

Abstract
Mutations in the MYO7A gene, encoding the motor protein myosin VIIa, can cause Usher 1B, a deafness/blindness syndrome in humans, and the shaker-1 phenotype, characterized by deafness, head tossing, and circling behavior, in mice. Myosin VIIa is responsible for tension bearing and the transduction mechanism in the stereocilia and for melanosome transport in the retina, in line with the phenotypic outcomes observed in mice. However, the effect of the shaker-1 mutation, a R502P amino acid substitution, on the motor function is unclear. To explore this question, we determined the kinetic properties and the effect on the filopodial tip localization of the recombinant mouse myosin VIIa-5IQ-SAH R502P (myoVIIa-sh1) construct. Interestingly, although residue 502 is localized to a region thought to be involved in interacting with actin, the kinetic parameters for actin binding changed only slightly for the mutant construct. However, the rate constant for ATP hydrolysis (k+H + k−H) was reduced by ∼200-fold from 12 s−1 to 0.05 s−1, making the hydrolysis step the rate-limiting step of the ATPase cycle in the presence and absence of actin. Given that wild-type mouse myosin VIIa is a slow, high-duty ratio, monomeric motor, this altered hydrolysis rate would reduce activity to extremely low levels. Indeed, the translocation to the filopodial tips was hampered by the diminished motor function of a dimeric construct of the shaker-1 mutant. We conclude that the diminished motor activity of this mutant is most likely responsible for impaired hearing in the shaker-1 mice.
Keywords: actin, ATPase, enzyme kinetics, fluorescence, molecular motor, myosin
Introduction
Myosins are a large family of mechanochemical motor proteins that are well-known for their role in muscle contraction and other cellular processes, such as cargo transport, cell division, phagocytosis, etc. (1–3). Myosins have a motor domain that contains the nucleotide- and actin-binding sites, a neck domain that binds the light chains, and a tail domain that has a variety of functions, including cargo binding (4). They hydrolyze ATP to ADP and Pi to provide the energy for muscle contraction (5), cellular motility, cargo transport, etc. (6). About 35 classes of myosins (7) have been broadly categorized as either conventional or unconventional myosins. Conventional myosins, such as skeletal myosin, usually have a role in muscle contraction, and unconventional myosins have a variety of roles, including intracellular transport, cell division, and cell motility (8–11).
Myosin VIIa is an unconventional myosin that has a motor domain, a 5IQ neck domain, a single-α-helix (SAH)2 domain, two large repeats of a myosin tail homology 4 (MyTH4) domain, an SH3 domain, and two 4.1-ezrin-radixin-moesin (FERM) domains that may function as a cargo-binding site (12, 13). It has been shown that the SAH domain can extend the functional length of the neck or 6IQ domain of myosin Va (14). Recently, the 5IQ neck domain and the SAH domain of myosin VIIa were crystallized with apo-calmodulin bound to the IQ domains. The structure shows that the predicted SAH domain forms a stable single α helix that can extend the lever arm (15). Myosin VIIa is expressed in a number of mammalian tissues, including testis, kidney, lung, inner ear, retina, and the ciliated epithelium of the nasal mucosa (16, 17). It has a role in cargo transport (18), endocytosis, cell adhesion (19), and tension bearing (20). In the hair cells of the inner ear, it is localized to lateral links and by interaction with vezatin to the fibrillar links that connect the bases of adjacent stereocilia (20, 21). Myosin VIIa influences actin treadmilling and therefore regulates the height of stereocilia (22). Actin assembly might also be regulated by the cargo protein twifilin-2, which binds to myosin VIIa and caps and severs actin (23). Grati and Kachar (24) showed that myosin VIIa and sans, a myosin VIIa–interacting protein, cluster at the upper tip-link density of the stereocilia, where they form a complex with harmonin-b. In this complex, myosin VIIa is the likely motor that pulls on the cadherin-23 protocadherin-15 complex and is directly linked to the mechanotransduction channel, which indicates a possible role for myosin VIIa in the transduction/adaptation mechanism. In the retina, myosin VIIa is present in the apical retinal pigmented epithelium, where it is involved in the transport of melanosomes by forming a complex with MyRIP and Rab27a (25–27). In photoreceptor cells, it is associated with the ciliary and periciliary plasma membranes and transports opsin between the inner and outer segment (27).
Patients carrying MYO7A mutations suffer from Usher syndrome type 1B, which presents as hereditary deafness and progressive retinal degeneration (retinitis pigmentosa) (28), and two autosomal recessive disorders associated with non-syndromic hearing loss, DFNB2 and DFNA11 (29–31).
Usher syndrome, an autosomal recessive inherited disease, is the most common condition that affects both hearing and vision (28, 32). The prevalence of Usher syndrome in the United States is about 1 in 23,000 live births (33). It is clinically classified into three subtypes. About half of the cases of Usher syndrome belong to the most severe Usher1B subtype. Mutations in the MYO7A gene lead to the shaker-1 phenotype in mice that partially replicate the symptoms of Usher syndrome (34). The “original” shaker-1 (34, 35) mice carry a missense mutation in the MYO7A gene, which results in the R502P amino acid substitution in the motor domain, leading to deafness, head tossing, circling behavior, and a mild retinal phenotype (34). Developmental and electrophysiological studies have shown that the shaker-1 mice have minor (36) anomalies, such as the predominance of outer hair cells with only two rows of stereocilia instead of the more usual three. These mice showed increased cochlear responses, which is abnormal and may interfere with the cochlear function (36).
Previously, we found that the wild-type mouse myosin VIIa is a slow, high-duty ratio, monomeric motor protein (37). To understand the motor biochemistry of the shaker-1 myosin VIIa in the present study, we used the baculoviral expressed truncated shaker-1 (myoVIIa-sh1) construct to analyze the mechanochemical cycle of this mutant. The Arg-502 residue is in a loop between the Q and R helices that has been called the activation loop (38) or supporting loop (39) that interacts with the N-terminal segment of actin in many myosins. In the shaker-1 mutant, the sequence of this loop is changed from NRPM to NPPM. This is likely to alter the conformation of the Q helix–loop–R helix structure, which is relatively straight in all myosins. Fig. 1A shows the location of the Arg-502 residue superimposed on the crystal structure of the chicken myosin V (40). Fig. 1B shows the alignment of this loop for several myosins.
Figure 1.

Location of the shaker-1 mutation. A, map of the myosin VIIa shaker-1 mutation on the crystal structure of chicken myosinV-ADP-BeFX. Arg-502 resides in the activation loop/supporting loop (red). Switch 1 loop (SW1) (yellow), the P-loop (purple), switch 2 loop (SW2) (yellow), the relay loop (blue), the relay helix (blue), the helix-loop-helix + activation/supporting loop (blue), and essential light chain (eLC) (pink) are shown. B, alignment of the activation/supporting loop for several myosins. Note that the flanking residues are alanine and asparagine for myosin VIIa but glutamic acid and glycine for all of the other myosins. The mutated proline residue for the shaker-1 myosin VIIa mutant is circled in red.
To better understand the physiological changes produced by this mutation, we have performed a detailed study of the ATP hydrolysis mechanism to determine which steps are altered by the R502P shaker-1 mutation.
Results
Steady-state basal and actin-activated ATPase activity of myoVIIa-sh1
The basal and actin-activated ATPase activities were measured by an NADH-coupled assay. The basal steady-state rate of 0.03 s−1 was activated ∼1.8-fold by actin filaments, which resulted in a rate of 0.054 s−1 (Fig. 2). The actin concentration at half-maximum of the steady-state ATPase rate was 2.74 ± 0.2 μm. There was a 10-fold decrease in the basal steady-state ATPase activity from 0.2 to 0.03 s−1 for the myoVIIa-sh1 compared with myoVIIa-WT (wild type) (37). Similarly, the actin-activated steady-state ATPase of myosin VIIa-sh1 decreased more than 10-fold from 0.7 s−1 to 0.054 s−1. The lower Kactin measured for myoVIIa-sh1 compared with the myoVIIa-WT is probably due to the lower maximum rate, not tighter binding of the myosin VIIa-sh1-ADP-Pi complex to actin filaments. These results (summarized in Table 1) suggest a significantly reduced motor function for the myoVIIa-sh1 mutant for which there is little if any actin activation of the basal steady-state ATPase.
Figure 2.

NADH coupled actin activated ATPase assays. Actin-activated ATPase experiments were measured by an NADH-coupled assay at 25 °C using 2 μm myoVIIa-sh1 in a buffer containing 10 mm MOPS (pH 7.5), 3 mm MgCl2, 1 mm DTT, 2 mm ATP, 0.15 mm EGTA, 2 μm CaM, 40 units/ml lactate dehydrogenase, 200 units/ml pyruvate kinase, 1 mm phosphoenolpyruvate, and 200 μm NADH. The actin-activated ATPase rate had a Vmax of 0.054 ± 0.01 s−1 and a Kactin of 2.74 ± 0.2 μm. Error bars, S.E.
Table 1.
Steady-state kinetic parameters
Values are shown ± S.E.
| Parameter | WTa | Shaker-1 |
|---|---|---|
| kbasal (s−1) | 0.2 ± 0.02 | 0.03 ± 0.001 |
| Vmax (s−1) | 0.7 ± 0.02 | 0.054 ± 0.01 |
| Kactin (μm) | 10.5 ± 1.7 | 2.74 ± 0.2 |
a The values listed for WT myosin VIIa are from Haithcock et al. (37).
To investigate which step(s) of the ATP hydrolysis cycle are affected by the shaker-1 mutation, we have done a detailed kinetic study using stopped-flow fluorescence and quenched flow. Scheme 1 is a schematic representation of the ATPase cycle.
Scheme 1.
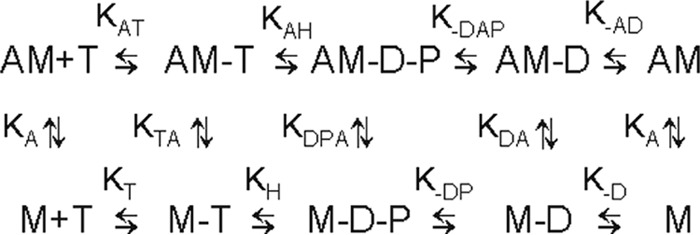
Minimal kinetic scheme of the ATPase cycle. A, actin; M, myoVIIa-sh1; T, ATP; D, ADP; P, phosphate. Positive subscripts denote the binding of the last ligand in the subscript, and negative subscripts denote dissociation (e.g. k−AD is the dissociation of ADP from actomyoVIIa-sh1-ADP).
Actin binding; myoVIIa-sh1 binding to actin in the presence and absence of ADP (A + M ⇄ AM; A + M-ADP ⇄ AM-ADP)
To investigate the kinetics of myoVIIa-sh1 binding to actin, we performed single mixing stopped-flow experiments by mixing myosin with actin in the presence or absence of ADP. The change in light scattering was recorded, and time courses were fit to a biexponential equation (Fig. 3A). The fast phase represents the kinetics of actin binding to myoVIIa-sh1 or myoVIIa-sh1-ADP and depends linearly on the actin concentration (Fig. 3B). The second phase was erratic and did not show a dependence upon actin concentrations and was not plotted. The apparent second order rate constant for myoVIIa-sh1 binding to actin is kA = 2.2 μm−1 s−1 in the absence and kDA = 0.5 μm−1 s−1 in the presence of ADP (Fig. 3B). Compared with our previously published data using wild-type myosin VIIa, there is a ∼50% reduction in the second order rate constant for actin binding to myosin VII-sh1 in the absence of ADP and a ∼5-fold reduction in the presence of ADP.
Figure 3.

MyoVIIa-sh1 binding to actin filaments. A, observed change in light scattering upon binding of myoVIIa-sh1 to actin. Experimental conditions were as follows: 0.8 μm myoVIIa-sh1 and 2.5 μm actin, 4 μm CaM, 10 mm MOPS, 3 mm MgCl2, 1 mm EGTA, 25 mm KCl, 1 mm DTT, pH 7.5, 20 °C. Three traces were averaged, and the solid line was fit to a biexponential equation: I(t) = 0.05e−4.7t + 0.03e−0.3t + C. B, concentration dependence of the observed fast rate of myoVIIa-sh1 binding to actin in the presence (■) and absence (□) of 20 μm ADP. Experimental conditions are the same as in A except that the actin concentration was varied between 1.4 and 7.2 μm. The data were fit to a linear equation and resulted in a second order rate constant of kA = 2.2 ± 0.16 μm−1 s−1 and kDA = 0.5 ± 0.05 μm−1 s−1, respectively.
ATP binding and hydrolysis
dmantATP binding to myosin VIIa-sh1 and actomyosin VIIa-sh1 (M + ATP ⇄ M-ATP; AM + ATP ⇄ AM-ATP)
The binding of dmantATP to myoVIIa-sh1 and actomyoVIIa-sh1 was measured by mixing dmantATP and myoVIIa-sh1 or actomyoVIIa-sh1 in the single mixing stopped flow (Fig. 4, A and B). The fluorescence increase was fit to a single exponential equation. The data showed a linear dependence on dmantATP concentration and resulted in an apparent second order rate constant of 1.7 μm−1 s−1 for ATP binding (kT) to myoVIIa-sh1 and a rate constant of 1.2 μm−1 s−1 for ATP binding (kAT) to actomyoVIIa-sh1 (Fig. 4C). These rate constants were not significantly different from those observed for dmantATP binding to wild-type myosin VIIa and actomyosin VIIa (37).
Figure 4.

dmantATP binding to myoVIIa-sh1 and actomyoVIIa-sh1. 1.1 μm myoVIIa-sh1 (A) and 0.7 μm actomyoVIIa-sh1 (0.7 μm myosin and 1.4 μm actin) (B) were mixed with 7.2 and 10.7 μm dmantATP, respectively. The increase in fluorescence was recorded, and three traces were averaged and fit to a single exponential equation, resulting in a kobs of 10.7 s−1 (A) and 17.7 s−1 (B). Experimental conditions in the cell were as follows: 1.1 μm myoVIIa-sh1 (A), 0.7 μm myosin and 1.4 μm actin (B), 5.5 μm CaM (A) and 3.5 μm CaM (B), 10 mm MOPS, 3 mm MgCl2, 1 mm EGTA, 25 mm KCl, 1 mm DTT, pH 7.5, 20 °C. C, experimental conditions were the same as in A and B except that 1.8–18 μm dmantATP was used. The data were fit to a linear equation and resulted in apparent second order association rate constants of kT = 1.7 μm−1 s−1 ± 0.07 and kAT = 1.2 μm−1 s−1 ± 0.14 for dmantATP binding to myoVIIa-sh1 (■) and actomyoVIIa-sh1 (□), respectively.
Quenched-flow measurements of pre-steady-state hydrolysis—
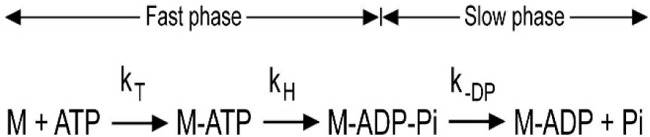 |
We used quenched flow to determine the rate of the hydrolysis step (Reaction 1). Reaction 1 describes the steps in the ATP hydrolysis measured by quenched flow. There is an initial fast phase, which is a measure of ATP binding and hydrolysis, and a slow phase, which is limited by phosphate dissociation. In these experiments, myoVIIa-sh1 was mixed with substoichiometric amounts of ATP, and the data were best fit by a single exponential equation resulting in a rate of 0.05 s−1 (Fig. 5), which is only slightly faster than the basal steady-state ATPase rate, 0.03 s−1, measured in Fig. 2. We also carried out stopped-flow experiments to measure the rate of the increase in tryptophan fluorescence by mixing myoVIIa-sh1 with ATP and observed a signal with only a 2% fluorescence increase (data not shown). The smaller signal for the shaker-1 mutant may be due to M-ATP being the primary steady-state intermediate rather than M-ADP-Pi, as was observed for the wild-type myosin VIIa. The rate of ATP binding was determined by the initial slope of 0.5 μm−1 s−1, which is 4-fold slower than was measured for the wild-type protein. This indicates that ATP binding is faster than the rate measured for the hydrolysis, which is 200-fold slower than measured for the wild-type myosin VIIa. This interpretation of the mechanism is consistent with the small actin activation of the steady-state ATPase rate to a value equal to that of the hydrolysis rate shown in Fig. 5. It was surprising that the primary effect of the mutation is on the hydrolysis step and not on the actin binding, as the R502P shaker-1 mutation is part of the actin-binding interface, which is on the other side of the myosin molecule from the ATP-binding site (Fig. 1A).
Figure 5.

Quenched-flow measurements of presteady-state ATP hydrolysis. 8 μm myoVIIa-sh1 was mixed with 2 μm ATP and allowed to react for the indicated amount and then quenched with acid. Experimental conditions were as follows: 50 mm KCl, 10 mm MOPS, 3 mm MgCl2, 20 μm CaM, 1 mm EGTA, pH 7.5, 20 °C. Final concentrations in the cell were as follows: 4 μm myosin VIIa-sh1 and 1 μm ATP. The solid lines through the data fit best to a single exponential equation, I(t) = 0.67e−0.05 + C.
Dissociation of myoVIIa-sh1 from actin upon ATP binding (AM + ATP ⇄ AM-ATP → A + M-ATP)
ATP binding to rigor myosin is followed by a change in the conformation of the myosin that results in reduced affinity and rapid dissociation of the M-ATP from actin. The rate of dissociation of myoVIIa-sh1 from actin in the presence of ATP was determined by measuring the change in light scattering (Fig. 6). ActomyoVIIa-sh1 was mixed with increasing concentrations of ATP in the stopped flow. The change in light scattering was recorded, and the average of the fast phase from three traces was fit to a single exponential equation. The observed rate constants were plotted against the ATP concentrations, and a maximal rate (k−TA) of 420 s−1 and a kAT of 1.2 μm−1 s−1 were obtained (Fig. 6B). There was a ∼1.3-fold difference between the maximal rates of dissociation from actin measured for the myoVIIa-sh1 when compared with myoVIIa-WT, which are the same within the error of measurement of such rapid steps (37).
Figure 6.

Dissociation of myoVIIa-sh1 from actin upon ATP binding. Experimental conditions in the cell were as follows: actomyoVIIa-sh1 (0.7 μm myosin + 1.4 μm actin) and 3.5 μm CaM, 0.72–715 μm ATP, 25 mm KCl, 10 mm MOPS, 3 mm MgCl2, 1 mm EGTA, pH 7.5, 20 °C. A, the decrease in light scattering was observed upon mixing actomyosin VIIa-sh1 with MgATP. Three traces for each ATP concentration were averaged and fit to a single exponential equation. A representative trace at 21.5 μm ATP yielded I(t) = 0.13e−26.8 + C. B, the dependence of kobs upon ATP concentration was fit to a hyperbola, resulting in a maximal kobs of 420.4 ± 17.4 s−1 and a Kapp = 293 ± 23.5 μm. Inset, data from 0–50 μm ATP. The rate of ATP binding to actomyosin calculated from the initial slope is 1.2 ± 0.01 μm−1 s−1.
ADP binding and dissociation
dmantADP binding to myoVIIa-sh1 and actomyoVIIa-sh1 (M + ADP ⇄ M-ADP; AM + ADP ⇄ AM-ADP)
The rate of dmantADP binding was measured by mixing myoVII-sh1 or actomyoVIIa-sh1 with increasing amounts of dmantADP (Fig. 7). Time courses were fit by single exponentials (Fig. 7, A and B), and a linear fit to the observed rate constants resulted in a second order rate constant of kD = 0.4 μm−1 s−1 for dmantADP binding to myoVIIa-sh1, which is one-third the rate measured for the wild type, and kAD = 1.8 μm−1 s−1 for dmantADP binding to actomyoVIIa-sh1, which is similar to the wild-type myosin VIIa (Fig. 7C).
Figure 7.

Kinetics of dmantADP binding to myoVIIa-sh1 and actomyoVIIa-sh1. 1.2 μm myoVIIa-sh1 (A) or 1.2 μm myoVIIa-sh1 + 2.4 μm actin (B) was mixed with dmantADP in single mixing stopped-flow fluorescence experiments. Experimental conditions were as follows: 25 mm KCl, 10 mm MOPS, 6 μm calmodulin, 3 mm MgCl2, 1 mm EGTA, pH 7.5, 20 °C. A, shows the average of three traces of the stopped-flow recording of 1.2 μm myoVIIa-sh1 binding to 7.2 μm dmantADP. The solid line was fit to a single exponential equation, resulting in a kobs of 4.2 s−1. B, average of three traces of the stopped-flow recording of 1.2 μm actomyosin VIIa-sh1 binding to 3.6 μm dmantADP. The solid line was best fit to a single exponential equation with a kobs of 7.4 s−1. C, experimental conditions are the same as in A and B except that 1.2 μm myoVIIa-sh1 or 1.2 μm actomyoVIIa-sh1 was mixed with increasing concentrations (3.6–17.8 μm) of dmantADP. The second order rate constant for dmantADP binding (kD) to myoVIIa-sh1 was 0.4 ± 0.04 μm−1 s−1, and the value for binding to actomyoVIIa-sh1 (kAD) was 1.8 ± 0.08 μm−1 s−1. The rate of ADP dissociation calculated from the intercept was 1.6 ± 0.43 s−1 for myosin VIIa-sh1 and 1.8 ± 0.2 s−1 for actomyosin VIIa-sh1.
Dissociation of ADP from actomyosin VIIa-sh1 (AM-ADP + ATP ⇄ AM + ATP → A + M-ATP)
The rate of ADP dissociation from actomyoVIIa-sh1-ADP was measured by mixing actomyosin VIIa-ADP with increasing concentrations of ATP and recording the change in light scattering (Fig. 8). The observed rate constants can be used to measure the rate of ADP dissociation from actomyoVIIa-ADP because the dissociation of myosin from actin by ATP is limited by the rate of ADP dissociation. Time courses were monophasic and were fit to single exponential equations (Fig. 8A). The observed rate constants showed a hyperbolic dependence on the ATP concentration, resulting in a maximal rate of k−AD = 1 s−1 (Fig. 8B) that is similar to what was obtained for the wild-type myosin VIIa construct (37).
Figure 8.

Dissociation of myoVIIa-sh1 ADP from actin upon ATP binding. 0.74 μm actomyoVIIa-sh1 (0.74 μm myosin + 1.48 μm actin) was mixed with ATP in the presence of ADP. The decrease in light scattering was observed upon mixing the actomyosin ADP complex with MgATP. Experimental conditions in the cell were as follows: 0.74 μm actomyoVIIa-sh1 (0.74 μm actomyoVIIa-sh1 + 1.48 μm actin), 35.7 μm ADP, 3.6 μm CaM, 1.07–714.3 μm ATP, 25 mm KCl, 10 mm MOPS, 3 mm MgCl2, 1 mm EGTA, pH 7.5, 20 °C. Three traces for each ATP concentration were averaged and fit to a single exponential equation. A, representative trace at 35.7 μm ATP yielded I(t) = 0.39e−0.84 + C. B, the dependence of kobs upon ATP concentration was fit to a hyperbola, resulting in a maximal kobs of 1 ± 0.01 s−1 and a Kapp = 2.63 ± 0.27 μm.
Phosphate release (M-ADP-Pi + A ⇄ AM-ADP-Pi → AM-ADP + Pi)
A fluorescently labeled phosphate-binding protein (MDCC-PBP) was used to monitor inorganic phosphate dissociation from the actomyoVIIa-sh1-ADP-Pi complex. In a double mixing stopped-flow experiment, myoVIIa-sh1 was first mixed with substoichiometric ATP and incubated for 10 s to allow ATP binding and hydrolysis to occur, followed by mixing with increasing concentrations of actin filaments to measure the rate of phosphate release from actomyoVIIa-sh1-ADP-Pi. Time courses were fit to single exponential equations (Fig. 9A). The maximal rate constant of phosphate dissociation (k−DAP) was ≥0.05 s−1 (Fig. 9C). The observed rates of phosphate dissociation were not dependent on the actin concentration and were the same within experimental error as the rates observed by quenched flow, 0.05 s−1, for the hydrolysis in Fig. 5. Double exponential fits of the signal obtained in these experiments (Fig. 9B) show that there is a small component (5–20% amplitude) with a rate constant of ∼0.2–0.7 s−1, which is faster than the actin-activated steady-state rate but still ∼20-fold slower than the rate observed for phosphate dissociation from wild-type actomyosin VIIa-ADP-Pi (37). This is consistent with the ATP hydrolysis being the rate-limiting step, because, based on these observations, M-ADP-Pi comprise only ∼10% of the intermediates, and ∼90% of the steady-state intermediates are in the M-ATP state. This is very different from what was observed for the myosin VIIa-WT, for which the amplitude of the fast phase was ∼50% and the extrapolated maximal rate of phosphate dissociation was >12 s−1. The transient kinetic data are summarized in Table 2.
Figure 9.

Phosphate dissociation from the actomyoVIIa-sh1-ADP-Pi complex. Double mixing stopped-flow experiments were performed using MDCC-PBP. MyoVIIa-sh1 was first mixed with ATP, held in a delay line for 10 s, and then mixed with actin to accelerate Pi release. Final concentrations in the flow cell were 0.8 μm myoVIIa-sh1, 4 μm CaM, 0.4 μm ATP, 0–44 μm actin, 1 mm EGTA, 3 mm MgCl2, 10 mm MOPS, 25 mm KCl, 10 μm MDCC-PBP, 0.1 mm 7-methylguanosine, and 0.01 unit/ml purine nucleoside phosphorylase, pH 7.5, 20 °C. A, phosphate dissociation from the 0.8 μm shaker-1-ADP-Pi complex in the presence of 20 μm actin. A single exponential equation fit through the data resulted in I(t) = 0.28e−0.06 + C. B, a biexponential fit to the data resulted in I(t) = 0.03e−0.29 + 0.26e−0.05. C, dependence of the kobs of the slow phase on actin concentrations resulted in a k−DAP ≥ 0.05 s−1.
Table 2.
Transient kinetic parameters
Values are shown ± S.E. ND, not determined.
| Parameter | Value |
Nucleotide | Method | |
|---|---|---|---|---|
| WTa | sh1 | |||
| ATP binding | ||||
| kT (μm−1 s−1) | 2.5 ± 0.04 | 1.7 ± 0.07 | dmantATP | Stopped-flow fluorescence |
| kAT (μm−1 s−1) | 0.5 ± 0.04 | 1.2 ± 0.14 | dmantATP | Stopped-flow fluorescence |
| kAT (μm−1 s−1) | 1.2 ± 0.03 | 1.2 ± 0.01 | ATP | Light scattering |
| k−TA (s−1) | 350 ± 19 | 420 ± 17.4 | ATP | Light scattering |
| Hydrolysis | ||||
| kH + k−H (s−1) | 12 ± 0.9 | 0.05 ± 0.01 | ATP | Quenched flow |
| KH | 1 | ND | ATP | Quenched flow |
| ADP binding | ||||
| kD (μm−1 s−1) | 1.2 ± 0.06 | 0.4 ± 0.04 | dmantADP | Stopped-flow fluorescence |
| k−D (s−1) | 2.1 ± 0.06 | 1.6 ± 0.43 | dmantADP | Stopped-flow fluorescence |
| KD (μm) | 1.75 | 4 | dmantADP | k−D/kD |
| kAD (μm−1 s−1) | 1.5 ± 0.09 | 1.8 ± 0.08 | dmantADP | Stopped-flow fluorescence |
| k−AD (s−1) | 1.7 ± 0.2 | 1.8 ± 0.2 | dmantADP | Light scattering |
| k−AD (s−1) | 1.28 ± 0.09 | 1 ± 0.01 | ADP | Light scattering |
| KAD (μm) | 1 | 0.8 | dmantADP | k−AD/kAD |
| Pi release | ||||
| k−DP (s−1) | 0.26 ± 0.07 | 0.05 | ATP | MDCC-PBP |
| k−DAP (s−1) | 12 ± 0.03 | ≥0.05; >0.2b | ATP | MDCC-PBP |
| Actin binding | ||||
| kA (μm−1 s−1) | 4.7 ± 0.25 | 2.2 ± 0.2 | Light scattering | |
| kDA (m−1 s−1) | 2.6 ± 0.25 | 0.5 ± 0.05 | Light scattering | |
a The rate constants for the WT myosin VIIa are from Haithcock et al. (37).
b A biexponential fit to the data resulted in rates >0.2 s−1 for the fast component. The amplitude of the fast phase was 5–20% of the total amplitude.
Localization of myoVIIa-WT and myoVIIa-sh1 in HeLa cells
To further understand the motor function of this mutant and how the shaker-1 mutation affects localization of the myosin VIIa to the filopodial tips, we transfected HeLa cells with pEGFP-C1-myoVIIa-WT-GCN4 or pEGFP-C1-myoVIIa-sh1-GCN4 constructs. An accumulation of the myosin VIIa at the tip of filopodia was only observed by transfection with pEGFP-C1-myoVIIa-WT-GCN4 but not pEGFP-C1-myoVIIa-sh1-GCN4 (Fig. 10). In addition, we did not observe any filopodial tip localization with either the wild-type or shaker-1 monomeric constructs (data not shown).
Figure 10.

Localization of myoVIIa-WT and myoVIIa-sh1 in HeLa cells. HeLa cells were transfected with pEGF-C1-myoVIIa-WT-GCN4 (A) or pEGFP-C1-myoVIIa-sh1-GCN4 (B) and then fixed with 4% paraformaldehyde and stained with 500 nm Texas Red phalloidin 14 h after transfection. C, percentage of cells with myoVIIa accumulation at the tip of filopodia. Left bar, pEGFP-C1-myosin VIIa WT-GCN4; right bar, pEGFP-C1-myosin VIIa shaker-1-GCN4. n = 3; mean ± S.E. (error bars).
The lack of accumulation of the shaker-1 protein at the filopodial tips indicates that the motor function is essential in this process, as was observed by Sakai et al. (25).
Discussion
Mutations in the MYO7A gene cause Usher syndrome type IB, which is an inherited deafness/blindness disorder (28). The shaker-1 mice, the mouse model for Usher 1B, exhibit circling behavior, head tossing, deafness, and mild retinal phenotype (34, 41, 42). We have previously published the kinetic analysis of the wild-type mouse myosin VIIa (37). The WT myosin VIIa has a steady-state ATPase rate of 0.7 s−1 and is a high-duty ratio motor (0.6). The rate-limiting step of the ATP hydrolysis mechanism is partially the ADP dissociation from actomyosin ADP and partially the attached hydrolysis at high actin concentrations. A point mutation in the myosin VIIa sequence is responsible for the arginine to proline substitution of residue 502 in the shaker-1 mice.
In this study, we have analyzed the motor biochemistry of the baculovirus-expressed myosin VIIa-5IQ-SAH shaker-1 mutant (R502P). The maximal rates of the basal steady-state ATPase and the actin-activated steady-state ATPase are 0.030 and 0.054 s−1, respectively, indicating that there is almost no actin activation of the basal rate. These rates are 6–15-fold slower than we observed for the wild-type construct (37). The major finding is that the observed rate of the ATP hydrolysis step, 0.05 s−1, measured directly by quenched flow, is ∼200-fold slower than we obtained for the wild-type myosin VIIa. Because there was <2-fold actin activation of the steady-state ATPase, we used Equation 1 to calculate the duty ratio of the shaker-1 construct (43) to be 0.04, which is ∼15-fold lower than we obtained for the wild-type myosin VIIa (0.6).
| (Eq. 1) |
Consistent with the hampered motor function of the shaker-1 mutant, there was no filopodial tip localization of the EGFP-tagged dimeric construct.
In the kinetic mechanism shown in Scheme 2, the rapid ATP binding to myosin VIIa is followed by slow and rate-limiting ATP hydrolysis. Actin accelerates phosphate dissociation, but the steady-state rate only increases 2-fold, as it is limited by the slow hydrolysis step. The small amplitudes of the phosphate dissociation measured in double mixing experiments in Fig. 9 are due to the slow hydrolysis rate and similar rate of breakdown of M-ADP-Pi to products in the absence of actin. A mechanism in which the hydrolysis step is reversible and the phosphate dissociation rates are more rapid would produce similar experimental results, but our data cannot distinguish between the two mechanisms. The most surprising result is the large effect of the mutation on the rate of the hydrolysis step that becomes the rate-limiting step of the mechanism, considering the distance between the site of the mutation and the active site (Fig. 1A). The specificity of reduction of the hydrolysis rate can be compared with the modest changes in the rates of ATP dissociation of actomyosin and the rate of ADP dissociation from actomyosin. Bar graphs show the comparison between the rate constants of the shaker-1 mutant and wild-type myosin VIIa constructs. It has been previously observed by White et al. (44) that actin binding to myosin decreases the rate of the hydrolysis step, which suggests that the mutation might mimic the effect of actin binding on the catalytic site of myosin. As it was shown in Fig. 1A, the shaker-1 mutation is in a loop (activation/supporting loop) of a helix-loop-helix structure in the actin-binding interface in which two short concentrically aligned helices (Q and R helices) are connected by 3–6 amino acids that may contain as many as three prolines in some myosins. The sequence of the loop and the flanking sequences are unique to myosin VIIa when compared with several other myosins (Fig. 1B). Previous work showed that the activation loop has a role in actin activation (38).
Scheme 2.

ATP hydrolysis mechanism of the myosin VIIa shaker-1 mutant construct. A, actin; M, myosin. Bar graphs show the comparison between the rate constants for wild-type and shaker-1 myosin.
Our results indicate that besides actin binding, the “activation loop/supporting loop” has a significant role in the detached hydrolysis step. A question that remains to be answered is how a mutation near the actin-binding site, which is on the opposite side of the myosin head from the ATP-binding site (Fig. 1A) has only modest effects (<2-fold) on the rates of actin, ADP, and ATP binding to myosin VIIa-sh1 but has such a large effect (∼200-fold) on the rate of ATP hydrolysis (Table 2). Fig. 1A shows the direct connection via the relay loop and relay helix between R502P and switch 2 of the active site. Along this pathway in the relay loop is Trp-484, which has a fluorescence enhancement that is coupled to ATP hydrolysis in the active site. It is thought that this fluorescence enhancement is produced by a conformational change that precedes hydrolysis of ATP (38, 45). The amplitude of the fluorescence enhancement is decreased by a factor of 2 in the shaker mutant, which suggests that the conformational change that occurs before ATP hydrolysis has been significantly altered by the mutation. We therefore propose that the mutation R502P in the activation loop alters the movement that is required for the conformational change that precedes hydrolysis. Future structure–function studies are needed to determine whether this mechanism is correct.
Experimental procedures
Construction of the shaker-1 mutant cDNA and expression and purification of the shaker-1 protein
The cDNA containing the shaker-1 (R502P) mutation (myoVIIa-sh1) was engineered by site-directed mutagenesis of the wild-type mouse pFastBac 1 myosin VIIa-5IQ-SAH construct, which contains the head domain, the neck domain, and an SAH domain with a C-terminal FLAG tag (DYKDDDK) (37).
The myoVIIa-sh1 construct was transformed into DH10Bac cells (Invitrogen). The Bacmid DNA was then transfected into SF9 insect cells to produce the low-titer myoVIIa-sh1 virus, which was amplified by several passages to obtain the high-titer virus stock. The high-titer baculovirus containing myoVIIa-sh1 was co-infected with calmodulin (CaM) virus into SF9 cells for protein expression. FLAG affinity chromatography was used to purify the myoVIIa-sh1 protein. For further purification and concentration of the protein, a Q-Sepharose column and a step salt gradient (100–500 mm KCl) were used. The protein concentration was determined from A280 − 1.5 A320 (A320 was subtracted to correct for light scattering) using the extinction coefficient per active site of ϵ280 = 0.099 μm−1 cm−1. All protein preparations were analyzed by SDS-polyacrylamide gel and active site titration with dmantATP.
Plasmids for expression of the myoVIIa-WT and myoVIIa-sh1 in HeLa cells were engineered by subcloning the myoVIIa-WT-5IQ-SAH and myoVIIa-sh1–5IQ-SAH cDNA with and without a GCN4 sequence into the pEGFP-C1 expression vector using BglII and BamHI and the Infusion HD cloning kit (Clontech) to obtain both monomeric and dimeric constructs (Fig. 9).
Reagents
Actin was purified from rabbit skeletal muscle (46). Rat calmodulin was expressed in BL21 (DE3) cells using the manufacturer's protocol (Invitrogen) and purified using Sepharose CL-4B (47, 48). ATP and ADP were purchased from Sigma-Aldrich. MDCC-PBP was synthesized according to Brune et al. (49). N-Methylanthraniloyl derivatives of the 2′-deoxynucleotides 3′-O-(N-methylanthraniloyl)-2′-deoxy-adenosine 5′-diphosphate (dmantADP) and 3′-O-(N-methylanthraniloyl)-2′-deoxy-adenosine 5′-triphosphate (dmantATP) were synthesized and purified according to the method of Hiratsuka et al. (50). 7-Methylguanosine and purine-nucleoside phosphorylase were purchased from Sigma-Aldrich.
Actin-activated steady-state ATPase measurements
Experiments were carried out in a buffer containing 10 mm MOPS, 3 mm MgCl2, 1 mm EGTA, pH 7.5, 25 °C. Steady-state ATPase activity was measured by an NADH-coupled assay as described by Haithcock et al. (37). The ATPase activity of the blanks containing actin filaments was subtracted from the actomyosin data.
Quenched-flow experiments
Chemical quench measurements were done using a computer-controlled stepper motor apparatus built in our laboratory (44) to drive syringes in a mixing unit from KinTek Corp. (Austin, TX). Solutions of myoVIIa-sh1 (8 μm) and ATP (2 μm) containing 100,000 dpm of [γ-32P]ATP) were mixed, held in a delay line for the desired time, and then quenched by a second mix with 0.35 m KH2PO4, 2 n HCl to give a final volume of 1.0 ml. The total radioactivity in each sample was determined by counting 0.3 ml of the sample directly. A 0.6-ml portion of the sample was mixed with an equal volume of a 10% (w/v) charcoal slurry and spun for 3 min at 10,000 rpm in a tabletop Eppendorf centrifuge to remove unhydrolyzed ATP by binding to charcoal, and 0.6 ml of the supernatant solution was counted. The percent hydrolysis was obtained from the ratio of the radioactivity in charcoal treated to directly counted samples after subtracting the background from each. The data were then fit to single exponential or biexponential equations using Simplex fitting routines in Scientist (Micromath, Ogden, UT) to obtain amplitude and rate information.
Stopped-flow experiments
All stopped-flow measurements were done at 20 °C using an SF-2001 stopped-flow apparatus (KinTek Corp.) fit with two 2-ml syringes and one 5-ml syringe. In double mixing experiments, myosin and ATP were mixed, allowed to incubate for the desired time, and then mixed with actin to give a 2:9 dilution of myosin VIIa and nucleotide and a 5:9 dilution of actin in the flow cell. Single mixing experiments resulted in a 2:7 dilution of myosin and 5:7 dilution of nucleotide or actin in the flow cell. The excitation light from a 75-watt xenon lamp was selected by using a 0.2-m monochromator (Photon Technology International, South Brunswick, NJ). All experiments were carried out in a buffer containing 10 mm MOPS, 3 mm MgCl2, 1 mm EGTA, 25 mm KCl, pH 7.5, at 20 °C. Light scattering was measured by using an excitation wavelength of 450 nm and a 400-nm long-pass filter. dmantATP was excited at 360 nm, and the emission was selected using a 400-nm long-pass filter. Phosphate dissociation from the actomyosin ADP-Pi complex was measured using MDCC-PBP, as described by White et al. (44). Background Pi was removed by including a phosphate mop consisting of 10 mm 7-methylguanosine and 0.01 units/ml purine-nucleoside phosphorylase in all of the reaction solutions. For the experiments with MDCC-PBP, the excitation wavelength was 430 nm, and the emitted light was selected by a 450-nm long-pass filter.
Data analysis and kinetic simulation
Three to four data traces of 1000 points were averaged, and the observed rate constants were obtained by fitting single exponential or biexponential terms to the data using the software package included with the KinTek stopped-flow instrument. Scientist (Micromath) software was used to replot the data for publication and for kinetic simulation.
Transfection of myoVIIa-WT and myoVIIa-sh1 and staining of actin in HeLa cells
HeLa cells were seeded on glass coverslips in DMEM (Gibco) with 10% fetal bovine serum in 5% CO2 at 37 °C overnight and then transfected with the myosin VIIa constructs using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. Fourteen hours after transfection, HeLa cells cultured on glass coverslips were fixed with 4% paraformaldehyde in PBS for 15 min at room temperature, washed three times with PBS, and then permeabilized with 0.1% Triton X-100 in PBS for 15 min at room temperature. Following blocking with 5% BSA in PBS for 15 min, the cells were incubated with 500 nm Texas Red phalloidin (Invitrogen) for 30 min at room temperature for actin staining. One drop of mounting buffer (Prolong Gold Antifade reagent with DAPI (Invitrogen)) was placed on the slide, and coverslips with cells were carefully put onto the mounting medium with the cell side down. Images were taken using a BX50 microscope and DP70 camera equipped with a DP controller software using a 100× objective (Olympus). The number of GFP-myosin VIIa at the tip of the filopodia were counted manually.
Author contributions
A. X. (Pi release, stopped-flow fluorescence), J. H. (cloning, steady-state ATPase), Y. L. (immunofluorescence), M. M. (ADP release), B. B. (quenched flow), L. B. (transient kinetic experiments), and E. F. (transient kinetic experiments) carried out the experiments, and A. X., H. D. W., and E. F. wrote the paper.
Acknowledgment
We thank Donald Winkelmann for help with the preparation of the structure figure.
This work was supported by NIDCD, National Institutes of Health RO3 Grants R03DC009335 and 3RO3DC009335-02S1 (to E. F.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- SAH
- single-α-helix
- dmantADP
- 3′-O-(N-methylanthraniloyl)-2′-deoxy-adenosine 5′-diphosphate
- dmantATP
- 3′-O-(N-methylanthraniloyl)-2′-deoxyadenosine 5′-triphosphate
- MDCC-PBP
- 7-diethylamino-3-(((2maleimidyl)ethyl)amino)carbonyl)coumarin-labeled phosphate-binding protein
- GCN4
- leucine zipper dimer
- myoVIIa
- myosin VIIa
- actomyoVIIa
- actomyosin VIIa
- EGFP
- enhanced GFP
- CaM
- calmodulin.
References
- 1. Chavrier P. (2002) May the force be with you: myosin-X in phagocytosis. Nat. Cell Biol. 4, E169–E171 10.1038/ncb0702-e169 [DOI] [PubMed] [Google Scholar]
- 2. Ebrahim S., Avenarius M. R., Grati M., Krey J. F., Windsor A. M., Sousa A. D., Ballesteros A., Cui R., Millis B. A., Salles F. T., Baird M. A., Davidson M. W., Jones S. M., Choi D., Dong L., et al. (2016) Stereocilia-staircase spacing is influenced by myosin III motors and their cargos espin-1 and espin-like. Nat. Commun. 7, 10833 10.1038/ncomms10833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Li J., Lu Q., and Zhang M. (2016) Structural basis of cargo recognition by unconventional myosins in cellular trafficking. Traffic 17, 822–838 10.1111/tra.12383 [DOI] [PubMed] [Google Scholar]
- 4. Sellers J. R. (2000) Myosins: a diverse superfamily. Biochim. Biophys. Acta 1496, 3–22 10.1016/S0167-4889(00)00005-7 [DOI] [PubMed] [Google Scholar]
- 5. Lymn R. W., and Taylor E. W. (1971) Mechanism of adenosine triphosphate hydrolysis by actomyosin. Biochemistry 10, 4617–4624 10.1021/bi00801a004 [DOI] [PubMed] [Google Scholar]
- 6. Heissler S. M., and Sellers J. R. (2016) Kinetic adaptations of myosins for their diverse cellular functions. Traffic 17, 839–859 10.1111/tra.12388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Odronitz F., and Kollmar M. (2007) Drawing the tree of eukaryotic life based on the analysis of 2,269 manually annotated myosins from 328 species. Genome Biol. 8, R196 10.1186/gb-2007-8-9-r196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hartman M. A., Finan D., Sivaramakrishnan S., and Spudich J. A. (2011) Principles of unconventional myosin function and targeting. Annu. Rev. Cell Dev. Biol. 27, 133–155 10.1146/annurev-cellbio-100809-151502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hartman M. A., and Spudich J. A. (2012) The myosin superfamily at a glance. J. Cell Sci. 125, 1627–1632 10.1242/jcs.094300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Walklate J., Ujfalusi Z., and Geeves M. A. (2016) Myosin isoforms and the mechanochemical cross-bridge cycle. J. Exp. Biol. 219, 168–174 10.1242/jeb.124594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bloemink M. J., and Geeves M. A. (2011) Shaking the myosin family tree: biochemical kinetics defines four types of myosin motor. Semin. Cell Dev. Biol. 22, 961–967 10.1016/j.semcdb.2011.09.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yang Y., Baboolal T. G., Siththanandan V., Chen M., Walker M. L., Knight P. J., Peckham M., and Sellers J. R. (2009) A FERM domain autoregulates Drosophila myosin 7a activity. Proc. Natl. Acad. Sci. U.S.A. 106, 4189–4194 10.1073/pnas.0808682106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Umeki N., Jung H. S., Watanabe S., Sakai T., Li X. D., Ikebe R., Craig R., and Ikebe M. (2009) The tail binds to the head-neck domain, inhibiting ATPase activity of myosin VIIA. Proc. Natl. Acad. Sci. U.S.A. 106, 8483–8488 10.1073/pnas.0812930106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Baboolal T. G., Sakamoto T., Forgacs E., White H. D., Jackson S. M., Takagi Y., Farrow R. E., Molloy J. E., Knight P. J., Sellers J. R., and Peckham M. (2009) The SAH domain extends the functional length of the myosin lever. Proc. Natl. Acad. Sci. U.S.A. 106, 22193–22198 10.1073/pnas.0909851106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Li J., Chen Y., Deng Y., Unarta I. C., Lu Q., Huang X., and Zhang M. (2017) Ca2+-induced rigidity change of the myosin VIIa IQ motif-single α helix lever arm extension. Structure 25, 579–591.e4 10.1016/j.str.2017.02.002 [DOI] [PubMed] [Google Scholar]
- 16. Hasson T., Heintzelman M. B., Santos-Sacchi J., Corey D. P., and Mooseker M. S. (1995) Expression in cochlea and retina of myosin VIIa, the gene product defective in Usher syndrome type 1B. Proc. Natl. Acad. Sci. U.S.A. 92, 9815–9819 10.1073/pnas.92.21.9815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wolfrum U., Liu X., Schmitt A., Udovichenko I. P., and Williams D. S. (1998) Myosin VIIa as a common component of cilia and microvilli. Cell Motil. Cytoskeleton 40, 261–271 10.1002/(SICI)1097-0169(1998)40:3%3C261::AID-CM5%3E3.0.CO%3B2-G [DOI] [PubMed] [Google Scholar]
- 18. Schwander M., Lopes V., Sczaniecka A., Gibbs D., Lillo C., Delano D., Tarantino L. M., Wiltshire T., Williams D. S., and Muller U. (2009) A novel allele of myosin VIIa reveals a critical function for the C-terminal FERM domain for melanosome transport in retinal pigment epithelial cells. J. Neurosci. 29, 15810–15818 10.1523/JNEUROSCI.4876-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tuxworth R. I., Weber I., Wessels D., Addicks G. C., Soll D. R., Gerisch G., and Titus M. A. (2001) A role for myosin VII in dynamic cell adhesion. Curr. Biol. 11, 318–329 10.1016/S0960-9822(01)00097-5 [DOI] [PubMed] [Google Scholar]
- 20. Maniak M. (2001) Cell adhesion: ushering in a new understanding of myosin VII. Curr. Biol. 11, R315–R317 10.1016/S0960-9822(01)00173-7 [DOI] [PubMed] [Google Scholar]
- 21. Küssel-Andermann P., El-Amraoui A., Safieddine S., Nouaille S., Perfettini I., Lecuit M., Cossart P., Wolfrum U., and Petit C. (2000) Vezatin, a novel transmembrane protein, bridges myosin VIIA to the cadherin-catenins complex. EMBO J. 19, 6020–6029 10.1093/emboj/19.22.6020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Prosser H. M., Rzadzinska A. K., Steel K. P., and Bradley A. (2008) Mosaic complementation demonstrates a regulatory role for myosin VIIa in actin dynamics of stereocilia. Mol. Cell. Biol. 28, 1702–1712 10.1128/MCB.01282-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Schwander M., Kachar B., and Müller U. (2010) Review series: the cell biology of hearing. J. Cell Biol. 190, 9–20 10.1083/jcb.201001138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Grati M., and Kachar B. (2011) Myosin VIIa and sans localization at stereocilia upper tip-link density implicates these Usher syndrome proteins in mechanotransduction. Proc. Natl. Acad. Sci. U.S.A. 108, 11476–11481 10.1073/pnas.1104161108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sakai T., Umeki N., Ikebe R., and Ikebe M. (2011) Cargo binding activates myosin VIIA motor function in cells. Proc. Natl. Acad. Sci. U.S.A. 108, 7028–7033 10.1073/pnas.1009188108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lopes V. S., Ramalho J. S., Owen D. M., Karl M. O., Strauss O., Futter C. E., and Seabra M. C. (2007) The ternary Rab27a-Myrip-myosin VIIa complex regulates melanosome motility in the retinal pigment epithelium. Traffic 8, 486–499 10.1111/j.1600-0854.2007.00548.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Williams D. S., and Lopes V. S. (2011) The many different cellular functions of MYO7A in the retina. Biochem. Soc. Trans. 39, 1207–1210 10.1042/BST0391207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Weil D., Blanchard S., Kaplan J., Guilford P., Gibson F., Walsh J., Mburu P., Varela A., Levilliers J., and Weston M. D. (1995) Defective myosin VIIA gene responsible for Usher syndrome type 1B. Nature 374, 60–61 10.1038/374060a0 [DOI] [PubMed] [Google Scholar]
- 29. Liu X. Z., Walsh J., Mburu P., Kendrick-Jones J., Cope M. J., Steel K. P., and Brown S. D. (1997) Mutations in the myosin VIIA gene cause non-syndromic recessive deafness. Nat. Genet. 16, 188–190 10.1038/ng0697-188 [DOI] [PubMed] [Google Scholar]
- 30. Weil D., Küssel P., Blanchard S., Lévy G., Levi-Acobas F., Drira M., Ayadi H., and Petit C. (1997) The autosomal recessive isolated deafness, DFNB2, and the Usher 1B syndrome are allelic defects of the myosin-VIIA gene. Nat. Genet. 16, 191–193 10.1038/ng0697-191 [DOI] [PubMed] [Google Scholar]
- 31. Tamagawa Y., Ishikawa K., Ishikawa K., Ishida T., Kitamura K., Makino S., Tsuru T., and Ichimura K. (2002) Phenotype of DFNA11: a nonsyndromic hearing loss caused by a myosin VIIA mutation. Laryngoscope 112, 292–297 10.1097/00005537-200202000-00017 [DOI] [PubMed] [Google Scholar]
- 32. Weston M. D., Kelley P. M., Overbeck L. D., Wagenaar M., Orten D. J., Hasson T., Chen Z. Y., Corey D., Mooseker M., Sumegi J., Cremers C., Moller C., Jacobson S. G., Gorin M. B., and Kimberling W. J. (1996) Myosin VIIA mutation screening in 189 Usher syndrome type 1 patients. Am. J. Hum. Genet. 59, 1074–1083 [PMC free article] [PubMed] [Google Scholar]
- 33. Cohen M., Bitner-Glindzicz M., and Luxon L. (2007) The changing face of Usher syndrome: clinical implications. Int. J. Audiol. 46, 82–93 10.1080/14992020600975279 [DOI] [PubMed] [Google Scholar]
- 34. Gibson F., Walsh J., Mburu P., Varela A., Brown K. A., Antonio M., Beisel K. W., Steel K. P., and Brown S. D. (1995) A type VII myosin encoded by the mouse deafness gene shaker-1. Nature 374, 62–64 10.1038/374062a0 [DOI] [PubMed] [Google Scholar]
- 35. Hasson T., Walsh J., Cable J., Mooseker M. S., Brown S. D., and Steel K. P. (1997) Effects of shaker-1 mutations on myosin-VIIa protein and mRNA expression. Cell Motil. Cytoskeleton 37, 127–138 10.1002/(SICI)1097-0169(1997)37:2%3C127::AID-CM5%3E3.0.CO%3B2-5 [DOI] [PubMed] [Google Scholar]
- 36. Self T., Mahony M., Fleming J., Walsh J., Brown S. D., and Steel K. P. (1998) Shaker-1 mutations reveal roles for myosin VIIA in both development and function of cochlear hair cells. Development 125, 557–566 [DOI] [PubMed] [Google Scholar]
- 37. Haithcock J., Billington N., Choi K., Fordham J., Sellers J. R., Stafford W. F., White H., and Forgacs E. (2011) The kinetic mechanism of mouse myosin VIIA. J. Biol. Chem. 286, 8819–8828 10.1074/jbc.M110.163592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Varkuti B. H., Yang Z., Kintses B., Erdelyi P., Bardos-Nagy I., Kovacs A. L., Hari P., Kellermayer M., Vellai T., and Malnasi-Csizmadia A. (2012) A novel actin binding site of myosin required for effective muscle contraction. Nat. Struct. Mol. Biol. 19, 299–306 10.1038/nsmb.2216 [DOI] [PubMed] [Google Scholar]
- 39. von der Ecken J., Heissler S. M., Pathan-Chhatbar S., Manstein D. J., and Raunser S. (2016) Cryo-EM structure of a human cytoplasmic actomyosin complex at near-atomic resolution. Nature 534, 724–728 10.1038/nature18295 [DOI] [PubMed] [Google Scholar]
- 40. Coureux P. D., Sweeney H. L., and Houdusse A. (2004) Three myosin V structures delineate essential features of chemo-mechanical transduction. EMBO J. 23, 4527–4537 10.1038/sj.emboj.7600458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gibbs D., Diemer T., Khanobdee K., Hu J., Bok D., and Williams D. S. (2010) Function of MYO7A in the human RPE and the validity of shaker1 mice as a model for Usher syndrome 1B. Invest. Ophthalmol. Vis. Sci. 51, 1130–1135 10.1167/iovs.09-4032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Peng Y. W., Zallocchi M., Wang W. M., Delimont D., and Cosgrove D. (2011) Moderate light-induced degeneration of rod photoreceptors with delayed transducin translocation in shaker1 mice. Invest. Ophthalmol. Vis. Sci. 52, 6421–6427 10.1167/iovs.10-6557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Forgacs E., Sakamoto T., Cartwright S., Belknap B., Kovács M., Tóth J., Webb M. R., Sellers J. R., and White H. D. (2009) Switch 1 mutation S217A converts myosin V into a low duty ratio motor. J. Biol. Chem. 284, 2138–2149 10.1074/jbc.M805530200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. White H. D., Belknap B., and Webb M. R. (1997) Kinetics of nucleoside triphosphate cleavage and phosphate release steps by associated rabbit skeletal actomyosin, measured using a novel fluorescent probe for phosphate. Biochemistry 36, 11828–11836 10.1021/bi970540h [DOI] [PubMed] [Google Scholar]
- 45. Liu Y., White H. D., Belknap B., Winkelmann D. A., and Forgacs E. (2015) Omecamtiv Mecarbil modulates the kinetic and motile properties of porcine β-cardiac myosin. Biochemistry 54, 1963–1975 10.1021/bi5015166 [DOI] [PubMed] [Google Scholar]
- 46. Spudich J. A., and Watt S. (1971) The regulation of rabbit skeletal muscle contraction. I. Biochemical studies of the interaction of the tropomyosin-troponin complex with actin and the proteolytic fragments of myosin. J. Biol. Chem. 246, 4866–4871 [PubMed] [Google Scholar]
- 47. Lee S. H., Seo H. Y., Kim J. C., Heo W. D., Chung W. S., Lee K. J., Kim M. C., Cheong Y. H., Choi J. Y., Lim C. O., and Cho M. J. (1997) Differential activation of NAD kinase by plant calmodulin isoforms: the critical role of domain I. J. Biol. Chem. 272, 9252–9259 10.1074/jbc.272.14.9252 [DOI] [PubMed] [Google Scholar]
- 48. Black D. J., Tikunova S. B., Johnson J. D., and Davis J. P. (2000) Acid pairs increase the N-terminal Ca2+ affinity of CaM by increasing the rate of Ca2+ association. Biochemistry 39, 13831–13837 10.1021/bi001106+ [DOI] [PubMed] [Google Scholar]
- 49. Brune M., Hunter J. L., Corrie J. E., and Webb M. R. (1994) Direct, real-time measurement of rapid inorganic phosphate release using a novel fluorescent probe and its application to actomyosin subfragment 1 ATPase. Biochemistry 33, 8262–8271 10.1021/bi00193a013 [DOI] [PubMed] [Google Scholar]
- 50. Hiratsuka T. (1983) New ribose-modified fluorescent analogs of adenine and guanine-nucleotides available as substrates for various enzymes. Biochim. Biophys. Acta 742, 496–508 10.1016/0167-4838(83)90267-4 [DOI] [PubMed] [Google Scholar]