

Abstract
β-Arrestins are key regulators and signal transducers of G protein–coupled receptors (GPCRs). The interaction between receptors and β-arrestins is generally believed to require both receptor activity and phosphorylation by GPCR kinases. In this study, we investigated whether β-arrestins are able to bind second messenger kinase–phosphorylated, but inactive receptors as well. Because heterologous phosphorylation is a common phenomenon among GPCRs, this mode of β-arrestin activation may represent a novel mechanism of signal transduction and receptor cross-talk. Here we demonstrate that activation of protein kinase C (PKC) by phorbol myristate acetate, Gq/11-coupled GPCR, or epidermal growth factor receptor stimulation promotes β-arrestin2 recruitment to unliganded AT1 angiotensin receptor (AT1R). We found that this interaction depends on the stability lock, a structure responsible for the sustained binding between GPCRs and β-arrestins, formed by phosphorylated serine–threonine clusters in the receptor's C terminus and two conserved phosphate-binding lysines in the β-arrestin2 N-domain. Using improved FlAsH-based serine-threonine clusters β-arrestin2 conformational biosensors, we also show that the stability lock not only stabilizes the receptor–β-arrestin interaction, but also governs the structural rearrangements within β-arrestins. Furthermore, we found that β-arrestin2 binds to PKC-phosphorylated AT1R in a distinct active conformation, which triggers MAPK recruitment and receptor internalization. Our results provide new insights into the activation of β-arrestins and reveal their novel role in receptor cross-talk.
Keywords: angiotensin II, arrestin, bioluminescence resonance energy transfer (BRET), G protein-coupled receptor (GPCR), mitogen-activated protein kinase (MAPK), protein kinase C (PKC), receptor endocytosis
Introduction
The family of G protein–coupled receptors (GPCRs)3 consists of ∼800 members in humans and about 30% of modern drugs target these molecules (1). GPCRs respond to a wide variety of endogenous ligands, including hormones, neurotransmitters, and lipids. Despite their huge diversity, the signal transduction mechanisms of GPCRs share several common features: agonist binding is followed by the activation of a relatively small number of heterotrimeric G proteins, which initiate complex intracellular signaling cascades. Receptor responsiveness to further stimulation is attenuated by a multistep process, called desensitization (2). In the case of homologous desensitization, active GPCRs are phosphorylated by GPCR kinases (GRKs) followed by the recruitment of β-arrestin proteins (β-arrestin1 and β-arrestin2, also known as arrestin-2 and arrestin-3, respectively). β-Arrestins uncouple the receptors from G proteins and initiate receptor internalization (3), thereby serving as the key regulators of GPCRs' function. In contrast, heterologous desensitization is mediated by second messenger-activated kinases, such as protein kinase C (PKC), which can phosphorylate active and inactive receptors. Heterologous desensitization was originally thought to be independent of β-arrestins, however, some data have challenged this concept (4–6). In addition to their role in receptor desensitization and internalization, receptor-bound β-arrestins participate in signal transduction. In this function, β-arrestins serve as scaffolds for signal transducer proteins and initiate a broad range of signaling events. These include activation of mitogen-activated protein kinase (MAPK) signaling pathways, including ERK1/2, p38, and c-Jun N-terminal kinase-3, as well as c-Src family kinases, AKT, phosphatidylinositol 3-kinase, and phosphodiesterase 4 (7, 8).
Arrestins are elongated molecules, composed of two “cup-like” (N and C) domains. In inactive arrestins the C terminus is bound to the N-domain. Arrestins are activated by receptor docking, which takes place in two steps (9, 10). First, in a process called “C-terminal swap,” arrestin releases its C terminus, and its N-domain binds the phosphorylated receptor C terminus. Then the “finger loop” of arrestin interacts with the cavity in the transmembrane helix bundle that opens in the activated receptor. The interaction between receptor C terminus and β-arrestin determines the stability of the complex, based on which receptors were categorized as belonging to class A or class B (11). Class A-type binding is weak and transient, and can only be detected at the plasma membrane. In contrast, the tight and stable class B interaction results in co-internalization of receptor and β-arrestin into endosomes. The interaction stability has important functional consequences: class B-type binding is associated with stronger MAPK activation (12), and drives receptors to lysosomes instead of recycling endosomes after internalization (13). It is known that phosphorylated serine–threonine clusters in the receptor C terminus participate in the stabilization of the arrestin–receptor complex (14), however, the structural basis of stable β-arrestin–receptor coupling is still poorly understood.
In a recent study, it was shown that the state, in which β-arrestin interacts solely with the receptor C terminus, is much more prevalent in the case of class B than class A receptors (15). A likely explanation for this phenomenon may be the difference in lifetime of C-terminal binding. Although an active receptor state is thought to be a prerequisite for β-arrestin binding (16), the former finding suggests that β-arrestins may bind to inactive receptors as well, if a structural basis of stable interaction with phosphorylated receptor C terminus is present. Namely, the phosphorylation of receptor C termini in proper positions might be sufficient for β-arrestins to engage the receptors via their N-domain. GRKs, the main GPCR kinases, preferentially phosphorylate the active receptors. In contrast, receptor phosphorylation by other kinases, such as PKC, occurs regardless of receptor activation. Thus, β-arrestin recruitment induced solely by receptor phosphorylation may serve as a novel physiological mechanism of β-arrestin activation. In some cases it has been suggested that PKC-dependent phosphorylation regulates receptor function through β-arrestins (4–6, 17). It was reported that the PKC-induced internalization of the δ-opioid or the D2 dopamine receptors was suppressed by overexpression of dominant-negative β-arrestin, and the β-arrestin-dependent desensitization of the calcium sensing receptor required phosphorylation by PKC. But to the best of our knowledge, receptor–β-arrestin interaction induced by PKC has been directly demonstrated only once with the α1B-adrenergic receptor (6). In the case of the AT1 angiotensin receptor (AT1R), heterologous phosphorylation and desensitization was thought to be independent of β-arrestins (18).
Here we show that activation of PKC by phorbol ester, α1A-adrenergic receptor, or epidermal growth factor receptor stimulation, leads to β-arrestin2 (βarr2) recruitment to the AT1R. This recruitment is independent of AT1R activation, but relies on the stability lock, the interaction responsible for sustained binding between GPCRs and β-arrestin2. Using improved versions of recently described fluorescein arsenical hairpin binder (FlAsH)-BRET sensors, we show that β-arrestin2 activated by PKC-phosphorylated receptor undergoes different conformational changes than in the case of full engagement. Selective disruption of the interaction-stabilizing elements showed that the stability lock not only sustains the receptor–β-arrestin interaction, but also governs the structural rearrangements within β-arrestin2. β-Arrestin activated by PKC-phosphorylated GPCR binds MEK1 and ERK2 and initiates receptor endocytosis, resulting in a different fate of internalized receptor.
Results
PKC activation leads to the recruitment of β-arrestin2 to inactive AT1R
It has been demonstrated previously that PKC phosphorylation sites in the AT1R C terminus overlap with the GRK-target sites (19, 20). Therefore, we tested whether C-terminal phosphorylation by PKC, in the absence of receptor stimulation, can induce β-arrestin2 binding to the receptor. To this end, we performed co-precipitation experiments in HEK 293T cells co-expressing YFP- and biotin acceptor peptide (BAP)-tagged AT1R, β-arrestin2–Cerulean, and biotin ligase (BirA). We stimulated the cells with either AT1R agonist angiotensin II (AngII) or specific PKC activator phorbol myristate acetate (PMA) for 20 min, and co-precipitated β-arrestin2–Cerulean with biotin-labeled receptors using NeutrAvidin beads. Strikingly, we found that both AngII and PMA induced β-arrestin2 binding to AT1R (Fig. 1a). To determine the kinetics of heterologous β-arrestin2 binding in real time, we measured bioluminescence resonance energy transfer (BRET) between Rluc8-tagged AT1R and β-arrestin2–Venus (Fig. 1b). Both PMA and AngII led to an increase of the BRET signal, reflecting the interaction between AT1R and β-arrestin2 (Fig. 1c). However, the PMA-induced β-arrestin2 binding to AT1R was delayed, suggesting that β-arrestin associates slower with inactive phosphorylated receptors than with active phosphorylated receptors. Physiologically, PKC is activated by stimulation of Gq/11-coupled GPCRs or growth factor receptors (21). To test whether β-arrestin2 interacts with AT1R upon activation of a Gq/11-coupled GPCR, we co-expressed the BRET partners with untagged α1A-adrenergic receptor (α1AAR), a receptor that showed no detectable β-arrestin binding (22). Stimulation with the selective α1AAR agonist A61603 induced β-arrestin2 binding to AT1R with comparable kinetics and extent as PMA (Fig. 1d). Similar results were obtained with M3 muscarinic acetylcholine receptor (M3AChR) (Fig. 1e), suggesting that this mechanism is not specific for α1AAR. Furthermore, because receptor tyrosine kinases also activate PKC, we co-expressed epidermal growth factor receptor (EGFR) with AT1R. As expected, EGF stimulation promoted β-arrestin2 binding to AT1R (Fig. 1f).
Figure 1.

β-Arrestin2 binds to unliganded AT1R upon PKC activation. a, PKC activation triggers β-arrestin2 binding to AT1R. HEK 293T cells were co-transfected with plasmids encoding NES–BirA (biotin ligase), β-arrestin2–Cerulean, and/or AT1R–YFP–BAP, as indicated. After a 20-min stimulation with 100 nm AngII or 100 nm PMA, the cells were lysed and biotin-labeled AT1R was pulled down using NeutrAvidin beads. Left panel, YFP- and Cerulean fluorescence of the co-precipitated proteins. The values were normalized to the autofluorescence of the beads. n = 3, *, p < 0.05, analyzed with one-way ANOVA (repeated measures, versus control samples containing AT1R–YFP–BAP) with Bonferroni post hoc test. Scatter dot plots with columns (mean ± S.E.) are shown. Right panel, representative images show the YFP (top) and Cerulean (bottom) fluorescence of the pulled-down proteins bound to the surface of the beads, recorded with confocal microscope. Scale bar represents 100 μm. b, schematic representation of the used BRET set-ups with the applied stimuli. c–f, kinetics of β-arrestin2 binding to AT1R upon PKC activation. Intermolecular BRET was measured between AT1R–Rluc8 and β-arrestin2–Venus after AngII (c–f), PMA (c and d), or α1AAR agonist A61603 (1 μm, d), M3AChR agonist carbachol (10 μm, e), or EGF (100 ng/ml, f) treatments in every 70 s in cells co-expressing the indicated constructs. Stimulus-induced BRET ratio change was calculated by subtracting the corresponding vehicle-stimulated control. Kinetic curves are shown in c (n = 6) and d–f (n = 3), data are mean ± S.E. of independent biological replicates.
To prove the pivotal role of PKC in heterologous β-arrestin2 recruitment, we pretreated HEK 293T cells with a set of protein kinase inhibitors. Both the specific PKC inhibitor GF109203X and the broad-spectrum serine–threonine kinase inhibitor staurosporine (used at a concentration that does not inhibit GRKs considerably (23)) prevented the PMA or α1AAR stimulation-induced β-arrestin2 recruitment (Fig. 2a). Similarly, the β-arrestin2 binding to AT1R promoted by M3AChR or EGFR activation was also sensitive to PKC inhibition (Fig. S1, a and b). Although the C-tail of AT1R contains three known PKC phosphorylation sites, PKC may also act indirectly through activation of other kinases. It was reported that PKC can enhance the activity of GRK2 (24, 25), but inhibits GRK5 (26). Therefore, we tested whether PKC induces the AT1R–β-arrestin2 binding via GRK2. Pretreatment with the GRK2/3 inhibitor compound 101 (Cmpd101) did not alter the PMA-triggered AT1R–β-arrestin2 interaction, and it had only a slight effect in the case of α1AAR stimulation (Fig. 2b). Furthermore, inhibition of several other potential downstream protein kinases also did not prevent the heterologous β-arrestin binding. These results support the idea that the PKC effects are mainly direct. In addition, the MEK1/2 inhibitor PD98059 did not have any effect on heterologous recruitment either (Fig. S1, c and d), demonstrating that ERK cascade activation is not involved in the process, in contrast to the heterologous regulation of the CXCR4 receptor (27). Interestingly, the inhibition by Cmpd101 on the AngII-induced binding was only mild, but was significantly potentiated by staurosporine (Fig. 2b), which is in good agreement with former observations that GRK and PKC phosphorylation sites overlap in AT1R.
Figure 2.
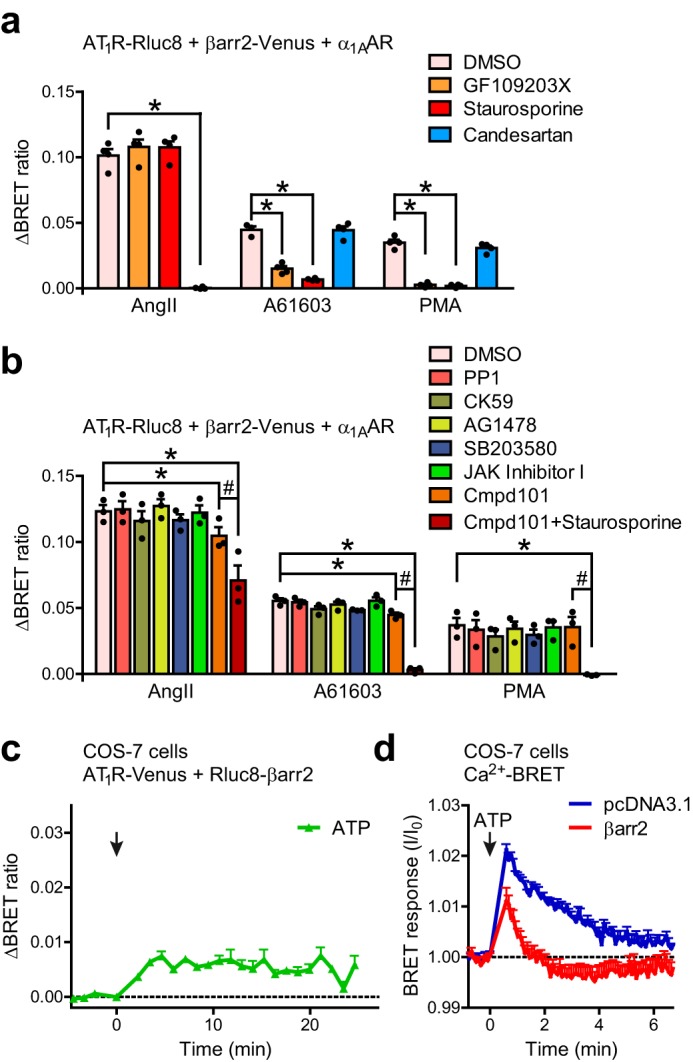
The heterologous β-arrestin2 recruitment to AT1R is PKC phosphorylation-dependent, but does not require the active state of the receptor. a and b, heterologous β-arrestin2 binding to inactive AT1R is dependent on PKC activity. HEK 293T cells, co-expressing AT1R-Rluc8, β-arrestin2–Venus, and untagged α1AAR were pretreated with the indicated inhibitors, and were stimulated with AngII, PMA, or A61603. Scatter dot plots with columns (mean ± S.E.) show the average BRET changes (area under curve) in the first 24 min after stimulation. In a, the cells were pretreated with PKC inhibitor GF109203X (2 μm), broad spectrum kinase inhibitor staurosporine (500 nm), AT1R inverse agonist candesartan (10 μm), or vehicle (DMSO) for 30 min. The PMA- or A61603-induced signals were hindered by PKC inhibition, but were not altered by the AT1R antagonist pretreatment. In b, the pretreatments were vehicle (DMSO), Src-kinase inhibitor PP1 (1 μm), Ca2+/calmodulin-dependent protein kinase II inhibitor CK-59 (20 μm); EGFR tyrosine kinase inhibitor AG-1478 (10 μm), p38 MAPK inhibitor SB203580 (20 μm), 1 μm JAK Inhibitor I (inhibitor of JAK kinases), GRK2/3 inhibitor Cmpd101 (30 μm), and co-pretreatment with Cmpd101 and staurosporine. The non-PKC inhibitors did not prevent the heterologous β-arrestin2 recruitment, suggesting that the PKC effects are mainly direct. The homologous binding was inhibited most efficiently by the co-pretreatment with Cmpd101 and staurosporine, which is in good agreement with the previous observations that the GRK and PKC target sites overlap, and suggests that phosphorylation of AT1R by either kinase is sufficient for β-arrestin recruitment. *, p < 0.05 means significant interaction between the effects of pretreatment and stimulation; #,p < 0.05 means significant interaction between Cmpd101 and staurosporine treatments, analyzed with two-way ANOVA. Scatter dot plots with columns (mean ± S.E.) are shown, n = 3–4. c, stimulation of endogenous Gq/11 protein-coupled receptors evoke β-arrestin2 binding to AT1R. COS-7 cells were co-transfected with AT1R–Venus and Rluc8–β-arrestin2, and BRET was measured 48 h after transfection. 50 μm ATP was used to stimulate the endogenous purinergic receptors. ATP induced a significant increase of the BRET signal (average BRET ratio was compared with non-stimulated control using paired two-sample two-tailed t test, p < 0.05, n = 3). d, overexpression of β-arrestin2 desensitizes the endogenous purinergic receptors in COS-7 cells. COS-7 cells were co-transfected with Cameleon D3 Ca2+ BRET biosensor and empty vector pcDNA3.1 or untagged β-arrestin2. The Ca2+ signal was lower and was terminated earlier in the case of β-arrestin2 co-expression. Data are mean ± S.E.
To test whether the PKC-induced β-arrestin2 binding is dependent on constitutive receptor activity, we incubated cells with the AT1R inverse agonist candesartan prior to PMA administration (Fig. 2a). Candesartan pretreatment completely inhibited the AngII-induced β-arrestin2 binding to AT1R, but did not prevent its heterologous stimulation, demonstrating that this process is independent of the activation state of the receptor.
Next, we examined whether stimulation of endogenous Gq/11 protein-coupled receptors could induce the observed phenomenon. Because we found unstable and low calcium signaling of endogenous GPCRs upon lysophosphatidic acid, histamine, bradykinin, carbachol, or ATP stimulation in our HEK cells (data not shown), we tested the effects of purinergic receptor activation in COS-7 cells. We found a small, but significant elevation of the BRET signal between AT1R–Venus and Rluc8–β-arrestin2 upon ATP stimulus (Fig. 2c). The lower and earlier maximum of the signal was expected, because β-arrestin2 overexpression desensitizes the endogenous purinergic receptors, as shown by calcium measurements using a BRET-based Ca2+ biosensor (Fig. 2d).
Formation of the stability lock is required for β-arrestin binding to inactive AT1R
To investigate the stability of the interaction, i.e. whether it is class A- or class B-type, we co-expressed Cerulean-tagged AT1R with β-arrestin2–Venus. Both AngII and PMA promoted translocation of β-arrestin2–Venus from diffuse cytoplasmic localization to the plasma membrane and then co-internalization to intracellular vesicles with AT1R (Fig. 3a, left panel). Similarly, α1AAR–mRFP stimulation resulted in intracellular co-localization of AT1R and β-arrestin2 (Fig. S2a). In contrast, the α1AAR-induced β-arrestin2 redistribution was missing in the absence of AT1R–Cerulean co-expression. Because PKC activity induced a class B-type interaction, we speculated that the residues, which are responsible for the stable receptor–β-arrestin interaction, play the decisive role in PKC-promoted β-arrestin2 binding.
Figure 3.

PKC-triggered β-arrestin2 recruitment to AT1R requires stable binding to the receptor C terminus. a, the PMA-induced stable binding between AT1R and β-arrestin2 depends on the interaction stabilizing elements. Images were taken with a confocal microscope from cells co-expressing wild type (WT) or phosphorylation-deficient (TSTS/A)-mutant, AT1R–Cerulean, and WT or phosphate binding-deficient (K2A)–mutant β-arrestin2–Venus, as indicated, before (control, top) and after 20 min stimulation with PMA (bottom) or after 20 min treatment with AngII (middle). Both PMA and AngII induced the co-translocation of AT1R–WT–Cerulean and WT–β-arrestin2–Venus to endosomes. In contrast, both TSTS/A and K2A mutations evoked only plasmalemmal binding after AngII treatment, and prevented the PMA-triggered interaction. Arrowheads mark β-arrestin2-enriched intracellular vesicles (class B-type binding), arrows point to plasma membrane-localized β-arrestin2 (class A interaction). Representative images are shown, n = 3. b, role of the stability lock in AT1R–β-arrestin2 binding. BRET titration experiments were performed. WT or TSTS/A-mutant AT1R–Rluc8 was co-transfected with increasing amounts of plasmid encoding WT- or K2A–β-arrestin2–Venus, as indicated. Stimulus (AngII, left panel, or PMA, right panel)-evoked the average BRET ratio changes (mean value of duplicate) are plotted as the function of fluorescence/luminescence ratio, n = 3. One-site specific binding curves were fitted on the values. The AngII-triggered β-arrestin2 recruitment was partially, and the PMA-induced recruitment was fully abolished by these mutations. c, disruption of the stability lock decreases the stability of AT1R–β-arrestin2 interaction. BRET was measured between WT or TSTS/A-mutant AT1R–Rluc8 and WT- or K2A–β-arrestin2–Venus in cells co-expressing dominant-negative dynamin, an internalization inhibitor. The interaction was induced by ∼20 min treatment with 100 nm AngII, thereafter 10 μm competitive antagonist candesartan (Cand) was added to displace AngII. Both TSTS/A and K2A mutations accelerated the BRET ratio drop, indicating faster dissociation of β-arrestin2 from AT1R. Values are presented as percentage of the peak AngII-induced signal in each experiment and each set-up as mean ± S.E., n = 3.
Previous studies showed that the stable β-arrestin–AT1R interaction is dependent on a serine–threonine cluster in the AT1R C terminus (residues Thr-332, Ser-335, Thr-336, and Ser-338 (TSTS) (12, 28, 29)) and on two lysines (Lys-11 and Lys-12) in the β-strand I of the N-domain of β-arrestin2 (30), which are conserved phosphate-binding residues in all arrestins (31–35). Thus, we set out to test whether the same receptor and arrestin elements are involved in the PMA-induced β-arrestin2 binding. To this end, we replaced the amino acids of interest with alanines (AT1R-TSTS/A and K2A–β-arrestin2, respectively), and expressed either AT1R–TSTS/A with wild type β-arrestin2 or wild type AT1R with K2A mutant β-arrestin2, respectively. In both cases, receptor activation by AngII promoted only transient β-arrestin2 binding, as reflected by the absence of β-arrestin2 in intracellular vesicles (Fig. 3a, middle and right panel). Moreover, BRET measurements showed that both mutations decreased the interaction to a similar extent (Fig. 3b). When the two mutants were co-expressed, no further decrease of the signal was observed, suggesting a direct interaction between the two regions. Furthermore, no detectable β-arrestin2 binding could be observed upon PMA treatment in cells expressing AT1R-TSTS/A and/or K2A-β-arrestin2 (Fig. 3, a and b). Conceivably, the same phosphorylation sites (TSTS) and phosphate-binding arrestin residues (Lys-11, Lys-12) are engaged during the PKC-induced β-arrestin2 translocation and in the canonical agonist-induced β-arrestin2 activation.
To examine whether the affinity between receptor and β-arrestin2 was affected by these mutations, and the observed phenomena are not due to altered internalization, we investigated the stability of this interaction under internalization-inhibited conditions using dominant-negative dynamin (36, 37). We induced β-arrestin2 binding by application of the AT1R agonist AngII, then added competitive antagonist candesartan to inactivate the receptors, and followed the rate of β-arrestin dissociation using BRET. Both K2A- and TSTS/A mutations accelerated the detachment of β-arrestin2 (Fig. 3c), proving that these mutations weakened the interaction. This effect was even more pronounced when we used the low-affinity agonist angiotensin IV (AngIV) (38) (Fig. S2b), which is displaced faster than AngII.
The role of Lys-11 and Lys-12 in AT1 receptor binding was in contrast with the results obtained with other GPCRs, like β2-adrenergic, D1 dopamine, and D2 dopamine receptors (32). We speculated that this distinction may explain the different β-arrestin binding phenotypes of these receptors, i.e. AT1R is a prototypic class B receptor, whereas the other three subtypes belong to the class A. These results suggested that Lys-11 and -12 in β-arrestin2 and distinct serine–threonine clusters of receptor C termini form a stability lock, which is responsible for the stable binding between GPCRs and β-arrestins. In accordance with that, the K2A mutation in β-arrestin2 prevented its intracellular appearance after stimulation of V2R (Fig. 4a), another prototypical class B GPCR. The phosphorylation-deficient V2R-AAA (S362A,S363A,S364A) mutant showed a similar class A interaction with β-arrestin2, as earlier described (13). Both V2R and β-arrestin2 mutations decreased the binding, but their co-expression resulted in only a slight additional decline (Fig. 4b, left panel). We observed no heterologous β-arrestin2 binding to V2R, probably because this receptor lacks target motifs for PKA or PKC (data not shown).
Figure 4.

Formation of the stability lock is necessary for the class B-type β-arrestin binding of GPCRs. a, stable binding between V2R and β-arrestin2 depends on Lys-11 and Lys-12 in β-arrestin2 and a receptor C-terminal serine–threonine cluster. Confocal images were taken from cells co-expressing Cerulean-tagged WT or phosphorylation-deficient AAA (S362A,S363A,S364A)-mutant V2R and WT- or K2A-β-arrestin2-Venus, as indicated, before and after a 20-min stimulation with 100 nm arginine vasopressin (AVP). Both AAA and K2A mutations changed the binding phenotype to class A. c, class B-type interaction of the phosphorylation site engineered β2AR mutant is reversed to class A by K2A mutation in β-arrestin2. Cells were co-transfected with plasmids encoding WT (class A)- or SSS (G361S,E362S,Q363S)-mutant (class B) β2AR–Cerulean, WT–, or K2A–β-arrestin2–Venus, and with untagged GRK2. Images were taken before and after 10 μm isoproterenol (ISO) stimulation for 20 min. K2A mutation prevented the appearance of β-arrestin2 in intracellular vesicles. Arrowheads mark β-arrestin2-enriched endosomes, arrows point to β-arrestin2 translocated to the plasma membrane. Representative images of 3 independent experiments are shown. Scale bars represent 5 μm. b, Lys-11 and Lys-12 participates in the β-arrestin2 binding of class B GPCRs. BRET titration experiments were performed to elucidate the role of Lys-11 and Lys-12 of β-arrestin2 in the binding to the following Sluc-tagged receptors: WT (class B) or phosphorylation-deficient (AAA, class A) mutant V2R (left panel) and WT (class A) or phosphorylation site-engineered (SSS, class B) β2AR (right panel). BRET acceptors were WT- or K2A–β-arrestin2–Venus, as indicated. GRK2 was co-expressed in the case of β2AR experiments. 100 nm V2R agonist arginine vasopressin and 10 μm β2AR agonist isoproterenol were used as stimuli, n = 3. One-site specific binding curves were fitted on the data values. K2A mutation decreased the binding of class B GPCRs substantially.
We tested whether the β-arrestin2 interaction with the class A β2-adrenergic receptor (β2AR) can be made Lys-11 and Lys-12-dependent. We used a mutant β2AR with engineered additional phosphorylation sites (β2AR-SSS; G361S,E362S,Q363S), which shows class B-type binding (39). The interaction phenotype of the mutant receptor was reversed to class A by K2A–β-arrestin2 (Fig. 4c). In agreement with these results, the BRET signal between the mutant receptor and wild type β-arrestin2 was elevated, but the increase was prevented by K2A mutation (Fig. 4b, right panel). These results indicate that formation of the stability lock between Lys-11 and Lys-12 in β-arrestin2 and phosphorylated Ser–Thr motifs in the receptor C termini, serves as a general mechanism for class B interaction.
Conformation of active β-arrestin2 is interaction-specific
Next, we investigated whether the β-arrestin2 conformation depends on the mode of receptor engagement. To follow the structural rearrangements of β-arrestin2, we generated a panel of intramolecular FlAsH–BRET reporters. In these constructs, BRET signal is measured between two parts of the same protein, so that changes in the signal indicate conformational rearrangements within the molecule. The sensors have been designed in a way that the positions of the FlAsH-binding cysteines match the previously published β-arrestin2 BRET and FRET sensors (40, 41) (Fig. 5a). CCPGCC binding motifs for the FlAsH were incorporated after residues 139, 154, 225, 263, or 410 (F139, F154, F225, F263, and F410 sensors, respectively) (where F used with position numbers in this paper refers to FlAsH). F139 and F154 are located within the N-domain of β-arrestin2, F225 and F263 are in the C-domain, whereas F410 is on the C terminus. The donor molecule was fused to the N terminus of β-arrestin2, but in contrast to the previous publication (40), we used a brighter version of Renilla luciferase, Rluc8. Rluc8 has been shown to be advantageous for BRET measurements, because it is 6 times brighter than conventional Renilla luciferase (42). This technical improvement prolonged the effective detection time, allowing us to detect the dynamics of BRET signal changes for at least 20 min. To verify the binding of the sensors to AT1 receptor, we co-expressed these chimeric proteins with AT1R–Venus in HEK 293T cells, and stimulated the receptors with AngII. In this setup, intermolecular BRET between the Rluc8 tag of β-arrestin2–FlAsH sensor and the Venus tag of AT1R is measured. As shown in Fig. S3a, all sensors bind AT1R–Venus similarly, with the same kinetics as the control unmodified Rluc8–β-arrestin2 (Fig. 5b). Thus, the ability of β-arrestin to interact with AT1R was not affected by the insertion of CCPGCC motifs in the selected positions. Next, we investigated the conformational changes within the β-arrestin2 sensors during their activation. In these experiments, we co-expressed untagged AT1R with different FlAsH sensors and followed the intramolecular BRET between Rluc8 and FlAsH labels. Upon stimulation with AngII, we detected the BRET signal increase with F154, F410, and decrease with F139, F225, and F263 (Fig. S3a). The direction of F139, F225, F263, and F410 signal changes were identical to those previously described using the similar FlAsH sensors with AT1R (40).
Figure 5.

Real time monitoring of recruitment and conformational changes of β-arrestin2. a, schematic representation of FlAsH-based β-arrestin2 BRET biosensors based on crystal structure of bovine β-arrestin2 (Protein Data Bank accession number 3P2D (56)). Rluc8 was fused to the N-terminal end of β-arrestin2 and CCPGCC amino acid motifs for FlAsH binding were inserted into rat β-arrestin2 after the indicated amino acid positions (F139, F154, F225, F263, and F410, the corresponding residues of bovine β-arrestin2 are highlighted, 410 is marked after the last amino acid (393) of the solved structure). b and c, dynamics of AT1R–β-arrestin2 interaction and β-arrestin2 conformational changes. Intermolecular BRET (interaction) was measured between AT1R–Venus and Rluc8–β-arrestin2 or FlAsH sensors, as indicated, upon AngII (b) or PMA (c) stimulation (F154, n = 4; others, n = 6). The intramolecular BRET (conformation) was followed in cells expressing untagged AT1R and β-arrestin2 biosensors labeled with FlAsH dye (AngII: n = 10, F139; n = 7, F154; n = 9, F225, F263, F410; PMA: n = 8, F139, F225, F263, F410; n = 6, F154). All experiments were performed in triplicate. BRET ratio changes are expressed as the percentage of the peak AngII-induced signal of each set-up. Values presented as mean ± S.E. The kinetics of interaction and conformational changes upon AngII stimulation were similar in the case of N-domain (F139, F154) and C-terminal (F410) sensors. In contrast, the C-domain (F225, F263) sensors showed an earlier peak in the conformational BRET change than in interaction, indicating that the conformational rearrangements in the C-domain are dynamic and constantly change. Upon PMA stimulation, N-domain, but not the C-domain sensors showed conformational changes, and their kinetics were similar to that of the binding.
To facilitate the kinetic comparison of inter- and intramolecular BRET, we presented the signal changes as the percentage of the peak signals (Fig. 5b), independently of the direction of BRET change. The kinetics of F139, F154, and F410 intramolecular BRET signals were similar to that of the receptor–β-arrestin2 intermolecular BRET, suggesting that the sensor-detected conformational changes occurred parallel with the interaction between the two proteins. However, in the case of the C-domain sensors (F225 and F263), the signals reached their maximum after a few minutes and then slowly decreased, in contrast to the AT1R–β-arrestin2 binding kinetics, which achieved a plateau and remained stable. These data suggest that the conformational rearrangements within the C-domain of β-arrestin2 do not end with receptor binding, but this part of the protein remains dynamic and constantly changes at least for the 20 min after stimulation.
To test which conformational changes are promoted by the PKC-induced interaction, we measured the conformational rearrangements in β-arrestin2 after PMA stimulation. As shown in Fig. 6, a and b, F139 and F154 BRET changes, when normalized to the magnitude of β-arrestin2 coupling (i.e. to intermolecular BRET), were similar to those after AngII stimulation, and their kinetics followed the β-arrestin2 binding to the receptor (Fig. 5c). In contrast, the F410 signal was reversed and lower, and the C-domain sensors (F225, F263) showed no changes compared with the unstimulated cells (Fig. 6b). Thus, the conformation of the PMA-activated β-arrestin2 bound to AT1R is static and significantly different from that induced by AngII.
Figure 6.

Structural rearrangements in β-arrestin2 are dependent on the mode of activation. a–d, patterns of conformational changes upon different modes of AT1R–β-arrestin2 binding. Because the number of interacting and activated β-arrestin molecules are different in case of the distinct activation modes, to facilitate the comparison, interaction-normalized relative FlAsH signals are shown (average conformational BRET ratio change divided by average interactional BRET change). The average inter- and intramolecular BRET ratio changes are presented in Fig. S3. a and b, conformational realignments in β-arrestin2 upon homologous and heterologous activation. Conformational BRET ratio changes were measured in cells expressing untagged wild type AT1R and wild type sensors labeled with FlAsH dye (AngII: n = 10, F139; n = 7, F154; n = 9, F225, F263, F410; PMA: n = 8, F139, F225, F263, F410; n = 6, F154). c and d, stability lock governs the conformational realignments in active receptor-bound β-arrestins. TSTS/A mutation in AT1R (c) and K2A mutation in β-arrestin2 (d) altered the FlAsH sensor activation profile upon AngII treatment similarly. Identical BRET set-ups were used as in panel a, with the indicated mutant constructs. n = 5 and n = 6 in the case of conformational BRET measurements with AT1R-TSTS/A and K2A-FlAsH biosensors, respectively, and interactional BRET was determined in 3 independent biological replicates. Scatter dot plots with columns (mean ± S.E.) are shown, *, p < 0.05, column means are significantly different from 0 value, analyzed with a one-sample t test. e, web of β-arrestin2 conformational changes induced by fully engaged interaction (AngII), only C-terminal interaction (PMA), or in the absence of stability lock formation (AT1R-TSTS/A or K2A–β-arrestin2). The relative average FlAsH signals were normalized to the AngII-induced signals in the case of wild type AT1R and biosensors, then the values were cube root transformed and plotted in radial diagram, as indicated. Patterns of at least 3 active conformations can be distinguished.
To explore the role of the stability lock in the conformational changes induced by the homologous activation, we used the AT1R and β-arrestin2 mutants. First, AT1R-TSTS/A was co-expressed with the intramolecular sensors. In this setup, FlAsH-154 and FlAsH-225 signals were abolished, whereas F263 and F410 signals were reversed upon AngII treatment (Fig. 6c). Signal changes of F225 and F410 are in good agreement with the previous observation, that activation of these constructs correlates with the stability of the receptor–β-arrestin2 complex (40). Next, we constructed β-arrestin2–FlAsH sensors with K11A and K12A mutations (F139–K2A, F154–K2A, F225–K2A, F263–K2A, and F410–K2A). Basal BRET ratios with F410–K2A were elevated compared with the wild type β-arrestin2 (Fig. S3c). The altered basal conformation of K2A–β-arrestin2 was expected, because it has been shown previously that the Lys-11,12 region binds the C-terminal part of arrestins to the N-domain in the inactive state (33, 43). Activation of β-arrestin2 results in the release of the C terminus (44, 45), which might be reflected by the BRET ratio change with F410 (Fig. 6a), and also by the altered basal BRET ratio with F410–K2A (Fig. S3c). Strikingly, when we stimulated the AT1R co-expressing cells with AngII, a similar pattern of structural rearrangements was observed to that of AT1R-TSTS/A and wild type β-arrestin2: F154 and F225 signals were missing, F410 was reversed and the F263 signal was abolished (Fig. 6d). Because changes in the structure of K2A–β-arrestin2 are comparable with that of wild type β-arrestin2 when AT1R-TSTS/A is expressed, these data indicate that the TSTS region in AT1R directly interacts with Lys-11 and Lys-12 in β-arrestin2.
As shown in the radial diagram of Fig. 6e, at least 3 active β-arrestin2 conformations can be clearly distinguished. The fully engaged conformation is different from the conformations induced by PKC or in the absence of the stability lock formation. These results also reveal the decisive role of the stability lock in structural rearrangements within β-arrestin2. Active stability lock is sufficient for promoting conformational changes and it has a prominent part in governing the full panel of structural realignments.
PKC-activated β-arrestin2 dictates distinct fate of internalized receptor
The confocal images showed that PMA treatment promoted the endocytosis of AT1R. Internalized receptors either recycle after dephosphorylation, or are degraded in lysosomes (46). It has been shown previously that receptor–β-arrestin interaction may regulate the receptor's intracellular processing (47), so we hypothesized that PKC-induced β-arrestin drives intracellular receptor trafficking differently compared with the fully engaged form. To test this idea we used bystander BRET measurements between AT1R–Rluc8 and one of the intracellular vesicle markers, Venus–Rab4, Venus–Rab5, Venus–Rab7, or Venus–Rab11, as described previously (48, 49). Rab5 is located in the early endosomes, Rab4 and Rab11 in the fast and slow recycling vesicles, respectively, whereas Rab7 is found in late endosomes, which fuse with lysosomes (46). Increased BRET signals between AT1R–Rluc8 and all four Rab constructs were detected after both AngII and PMA treatments (Fig. 7, a–d), as well as after α1AR stimulation (Fig. S4, a–d), demonstrating the redistribution of AT1R from the cell membrane to intracellular vesicles. To better visualize the intracellular trafficking pattern, we presented the BRET ratio changes after 60 min stimulation in a radial diagram (Fig. 7e and Fig. S4e). It shows that PKC-activated β-arrestin2 drives recycling with similar efficiency, but the degradation pathway is impaired compared with that of AngII. These data suggest that the PKC-induced β-arrestin2 activation promotes a distinct internalization pattern of AT1R, therefore receptor fate depends on the mode of β-arrestin2 engagement.
Figure 7.

Homologous and heterologous β-arrestin recruitment to AT1R governs the intracellular receptor fate differently. a–d, appearance of AT1R in Rab-enriched compartments triggered by homologous or heterologous β-arrestin2 activation. Cells were co-transfected with plasmids encoding wild type AT1R–Rluc8, Venus–Rab construct (Rab5, early endosome marker (a); Rab4, rapid recycling and early endosome marker (b); Rab7, multivesicular body/late endosome marker (c); Rab11, late recycling route marker (d)) and untagged wild type β-arrestin2. AngII or PMA were used as stimuli. Both PMA and AngII induced the appearance of AT1R in all the tested Rab-enriched compartments. β-Arrestin2 was co-transfected, because the relatively low level of endogenous β-arrestin2 limited the internalization of overexpressed AT1R–Rluc8. Values are presented as mean ± S.E., n = 3. e, patterns of intracellular AT1R redistribution after homologous and heterologous β-arrestin2 activation. BRET changes after 60 ± 2 min stimulation were normalized to the AngII-induced values, and are presented in a radial diagram. PMA-induced less trafficking of AT1R to Rab7-enriched vesicles.
Inactive receptor-bound β-arrestins recruit MAPKs
Following the binding to certain GPCRs, β-arrestin2 can recruit the members of the MAPK cascade (8). We investigated whether β-arrestins bound to an inactive receptor retain this ability. To test this, we designed a BRET assay in which energy transfer was detected between AT1R–Rluc8 and either ERK2–Venus or Venus–MEK1 (Fig. 8a). We measured the increase in the BRET ratios between AT1R–Rluc8 and ERK2–Venus or Venus–MEK1 upon stimulation with AngII. Strikingly, when β-arrestin2 was co-expressed with the BRET pairs, AngII induced a much more pronounced BRET signal elevation (Fig. 8b), suggesting that the proper stoichiometry of receptor, ERK, MEK, and β-arrestin2 is required for the correct assembly of the signaling complex. This is in agreement with previous observation where JNK3 translocation to AT1R was detected (50). Interestingly, the kinetics of MEK1 and ERK2 recruitment were similar to the dynamics of the receptor–β-arrestin2 interaction (Fig. 8c), and did not follow that of F263 (Fig. 5b), which was suggested previously to correlate with ERK1/2 activation (40). Strikingly, PMA stimulation led to a significant increase in the signal, although the amplitude was lower than that observed with AngII (Fig. 8b) and was proportional to the magnitude of β-arrestin2 recruitment (Fig. 8c). A similar effect was observed upon α1AAR activation (Fig. 8d). These data show that not only active receptors can participate in signal transduction, but inactive receptors can also serve as scaffold proteins for signaling complexes, representing a novel mechanism of receptor cross-talk.
Figure 8.

β-Arrestin2 bound to PKC-phosphorylated AT1R promotes the formation of AT1R–β-arrestin2–MAPK complexes. a, schematic representation of the experimental set-up for detection of MAPK recruitment to AT1R. b, intermolecular BRET was measured in cells co-expressing AT1R-Rluc8, Venus-tagged MAPK proteins (Venus–MEK1, left panel, or ERK2–Venus, right panel), and empty vector pcDNA3.1 or β-arrestin2 after AngII or PMA treatment. Scatter dot plots with columns (mean ± S.E.) of independent biological replicates (n = 6, AngII; n = 3, PMA) are shown, *, p < 0.05, stimulus-induced BRET signal is significantly different from vehicle-stimulated control, analyzed with paired two-tailed two-sample t test. c, kinetics of MAPK recruitment to AT1R. Intermolecular BRET ratio changes upon AngII or PMA stimulation in the case of the different BRET setups are shown as percentage of the maximal AngII-induced signal averaged from 6 experiments. The indicated BRET set-ups are AT1R–β-arrestin2 interactions (BRET between AT1R–Rluc8 and β-arrestin2–Venus) and the recruitment of MAPK to AT1R (BRET signal in cells co-expressing AT1R–Rluc8, untagged β-arrestin2, and Venus–MEK1 (left panel) or ERK2–Venus (right panel), as indicated, n = 6). The kinetics of β-arrestin2-dependent MAPK recruitment to AT1R was similar and the magnitude was proportional to the receptor–arrestin interaction. d, cells were co-transfected with plasmids encoding AT1R–Rluc8, α1AAR, Venus-MEK1 (left panel), or ERK2–Venus (right panel), and empty vector control (pcDNA3.1) or untagged β-arrestin2, as indicated. AngII and α1AAR agonist A61603 were used as stimuli. Average BRET ratio changes are presented in scatter dot plots with columns (mean ± S.E., n = 7). *, p < 0.05 (versus vehicle-stimulated control), analyzed with paired two-tailed two-sample t test.
Discussion
GPCRs contact β-arrestins via two non-exclusive interactions. One is the binding to the activated receptor's helix bundle, whereas the other is mediated by the receptor-attached phosphates. Active conformation of the receptor and subsequent GRK-mediated phosphorylation are generally thought to be prerequisite for β-arrestin binding (16), suggesting that the two main interactions occur simultaneously. Recent studies have shown that the phosphate and the core interactions can be separated in vitro or by using receptor chimeras and mutants (51–53). However, the question remained whether the distinct interactions could be independent in physiologically relevant situations.
Here we show that activation of PKC by PMA, stimulation of Gq/11-coupled GPCRs, or EGFR initiates β-arrestin2 recruitment to AT1R. Upon receptor overexpression or direct PKC stimulation with PMA, the PKC activity may be well over the levels that are physiologically observed, and may lead to phosphorylation of proteins that are normally not targets of this kinase. Therefore, we also provide evidence that endogenous purinergic receptors can exert the same effect, proving that the interaction can be triggered at physiological levels of PKC activation. Because there are three consensus PKC phosphorylation sites in the C-tail of AT1R (Ser-331, Ser-338, and Ser-348; Ser-338 is part of the TSTS motif) (20, 28), and inhibitors of other protein kinases did not prevent the heterologous AT1R–β-arrestin2 interaction, we assume that PKC acts mainly via direct AT1R phosphorylation. However, we cannot rule out that other, PKC-activated kinases may also participate in this process.
Although the homologous and heterologous mechanisms of β-arrestin recruitment to AT1R show common features, such as the dependence on the same phosphorylated residues in the receptor C terminus, there are important differences: in addition to the different participating kinases, the heterologous mechanism does not demand the active state of the receptor. This was surprising, because receptor binding of β-arrestins, in contrast to visual arrestin (54), was assumed to require receptor activity (16). These results demonstrate that β-arrestins can bind either to the phosphorylated receptor C terminus or to both sites.
We found that the interaction only with the C-terminal phosphates on the receptor, a hallmark of class B receptors (15), was dependent on the stability lock. The stability lock is formed between phosphorylated C-terminal serine–threonine clusters in the receptor and two conserved lysines in the β-arrestin2 N-domain. In the inactive state of β-arrestins these lysines are shielded by the β-arrestin C terminus. Receptor docking of arrestin induces the disruption of the “polar core” and displaces the β-arrestin C terminus (44, 45). Thereafter, Lys-11 and Lys-12 can interact with phosphorylated residues of the receptor C-tail, thereby completing and maintaining the C-terminal swap, or rebind the negatively charged β-arrestin C terminus. This competition dictates the receptor–β-arrestin interaction stability: the presence of phosphorylated serine–threonine cluster favors the receptor binding, and the formed bonds stabilize the interaction. This model is supported by the previous observation that C-terminal truncation of β-arrestin1 enhances the binding to weakly interacting receptors (14). A former study showed that arginine replacement of Lys-11 and Lys-12 transforms the interaction phenotype with AT1R to class A but does not alter it in the case of V2R (30). This discrepancy may be explained by that the highly charged arginines bind the β-arrestin C terminus with higher affinity and therefore do not allow the stable C-terminal swap in the case of AT1R. However, these lysines are not essential for the C-terminal swap, as there are receptors where β-arrestin binding is enhanced by phosphorylation, but the K2A mutation does not significantly alter their interaction (32). Our data do not suggest that other basic residues of β-arrestin may not participate in formation of stabile interaction, but our results clearly demonstrate that the formation of Lys-11 and Lys-12-dependent stability lock is necessary and sufficient to promote class B-type binding.
Moreover, we found that the engagement of Lys-11 and Lys-12 not only stabilizes the interaction with the receptor C-tail, but also dictates the conformation of β-arrestin. PKC phosphorylation was sufficient to induce conformational changes in the β-arrestin2 N-domain. This role of Lys-11 and Lys-12 is in good agreement with the results of a previous study, where phosphopeptide-induced β-arrestin1 conformational shifts were investigated (55). Interestingly, the C-domain sensors (F225, F263) in arrestin did not respond to the recruitment upon PKC phosphorylation of the receptor or in the absence of stability lock, indicating that both stability lock and receptor activity are required for the full set of structural rearrangements in the C-domain. The conformational change of the C-domain seems to be a dynamically regulated process. We could not correlate the kinetic pattern of C-domain activation and MEK1/ERK2 recruitment, however, it does not rule out the intriguing possibility that dynamical conformational changes may play a role in the temporal regulation of β-arrestin interactions with effectors. It is tempting to speculate that the kinetics of conformational rearrangement of the C-domain may reflect the dynamical change of posttranslational modifications, i.e. phosphorylation or ubiquitination (3) of β-arrestin2, but this idea needs to be experimentally tested. Our results also suggest that, besides the C-terminal phosphorylation pattern, the conformational state of the helix bundle also has a prominent role in governing the structural rearrangements in β-arrestins, thereby representing an important part of the receptor barcode.
These data directly demonstrate the existence of multiple active β-arrestin conformations as proposed earlier (31). To the best of our knowledge, this is the first example, where two mechanisms, i.e. the canonical homologous and the novel heterologous activation, induce β-arrestin binding to the same receptor in different conformations in a physiologically relevant setting. Previous studies suggested that the multiple active conformations of β-arrestins have different functions (55, 57, 58). This may explain our finding that PKC-induced β-arrestin2 recruitment directed the receptor to Rab7-enriched late endosomes to a lesser extent than AngII. Interestingly, the heterologous β-arrestin2 recruitment to the α1B-adrenergic receptor was shown to promote late endosomal trafficking, indicating that this type of regulation exhibits receptor specificity (59). Additionally, in a previous study we showed that the intracellular fate of AT1R is ligand-specific (48). These observations support the idea that different β-arrestin conformations could be biased toward particular effectors. However, we cannot rule out that the observed difference may arise from the decreased receptor–β-arrestin2 binding. Nonetheless, heterologous activated β-arrestin2 was also functionally active, capable to induce receptor internalization.
Accumulating data have emphasized the pivotal role of β-arrestin2 in mediating the MAPK signaling of AT1R (21, 60, 61). In this function, β-arrestin plays a role as a scaffold for members of the signaling cascade. In contrast to JNK3, direct interaction between receptor and β-arrestin is required for the activation of ERK (62, 63). Recent data has shown that fully engaged β-arrestin binding to the GPCR is not required for this function, and at least core interaction is dispensable (52). In our experiments, although the fully engaged form of β-arrestin2 was most efficient in binding MAPK cascade members, the PKC-activated form also resulted in the recruitment of MEK1 and ERK2 to the receptor, expanding this observation, and suggesting that the binding of β-arrestin to the receptor per se is critical for MEK/ERK recruitment. We also detected AT1R–β-arrestin2–MAPK complex formation upon α1AAR stimulation. The assembly of the AT1R–β-arrestin2–MEK–ERK complex induced by another receptor offers an intriguing mechanism by which a GPCR serves as a scaffolding protein rather than a signal initiator. The complex may serve as activator of ERK signaling or may simply change the localization of the cascade members. This mechanism, through AT1R or other GPCRs, may resolve the apparent discrepancy in previous studies showing that α1AAR does not interact with β-arrestin2, but activates ERK β-arrestin2-dependently (64). The data also show how the expression of one GPCR (e.g. α1AAR) with another (e.g. AT1R) can expand its signaling repertoire without the formation of receptor heteromers.
Although observing the phenomena described in this paper between endogenously expressed proteins would strengthen our claims, detection of the AT1R–β-arrestin interaction in cell lines expressing AT1R endogenously is currently not possible. The only way of experimental detection would be co-immunoprecipitation, but to the best of our knowledge, no reliably working AT1R antibody is available to date. Nonetheless, effects of PKC activation on the function of endogenous AT1R have been previously observed in rat cardiac fibroblasts, cardiomyocytes, and in a hepatic cell line (65–67). In these cells, activation of PKC by stimulation of vasopressin or lysophosphatidic receptors, or directly by PMA leads to the desensitization of AT1R through receptor phosphorylation. However, β-arrestin has never been demonstrated previously to bind to AT1R during heterologous desensitization.
In conclusion, we have shown that β-arrestin2 can bind to the same GPCR in distinct active conformations in physiologically relevant circumstances. The results improve our understanding of the mechanics of β-arrestin activation.
Experimental procedures
Materials
Molecular biology reagents and High Capacity NeutrAvidin-Agarose Resin were from Thermo Scientific (Waltham, MA). Cell culture reagents were from Invitrogen. Coelenterazine h was obtained from Regis Technologies (Morton Grove, IL). Fluorescein arsenical hairpin binder-ethanedithiol (FlAsH-EDT2) was from Santa Cruz Biotechnology (Dallas, TX). Biotin was from SERVA Electrophoresis GmbH (Heidelberg, Germany). Candesartan and compound 101 were from Tocris (Bristol, United Kingdom). Antibodies were from Cell Signaling Technologies (Danvers, MA). Protease inhibitor mixture (cOmplete) was from Roche Applied Science (Basel, Switzerland). Immobilon Western Chemiluminescent HRP substrate was purchased from Millipore (Billerica, MA). Unless otherwise stated, all other chemicals and reagents were purchased from Sigma.
Plasmid DNA constructs
The Renilla luciferase variant Rluc8 was a kind gift of Dr. Sanjiv Gambhir (68). To generate pRluc8-C1 and pRluc8-N1 plasmids, the coding sequence of Rluc8 was PCR amplified and EYFP was replaced with Rluc8 in Clontech pEYFP-C1 and pEYFP-N1 vectors using AgeI/BglII and AgeI/NotI restriction enzymes, respectively. β-Arrestin2-GFP, β-arrestin2 in pCMV5 vector, and dominant–negative (K44A-mutant) dynamin2A were kind gifts of Dr. Marc G. Caron, Dr. Stephen S. Ferguson, and Dr. Kazuhisa Nakayama, respectively. β-Arrestin2–Venus, β-arrestin2–Cerulean, Venus–Rab5, Super Renilla luciferase-tagged β2AR (β2AR-Sluc), EGFR, Cameleon D3 Ca2+ BRET biosensor, β2AR–Cerulean, V2R–Sluc, M3AChR, and α1AAR constructs were described previously (69–74). NES-BirA was produced by cloning the biotin ligase BirA from pcDNA3.1 MCS-BirA(R118G)–HA (obtained from Abcam (Cambridge, UK)) with a nuclear export signal (PubMed PMID 20972448, QVQAGELQGQLVDVH) attached to its N terminus and was placed in pEYFP-N1 vector between AgeI and NotI restriction sites. Subsequently, Gly-118 was mutated back to Arg with precise gene fusion PCR.
AT1R–Cerulean, AT1R–Rluc8, AT1R-TSTS/A–Rluc, and AT1R–TSTS/A-Cerulean were created by exchanging Rluc to Cerulean or Rluc8 in AT1R–Rluc and AT1R–TSTS/A–Rluc constructs (75), respectively. To generate AT1R–YFP–BAP, we amplified the coding sequence of YFP in-frame fused with BAP (MSGLNDIFEAQKIEWHE) by PCR, then Rluc was replaced with YFP-BAP in AT1R-Rluc. Untagged AT1R in pEYFP-C1 vector was constructed by replacing EYFP with the coding sequence of rat AT1A receptor without the untranslated regions. The cloning strategy was the same for untagged AT1R-TSTS/A, carrying the T332A, S335A, T336A, and S338A mutations. α1AAR–mRFP was constructed by subcloning the open reading frame of α1AAR with the GGGDPPVAT linker sequence into pmRFP-N1 backbone between KpnI and BamHI restriction sites. Precise gene fusion PCR was used to introduce S362A, S363A, S364A mutations into V2R-Sluc (V2R-AAA-Sluc) and G361S, E362S, and Q363S mutations into β2AR-Sluc (β2AR-SSS-Sluc). Cerulean-tagged versions of these mutants and wild type V2R were generated by replacing Sluc to Cerulean using AgeI/NotI restriction enzymes.
Venus–Rab4, Venus–Rab7, and Venus–Rab11 were created by replacing EYFP to monomeric Venus in YFP–Rab4, YFP–Rab7, and YFP–Rab11 constructs (48). To generate Rluc8–β-arrestin2, the coding sequence of rat β-arrestin2 was subcloned from YFP-β-arrestin2 (72) after Rluc8 with a short linker sequence (RSRAQACTR) into pRluc8-C1. Rluc8–β-arrestin2–FlAsH sensors (F139, F154, F225, F263, and F410) were constructed by inserting the CCPGCC amino acid motif after the 139th, 154th, 225th, 263rd, or 410th positions of β-arrestin2 using precise gene fusion PCR, the positions were selected based on earlier studies using similar sensors (40, 41). The K2A–β-arrestin2 mutants were made analogously to bovine K2A–β-arrestin2 (32); K11A and K12A mutations were introduced to rat β-arrestin2 by precise gene fusion PCR to create K2A–β-arrestin2–Venus and Rluc8–K2A–β-arrestin2. K2A mutation was inserted into F139, F154, F225, F263, F410 constructs by exchanging Rluc8 and the beginning of β-arrestin2 coding sequence using AgeI and PstI restriction enzymes with the identical sequence of Rluc8–K2A–β-arrestin2. ERK2–Venus and Venus–MEK1 constructs were kind gifts of Dr. Attila Reményi.
Cell culture and transfection
HEK 293T and COS-7 cells were from American Type Culture Collection (ATCC CRL-3216 and CRL-1651, respectively). The cells were cultured in DMEM supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin. For co-precipitation and BRET measurements, cells were transfected in suspension using Lipofectamine 2000 according to the manufacturer's protocol and plated on 10-cm cell culture dishes or white 96-well plates, respectively. In the case of HEK 293T cells, poly-l-lysine-coated plates were used. For immunoblotting and confocal microscopy, cells were plated the day before transfection, and the transfection was performed on adherent cells. Cells were not tested for mycoplasma contamination.
Co-precipitation experiments
HEK 293T cells were transfected in suspension with plasmids encoding NES–BirA (biotin ligase), β-arrestin2–Cerulean and/or AT1R–YFP–BAP. 24 h after transfection 150 μm biotin was added for 20–24 h to allow substantial biotinylation of AT1R–YFP–BAP. Serum and the excess biotin were removed by changing the media to DMEM supplemented with antibiotics and 1% bovine serum albumin. Cells were serum-starved for 2–4 h, then were stimulated for 20 min at 37 °C. Reactions were stopped by placing the dishes to ice and washing with ice-cold PBS solution. The washing step was repeated 5 times. Then the cells were lysed with RIPA buffer (50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1% Triton X-100, 0.1% SDS, 0.25% sodium deoxycholate, 1 mm EDTA) supplemented with cOmplete Protease Inhibitor mixture (Roche) and Phosphatase Inhibitor Mixture 3 (Sigma). Lysates were collected, rotated for 15 min at low speed, then centrifuged at 20,800 × g for 10 min. Supernatants were incubated with 30 μl of High Capacity NeutrAvidin-agarose resin for 30 min at 4 °C, then the beads were washed 3 times with ice-cold supplemented RIPA and once with PBS. The beads were resuspended in PBS. YFP and Cerulean fluorescence intensities were determined by exciting at 510 or 435 nm and measuring emission at 535 or 480 nm, respectively, using a Thermo Scientific Varioskan Flash multimode plate reader. Fluorescent images from the NeutrAvidin beads were made with Zeiss LSM 710 confocal laser-scanning microscope using a ×20 objective.
Bioluminescence resonance energy transfer measurements
All the BRET measurements were performed on adherent cells, the measurements were made 24–28 or 44–48 h post-transfection in the case of HEK 293T or COS-7 cells, respectively. The cells were washed, and the culture medium was changed to modified Kreb's-Ringer medium (120 mm NaCl, 10 mm glucose, 10 mm Na-HEPES, 4.7 mm KCl, 1.2 mm CaCl2, 0.7 mm MgSO4, pH 7.4). Intermolecular BRET was detected between Renilla luciferase (Sluc or Rluc8)- and Venus-tagged proteins. First, we measured Venus fluorescence intensity by exciting at 510 nm and measuring emission at 535 nm using a Thermo Scientific Varioskan Flash multimode plate reader. Luminescence intensity was recorded after addition of Renilla luciferase substrate coelenterazine h (5 μm) every 70 s for 24 min using 530- and 480-nm filters at 37 °C. To calculate the BRET ratio change, the vehicle-stimulated BRET ratio was subtracted from the stimulus-induced BRET ratio. For quantification, we determined the average BRET ratio change in the first 24 min after stimulation. The total luminescence was determined without filter. BRET titration experiments and candesartan-induced β-arrestin2 dissociation measurements were made in duplicate, all other BRET measurements were performed in triplicate.
To detect conformational changes of Rluc8–β-arrestin2–FlAsH sensors, the cells were washed three times with modified Kreb's-Ringer medium. Thereafter, an earlier described labeling procedure (76) was adapted for 96-well plate measurements. Briefly, FlAsH-EDT2 was incubated with EDT for 5 min at room temperature (1 and 12.5 mm diluted in DMSO, respectively), to assure that all FlAsH molecules are in FlAsH-EDT2 form. Then the cells were labeled with 500 nm FlAsH-EDT2 (together with 12.5 μm EDT and 0.1% (v/v) DMSO in modified Kreb's-Ringer solution) for 1 h at room temperature. After labeling, the excess and non-specifically bound FlAsH dye was removed by changing the labeling medium to Kreb's-Ringer solution containing 250 μm EDT for 10 min. Next, the cells were washed three times with Kreb's-Ringer medium, and the same BRET measurement procedure was used as for the intermolecular BRET experiments. Intramolecular BRET was measured between Rluc8 tag and FlAsH label of the sensor.
Because the amplitude of intramolecular BRET change is dependent on the number of the activated β-arrestins, we calculated relative FlAsH signals: the stimulus-induced intramolecular BRET change was normalized to the intermolecular BRET change (measured between Rluc8 tag of the sensor and Venus-tagged receptor in separate experiments), which is proportional to the quantity of activated molecules.
Expression of the wild type and mutant BRET constructs
The expression of the wild type and mutant donor or acceptor constructs was compared by the determination of the total luminescence or Venus fluorescence, respectively. The expression data are shown in Fig. S5.
Confocal laser-scanning microscopy
To obtain confocal images from living cells, HEK 293T cells were plated on poly-l-lysine-coated glass coverslips. The next day the cells were transfected with plasmids encoding fluorescently labeled receptors and β-arrestin2. 24 h after transfection, the medium was changed to modified Kreb's-Ringer and the localization of the probes was examined in living cells at 37 °C with Zeiss LSM 710 confocal laser-scanning microscope using a ×63 objective.
Immunoblotting
The reactions were stopped by placing the 6-well plates on ice and washing each well twice with ice-cold PBS. Cells were scraped into SDS sample buffer, briefly sonicated, and heated for 15 min at 95 °C. Proteins were separated by SDS-polyacrylamide gel electrophoresis and transferred to PVDF membranes. The membranes were blocked in 5% (w/v) fat-free milk powder in PBS with 0.05% (v/v) Tween 20 (PBST) for 1 h at room temperature and incubated with primary antibody (diluted in PBST containing 5% fat-free milk powder) overnight at 4 °C. The membranes were washed 3 times with PBST for 10 min, then incubated with HRP-linked secondary antibody (goat anti-mouse or goat anti-rabbit, 1:5000 diluted in PBST containing 5% fat-free milk powder) for 1 h at room temperature and washed 3 times. The antibodies were visualized using enhanced chemiluminescence. First the membranes were incubated with mouse anti-phospho-p44/42 MAPK (Thr-202, Tyr-204) primary antibody (number 9101, Cell Signaling) and developed, then the membranes were stripped using a guanidine hydrochloride-based stripping solution (77). Thereafter total ERK1/2 amount was determined using rabbit anti-p44/42 MAPK (number 9102, Cell Signaling) antibody.
Statistical analysis
No statistical methods were used to predetermine the sample size. The sample size (n) in figure legends refers to the number of independent experiments (biological replicates), all data points were included in the statistical analyses. The experiments were not randomized and the investigators were not blinded. GraphPad Prism software was used for graph construction, statistical analysis, and curve fitting. Unless otherwise stated, data are presented as mean ± S.E. Paired two-sample two-tailed Student's t tests or repeated measures one-way ANOVA with Bonferroni post hoc test were performed to compare the stimulus-promoted signals with the vehicle-stimulated controls. The effects of inhibitors on stimulus-induced signals were analyzed using two-way ANOVA with Bonferroni post hoc test, a significant inhibitory effect was concluded if the p value of interaction was less than 0.05. The binding ability of the wild type biosensors to AT1R was compared using one-way ANOVA with Bonferroni post hoc test. The same statistical test was used to compare the expression of all biosensors. The expression of the wild type and mutant receptors were compared with two-sample two-tailed Student's t tests. The effect of the K2A mutation on the basal intramolecular BRET ratio of the biosensors was analyzed with repeated measures one-way ANOVA with a Bonferroni post hoc test. The relative, interaction-normalized FlAsH signals were considered significant if the mean value was different from 0 (p < 0.05), analyzed with one-sample t test, paired two-sample two-tailed t tests were always performed simultaneously on the raw data.
Author contributions
A. D. T., S. P., and P. G. performed the experiments. L. H., V. V. G., and G. T. supervised the work. A. D. T., S. P., P. G., P. V., A. B., V. V. G., L. H., and G. T. analyzed the results, wrote the manuscript, and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We are grateful to Eszter Soltész-Katona for generation of α1AAR-mRFP construct. The technical assistance of Ilona Oláh and Eszter Halász is greatly appreciated.
This work was supported by Hungarian National Research, Development and Innovation Fund Grants NKFI NK100883, K116954, and NVKP_16-1-2016-0039 (to L. H.), the ÚNKP-17-3-III-SE-23 New National Excellence Program of the Ministry of Human Capacities (to A. D. T.), and National Institutes of Health Grant R35 GM122491 (to V. V. G.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S5.
- GPCR
- G protein–coupled receptor
- GRK
- GPCR receptor kinase
- AT1R
- AT1 angiotensin receptor
- BAP
- biotin acceptor peptide
- βarr2
- β-arrestin2
- AngII
- angiotensin II
- PMA
- phorbol myristate acetate
- BRET
- bioluminescence resonance energy transfer
- M3AChR
- M3 muscarinic acetylcholine receptor
- EGFR
- epidermal growth factor receptor
- Cmpd101
- compound 101
- β2AR
- β2-adrenergic receptor
- FlAsH
- fluorescein arsenical hairpin binder
- EDT
- ethanedithiol
- ANOVA
- analysis of variance
- TSTS
- Thr-332, Ser-335, Thr-336, and Ser-338
- α1AAR
- α1A-adrenergic receptor
- AT1R-TSTS/A
- AT1R harboring T332, S335, T336, S338 mutation
- β2AR-SSS
- G361S, E362S, Q363S mutant β2AR
- K2A–β-arrestin2
- β-arrestin2 harboring K11,12A mutation
- Rluc
- Renilla luciferase
- Sluc
- Super Renilla luciferase
- V2R
- V2 vasopressin receptor
- V2R-AAA
- S362,363,364A mutant V2R.
References
- 1. Overington J. P., Al-Lazikani B., and Hopkins A. L. (2006) How many drug targets are there? Nat. Rev. Drug Discov. 5, 993–996 10.1038/nrd2199 [DOI] [PubMed] [Google Scholar]
- 2. Kelly E., Bailey C. P., and Henderson G. (2008) Agonist-selective mechanisms of GPCR desensitization. Br. J. Pharmacol. 153, S379–388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Shenoy S. K., and Lefkowitz R. J. (2011) β-Arrestin-mediated receptor trafficking and signal transduction. Trends Pharmacol. Sci. 32, 521–533 10.1016/j.tips.2011.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Xiang B., Yu G. H., Guo J., Chen L., Hu W., Pei G., and Ma L. (2001) Heterologous activation of protein kinase C stimulates phosphorylation of δ-opioid receptor at serine 344, resulting in β-arrestin- and clathrin-mediated receptor internalization. J. Biol. Chem. 276, 4709–4716 10.1074/jbc.M006187200 [DOI] [PubMed] [Google Scholar]
- 5. Namkung Y., and Sibley D. R. (2004) Protein kinase C mediates phosphorylation, desensitization, and trafficking of the D2 dopamine receptor. J. Biol. Chem. 279, 49533–49541 10.1074/jbc.M408319200 [DOI] [PubMed] [Google Scholar]
- 6. Castillo-Badillo J. A., Sánchez-Reyes O. B., Alfonzo-Méndez M. A., Romero-Ávila M. T., Reyes-Cruz G., and García-Sáinz J. A. (2015) α1B-adrenergic receptors differentially associate with Rab proteins during homologous and heterologous desensitization. PLoS ONE. 10, e0121165 10.1371/journal.pone.0121165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shukla A. K., Xiao K., and Lefkowitz R. J. (2011) Emerging paradigms of β-arrestin-dependent seven transmembrane receptor signaling. Trends Biochem. Sci. 36, 457–469 10.1016/j.tibs.2011.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. DeWire S. M., Ahn S., Lefkowitz R. J., and Shenoy S. K. (2007) β-Arrestins and cell signaling. Annu. Rev. Physiol. 69, 483–510 10.1146/annurev.physiol.69.022405.154749 [DOI] [PubMed] [Google Scholar]
- 9. Kang Y., Zhou X. E., Gao X., He Y., Liu W., Ishchenko A., Barty A., White T. A., Yefanov O., Han G. W., Xu Q., de Waal P. W., Ke J., Tan M. H., Zhang C., et al. (2015) Crystal structure of rhodopsin bound to arrestin by femtosecond X-ray laser. Nature 523, 561–567 10.1038/nature14656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shukla A. K., Westfield G. H., Xiao K., Reis R. I., Huang L. Y., Tripathi-Shukla P., Qian J., Li S., Blanc A., Oleskie A. N., Dosey A. M., Su M., Liang C. R., Gu L. L., Shan J. M., et al. (2014) Visualization of arrestin recruitment by a G-protein-coupled receptor. Nature 512, 218–222 10.1038/nature13430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Oakley R. H., Laporte S. A., Holt J. A., Caron M. G., and Barak L. S. (2000) Differential affinities of visual arrestin, β-arrestin1, and β-arrestin2 for G protein-coupled receptors delineate two major classes of receptors. J. Biol. Chem. 275, 17201–17210 10.1074/jbc.M910348199 [DOI] [PubMed] [Google Scholar]
- 12. Wei H., Ahn S., Barnes W. G., and Lefkowitz R. J. (2004) Stable interaction between β-arrestin 2 and angiotensin type 1A receptor is required for β-arrestin 2-mediated activation of extracellular signal-regulated kinases 1 and 2. J. Biol. Chem. 279, 48255–48261 10.1074/jbc.M406205200 [DOI] [PubMed] [Google Scholar]
- 13. Oakley R. H., Laporte S. A., Holt J. A., Barak L. S., and Caron M. G. (1999) Association of β-arrestin with G protein-coupled receptors during clathrin-mediated endocytosis dictates the profile of receptor resensitization. J. Biol. Chem. 274, 32248–32257 10.1074/jbc.274.45.32248 [DOI] [PubMed] [Google Scholar]
- 14. Oakley R. H., Laporte S. A., Holt J. A., Barak L. S., and Caron M. G. (2001) Molecular determinants underlying the formation of stable intracellular G protein-coupled receptor-β-arrestin complexes after receptor endocytosis. J. Biol. Chem. 276, 19452–19460 10.1074/jbc.M101450200 [DOI] [PubMed] [Google Scholar]
- 15. Thomsen A. R. B., Plouffe B., Cahill T. J. 3rd, Shukla A. K., Tarrasch J. T., Dosey A. M., Kahsai A. W., Strachan R. T., Pani B., Mahoney J. P., Huang L., Breton B., Heydenreich F. M., Sunahara R. K., Skiniotis G., Bouvier M., and Lefkowitz R. J. (2016) GPCR-G protein-β-arrestin super-complex mediates sustained G protein signaling. Cell 166, 907–919 10.1016/j.cell.2016.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Krasel C., Bünemann M., Lorenz K., and Lohse M. J. (2005) β-Arrestin binding to the β2-adrenergic receptor requires both receptor phosphorylation and receptor activation. J. Biol. Chem. 280, 9528–9535 10.1074/jbc.M413078200 [DOI] [PubMed] [Google Scholar]
- 17. Lorenz S., Frenzel R., Paschke R., Breitwieser G. E., and Miedlich S. U. (2007) Functional desensitization of the extracellular calcium-sensing receptor is regulated via distinct mechanisms: role of G protein-coupled receptor kinases, protein kinase C and β-arrestins. Endocrinology 148, 2398–2404 10.1210/en.2006-1035 [DOI] [PubMed] [Google Scholar]
- 18. Hunyady L., Catt K. J., Clark A. J., and Gáborik Z. (2000) Mechanisms and functions of AT(1) angiotensin receptor internalization. Regul. Pept. 91, 29–44 10.1016/S0167-0115(00)00137-3 [DOI] [PubMed] [Google Scholar]
- 19. Smith R. D., Hunyady L., Olivares-Reyes J. A., Mihalik B., Jayadev S., and Catt K. J. (1998) Agonist-induced phosphorylation of the angiotensin AT1a receptor is localized to a serine/threonine-rich region of its cytoplasmic tail. Mol. Pharmacol. 54, 935–941 [DOI] [PubMed] [Google Scholar]
- 20. Qian H., Pipolo L., and Thomas W. G. (1999) Identification of protein kinase C phosphorylation sites in the angiotensin II (AT1A) receptor. Biochem. J. 343, 637–644 10.1042/bj3430637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hunyady L., and Catt K. J. (2006) Pleiotropic AT1 receptor signaling pathways mediating physiological and pathogenic actions of angiotensin II. Mol. Endocrinol. 20, 953–970 10.1210/me.2004-0536 [DOI] [PubMed] [Google Scholar]
- 22. Mustafa S., See H. B., Seeber R. M., Armstrong S. P., White C. W., Ventura S., Ayoub M. A., and Pfleger K. D. (2012) Identification and profiling of novel α1A-adrenoceptor-CXC chemokine receptor 2 heteromer. J. Biol. Chem. 287, 12952–12965 10.1074/jbc.M111.322834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Oppermann M., Freedman N. J., Alexander R. W., and Lefkowitz R. J. (1996) Phosphorylation of the type 1A angiotensin II receptor by G protein-coupled receptor kinases and protein kinase C. J. Biol. Chem. 271, 13266–13272 10.1074/jbc.271.22.13266 [DOI] [PubMed] [Google Scholar]
- 24. Chuang T. T., LeVine H. 3rd, and De Blasi A. (1995) Phosphorylation and activation of β-adrenergic receptor kinase by protein kinase C. J. Biol. Chem. 270, 18660–18665 10.1074/jbc.270.31.18660 [DOI] [PubMed] [Google Scholar]
- 25. Krasel C., Dammeier S., Winstel R., Brockmann J., Mischak H., and Lohse M. J. (2001) Phosphorylation of GRK2 by protein kinase C abolishes its inhibition by calmodulin. J. Biol. Chem. 276, 1911–1915 10.1074/jbc.M008773200 [DOI] [PubMed] [Google Scholar]
- 26. Pronin A. N., and Benovic J. L. (1997) Regulation of the G protein-coupled receptor kinase GRK5 by protein kinase C. J. Biol. Chem. 272, 3806–3812 10.1074/jbc.272.6.3806 [DOI] [PubMed] [Google Scholar]
- 27. Paradis J. S., Ly S., Blondel-Tepaz É., Galan J. A., Beautrait A., Scott M. G. H., Enslen H., Marullo S., Roux P. P., and Bouvier M. (2015) Receptor sequestration in response to β-arrestin-2 phosphorylation by ERK1/2 governs steady-state levels of GPCR cell-surface expression. Proc. Natl. Acad. Sci. U.S.A. 112, E5160–8 10.1073/pnas.1508836112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hunyady L., Bor M., Balla T., and Catt K. J. (1994) Identification of a cytoplasmic Ser-Thr-Leu motif that determines agonist-induced internalization of the AT1 angiotensin receptor. J. Biol. Chem. 269, 31378–31382 [PubMed] [Google Scholar]
- 29. Qian H., Pipolo L., and Thomas W. G. (2001) Association of β-arrestin 1 with the type 1A angiotensin II receptor involves phosphorylation of the receptor carboxyl terminus and correlates with receptor internalization. Mol. Endocrinol. 15, 1706–1719 10.1210/mend.15.10.0714, [DOI] [PubMed] [Google Scholar]
- 30. Shenoy S. K., and Lefkowitz R. J. (2005) Receptor-specific ubiquitination of β-arrestin directs assembly and targeting of seven-transmembrane receptor signalosomes. J. Biol. Chem. 280, 15315–15324 10.1074/jbc.M412418200 [DOI] [PubMed] [Google Scholar]
- 31. Gurevich E. V., and Gurevich V. V. (2006) Arrestins: ubiquitous regulators of cellular signaling pathways. Genome Biol. 7, 236 10.1186/gb-2006-7-9-236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gimenez L. E., Kook S., Vishnivetskiy S. A., Ahmed M. R., Gurevich E. V., and Gurevich V. V. (2012) Role of receptor-attached phosphates in binding of visual and non-visual arrestins to G protein-coupled receptors. J. Biol. Chem. 287, 9028–9040 10.1074/jbc.M111.311803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shukla A. K., Manglik A., Kruse A. C., Xiao K., Reis R. I., Tseng W. C., Staus D. P., Hilger D., Uysal S., Huang L. Y., Paduch M., Tripathi-Shukla P., Koide A., Koide S., Weis W. I., Kossiakoff A. A., Kobilka B. K., and Lefkowitz R. J. (2013) Structure of active β-arrestin-1 bound to a G-protein-coupled receptor phosphopeptide. Nature 497, 137–141 10.1038/nature12120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gimenez L. E., Babilon S., Wanka L., Beck-Sickinger A. G., and Gurevich V. V. (2014) Mutations in arrestin-3 differentially affect binding to neuropeptide Y receptor subtypes. Cell Signal. 26, 1523–1531 10.1016/j.cellsig.2014.03.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhou X. E., He Y., de Waal P. W., Gao X., Kang Y., Van Eps N., Yin Y., Pal K., Goswami D., White T. A., Barty A., Latorraca N. R., Chapman H. N., Hubbell W. L., Dror R. O., et al. (2017) Identification of phosphorylation codes for arrestin recruitment by G protein-coupled receptors. Cell 170, 457–469.e13 10.1016/j.cell.2017.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gáborik Z., Szaszák M., Szidonya L., Balla B., Paku S., Catt K. J., Clark A. J., and Hunyady L. (2001) β-Arrestin- and dynamin-dependent endocytosis of the AT1 angiotensin receptor. Mol. Pharmacol. 59, 239–247 [DOI] [PubMed] [Google Scholar]
- 37. Anborgh P. H., Seachrist J. L., Dale L. B., and Ferguson S. S. (2000) Receptor/β-arrestin complex formation and the differential trafficking and resensitization of β2-adrenergic and angiotensin II type 1A receptors. Mol. Endocrinol. 14, 2040–2053 10.1210/mend.14.12.0565, [DOI] [PubMed] [Google Scholar]
- 38. Le M. T., Vanderheyden P. M. L., Szaszák M., Hunyady L., and Vauquelin G. (2002) Angiotensin IV is a potent agonist for constitutive active human AT1 receptors. Distinct roles of the N-and C-terminal residues of angiotensin II during AT1 receptor activation. J. Biol. Chem. 277, 23107–23110 10.1074/jbc.C200201200 [DOI] [PubMed] [Google Scholar]
- 39. Zindel D., Butcher A. J., Al-Sabah S., Lanzerstorfer P., Weghuber J., Tobin A. B., Bünemann M., and Krasel C. (2015) Engineered hyperphosphorylation of the β2-adrenoceptor prolongs arrestin-3 binding and induces arrestin internalization. Mol. Pharmacol. 87, 349–362 10.1124/mol.114.095422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lee M. H., Appleton K. M., Strungs E. G., Kwon J. Y., Morinelli T. A., Peterson Y. K., Laporte S. A., and Luttrell L. M. (2016) The conformational signature of β-arrestin2 predicts its trafficking and signalling functions. Nature 531, 665–668 10.1038/nature17154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nuber S., Zabel U., Lorenz K., Nuber A., Milligan G., Tobin A. B., Lohse M. J., and Hoffmann C. (2016) β-Arrestin biosensors reveal a rapid, receptor-dependent activation/deactivation cycle. Nature 531, 661–664 10.1038/nature17198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kocan M., See H. B., Seeber R. M., Eidne K. A., and Pfleger K. D. (2008) Demonstration of improvements to the bioluminescence resonance energy transfer (BRET) technology for the monitoring of G protein-coupled receptors in live cells. J. Biomol. Screen. 13, 888–898 10.1177/1087057108324032 [DOI] [PubMed] [Google Scholar]
- 43. Vishnivetskiy S. A., Schubert C., Climaco G. C., Gurevich Y. V., Velez M. G., and Gurevich V. V. (2000) An additional phosphate-binding element in arrestin molecule. Implications for the mechanism of arrestin activation. J. Biol. Chem. 275, 41049–41057 10.1074/jbc.M007159200 [DOI] [PubMed] [Google Scholar]
- 44. Hanson S. M., Francis D. J., Vishnivetskiy S. A., Kolobova E. A., Hubbell W. L., Klug C. S., and Gurevich V. V. (2006) Differential interaction of spin-labeled arrestin with inactive and active phosphorhodopsin. Proc. Natl. Acad. Sci. U.S.A. 103, 4900–4905 10.1073/pnas.0600733103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhuo Y., Vishnivetskiy S. A., Zhan X., Gurevich V. V., and Klug C. S. (2014) Identification of receptor binding-induced conformational changes in non-visual arrestins. J. Biol. Chem. 289, 20991–21002 10.1074/jbc.M114.560680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gáborik Z., and Hunyady L. (2004) Intracellular trafficking of hormone receptors. Trends Endocrinol. Metab. 15, 286–293 10.1016/j.tem.2004.06.009 [DOI] [PubMed] [Google Scholar]
- 47. Moore C. A., Milano S. K., and Benovic J. L. (2007) Regulation of Receptor Trafficking by GRKs and Arrestins. Annu. Rev. Physiol. 69, 451–482 10.1146/annurev.physiol.69.022405.154712 [DOI] [PubMed] [Google Scholar]
- 48. Szakadáti G., Tóth A. D., Oláh I., Erdélyi L. S., Balla T., Várnai P., Hunyady L., and Balla A. (2015) Investigation of the fate of type I angiotensin receptor after biased activation. Mol. Pharmacol. 87, 972–981 10.1124/mol.114.097030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Namkung Y., Le Gouill C., Lukashova V., Kobayashi H., Hogue M., Khoury E., Song M., Bouvier M., and Laporte S. A. (2016) Monitoring G protein-coupled receptor and β-arrestin trafficking in live cells using enhanced bystander BRET. Nat. Commun. 7, 12178 10.1038/ncomms12178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zhan X., Perez A., Gimenez L. E., Vishnivetskiy S. A., and Gurevich V. V. (2014) Arrestin-3 binds the MAP kinase JNK3α2 via multiple sites on both domains. Cell. Signal. 26, 766–76 10.1016/j.cellsig.2014.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kumari P., Srivastava A., Ghosh E., Ranjan R., Dogra S., Yadav P. N., and Shukla A. K. (2017) Core engagement with β-arrestin is dispensable for agonist-induced vasopressin receptor endocytosis and ERK activation. Mol. Biol. Cell 28, 1003–1010 10.1091/mbc.E16-12-0818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kumari P., Srivastava A., Banerjee R., Ghosh E., Gupta P., Ranjan R., Chen X., Gupta B., Gupta C., Jaiman D., and Shukla A. K. (2016) Functional competence of a partially engaged GPCR-β-arrestin complex. Nat. Commun. 7, 13416 10.1038/ncomms13416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Cahill T. J. 3rd, Thomsen A. R., Tarrasch J. T., Plouffe B., Nguyen A. H., Yang F., Huang L. Y., Kahsai A. W., Bassoni D. L., Gavino B. J., Lamerdin J. E., Triest S., Shukla A. K., Berger B., Little J., et al. (2017) Distinct conformations of GPCR-β-arrestin complexes mediate desensitization, signaling, and endocytosis. Proc. Natl. Acad. Sci. U.S.A. 114, 2562–2567 10.1073/pnas.1701529114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Gurevich V. V., and Benovic J. L. (1992) Cell-free expression of visual arrestin: truncation mutagenesis identifies multiple domains involved in rhodopsin interaction. J. Biol. Chem. 267, 21919–21923 [PubMed] [Google Scholar]
- 55. Yang F., Yu X., Liu C., Qu C. X., Gong Z., Liu H. D., Li F. H., Wang H. M., He D. F., Yi F., Song C., Tian C. L., Xiao K. H., Wang J. Y., and Sun J. P. (2015) Phospho-selective mechanisms of arrestin conformations and functions revealed by unnatural amino acid incorporation and 19F-NMR. Nat. Commun. 6, 8202 10.1038/ncomms9202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zhan X., Gimenez L. E., Gurevich V. V., and Spiller B. W. (2011) Crystal structure of arrestin-3 reveals the basis of the difference in receptor binding between two non-visual subtypes. J. Mol. Biol. 406, 467–478 10.1016/j.jmb.2010.12.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kim J., Ahn S., Ren X. R., Whalen E. J., Reiter E., Wei H., and Lefkowitz R. J. (2005) Functional antagonism of different G protein-coupled receptor kinases for β-arrestin-mediated angiotensin II receptor signaling. Proc. Natl. Acad. Sci. U.S.A. 102, 1442–1447 10.1073/pnas.0409532102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Nobles K. N., Xiao K., Ahn S., Shukla A. K., Lam C. M., Rajagopal S., Strachan R. T., Huang T. Y., Bressler E. A., Hara M. R., Shenoy S. K., Gygi S. P., and Lefkowitz R. J. (2011) Distinct phosphorylation sites on the β2-adrenergic receptor establish a barcode that encodes differential functions of β-arrestin. Sci. Signal. 4, ra51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Alfonzo-Méndez M. A., Hernández-Espinosa D. A., Carmona-Rosas G., Romero-Ávila M. T., Reyes-Cruz G., and García-Sáinz J. A. (2017) Protein kinase C activation promotes α1B-adrenoceptor internalization and late endosome trafficking through Rab9 interaction: role in heterologous desensitization. Mol. Pharmacol. 91, 296–306 10.1124/mol.116.106583 [DOI] [PubMed] [Google Scholar]
- 60. Luttrell L. M., Roudabush F. L., Choy E. W., Miller W. E., Field M. E., Pierce K. L., and Lefkowitz R. J. (2001) Activation and targeting of extracellular signal-regulated kinases by β-arrestin scaffolds. Proc. Natl. Acad. Sci. U.S.A. 98, 2449–2454 10.1073/pnas.041604898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Wei H., Ahn S., Shenoy S. K., Karnik S. S., Hunyady L., Luttrell L. M., and Lefkowitz R. J. (2003) Independent β-arrestin 2 and G protein-mediated pathways for angiotensin II activation of extracellular signal-regulated kinases 1 and 2. Proc. Natl. Acad. Sci. U.S.A. 100, 10782–10787 10.1073/pnas.1834556100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Coffa S., Breitman M., Hanson S. M., Callaway K., Kook S., Dalby K. N., and Gurevich V. V. (2011) The effect of arrestin conformation on the recruitment of c-Raf1, MEK1, and ERK1/2 activation. PLoS ONE 6, e28723 10.1371/journal.pone.0028723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Breitman M., Kook S., Gimenez L. E., Lizama B. N., Palazzo M. C., Gurevich E. V., and Gurevich V. V. (2012) Silent scaffolds: inhibition of c-Jun N-terminal kinase 3 activity in cell by dominant-negative arrestin-3 mutant. J. Biol. Chem. 287, 19653–19664 10.1074/jbc.M112.358192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Perez-Aso M., Segura V., Montó F., Barettino D., Noguera M. A., Milligan G., and D'Ocon P. (2013) The three α1-adrenoceptor subtypes show different spatio-temporal mechanisms of internalization and ERK1/2 phosphorylation. Biochim. Biophys. Acta 1833, 2322–2333 10.1016/j.bbamcr.2013.06.013 [DOI] [PubMed] [Google Scholar]
- 65. Meszaros J. G., Raphael R., Lio F. M., and Brunton L. L. (2000) Protein kinase C contributes to desensitization of ANG II signaling in adult rat cardiac fibroblasts. Am. J. Physiol. Cell Physiol. 279, C1978–85 [DOI] [PubMed] [Google Scholar]
- 66. Zhang M., Turnbaugh D., Cofie D., Dogan S., Koshida H., Fugate R., and Kem D. C. (1996) Protein kinase C modulation of cardiomyocyte angiotensin II and vasopressin receptor desensitization. Hypertension 27, 269–275 10.1161/01.HYP.27.2.269 [DOI] [PubMed] [Google Scholar]
- 67. García-Caballero A., Olivares-Reyes J. A., Catt K. J., and García-Saínz J. A. (2001) Angiotensin AT1 receptor phosphorylation and desensitization in a hepatic cell line: roles of protein kinase C and phosphoinositide 3-kinase. Mol. Pharmacol. 59, 576–585 [DOI] [PubMed] [Google Scholar]
- 68. De A., Loening A. M., and Gambhir S. S. (2007) An improved bioluminescence resonance energy transfer strategy for imaging intracellular events in single cells and living subjects. Cancer Res. 67, 7175–7183 10.1158/0008-5472.CAN-06-4623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Gyombolai P., Boros E., Hunyady L., and Turu G. (2013) Differential β-arrestin2 requirements for constitutive and agonist-induced internalization of the CB1 cannabinoid receptor. Mol. Cell Endocrinol. 372, 116–127 10.1016/j.mce.2013.03.013 [DOI] [PubMed] [Google Scholar]
- 70. Tóth D. J., Tóth J. T., Gulyás G., Balla A., Balla T., Hunyady L., and Várnai P. (2012) Acute depletion of plasma membrane phosphatidylinositol 4,5-bisphosphate impairs specific steps in endocytosis of the G-protein-coupled receptor. J. Cell Sci. 125, 2185–2197 10.1242/jcs.097279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Gulyás G., Tóth J. T., Tóth D. J., Kurucz I., Hunyady L., Balla T., and Várnai P. (2015) Measurement of inositol 1,4,5-trisphosphate in living cells using an improved set of resonance energy transfer-based biosensors. PLoS ONE 10, e0125601 10.1371/journal.pone.0125601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Tóth A. D., Gyombolai P., Szalai B., Várnai P., Turu G., and Hunyady L. (2017) Angiotensin type 1A receptor regulates β-arrestin binding of the β2-adrenergic receptor via heterodimerization. Mol. Cell Endocrinol. 442, 113–124 10.1016/j.mce.2016.11.027 [DOI] [PubMed] [Google Scholar]
- 73. Szalai B., Hoffmann P., Prokop S., Erdélyi L., Várnai P., and Hunyady L. (2014) Improved methodical approach for quantitative BRET analysis of G protein-coupled receptor dimerization. PLoS ONE. 9, e109503 10.1371/journal.pone.0109503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Turu G., Várnai P., Gyombolai P., Szidonya L., Offertaler L., Bagdy G., Kunos G., and Hunyady L. (2009) Paracrine transactivation of the CB1 cannabinoid receptor by AT1 angiotensin and other Gq/11 protein-coupled receptors. J. Biol. Chem. 284, 16914–16921 10.1074/jbc.M109.003681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Balla A., Tóth D. J., Soltész-Katona E., Szakadáti G., Erdélyi L. S., Várnai P., and Hunyady L. (2012) Mapping of the localization of type 1 angiotensin receptor in membrane microdomains using bioluminescence resonance energy transfer-based sensors. J. Biol. Chem. 287, 9090–9099 10.1074/jbc.M111.293944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Hoffmann C., Gaietta G., Zürn A., Adams S. R., Terrillon S., Ellisman M. H., Tsien R. Y., and Lohse M. J. (2010) Fluorescent labeling of tetracysteine-tagged proteins in intact cells. Nat. Protoc. 5, 1666–1677 10.1038/nprot.2010.129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Yeung Y. G., and Stanley E. R. (2009) A solution for stripping antibodies from polyvinylidene fluoride immunoblots for multiple reprobing. Anal. Biochem. 389, 89–91 10.1016/j.ab.2009.03.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.