrogen bond with residue Asn155, stabilizing the closed conformation of the C-terminal tail that takes a novel orientation. Mutating Arg24 to threonine significantly reduced the catalytic rate and moderately decreased dUTP binding affinity to result in a 30-fold reduction of catalytic efficiency (Table 1 and Fig. S6), supporting its important role in catalysis. Therefore, Arg24 subrogates the conserved arginine/lysine to maintain a proper interaction network in the second-shell region of wDUT (Fig. 6B). Why wDUT adopts an arginine configuration different from other dUTPase was unclear. We wondered whether this reconfiguration is related to the unusual structural organization of wDUT.
First, we checked whether wDUT can use a classic arginine configuration. We mutated Ser115 of wDUT to an arginine residue, as in other dUTPases. The mutant wDUTS115R showed a catalytic efficiency of only about 13% of that of the wild type (Table 1 and Fig. S6). Probably, Arg115 is too close to Arg24 in space, resulting in an improper hydrogen-bonding network and thus a rather low enzymatic activity. Accordingly, we prepared a double mutant, replacing Ser115 and Arg24 of wDUT to an arginine and a threonine, respectively, thereby mimicking the residue combination in hDUT. Surprisingly, the catalytic efficiency of wDUTR24T/S115R is even lower than that of the single mutant, only about 1% of that of the wild type (Table 1 and Fig. S6). Apparently, a proper interaction network was not built either in this double mutant. In the vicinity of Ser115 and Arg24, wDUT contains residue Asp170, which is usually a glycine residue at the same position in the known dUTPases (except for Mason–Pfizer monkey virus, where it is also an aspartate (Fig. 1)). In the wDUT complex structure, Asp170 forms a salt bridge with Arg71 (Fig. 6B). Deleting Asp170-Asn171 or mutating Asp170 to leucine greatly decreased the catalytic efficiency, whereas mutating Asp170 to the common glycine only slightly decreased enzyme activity (Table 1 and Fig. S6). In wDUTR24T/S115R, Asp170 could alternatively form a salt bridge with Arg115 to seriously disturb the interaction network, thereby leading to a very low enzymatic activity. Therefore, we prepared a triple mutant, wDUTR24T/S115R/D170G, to mimic the classic dUTPase better. Its catalytic efficiency is about 20% of that of the wild type, much higher than that of the single or double mutant, suggesting that a relatively well-functioning interaction network in this region is restored. Nevertheless, the enzyme's power was not restored to the level of the wild type. Compared with the wild-type wDUT, the hydrogen bond between Arg24 and Asn155 (Fig. 4A), which facilitates the ordering of the C-terminal region, is missing in wDUTR24T/S115R/D170G, probably explaining the inferior activity of the triple mutant. We conclude that the wDUT arginine configuration is superior to the classic arginine configuration for wDUT; besides maintaining a proper hydrogen-bonding network at the second shell of the active site, the wDUT arginine configuration can facilitate the ordering of the C-terminal region in its noncanonical orientation, which cannot be provided by the classic configuration.
Second, we checked whether the wDUT arginine configuration can be used by classic dUTPase. We prepared the eDUTT22R/R116S mutant to mimic the wDUT arginine configuration. Surprisingly, this double mutant exhibited a catalytic efficiency of only 2.5% of that of the wild-type eDUT (Table 1 and Fig. S6). Apparently, Arg22 in eDUTT22R/R116S is not capable of subrogating the conserved arginine Arg116 as wDUT Arg24 does. Because we were not able to obtain a crystal structure of eDUTT22R/R116S for a direct structural explanation, we attempted to find a clue from the wDUT structures. Whereas Arg24 makes hydrogen bonds with Asp34 and Thr169 in the wDUT-dU-PPi-Mg2+ structure (Fig. 6B), it is located at the surface of the protein in the ligand-free structure, showing various conformations in all monomers that are located in different crystallographic environments. In all cases, its side chain points away from the active position (Fig. 6C). Obviously, the interaction network in this region is not preformed in the ligand-free wDUT. To achieve full catalytic power, Arg24 must be ordered to form a proper hydrogen-bonding network in the second-shell region. Is its ordering induced by ligand binding? Within our project, we also determined the structure of the wDUTD88N/R158E-dUTP-Mg2+ complex, where dUTP clearly bound to the active site, but the C-terminal region was still disordered and not defined by electron density. In this structure, Arg24 deviates from its proper position as well (Fig. 6D), suggesting that ligand binding cannot directly induce Arg24 ordering. Indeed, it is the ordering of the C-terminal region upon ligand binding and its subsequent interaction with Arg24 that are required for positioning Arg24 into a functional state (Fig. 6E). In contrast, in eDUTT22R/R116S, Arg22 could be always in a disordered state even if the C-terminal segment becomes ordered upon ligand binding, as the C-terminal segment takes the classic route and cannot interact with Arg22 to induce its ordering. This gives a plausible explanation for the low activity of eDUTT22R/R116S. Therefore, whereas the wDUT arginine configuration is optimal in wDUT due to the novel C-terminal orientation, it is not a good option for classic dUTPases. From the above analysis, we conclude that reconfiguration of the residues in the second-shell region of the active site in wDUT facilitates the ordering of its C-terminal region in the novel orientation, which in turn promotes a proper interaction network in the second-shell region.
Deletion of the pre-V insert significantly reduces the enzymatic activity and thermal stability of wDUT
From the above analysis, the pre-V insert (134YINETTGERTIIDSSSKKDN153; Fig. 1) is the key factor to endow wDUT with a new structural form. We wondered whether deletion of this extra sequence could convert wDUT into a classic form. We prepared a truncated version, wDUTΔ134–153, which was supposed to take a classic form, restricted by the length of the C terminus. However, its enzymatic efficiency was extremely low, less than 1% of that of wild type (Table 1 and Fig. S6). In this respect, the pre-V insert is different from the known motifs VI, which are dispensable for the dUTPase activity (22, 25). As shown above, the residue configuration in the second shell also plays a significant role in enzyme activity. We propose that the enzymatic activity might be improved by introducing the classic arginine configuration into wDUTΔ134–153. Unfortunately, we could not test this hypothesis, because the mutant protein could not be produced in a soluble form. Meanwhile, we also monitored the thermal stability of wDUTΔ134–153. Its Tm values were about 29 and 23 °C lower than that of wild type, or about 19 and 15 °C lower than that of eDUT, estimated from the Thermofluor assay and the thermal inactivation assay, respectively (Fig. S10). The reduced thermal stability may be partially responsible for the low activity of wDUTΔ134–153 as well. To convert wDUT into a well-functioning classic form, more amino acid substitutions should be introduced to counteract the adverse effect of the pre-V deletion on both enzyme activity and thermal stability.
Discussion
In this report, we present the crystal structures of wDUT in both ligand-free and ligand-bound forms. wDUT shares a similar structure fold with the known trimeric dUTPases, but its C-terminal segment takes an alternative orientation, which is largely attributed to a sequence insert, called the pre-V insert in this study. This insert forms a β-hairpin reversing the C-terminal orientation and resulting in a noncanonical two-subunit active site in wDUT, in contrast to the three-subunit active site in classic dUTPases (Fig. S7). Despite the unusual structural organization, wDUT has a local structure at the active site similar to that of the classic dUTPases, implicating a similar catalytic mechanism. Nevertheless, wDUT reconfigures the residues in the second-shell region of the active site as an adaption to its novel C-terminal orientation. We also show that deletion of the pre-V insert solely is not able to convert wDUT back to a fully functional classic form. It is worthy of mention that the full-length dUTPase of WSSV contains a 290-residue C-terminal tail continuing motif V. This tail may not change the essential structural features of the dUTPase domain as observed in this study; however, it can modulate the dUTPase activity via affecting motif V organization.
Sequence insertion and deletion (indel), along with amino acid substitution, is a major way for protein evolution, even more powerful than the latter, to achieve novel structure and function (33, 34). Recent studies show that a sequence insert, often named motif VI, is an important factor in acquiring new functions in the dUTPase family (24). This extra motif can be located at the N-terminal, the middle, or the C-terminal position of dUTPase. wDUT is unique by harboring a 20-residue insert preceding the C-terminal motif V, the pre-V insert. Although a five-residue insert at a similar position is reported in Mycobacterium tuberculosis dUTPase (25), such a “long” insert is absent in any other trimeric dUTPases. This pre-V insert can be regarded as a motif VI of wDUT; however, it is distinct from other known motifs VI by having an obvious structural role. It forms a β-hairpin, dramatically affecting the orientation of the following C-terminal region and resulting in a two-subunit active site in wDUT. In general, most inserts in proteins just loop out in situ with limited local structure alteration. Only in few characterized structures can a sequence insert cause dramatic changes in the neighboring region to obviously drive protein evolution (35). An outstanding example is found for myosin VI. Compared with other myosin molecules, myosin VI contains an extra sequence insertion forming a helix, which changes the direction of the following level arm and introduces an unusual backstroke function (36). Our study adds another showcase, being the first report that a short sequence insert plays a role in reorganizing the active site.
A remarkable discovery of this study is that wDUT adopts a noncanonical two-subunit active site. The trimeric dUTPases have been intensively studied for decades, and it is widely accepted that their active sites are formed by all three subunits. It is therefore surprising to find an exception in wDUT. Indeed, the isolated dUTPase domain of Mason–Pfizer monkey virus (MPMV) was suggested to be an exception to the rule as well (37). The initial orientation of the C-terminal region observed in the MPMV dUTPase structure and a calculation suggested that this dUTPase uses a two-subunit active site. However, the complete active site was not shown, because motif V is disordered even in the ligand-bound crystal structure. The hypothesis was supported by molecular dynamics and other experiments. Here we present definite structural evidence to support the presence of a two-subunit active site in the trimeric dUTPase family. Notably, the structural solutions to form a two-subunit active site are distinct in WSSV and MPMV dUTPases. In MPMV dUTPase, it results from a shortage of residues between motif IV and V and a lack of the N-terminal β-strand that is necessary for domain swapping (Fig. S11A). By contrast, in wDUT, it is attributed to a sequence insert, which forms a β-hairpin to reverse the following C terminus (Fig. S11B). Comparison of the two structures side by side shows that the missing C-terminal region of MPMV dUTPase might take a similar route as in wDUT to reach the active site (Fig. S11). For the moment, the two-subunit active site form seems a rare variation in the dUTPase family. However, this form may become more populated when more dUTPase sequences and structures are studied.
How nature evolves such a new structural form is suggested in our study. Despite using distinct solutions to organize their active sites, wDUT and the classic dUTPases share similar overall and active-site structures, suggesting that the former evolved from the latter. Obviously, the pre-V insert plays a key role in the evolution, by forming the β-hairpin to reverse the C-terminal orientation, finally leading to a two-subunit active site. Besides the sequence insert, we found that a conserved arginine/lysine located in motif IV of the classic dUTPases is functionally substituted by another arginine (residue Arg24) in wDUT (Figs. 1 and 6). We show that such a reconfiguration in the second-shell region of the active site is an adaption to the novel C-terminal orientation of wDUT and required for maintaining full enzyme activity. Eventually, the evolution of wDUT should be considered the combined result of sequence insertion and amino acid substitution.
Why wDUT adopts this unusual structural form is an interesting question. The catalytic power of wDUT is comparable with that of the traditional members of the dUTPase family. Apparently, the new structural form does not constitute a benefit for the virus regarding the dUTPase activity. Compared with the prototypic hDUT, wDUT contains a long β-hairpin, largely derived from its extra pre-V insert. It protrudes from the trimeric body as a bulge (Bulge 1 in Fig. 7A) and can be an excellent structural element to mediate intermolecular interactions (38). Furthermore, in the vicinity, there is another bulge formed by residues Pro14–Glu17 (Bulge 2 in Fig. 7A), which is also not commonly seen in other dUTPase structures. These two bulges together form an extended surface with negative charges (Fig. 7B), serving as a potential region to mediate intermolecular interaction. Interestingly, a recent study showed that pseudorabies virus UL50, a monomeric family dUTPase, can inhibit IFNα signaling independent of its enzymatic activity. In this protein, a sequence insert of about 30 residues at a position similar to that of the pre-V insert plays a key role (39). Analogously, by harboring the extra pre-V insert, wDUT might benefit the virus in certain aspects beyond its dUTPase activity. Nevertheless, the biological meaning of this new form remains to be investigated.
Figure 7.

Structural features of wDUT. A, superposition of the trimers of wDUT and hDUT reveals extra bulges in wDUT. Only one pair of bulges is indicated. B, charge distribution on the surface of wDUT. The positive, negative, and neutrally charged residues are colored blue, red, and white, respectively.
Last but not least, our structures would facilitate structure-based antiviral design against WSSV. WSSV is a serious pathogen in shrimp aquaculture, causing $1 billion economic loss every year since its emergence more than 20 years ago (28). Until now, there has been no efficient treatment available, and development of a suitable antiviral drug is an urgent task. A previous study shows that WSSV dUTPase can be related to virus replication and thus serve as a potential antiviral target (30). Our study shows that the active site of wDUT shares high similarity with those of known dUTPases, indicating that inhibitors designed to target the active site of general dUTPases should be applicable for the inhibition of wDUT as well. Notably, such inhibitors could have high cytosolic toxicity, by acting on host dUTPase. On the other hand, unique structural features of wDUT can be explored to design specific inhibitors. For example, the C-terminal tail of wDUT binds the trimer body at a surface completely different from that of classic dUTPases; several small pockets present at this surface can serve as potential “hot spots” for drug design.
In summary, our study discovers a novel structural form of dUTPase and demonstrates how sequence insertion and amino acid substitution in concert drive protein evolution. The structural information is valuable for antiviral drug design against the white spot syndrome virus.
Experimental procedures
Gene cloning and protein production
The DNA sequence encoding wDUT (comprising the N-terminal 171 residues of WSV112) was amplified by PCR with primers 5′-TACTTCCAATCCAATGCCATGGACTCATCTGCATCTGTC-3′ (forward) and 5′- TTATCCACTTCCAATGCTATCAGTTATCTGTAGATCCAAAT-3′ (reverse), using the WSSV genomic DNA as template. The PCR product was inserted into a modified pET30 vector using a ligation-independent cloning protocol (40). The DNA encoding eDUT was amplified by PCR with primers 5′-CATGCCATGGCGATGAAAAAAATCGACGTTAAG-3′ (forward) and 5′-CCGCTCGAGTCACTGACGACCAGAGTGACCAAA-3′ (reverse), using E. coli (K12-MG1655) genomic DNA as template. The PCR product was inserted into vector pETM11 (EMBL) using NcoI and XhoI sites. Site-directed mutagenesis of wDUT or eDUT was performed using the QuikChange method (Stratagene). All recombinant proteins were expected to contain an N-terminal His6 tag cleavable by tobacco etch virus protease (TEV), and the final protein products carry a few residues derived from the vector sequence (SNA for wDUT/mutants or GAMA for eDUT/mutants). Similar protocols were used for protein production, as described below.
The sequence-verified gene construct was transformed into the E. coli strain Rossetta (DE3). The culture was grown in Luria–Bertani medium with vigorous shaking at 37 °C until A600 reached 0.8, and then 0.2 mm isopropyl β-d-l-thiogalactopyranoside was added to induce protein overexpression, with further incubation at 16 °C for 16 h. Cells were harvested by centrifugation and lysed by sonication in buffer containing 50 mm Tris-HCl, pH 8.0, 500 mm NaCl. The lysate was clarified by centrifugation, and the supernatant was applied onto a nickel-chelating Sepharose affinity chromatography column (GE Healthcare). After washing with washing buffer (50 mm Tris-HCl, pH 8.0, 500 mm NaCl, 10 mm imidazole), the protein was eluted with elution buffer (50 mm Tris-HCl, pH 8.0, 500 mm NaCl, 500 mm imidazole). Then the protein solution was changed to 50 mm Tris-HCl, pH 8.0, 500 mm NaCl using a PD-10 column (GE Healthcare) for TEV digestion. The TEV-digested protein was reloaded onto the nickel column to remove the uncleaved protein and TEV. The flow-through containing untagged protein was collected, concentrated, and finally purified with size-exclusion chromatography using a HiLoad 16/60 Superdex 200 column (GE Healthcare) equilibrated in buffer that was optimized according to protein usage. For protein that was used for crystallization, 20 mm Tris-HCl, pH 8.0, 500 mm NaCl, and 1 mm DTT was applied to wDUT, whereas 20 mm Tris-HCl, pH 8.0, 150 mm NaCl, and 1 mm DTT was applied to wDUTD88N/R158E. When protein was used for the enzymatic assay, the buffer was 10 mm HEPES, pH 7.5, 150 mm NaCl, 5 mm MgCl2, 1 mm DTT. The purified protein was concentrated to about 10 mg/ml, as determined by absorbance at 280 nm. Protein was either used freshly or frozen at −80 °C for later use.
Crystallization and structure determination
Crystallization was conducted with the sitting-drop vapor-diffusion method at 20 °C. Crystals of free wDUT were grown in drops containing 1 μl of protein solution (6.7 mg/ml in a buffer containing 20 mm Tris-HCl, pH 8.0, 500 mm NaCl, and 1 mm DTT) and 1 μl of reservoir solution containing 30% (w/v) PEG 1500. Crystals of the wDUT-dU-PPi-Mg2+ complex were grown in drops containing 1 μl of protein solution (10 mg/ml protein + 10 mm dUTP + 8 mm MgCl2 incubated for 1 h at 4 °C) and 1 μl of reservoir solution (0.2 m sodium acetate, 0.1 m Tris-HCl, pH 8.1, 28% (w/v) PEG 3350). The solution (0.2 m sodium acetate, 0.1 m Tris-HCl, pH 8.1, 35% (w/v) PEG 3350) was used as cryoprotectant. Crystals of wDUTD88N/R158E-dUTP-Mg2+ were grown in a drop containing 1 μl of protein solution (10 mg/ml protein + 10 mm dUTP + 8 mm MgCl2 incubated for 1 h at 4 °C) and 1 μl of reservoir solution of 25% (w/v) PEG 1500. 35% (w/v) PEG 1500 was used as cryoprotectant.
Crystals were frozen by plunging into liquid nitrogen. The diffraction data were collected at 100 K on beamline BL17U1 (41) at the Shanghai Synchrotron Radiation Facility and were processed using autoPROC (42, 43) or XDS (44).
The ligand-free structure of wDUT was solved by molecular replacement using the Phaser program (45), with the trimeric dUTPase of chlorella virus (PDB code 3C3I) as a search model (46). Structure refinement of atomic coordinates, B-factors, and TLS parameters with Refmac5 (47) or autoBUSTER (48) and model building with Coot (49) were carried out alternatively. For the ligand-bound forms, the structures were solved by molecular replacement, using the refined ligand-free structure as a search model. Ligands were modeled at late stage of refinement, and the geometry restraints of ligands were generated by the GRADE server (http://grade.globalphasing.org).3 Refinement and model building were similar to that for the ligand-free structure. Crystallographic statistics are summarized in Table S1.
Sequence alignment was performed on the ESPript server (50). Protein-protein interactions were analyzed with PISA (51), whereas protein-ligand interactions were analyzed with LigPlot+ (52). The MolProbity server (53) and other CCP4 programs (54) were used for model analysis as well. Structural alignments were performed with Align (55). If not specified, ligand-bound DUTs that have an ordered C terminus are used in structural comparisons. Figures for structures were prepared using PyMOL (Schrödinger, LLC).
Enzyme kinetics measurement
The enzyme activities of dUTPases and their mutants were measured by a phenol red indicator assay (56), in which protons released in the dUTPase reaction were detected. The measurements were performed in 1 mm HEPES, pH 7.5, buffer containing 150 mm KCl, 40 μm phenol red, 5 mm MgCl2, and 1–120 μm dUTP on a UV-1801 spectrophotometer (Rayleigh) with 10-mm path length cuvettes at 20 °C. Absorbance was recorded at 559 nm. The Michaelis–Menten equation was fitted to the steady-state curves using Origin version 8.0 (OriginLab Corp.).
HPLC assay
HPLC analysis of the dUTPase reaction products was performed on a Wufeng HPLC system equipped with a 5-μm WondaCract ODS-2 C18 column (GL Sciences) and a Wonda guard column (GL Sciences). Reactions were started by adding 25 mm enzyme (final concentration) into reaction buffer (10 mm HEPES, pH 7.5, 150 mm NaCl, 5 mm MgCl2, 1 mm DTT, 1 mm dUTP) with a final volume of 0.2 ml. After incubation at 20 °C for 30 min, the reaction solution was cleared by centrifugation at 13,800 × g for 5 min at 4 °C, and 20 μl of supernatant was analyzed on the HPLC system using 0.1 m KH2PO4 (pH 6.0) and 12% (v/v) methanol as mobile phase at a flow rate of 0.4 ml/min. The run was performed isocratically for 25 min at 20 °C, and the eluent was monitored by absorbance at 254 nm. dUTP, dUMP, and dU (all from Sigma) were used as controls, where no enzyme was added. To test dUMP stability, 1 mm dUMP was incubated in 0.2 ml of reaction buffer at 20 °C for 24 h.
Thermal stability assays
Thermofluor assay
The Thermofluor assay (57) was performed on a quantitative PCR instrument (CFX96, Bio-Rad). The fluorescence (excitation wavelength at 498 nm and emission wavelength at 610 nm) of the protein solution (100 μm protein in 10 mm HEPES, pH 7.5, 150 mm NaCl, 5 mm MgCl2, 1 mm DTT, plus 10× SYPRO Orange dye (Sigma) in a final volume of 25 μl) was measured as a function of temperature at a climbing rate of 1 °C/min from 25 to 95 °C. The data were analyzed using the Boltzmann equation in Origin version 8.0 (OriginLab Corp.).
Thermal inactivation assay
The thermal inactivation assay was conducted in the following manner. Protein samples were incubated for 10 min in buffer (10 mm HEPES, pH 7.5, 150 mm NaCl, 5 mm MgCl2, 1 mm DTT) at various temperatures (37.5–80 °C) in a PCR instrument (Thermal cycler 2720, Applied Biosystems). After being cooled on ice for 5 min and centrifuged at 13,800 × g for 5 min at 4 °C, enzyme activity was measured at 20 °C with the phenol red indicator assay to acquire the initial velocity.
Accession numbers
The coordinates and diffraction data of free wDUT, wDUT-dU-PPi-Mg2+, and wDUTD88N/R158E-dUTP-Mg2+ have been deposited in the PDB with accession numbers 5Y5O, 5Y5P, and 5Y5Q, respectively.
Author contributions
K. Z. and Q. M. designed, performed, and analyzed the experiments and wrote the paper. F. L. contributed new reagent. Q. M. conceived and supervised the study. All authors reviewed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We thank Prof. Rolf Hilgenfeld, Prof. Jianhai Xiang, Prof. Ying Zhong Tang, Prof. Ka Yin Leung, and Dr. Ian R. Jenkinson for critical reading of the manuscript and suggestions. We thank Dr. Xue Yang, Prof. Yuequan Shen, and the staff from the BL17U1 beamline at the Shanghai Synchrotron Radiation Facility for assistance in data collection.
This work was supported by National Natural Science Foundation of China Grant 31572660, the 1000 Talents Program of China, the 100 Talents Program of the Chinese Academy of Sciences, and the AoShan Talents Program of Qingdao National Laboratory for Marine Science and Technology Grant 2015ASTP (to Q. M.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Table S1 and Figs. S1–S11.
The atomic coordinates and structure factors (codes 5Y5O, 5Y5P, and 5Y5Q) have been deposited in the Protein Data Bank (http://wwpdb.org/).
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- eDUT
- E. coli dUTPase
- hDUT
- human dUTPase
- WSSV
- white spot syndrome virus
- wDUT
- WSSV dUTPase
- bDUT
- B. subtilis dUTPase
- dUPNPP
- α,β-imido-deoxyuridine triphosphate
- MPMV
- Mason-Pfizer monkey virus
- RMSD
- root mean square deviation
- PDB
- Protein Data Bank
- TEV
- tobacco etch virus.
References
- 1. Vértessy B. G., and Tóth J. (2009) Keeping uracil out of DNA: physiological role, structure and catalytic mechanism of dUTPases. Acc. Chem. Res. 42, 97–106 10.1021/ar800114w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gadsden M. H., McIntosh E. M., Game J. C., Wilson P. J., and Haynes R. H. (1993) dUTP pyrophosphatase is an essential enzyme in Saccharomyces cerevisiae. EMBO J. 12, 4425–4431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kouzminova E. A., and Kuzminov A. (2004) Chromosomal fragmentation in dUTPase-deficient mutants of Escherichia coli and its recombinational repair. Mol. Microbiol. 51, 1279–1295 10.1111/j.1365-2958.2003.03924.x [DOI] [PubMed] [Google Scholar]
- 4. Wilson P. M., Fazzone W., LaBonte M. J., Deng J., Neamati N., and Ladner R. D. (2008) Novel opportunities for thymidylate metabolism as a therapeutic target. Mol. Cancer Ther. 7, 3029–3037 10.1158/1535-7163.MCT-08-0280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Miyahara S., Miyakoshi H., Yokogawa T., Chong K. T., Taguchi J., Muto T., Endoh K., Yano W., Wakasa T., Ueno H., Takao Y., Fujioka A., Hashimoto A., Itou K., Yamamura K., et al. (2012) Discovery of highly potent human deoxyuridine triphosphatase inhibitors based on the conformation restriction strategy. J. Med. Chem. 55, 5483–5496 10.1021/jm300416h [DOI] [PubMed] [Google Scholar]
- 6. Nyíri K., and Vértessy B. G. (2017) Perturbation of genome integrity to fight pathogenic microorganisms. Biochim. Biophys. Acta 1861, 3593–3612 10.1016/j.bbagen.2016.05.024 [DOI] [PubMed] [Google Scholar]
- 7. Tarbouriech N., Buisson M., Seigneurin J. M., Cusack S., and Burmeister W. P. (2005) The monomeric dUTPase from Epstein-Barr virus mimics trimeric dUTPases. Structure 13, 1299–1310 10.1016/j.str.2005.06.009 [DOI] [PubMed] [Google Scholar]
- 8. Harkiolaki M., Dodson E. J., Bernier-Villamor V., Turkenburg J. P., González-Pacanowska D., and Wilson K. S. (2004) The crystal structure of Trypanosoma cruzi dUTPase reveals a novel dUTP/dUDP binding fold. Structure 12, 41–53 10.1016/j.str.2003.11.016 [DOI] [PubMed] [Google Scholar]
- 9. Cedergren-Zeppezauer ES, Larsson G., Nyman P. O., Dauter Z., and Wilson K. S. (1992) Crystal-structure of a dUTPase. Nature 355, 740–743 10.1038/355740a0 [DOI] [PubMed] [Google Scholar]
- 10. Mol C. D., Harris J. M., McIntosh E. M., and Tainer J. A. (1996) Human dUTP pyrophosphatase: uracil recognition by a beta hairpin and active sites formed by three separate subunits. Structure 4, 1077–1092 10.1016/S0969-2126(96)00114-1 [DOI] [PubMed] [Google Scholar]
- 11. Varga B., Barabás O., Kovári J., Tóth J., Hunyadi-Gulyás E., Klement E., Medzihradszky K. F., Tölgyesi F., Fidy J., and Vértessy B. G. (2007) Active site closure facilitates juxtaposition of reactant atoms for initiation of catalysis by human dUTPase. FEBS Lett. 581, 4783–4788 10.1016/j.febslet.2007.09.005 [DOI] [PubMed] [Google Scholar]
- 12. Varga B., Barabás O., Takács E., Nagy N., Nagy P., and Vértessy B. G. (2008) Active site of mycobacterial dUTPase: structural characteristics and a built-in sensor. Biochem. Biophys. Res. Commun. 373, 8–13 10.1016/j.bbrc.2008.05.130 [DOI] [PubMed] [Google Scholar]
- 13. Tchigvintsev A., Singer A. U., Flick R., Petit P., Brown G., Evdokimova E., Savchenko A., and Yakunin A. F. (2011) Structure and activity of the Saccharomyces cerevisiae dUTP pyrophosphatase DUT1, an essential housekeeping enzyme. Biochem. J. 437, 243–253 10.1042/BJ20110304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. García-Nafría J., Timm J., Harrison C., Turkenburg J. P., and Wilson K. S. (2013) Tying down the arm in Bacillus dUTPase: structure and mechanism. Acta Crystallogr. D Biol. Crystallogr. 69, 1367–1380 10.1107/S090744491300735X [DOI] [PubMed] [Google Scholar]
- 15. Nagy G. N., Suardíaz R., Lopata A., Ozohanics O., Vékey K., Brooks B. R., Leveles I., Tóth J., Vértessy B. G., and Rosta E. (2016) Structural characterization of arginine fingers: identification of an arginine finger for the pyrophosphatase dUTPases. J. Am. Chem. Soc. 138, 15035–15045 10.1021/jacs.6b09012 [DOI] [PubMed] [Google Scholar]
- 16. Takács E., Nagy G., Leveles I., Harmat V., Lopata A., Tóth J., and Vértessy B. G. (2010) Direct contacts between conserved motifs of different subunits provide major contribution to active site organization in human and mycobacterial dUTPases. FEBS Lett. 584, 3047–3054 10.1016/j.febslet.2010.05.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Madrid A. S., and Ganem D. (2012) Kaposi's sarcoma-associated herpesvirus ORF54/dUTPase downregulates a ligand for the NK activating receptor NKp44. J. Virol. 86, 8693–8704 10.1128/JVI.00252-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ariza M. E., Glaser R., and Williams M. V. (2014) Human herpesviruses-encoded dUTPases: a family of proteins that modulate dendritic cell function and innate immunity. Front. Microbiol. 5, 504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tormo-Más M. A., Mir I., Shrestha A., Tallent S. M., Campoy S., Lasa I., Barbé J., Novick R. P., Christie G. E., and Penadés J. R. (2010) Moonlighting bacteriophage proteins derepress staphylococcal pathogenicity islands. Nature 465, 779–782 10.1038/nature09065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tormo-Más M. A., Donderis J., García-Caballer M., Alt A., Mir-Sanchis I., Marina A., and Penadés J. R. (2013) Phage dUTPases control transfer of virulence genes by a proto-oncogenic G protein-like mechanism. Mol. Cell 49, 947–958 10.1016/j.molcel.2012.12.013 [DOI] [PubMed] [Google Scholar]
- 21. Szabó J. E., Németh V., Papp-Kádár V., Nyíri K., Leveles I., Bendes A. Á., Zagyva I., Róna G., Pálinkás H. L., Besztercei B., Ozohanics O., Vékey K., Liliom K., Tóth J., and Vértessy B. G. (2014) Highly potent dUTPase inhibition by a bacterial repressor protein reveals a novel mechanism for gene expression control. Nucleic Acids Res. 42, 11912–11920 10.1093/nar/gku882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Maiques E., Quiles-Puchalt N., Donderis J., Ciges-Tomas J. R., Alite C., Bowring J. Z., Humphrey S., Penadés J. R., and Marina A. (2016) Another look at the mechanism involving trimeric dUTPases in Staphylococcus aureus pathogenicity island induction involves novel players in the party. Nucleic Acids Res. 44, 5457–5469 10.1093/nar/gkw317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Williams D., Norman G., Khoury C., Metcalfe N., Briard J., Laporte A., Sheibani S., Portt L., Mandato C. A., and Greenwood M. T. (2011) Evidence for a second messenger function of dUTP during Bax mediated apoptosis of yeast and mammalian cells. Biochim. Biophys. Acta 1813, 315–321 10.1016/j.bbamcr.2010.11.021 [DOI] [PubMed] [Google Scholar]
- 24. Penadés J. R., Donderis J., García-Caballer M., Tormo-Más M. Á., and Marina A. (2013) dUTPases, the unexplored family of signalling molecules. Curr. Opin. Microbiol. 16, 163–170 10.1016/j.mib.2013.02.005 [DOI] [PubMed] [Google Scholar]
- 25. Pecsi I., Hirmondo R., Brown A. C., Lopata A., Parish T., Vertessy B. G., and Tóth J. (2012) The dUTPase enzyme is essential in Mycobacterium smegmatis. PLoS One 7, e37461 10.1371/journal.pone.0037461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chu R., Lin Y., Rao M. S., and Reddy J. K. (1996) Cloning and identification of rat deoxyuridine triphosphatase as an inhibitor of peroxisome proliferator-activated receptor α. J. Biol. Chem. 271, 27670–27676 10.1074/jbc.271.44.27670 [DOI] [PubMed] [Google Scholar]
- 27. Lo C. F., Ho C. H., Peng S. E., Chen C. H., Hsu H. C., Chiu Y. L., Chang C. F., Liu K. F., Su M. S., Wang C. H., and Kou G. H. (1996) White spot syndrome baculovirus (WSBV) detected in cultured and captured shrimp, crabs and other arthropods. Dis. Aquat. Org. 27, 215–225 10.3354/dao027215 [DOI] [Google Scholar]
- 28. Verbruggen B., Bickley L. K., van Aerle R., Bateman K. S., Stentiford G. D., Santos E. M., and Tyler C. R. (2016) Molecular mechanisms of white spot syndrome virus infection and perspectives on treatments. Viruses 8, E23 10.3390/v8010023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yang F., He J., Lin X., Li Q., Pan D., Zhang X., and Xu X. (2001) Complete genome sequence of the shrimp white spot bacilliform virus. J. Virol. 75, 11811–11820 10.1128/JVI.75.23.11811-11820.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Liu X., and Yang F. (2005) Identification and function of a shrimp white spot syndrome virus (WSSV) gene that encodes a dUTPase. Virus Res. 110, 21–30 10.1016/j.virusres.2005.01.003 [DOI] [PubMed] [Google Scholar]
- 31. Vertessy B. G. (1997) Flexible glycine rich motif of Escherichia coli deoxyuridine triphosphate nucleotidohydrolase is important for functional but not for structural integrity of the enzyme. Proteins 28, 568–579 10.1002/(SICI)1097-0134(199708)28:4%3C568::AID-PROT10%3E3.0.CO%3B2-E [DOI] [PubMed] [Google Scholar]
- 32. Pécsi I., Szabó J. E., Adams S. D., Simon I., Sellers J. R., Vértessy B. G., and Tóth J. (2011) Nucleotide pyrophosphatase employs a P-loop-like motif to enhance catalytic power and NDP/NTP discrimination. Proc. Natl. Acad. Sci. U.S.A. 108, 14437–14442 10.1073/pnas.1013872108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Grishin N. V. (2001) Fold change in evolution of protein structures. J. Struct. Biol. 134, 167–185 10.1006/jsbi.2001.4335 [DOI] [PubMed] [Google Scholar]
- 34. Hormozdiari F., Salari R., Hsing M., Schönhuth A., Chan S. K., Sahinalp S. C., and Cherkasov A. (2009) The effect of insertions and deletions on wirings in protein-protein interaction networks: a large-scale study. J. Comput. Biol. 16, 159–167 10.1089/cmb.2008.03TT [DOI] [PubMed] [Google Scholar]
- 35. Kim R., and Guo J. T. (2010) Systematic analysis of short internal indels and their impact on protein folding. BMC Struct. Biol. 10, 24 10.1186/1472-6807-10-24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Park H., Li A., Chen L. Q., Houdusse A., Selvin P. R., and Sweeney H. L. (2007) The unique insert at the end of the myosin VI motor is the sole determinant of directionality. Proc. Natl. Acad. Sci. U.S.A. 104, 778–783 10.1073/pnas.0610066104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Németh-Pongrácz V., Barabás O., Fuxreiter M., Simon I., Pichová I., Rumlová M., Zábranská H., Svergun D., Petoukhov M., Harmat V., Klement E., Hunyadi-Gulyás E., Medzihradszky K. F., Kónya E., and Vértessy B. G. (2007) Flexible segments modulate co-folding of dUTPase and nucleocapsid proteins. Nucleic Acids Res. 35, 495–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Robinson J. A. (2008) β-Hairpin peptidomimetics: design, structures and biological activities. Acc. Chem. Res. 41, 1278–1288 10.1021/ar700259k [DOI] [PubMed] [Google Scholar]
- 39. Zhang R., Xu A., Qin C., Zhang Q., Chen S., Lang Y., Wang M., Li C., Feng W., Zhang R., Jiang Z., and Tang J. (2017) Pseudorabies virus dUTPase UL50 induces lysosomal degradation of type I interferon receptor 1 and antagonizes the α interferon response. J. Virol. 91, e01148–17 10.1128/JVI.01148-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Aslanidis C., and de Jong P. J. (1990) Ligation-independent cloning of PCR products (LIC-PCR). Nucleic Acids Res. 18, 6069–6074 10.1093/nar/18.20.6069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wang Q. S., Yu F., Huang S., Sun B., Zhang K. H., Liu K., Wang Z. J., Xu C. Y., Wang S. S., Yang L. F., Pan Q. Y., Li L., Zhou H., Cui Y., and Xu Q., et al. (2015) The macromolecular crystallography beamline of SSRF. Nucl. Sci. Tech. 26, 12–17 10.13538/j.1001-8042/nst.26.010102 [DOI] [Google Scholar]
- 42. Kabsch W. (2010) Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr. D Biol. Crystallogr. 66, 133–144 10.1107/S0907444909047374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Vonrhein C., Flensburg C., Keller P., Sharff A., Smart O., Paciorek W., Womack T., and Bricogne G. (2011) Data processing and analysis with the autoPROC toolbox. Acta Crystallogr. D Biol. Crystallogr. 67, 293–302 10.1107/S0907444911007773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 12 10.1107/S09074449090473375–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 10.1107/S0021889807021206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Homma K., and Moriyama H. (2009) Crystallization and crystal-packing studies of Chlorella virus deoxyuridine triphosphatase. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 65, 1030–1034 10.1107/S1744309109034459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Murshudov G. N., Vagin A. A., and Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 10.1107/S0907444996012255 [DOI] [PubMed] [Google Scholar]
- 48. Smart O. S., Womack T. O., Flensburg C., Keller P., Paciorek W., Sharff A., Vonrhein C., and Bricogne G. (2012) Exploiting structure similarity in refinement: automated NCS and target-structure restraints in BUSTER. Acta Crystallogr. D Biol. Crystallogr. 68, 368–380 10.1107/S0907444911056058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 10.1107/S0907444904019158 [DOI] [PubMed] [Google Scholar]
- 50. Robert X., and Gouet P. (2014) Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 42, W320–W324 10.1093/nar/gku316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Krissinel E., and Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 10.1016/j.jmb.2007.05.022 [DOI] [PubMed] [Google Scholar]
- 52. Laskowski R. A., and Swindells M. B. (2011) LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 51, 2778–2786 10.1021/ci200227u [DOI] [PubMed] [Google Scholar]
- 53. Davis I. W., Leaver-Fay A., Chen V. B., Block J. N., Kapral G. J., Wang X., Murray L. W., Arendall W. B. 3rd, Snoeyink J., Richardson J. S., and Richardson D. C. (2007) MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 35, W375–W383 10.1093/nar/gkm216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., et al. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 10.1107/S0907444910045749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Satow Y., Cohen G. H., Padlan E. A., and Davies D. R. (1986) Phosphocholine binding immunoglobulin Fab McPC603: an X-ray diffraction study at 2.7 Å. J. Mol. Biol. 190, 593–604 10.1016/0022-2836(86)90245-7 [DOI] [PubMed] [Google Scholar]
- 56. Vertessy B. G., Persson R., Rosengren A. M., Zeppezauer M., and Nyman P. O. (1996) Specific derivatization of the active site tyrosine in dUTPase perturbs ligand binding to the active site. Biochem. Biophys. Res. Commun. 219, 294–300 10.1006/bbrc.1996.0226 [DOI] [PubMed] [Google Scholar]
- 57. Niesen F. H., Berglund H., and Vedadi M. (2007) The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat. Protoc. 2, 2212–2221 10.1038/nprot.2007.321 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.