

Abstract
Clostridium difficile infection is the leading cause of hospital-acquired diarrhea and is mediated by the actions of two toxins, TcdA and TcdB. The toxins perturb host cell function through a multistep process of receptor binding, endocytosis, low pH–induced pore formation, and the translocation and delivery of an N-terminal glucosyltransferase domain that inactivates host GTPases. Infection studies with isogenic strains having defined toxin deletions have established TcdB as an important target for therapeutic development. Monoclonal antibodies that neutralize TcdB function have been shown to protect against C. difficile infection in animal models and reduce recurrence in humans. Here, we report the mechanism of TcdB neutralization by PA41, a humanized monoclonal antibody capable of neutralizing TcdB from a diverse array of C. difficile strains. Through a combination of structural, biochemical, and cell functional studies, involving X-ray crystallography and EM, we show that PA41 recognizes a single, highly conserved epitope on the TcdB glucosyltransferase domain and blocks productive translocation and delivery of the enzymatic cargo into the host cell. Our study reveals a unique mechanism of C. difficile toxin neutralization by a monoclonal antibody, which involves targeting a process that is conserved across the large clostridial glucosylating toxins. The PA41 antibody described here provides a valuable tool for dissecting the mechanism of toxin pore formation and translocation across the endosomal membrane.
Keywords: antibody, bacterial pathogenesis, bacterial toxin, electron microscopy (EM), membrane transport, toxin, X-ray crystallography, neutralization
Introduction
Clostridium difficile has become a major global healthcare problem, causing nearly half a million infections each year in the United States alone (1, 2). C. difficile colonizes and infects the human colon following antibiotic-induced dysbiosis of the gut microbiota. Disease symptoms range from mild gastritis to recurrent diarrhea and can include severe outcomes such as pseudomembranous colitis, sepsis, and death (1). Traditional treatments for C. difficile infections (CDIs)4 involve the prescription of antibiotics such as metronidazole and vancomycin, but problems with recurrence are common, and new therapeutic strategies are needed (1). Blocking virulence factors responsible for the pathology of CDI, alone or in conjunction with traditional treatments that target the pathogen, may be an effective therapeutic approach.
The pathogenesis of C. difficile is mediated by two large, homologous, multidomain toxins, TcdA and TcdB, with TcdB often linked with severe disease phenotypes (3, 4). Although most pathogenic strains express both toxins, clinical isolates that express only TcdB (A−B+ strains) have also been observed (5). Despite their homology, TcdA and TcdB engage different receptors on the colonic epithelium. TcdA has been proposed to bind glycosylated receptors through its C-terminal combined repetitive oligopeptides (CROPs) domain (6, 7). TcdB engages multiple receptors on the host cell surface, but the binding interactions are not restricted to the CROPs (8–10). Upon binding, the toxins are endocytosed and transported to acidified endosomes where they undergo a conformational change that is thought to lead to membrane insertion and formation of a “pore” (11–14). The term pore refers to the ion conductivity and presumed protein-conducting properties associated with this membrane-associated state, although the structural character and stability of such a pore have not yet been clearly defined. The cargo of the toxins, an enzymatically active glucosyltransferase domain (GTD), is translocated through the newly formed pore into the cytosol of the host cell. Binding of inositol hexakisphosphate (InsP6) to the autoprotease domain of the toxin facilitates cleavage and release of the GTD into the cell (15). The GTD binds and inactivates host Rho family GTPases (16). Functional effects of GTPase inactivation include dysregulation of the actin cytoskeleton and altered signaling events, which are linked to the cell rounding and apoptotic cell death phenotypes of intoxication.
The mechanism of cellular intoxication provides multiple targets for therapeutic intervention. Several studies have demonstrated the effectiveness of toxin-specific monoclonal antibodies (mAbs) in preventing mortality in animal models of CDI (4, 17–20). Recently, the mechanisms of neutralization for two such mAbs, actoxumab (anti-TcdA) and bezlotoxumab (anti-TcdB), have been reported. Both of the mAbs bind the C-terminal CROPs domain of their respective toxins and neutralize toxicity in cell culture by blocking cell-surface binding (21, 22). Treatment with bezlotoxumab alone has been shown to reduce recurrence of CDI in Phase III clinical trials (23) and has been approved by the United States Food and Drug Administration (24), validating the use of anti-TcdB antibodies as supportive therapy.
Given that some of the reported TcdB receptors bind outside of the CROPS domain (8–10), there is a potential that neutralizing antibodies against other toxin domains could provide comparable or enhanced efficacy. A survey of other anti-toxin antibodies reveals that the GTD-containing N-terminal portion of TcdB can also present neutralizing epitopes (20, 25–28). This led us to characterize the mechanism of TcdB neutralization by the humanized monoclonal antibody PA41, which targets the N-terminal region of the toxin. PA41 has previously been shown to effectively neutralize TcdB from multiple historical and epidemic strains of C. difficile and, in combination with an anti-TcdA antibody, prevents mortality in an animal model of CDI (20). Using structural and biochemical approaches, we show that PA41 binds a single, highly conserved epitope within the TcdB-GTD but does not affect its enzymatic function. Functional assays, examining each stage of intoxication in epithelial cells, show that PA41 neutralizes TcdB by interfering with the delivery of the GTD across the endosomal membrane.
Results
PA41 binds a single site on TcdB near the GTD as shown by electron microscopy
To address whether PA41 accesses a single binding site on TcdB, we imaged TcdB in complex with PA41 Fab using negative-stain electron microscopy. A previous study indicated that the TcdB CROPs is flexible relative to the rest of TcdB (29), so we used TcdB(1–1810), a construct that does not contain the CROPs domain, to allow clear identification of bound antibodies. Two-dimensional reference-free class averaging was performed on a data set of 8,301 particles of the TcdB(1–1810)-PA41 Fab complex, and the resulting classes revealed multiple views of a single Fab bound to one end of the toxin (Figs. 1, A and B, and S1). A homology model of TcdB(1–1810) was generated from the crystal structure of TcdA(1–1832) (30) and superimposed onto a representative class average (Fig. 1C). The Fab binds near the TcdB-GTD, consistent with the previous report of a single binding site within the N-terminal region of TcdB (20).
Figure 1.

PA41 binds a single site on TcdB as evaluated by negative-stain electron microscopy. Top, domain organization of TcdB holotoxin, including residue ranges for each domain (74). A, panel of four representative two-dimensional class averages of TcdB(1–1810) bound by PA41 Fab (with number of particles in each class in white). B, schematic for TcdB (white) and PA41 Fab (red) for each of the representative classes. C, overlay of a model of TcdB(1–1810) onto one of the class averages from A (indicated with an asterisk) with the GTD in red, autoprotease domain (APD) in blue, and delivery domain in yellow.
The crystal structure of TcdB-GTD in complex with PA41 Fab identifies a distinct and highly conserved epitope
PA41 was shown previously to effectively neutralize TcdB from multiple strains of C. difficile, including VPI 10463 (ribotype 003) and a set of epidemic and non-epidemic ribotype 027 strains (20). One strain within the 027 ribotype has been characterized as having higher rates of GTD release during intoxication and higher cytotoxicity (31). There are minor differences in protein sequence between the VPI 10463 and ribotype 027 GTDs, so crystallization studies were performed with the GTD sequence from R20291, an 027/BI/NAP1 strain (32) (TcdB-027-GTD). Purified GTD was crystallized in complex with PA41 Fab, and the structure was determined to 2.8-Å resolution (Table 1). The Fab binds to a single site on a small subdomain of the GTD composed of several small helices connected by large loops (GTD residues 290–360) (Fig. 2A) with no interaction with the GTD core structure containing the active site. The interface between PA41 and the GTD is flat and encompasses a relatively small surface area on the GTD (806 Å2). It includes the helix containing GTD residues 340–351 and the end of the helix containing residues 322–325 (Figs. 2B and S2). All of the complementarity-determining regions (CDRs) from the heavy chain (HC) and light chain (LC) of PA41 contribute to binding with CDR-H3 and CDR-L1 predominating (Table S1). One salt bridge is located within the interface (HC Arg-50/GTD Glu-325) and could contribute to binding specificity (Fig. 2B). GTD Glu-347 is centrally located within the epitope and potentially shares hydrogen bonds with residues from both the HC (Ser-103) and LC (Ser-91 and Ser-32) of the Fab (Fig. 2B).
Table 1.
Crystallographic data collection and refinement statistics
Values in parentheses represent the highest-resolution shell. r.m.s.d., root mean square deviation.
| Protein complex | TcdB-GTD + PA41 Fab |
| Crystallization | 0.1 m Bis-Tris, pH 5.5, 1 m NaCl, 11% PEG 3350 |
| Data collection | |
| Space group | C2221 |
| Cell dimensions | |
| a, b, c (Å) | 96.1, 251.7, 224.5 |
| α, β, γ (°) | 90.0, 90.0, 90.0 |
| Resolution (Å) | 60.9–2.8 (2.84–2.79) |
| Rmerge | 0.161 (1.526) |
| Rmeas | 0.171 (1.665) |
| CC1/2 | 0.996 (0.569) |
| I/σI | 7.5 (1.0) |
| Completeness (%) | 99.9 (99.9) |
| Redundancy | 8.5 (6.3) |
| Wavelength (Å) | 0.97872 |
| Total observations | 578,506 (21,380) |
| Unique observations | 68,012 (3,378) |
| Refinement | |
| Resolution (Å) | 60.9–2.8 |
| No. of reflections | 67,920 |
| Rwork/Rfree | 0.241/0.292 |
| No. of non-hydrogen atoms | |
| Protein | 15,152 |
| Water | 146 |
| Total | 15,298 |
| Average B-factor (Å2) | 98.2 |
| r.m.s.d. values | |
| Bond lengths (Å) | 0.002 |
| Angles (°) | 0.455 |
| Ramachandran plot (%) | |
| Most favored | 92.7 |
| Allowed | 6.2 |
| Disallowed | 1.2 |
Figure 2.
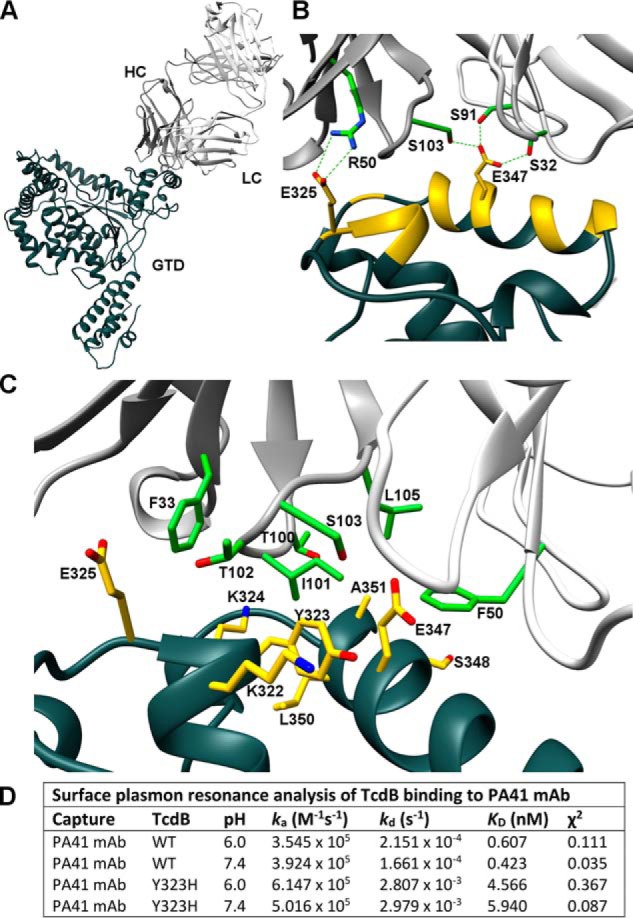
X-ray crystal structure of TcdB-027-GTD + PA41 Fab and structural determinants of epitope interaction. A, wide view of complex with TcdB-027-GTD in dark gray, PA41-HC in gray, and PA41-LC in white. B, residues within the epitope on the GTD are colored yellow with critical PA41 residues in green. Side-chain carbons (red) and nitrogens (blue) are also indicated. A salt bridge is located at one end of the epitope (Glu-325–Arg-50) with GTD Glu-347 potentially mediating hydrogen bonds with both Fab chains. C, hydrophobic pocket accommodating Ile-101 of PA41-HC with residues forming all sides of site labeled (GTD, yellow; Fab, green). D, summary of kinetic rate and dissociation constants for the interactions of TcdB(WT) and TcdB(Y323H) with PA41 mAb measured by SPR.
The core of the TcdB-PA41 interaction involves a shallow hydrophobic pocket formed between the GTD and PA41-HC (Fig. 2C). Ile-101 of CDR-H3 is oriented toward the cleft between the GTD helices with GTD Leu-350 forming the floor of the pocket. HC Phe-33 and LC Phe-50 could interact with the side-chain backbones of several residues (Lys-324/Glu-325 and Glu-347/Ser-348, respectively) to stabilize either end of the site. Several other interactions surround Ile-101 to create the pocket, including possible backbone hydrogen bonds between Lys-322 and Lys-324 of the GTD and Thr-102 and Thr-100, respectively, of PA41-HC (Fig. 2C).
Tyr-323 is in the center of the epitope and forms one wall of the hydrophobic pocket. The phenol group is oriented toward the side chains of Lys-322 and Glu-347 with the aromatic ring positioned between the GTD helices (Fig. 2C). Comparison of the TcdB sequences from multiple C. difficile strains, including those from the previous PA41 study (20) (Table S2), reveals Tyr-323 to be the only epitope residue mutated between strains. The tyrosine is changed to a histidine in the one strain that was not neutralized by PA41 (ribotype 017 strain F1470) and in strain CH6223 (Table S2). To address whether this single mutation could have an effect on the ability of PA41 to bind to TcdB, surface plasmon resonance binding experiments were performed. Wild-type TcdB holotoxin (VPI 10463) bound PA41 mAb with subnanomolar affinity at pH 7.5 and 6.0 (0.42 and 0.61 nm, respectively), whereas the Y323H mutant had an 8–10-fold weaker KD at both pH values (4.6 nm at pH 7.5 and 6.0 nm at pH 6.0) (Fig. 2D). The association rates (ka) for both wild-type and mutant TcdB were comparable, so the weaker affinity results from a faster dissociation rate (kd) of the complexes with the Tyr-to-His mutation (Fig. 2D).
PA41 prevents Rac1 glucosylation by TcdB in cells but not in vitro
The crystal structure reveals that PA41 does not interact directly with the enzymatic active site of the TcdB-GTD, but the presence of the antibody could indirectly affect the glucosyltransferase activity. To test this, we examined the ability of TcdB-027-GTD to transfer [14C]glucose onto purified Rac1 in vitro in the presence of PA41. Preincubation of the GTD with increasing concentrations of PA41 mAb had no effect on its ability to glucosylate Rac1 in a cell-free system (Fig. 3A). However, glucosylation assays performed in Caco-2 cells, a colonic epithelial cell line, show a complete lack of Rac1 modification by TcdB in the presence of PA41 mAb (Fig. 3, B and C) or Fab (Fig. 3, D and E). Taken together, our findings indicate that PA41 does not interfere with the enzymatic activity of the GTD but prevents TcdB from gaining access to its substrates within the host cell. Interestingly, in the same cell-based glucosylation assays, TcdB(Y323H) modifies the majority of the Rac1 within the cells irrespective of the presence of PA41 mAb or Fab (Fig. 3, B–E), indicating that this point mutation is sufficient to allow TcdB to escape neutralization by PA41.
Figure 3.

PA41 inhibits TcdB-induced Rac1 glucosylation in Caco-2 cells but not in vitro. A, in vitro glucosylation of GST-Rac1 (2 μm) by TcdB-027-GTD (10 nm) in the presence of [14C]UDP-glucose (24 μm) and increasing concentrations of PA41 mAb. Controls are shown in lane 1 (GTD with Rac1) and lane 2 (Rac1 only). B, Rac1 glucosylation by TcdB(WT) or TcdB(Y323H) in the presence of isotype control or PA41 mAb was assessed in Caco-2 cells. Cells that did not receive toxin or antibody were used as controls. C, experiments in B were quantified by densitometry, and the extent of Rac1 glucosylation was determined by normalizing the unglucosylated and total Rac1 levels. Means ± S.D. (n = 3) are shown. D and E, similar experiment performed with Fab fragments. The antibody concentration was doubled to keep the available number of antigen-binding sites similar between the mAb and Fab experiments. Error bars represent S.D.
PA41 does not block TcdB from binding or entering the host cell
To gain access to its GTPase substrates, the toxin must bind the host cell, enter, insert into the endosomal membrane, and translocate the GTD into the host cytosol. It is likely that PA41 interferes with one or more of these cell intoxication mechanisms, which could lead to the observed defect in downstream modification of Rac1 by TcdB. To test the impact of PA41 on the binding and uptake of TcdB, we developed a toxin entry assay where the bound and internalized toxins can be distinguished by using a surface-stripping procedure. We used 3xFLAG-TcdB(C698A), a mutant that is deficient in autoprocessing but can still translocate across the endosomal membrane and glucosylate Rac1 in cells (33), and detected the toxin using an anti-FLAG antibody. The 3xFLAG tag is at the N terminus, which would typically get cleaved and released into the cytosol upon toxin entry and thus result in two detectable toxin bands (uncleaved holotoxin and free GTD) in the cell lysates. In contrast, the autoprocessing mutant will be detected as a single holotoxin band, which simplifies quantification of the total internalized toxin within the cells. Cell viability assays confirmed that the 3xFLAG tag does not affect TcdB function (Fig. S3, Supporting Methods). Additionally, inhibition of entry using Dynasore, an inhibitor of dynamin GTPases, prevented the uptake of 3xFLAG-TcdB(C698A) and the subsequent toxin-induced Rac1 glucosylation in cells (Fig. S4, Supporting Methods). These results, consistent with the known role of dynamin in TcdB entry (34, 35), show that the assay is capable of differentiating between cell surface–bound and internalized toxin.
We then performed this assay in the presence of PA41 mAb (Fig. 4A) or Fab (Fig. 4E), which enabled us to simultaneously monitor toxin binding, entry, and toxin-induced Rac1 glucosylation within cells. Our results show that preincubation of 3xFLAG-TcdB(C698A) with either PA41 mAb or Fab has no effect on toxin binding to the cell surface (Fig. 4, B and F) or uptake into the cell (Fig. 4, C and G), but toxin-induced Rac1 glucosylation is still completely inhibited (Fig. 4, D and H). These data indicate that the lack of Rac1 modification by TcdB in the presence of PA41 is not due to defects in toxin binding or uptake.
Figure 4.

PA41 does not affect binding or entry of TcdB in Caco-2 cells, but it does inhibit Rac1 modification. A, TcdB binding and entry in the presence of isotype control or PA41 mAb were assessed in toxin entry assays. Cells that did not receive any toxin or antibody were used as controls. Comparison of bound toxin levels between −strip (lanes 2 and 3) and +strip (lanes 4 and 5) conditions confirms efficient removal of surface-bound toxin by this procedure. B, experiments in A were quantified by densitometry, and the relative binding of TcdB to cells was determined by normalizing bound TcdB levels to that of GAPDH (lanes 2 and 3). Means ± S.D. (n = 3) are shown and were analyzed using two-tailed t test. ns, not significant. C, the internalized TcdB signal (lanes 6–9) was normalized to the corresponding GAPDH levels to obtain the relative entry signal, which was then normalized to the relative bound toxin levels and expressed as the percentage of internalized TcdB. Means ± S.D. (n = 3) are shown and were analyzed using two-tailed t test. ns, not significant. D, the extent of Rac1 glucosylation by TcdB was determined by normalizing the unglucosylated and total Rac1 levels. Means ± S.D. (n = 3) are shown and were analyzed using one-way analysis of variance. p values were generated using Dunnett's multiple comparisons test. ***, p < 0.0005. E–H, similar experiments performed with the Fab fragments. Error bars represent S.D.
PA41 inhibits low pH–induced rubidium release by TcdB and the cargo delivery into the host cytosol
We hypothesized that PA41 was restricting TcdB's access to its substrates by interfering with pore formation and/or translocation across the endosomal membrane. Such a mechanism is possible given that PA41 remains bound to TcdB-GTD in the surface plasmon resonance (SPR) and crystallization studies where the pH conditions used (6.0 and 5.5, respectively) resemble that within an endosome. To investigate whether PA41 interferes with TcdB pore formation, we acid-activated toxin only or toxin-PA41 complexes that were bound to Chinese hamster ovary (CHO)-K1 cells preloaded with rubidium (86Rb+). PA41 mAb and Fab did not affect TcdB binding to CHO-K1 cells (Fig. S5, Supporting Methods) but significantly reduced low pH–induced 86Rb+ release by surface-bound TcdB (Fig. 5A). We next examined the effect of PA41 on delivery of the GTD into the host cell, a process that requires pore formation and translocation. Toxin cleavage assays in Caco-2 cells performed using 3xFLAG-TcdB(WT) show a reduction in the fraction of the internalized holotoxin that is processed and released into the cytosol when the toxin was preincubated with either PA41 mAb (Fig. 5, B and C) or Fab (Fig. 5, D and E), and this reduction in toxin processing correlated with a decrease in Rac1 modification by the toxin (Fig. 5, B and D). A reduction in GTD cleavage and release could result from either an inhibition of translocation or a defect in the InsP6-dependent autoprocessing of the toxin within the cytosol. We performed in vitro autoprocessing assays and observed no difference in the InsP6-induced autocatalytic cleavage of TcdB in the presence of PA41 (Fig. S6, Supporting Methods), suggesting that this antibody inhibits GTD release by blocking transport of the enzyme across the endosomal membrane. In sum, our findings support a neutralization mechanism for PA41 wherein the antibody binds the TcdB-GTD and is carried along with the toxin into the endosome where it prevents pore formation and/or translocation of the GTD across the membrane (Fig. 6).
Figure 5.

PA41 inhibits pore formation and the subsequent delivery of the GTD into the cytosol. A, pore formation on biological membranes. TcdB holotoxin alone (1 nm) or preincubated with PA41 mAb or Fab (10 nm) was applied to CHO-K1 cells preloaded with 86Rb+, and rubidium release was compared at pH 7.5 and 4.5. Means ± S.D. (n = 6) are shown. B, Caco-2 monolayers were intoxicated with 3xFLAG-TcdB (25 nm) preincubated with equimolar (25 nm) amounts of control or PA41 mAb. Toxin cleavage assays were performed as described under “Experimental procedures.” Blots were probed with antibodies against the toxin (anti-FLAG; detects both internalized holotoxin and free GTD), unglucosylated and total Rac1, and GAPDH. C, the fraction of the internalized toxin that is cleaved and released in each condition was determined by normalizing the free GTD signal to the corresponding internalized holotoxin signal. For each time point, the fractional GTD release measurements were normalized to that of the isotype mAb controls to obtain the relative GTD cleavage. D and E, similar experiments performed with the Fab fragments. Means ± S.D. (n = 3) are shown. Error bars represent S.D.
Figure 6.
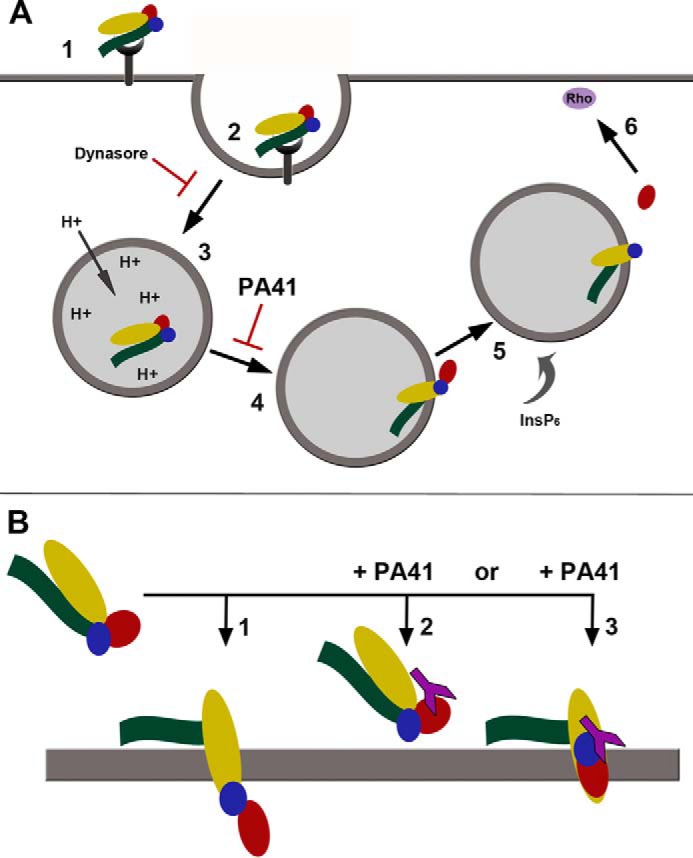
Proposed models of cellular intoxication by TcdB. A, schematic summarizing the individual steps involved in toxin uptake, including cell surface binding (1), endosome formation (2) and maturation (3), pH-dependent pore formation and translocation (4), toxin autoprocessing and GTD release (5), and targeting of Rho-family GTPases (6). In contrast to Dynasore, which directly blocks endocytosis of toxin, PA41 targets the processes involved in pore formation and/or translocation. B, a comparison of the two potential mechanisms through which PA41 could prevent cargo transport during intoxication (1). TcdB normally forms a channel through the endosomal membrane, allowing translocation of the enzymatic toxin domains (2). In the presence of PA41, formation of the pore could be directly blocked through steric hindrance of conformational changes within the toxin. Although an Rb+-conductive pore can be formed in the absence of the GTD (14), binding of PA41 may restrict individual domain movements or overall toxin conformation (3). PA41 could also prevent unfolding of the GTD for transport, which would trap the GTD at some point within the toxin pore. Either of these mechanisms could result in the decreased cargo transport observed in the presence of PA41. TcdB domains are colored as shown previously (GTD, red; autoprotease domain, blue; delivery domain, yellow; CROPs, green) with PA41 in purple.
Discussion
We have defined the TcdB epitope for the neutralizing monoclonal antibody PA41 and the mechanism of neutralization. Electron microscopy confirmed that PA41 recognizes a single site on the TcdB-GTD, and the crystal structure of the complex details the structural features mediating the interaction of PA41 with a small subdomain of the GTD. Cell assays in epithelial cells were used to probe each functional step along the TcdB intoxication pathway and to identify the mechanism of PA41 neutralization. The antibody does not inhibit cell binding, cell entry, or the enzymatic activities of TcdB (glucosyltransferase or toxin autoprocessing). Notably, PA41 significantly reduces both TcdB-dependent rubidium release on biological membranes and the release of free GTD in intoxicated cells. The molecular events involved in formation of a conductive pore and/or physical translocation of the GTD across the endosomal membrane are the apparent targets of neutralization by PA41.
Based on the current paradigm for AB pore-forming toxins (36, 37), cell intoxication by TcdB would rely on pH-dependent conformational changes within the toxin structure that facilitate the formation of a membrane-spanning structure through which the GTD can be delivered and processed for release (Fig. 6). Although it remains unclear whether TcdB forms a defined transmembrane structure, rubidium release experiments imply formation of a functional pore, one where the ion conductivity properties correlate with conditions that promote protein translocation across the endosomal membrane (12–14). Although several residues and structural regions within the TcdB delivery domain have been implicated in membrane insertion and/or cargo delivery processes (12, 14, 38, 39), it is not immediately apparent how binding of the TcdB-GTD by PA41 could block delivery of the toxin cargo. A GTD-less TcdB construct has been shown to be competent in forming a pore in the rubidium release assay (14), suggesting that the GTD itself is not required for this process. For other AB pore-forming toxins (such as anthrax toxin, botulinum neurotoxin, and diphtheria toxin), the cargo must be at least partially unfolded for translocation to occur (40–42). The GTD subdomain containing the PA41 epitope may be involved in essential unfolding events, and PA41 could directly stabilize the tertiary structure of the protein. A similar model for antibody-mediated stabilization of a target antigen has been proposed for neutralizing single-domain antibodies against the catalytic domain of ricin (43).
The rubidium release assay is a standard technique used for monitoring toxin-mediated pore formation in biological membranes. Although the absence of rubidium release in the presence of antibody could suggest that pore formation has been inhibited, it is also possible that the decrease in signal is due to occlusion of the pore. Studies on botulinum and anthrax toxins using antibody Fabs, cross-linking, or biochemical modification have demonstrated that trapping the toxin cargo in the pore during translocation can lead to a loss of conductance through the pore (44–47). GTD trapped within the TcdB pore during translocation, due to either incomplete unfolding or the presence of bound antibody, could result in reduction of both rubidium release and cytosolic GTD delivery. Because we know that the GTD is not required for pore formation (14), we favor a model where PA41 is preventing translocation of the GTD across the membrane.
For PA41 to obstruct processes within the endosome, the antibody needs to remain bound to TcdB through the pH changes inherent to endosomal maturation. The complex of TcdB with PA41 retains stable, high affinity in the pH range 5.5–6.0 as illustrated by the crystal structure and SPR binding experiments (Fig. 2), conditions consistent with early-to-late endosomes (48). Although we could not assess the binding affinity at lower pH values due to TcdB aggregation, we note that the antibodies blocked Rb+ release when cells were exposed to pH 4.5 (Fig. 5A). We therefore anticipate that PA41 will remain bound to TcdB even at the pH of late endosomes. Given the theoretical pKa of 6.8 of histidine, we considered a mechanism of pH-dependent release of PA41 for the TcdB(Y323H) mutant inside the endosome. The binding data, however, indicate that the affinity of the mutant complex is effectively unchanged in moving from pH 7.4 (KD = 5.9 nm) to pH 6.0 (KD = 4.7 nm) (Fig. 2D). In both cases, the KD is 10–15-fold weaker than what was observed with the wild-type sequence. The ability of the TcdB(Y323H) mutant to glucosylate Rac1, even in the presence of PA41 (Fig. 3), suggests that a 5–6 nm affinity interaction is below the affinity threshold for what is needed to neutralize TcdB function, at least in cells.
Within the pore-forming toxin family, there are other examples of antibodies that neutralize at discrete mechanistic steps (49). Specific antibodies against α-hemolysin from Staphylococcus aureus and Shiga toxin from Escherichia coli have been isolated that halt the toxins at the plasma membrane of the cell (50, 51). The prepore-to-pore transition of anthrax toxin is inhibited by an antibody against the protective antigen component of the toxin (52). Stabilization of the conformational changes required for endocytosis is the proposed mechanism for anti-ricin antibodies (43). Remarkably, a mAb against S. aureus α-toxin has been shown to inhibit not only membrane binding through receptor blockade but also the heptameric oligomer association needed for pore formation, providing a dual mechanism and potent neutralizing activity (53, 54).
When considering how antibodies against the C. difficile toxins neutralize their targets, PA41 occupies a unique mechanistic niche. The anti-TcdA mAb actoxumab binds the TcdA CROPs and interferes with toxin binding to cells, most likely through receptor blockade (21). Bezlotoxumab recognizes the CROPs domain of TcdB and blocks Vero cell binding (22). In light of the multiplicity of cellular receptors for TcdB and their distribution of binding sites, it is possible that some toxin will escape neutralization from an anti-CROPs antibody. The more general strategy of blocking toxin translocation from within the endosome provides an additional mechanism to exploit in the effort to treat and prevent CDI. In addition, the PA41 antibody provides a novel tool for dissecting the mechanism of toxin pore formation and translocation across the endosomal membrane.
Experimental procedures
Expression and purification of recombinant TcdB constructs
Constructs for full-length TcdB(WT) (pBL377) and N-terminal 3xFLAG-TcdB(WT) (pBL705) (corresponding to the gene from C. difficile strain VPI 10463) were expressed in Bacillus megaterium with C-terminal His8 tags and purified as described previously (8, 29). TcdB(WT) and N-terminal 3xFLAG-TcdB(WT) were used as templates for generating TcdB(Y323H) (pBL833) and the 3xFLAG-TcdB(C698A) autoprocessing mutant (pBL751), respectively, by site-directed mutagenesis. The construct for TcdB(1–1810) (pBL832), which does not contain the CROPs domain, was made by loop-out mutagenesis on the TcdB(WT) gene using the primers 5′-GAGGATGGATTGATTATTAGTGAAGGTACCCATC-3′ and 5′-GATGGGTACCTTCACTAATAATCAATCCATCCTC-3′. The sequence for the TcdB-027-GTD construct is from C. difficile 027/BI/NAP1 (32) and was subcloned from the full-length toxin construct into the B. megaterium expression vector pHis1622 (BMEG20, MoBiTec, Göttingen, Germany) with a C-terminal His6 tag (pBL834). The TcdB-027-GTD was expressed and purified using the same protocol as for the full-length toxin. Final proteins were analyzed for purity by SDS-PAGE, and concentrations were determined by absorbance (TcdB holotoxin: ϵ0.1%,280 nm, 1.067; molecular weight, 269,710; TcdB(Y323H) holotoxin: ϵ0.1%,280 nm, 1.061; molecular weight, 269,686; TcdB(1–1810): ϵ0.1%,280 nm, 0.886; molecular weight, 205,728; TcdB-027-GTD: ϵ0.1%, 280 nm, 1.124; molecular weight, 62,903). Proteins for structural studies were stored in 20 mm Tris, pH 8.0, 100 mm NaCl, and proteins for cellular studies were stored in 20 mm HEPES, pH 6.9, 50 mm NaCl.
Construction, expression, and purification of PA41 mAb and PA41 Fab
DNA constructs for the light chain and heavy chain of PA41 were cloned into a CMV promotor–driven expression vector with secretion controlled by the native immunoglobulin light chain signal peptide to produce both the PA41 mAb and Fab. For the Fab, the heavy chain consists of the variable region, the CH1 domain, and a partial hinge region (including the EPKSC sequence). The antibodies were transiently expressed in a CHO-derived cell line. Cell cultures were fed with a nutrient mixture until expression was terminated 10–13 days post-transfection (55). Fabs were purified by κ light chain affinity purification (KappaSelect resin, 17-5458-02, GE Healthcare), whereas mAb was isolated by protein A affinity (HiTrap Protein A HP, 17-0403-01, GE Healthcare) according to the manufacturer's instructions.
Electron microscopy of TcdB(1–1810) with PA41 Fab
Complexes of TcdB(1–1810) (100 nm) with PA41 Fab (100 nm) were set up in 20 mm Tris, pH 8.0, 100 mm NaCl and incubated for 30 min at room temperature. Samples were diluted in the same buffer to 5 ng/μl toxin and immediately applied to glow-discharged, carbon-coated copper grids. Grids were stained with 0.75% uranyl formate for negative-stain imaging (56). Micrographs for two-dimensional class averaging of TcdB in complex with PA41 Fab were collected at ×62,000 magnification on a Tecnai F20 (200 keV; FEI) transmission electron microscope equipped with a Gatan US4000 charge-coupled device camera. Images were converted to mixed raster content and binned by 2, giving a final value of 3.5 Å/pixel. Individual particles were manually selected in Boxer/EMAN (57) (box size, 128 × 128 pixels), and image stacks were generated in SPIDER (58). Reference-free two-dimensional class averaging was performed in Scipion (59) using the Xmipp3-CL2D algorithm (60).
X-ray crystallography of TcdB-027-GTD with PA41 Fab
TcdB-027-GTD (60 μm) was incubated with a 1.1-fold molar excess of PA41 Fab (66 μm) in 20 mm Tris, pH 8.0, 100 mm NaCl for 30 min at room temperature. The complex was isolated from unbound Fab over a Sephadex 200 size exclusion column (1 × 25 cm; GE Healthcare) in the same buffer. The resulting pool was concentrated to 8.6 mg/ml complex and set up in broad matrix crystallization screens to identify crystal growth conditions. After optimization, final crystallization conditions were in 0.1 m Bis-Tris, pH 5.5, 1 m NaCl, 11% polyethylene glycol 3350, and crystals were harvested and cryoprotected in mother liquor with 20% ethylene glycol. X-ray diffraction data were collected at the LS-CAT beamline (Advanced Photon Source, Argonne National Laboratory). Data were processed with Xia2 through the XDS pipeline using programs within CCP4 (Pointless/Aimless) for indexing and scaling (61–64). Molecular replacement was performed in Phaser (65) with the structures of the TcdB-GTD from C. difficile VPI 10463 (Protein Data Bank code 2BVM) and a humanized antibody Fab fragment (Protein Data Bank code 3L5X) as search models to phase the data. The electron density maps allowed construction of a model containing residues 2–529 of the GTD and 1–222 and 1–214 of the Fab HC and LC, respectively. The Fab HC loops containing residues 135–140 were not included in the model due to poorly resolved electron density. Refinement was performed in Phenix (66), including TLS (translation-libration-screw-rotation) refinement and NCS (non-crystallographic symmetry) restraints in the strategy. Coot was used for model building (67). All structure figures were made in UCSF Chimera (68) or PyMOL (The PyMOL Molecular Graphics System, version 1.8, Schrödinger, LLC). Software was curated by SBGrid (69). Epitope characteristics in the final refined crystal structure were analyzed with ePISA (70) and PDBSum (71).
SPR assays
Kinetic rate constants (ka and kd) for binding of PA41 to purified TcdB(WT) and TcdB(Y323H) were measured using an IgG capture assay on a BIAcore T200 instrument. Mouse anti-human IgG-Fc was immobilized on a CM4 sensor chip with a final surface density of 1700 resonance units. A reference flow cell surface was also prepared on this sensor chip by use of an identical immobilization protocol. PA41 mAb was prepared at 5 nm in instrument buffer (HBS-EP buffer (0.01 m HEPES, pH 7.4, 0.15 m NaCl, 3 mm EDTA, 0.005% P-20) or MES-EP buffer (0.05 m MES, pH 6.0, 0.15 m NaCl, 3 mm EDTA, 0.05% Tween 20)) along with 3-fold serial dilutions of TcdB(WT) and TcdB(Y323H) (0.0025–50 nm) in instrument buffer. A sequential approach was utilized for kinetic measurements. PA41-human IgG1 was first injected over the capture surface at a flow rate of 10 μl/min. Once the binding of the captured PA41 stabilized, a single concentration of TcdB was injected over both surfaces at a flow rate of 75 μl/min. The resulting binding-response curves yielded the association phase data. Following the injection of TcdB, the flow was switched back to instrument buffer for 10 min to permit the collection of dissociation-phase data with a subsequent 1-min pulse of 3 m MgCl2 to regenerate the IgG capture surface on the chip. Binding responses from duplicate injections of each concentration of TcdB were recorded against PA41. In addition, several buffer injections were interspersed throughout the injection series. Select buffer injections were used along with the reference cell responses to correct the raw data sets for injection artifacts and/or nonspecific binding interactions, commonly referred to as “double referencing.” Fully corrected binding data were then globally fit to a 1:1 binding model (Biacore T200 Evaluation 2.0 software, GE Healthcare) that included a term to correct for mass transport–limited binding should it be detected. These analyses determined the kinetic rate constants ka and kd from which the apparent dissociation constant (KD) was calculated as kd/ka.
In vitro glucosylation assay
Assays were performed as described previously (72) with minor modifications using TcdB-027-GTD (10 nm) and GST-Rac1 (2 μm) in the presence of [14C]UDP-glucose (24 μm final concentration; PerkinElmer Life Sciences, NEC403050UC). GST-tagged Rac1 (pBL436) was expressed in BL21-CodonPlus-RIL E. coli (Agilent Technologies) and purified as described previously over glutathione-Sepharose and by size-exclusion chromatography (72). TcdB-027-GTD was preincubated with increasing concentrations of PA41 mAb (50–250 nm final) at room temperature for 30 min, and reactions were set up in glucosylation buffer (50 mm HEPES, pH 7.5, 100 mm KCl, 2 mm MgCl2,1 mm MnCl2, 0.1 mg/ml BSA) and incubated at 37 °C for 1 h. Reactions were quenched by addition of hot sample buffer and boiling for 5 min, and proteins were separated by reducing SDS-PAGE on a 12% polyacrylamide gel. Radiolabeled Rac1 was visualized after 42-h phosphorimaging screen exposure by autoradiography on a phosphorimaging system (FLA700 Typhoon, GE Healthcare). Three independent replicates were performed with a representative experiment shown in the figure.
Cell culture
Caco-2 cells (ATCC HTB-37) were maintained in minimum essential medium supplemented with 10% fetal bovine serum (FBS; Atlanta Biologicals), 1% minimum essential medium non-essential amino acids (M7145, Sigma), 1% HEPES buffer (15630080, Gibco), and 1% sodium pyruvate (S8636, Sigma). CHO-K1 (ATCC CCL-61) cells were grown in Ham's F-12 medium supplemented with 10% FBS.
Cell-based Rac1 glucosylation assay
Caco-2 cells were seeded at a density of 500,000 cells/well in 35-mm dishes and incubated at 37 °C for 48 h. For the glucosylation assay, Caco-2 monolayers were switched to 4 °C for 1 h and then intoxicated with 10 nm TcdB (wild type or Y323H mutant) that was preincubated for 30 min at room temperature with equimolar (10 nm) concentrations of isotype control mAb or PA41 mAb. For assays with Fab fragments, the antibody concentration was doubled to keep the available number of antigen-binding sites similar between the mAb and Fab experiments. Toxins were allowed to bind at 4 °C for 1 h and then to internalize at 37 °C for 20 min. Cells that did not receive toxin or antibody were used as controls. Cells were harvested, and cell pellets were homogenized in lysis buffer (10 mm Tris, pH 7.4, 250 mm sucrose, 3 mm imidazole) supplemented with protease inhibitor mixture (1:100; Sigma, P8340). Lysates were subjected to SDS-PAGE and Western blot analyses. The blot was probed with antibodies against the unglucosylated Rac1 (610650, BD Biosciences), total Rac1 (05-389, Millipore), and GAPDH. Binding of anti-mouse (7076S, Cell Signaling Technology) or anti-rabbit (7074S, Cell Signaling Technology) HRP-linked secondary antibodies was detected using ECL Western Blotting Substrate (32106, Pierce). Densitometry quantifications were performed using Fiji (73).
Toxin entry and cleavage assays in cells
Caco-2 cells were seeded at a density of 500,000 cells/well in 35-mm dishes and incubated at 37 °C for 48 h. For entry assays, cells were moved to 4 °C for 1 h and then intoxicated with 25 nm 3xFLAG-tagged TcdB(C698A) preincubated with 25 nm mAb or 50 nm Fab for 30 min at room temperature. For assays with Fab fragments, the antibody concentration was doubled to keep the available number of antigen-binding sites similar between the mAb and Fab experiments. Toxin binding occurred at 4 °C for 1 h. To allow toxin entry, medium containing unbound toxin was removed and replaced with fresh medium containing antibody, and cells were then switched to 37 °C for the indicated time points. To assess the bound toxin levels, cells were washed twice with PBS, dislodged using a cell scraper, collected, and homogenized to obtain lysates. To assess internalized toxin levels, the medium was removed, and cells were washed once with PBS and then incubated with an acid solution (0.2 m acetic acid, 0.5 m NaCl in PBS) for 45 s. The acid solution was then removed, and cells were washed twice with PBS and further treated with trypsin solution to facilitate proteolysis and removal of any toxin still bound to the surface post-acid treatment. Cells were then collected, and lysates were prepared for SDS-PAGE and Western blotting. The blot was probed with antibodies against the toxin (anti-FLAG), unglucosylated and total Rac1, and GAPDH. The efficiency of the stripping procedure was assessed by comparing the total bound toxin levels before and after acid wash and trypsin treatment. For cleavage assays, cells were intoxicated with 3xFLAG-TcdB(WT) preincubated with various antibodies. To determine the levels of internalized holotoxin and cleaved GTD, cells were subjected to the surface-stripping procedure described above, and lysates were prepared for SDS-PAGE and Western blotting. Because the 3xFLAG tag is at the N terminus, the anti-FLAG primary antibody can detect the holotoxin and the cleaved GTD in cell lysates simultaneously.
Rubidium release assay
The 86Rb+ release assay was performed as reported previously (12) with slight modifications. Briefly, CHO-K1 cells were seeded in 24-well plates in medium (Ham's F-12 with 10% FBS) supplemented with 1 μCi/ml 86Rb+ (PerkinElmer Life Sciences) at a density of 8 × 104 cells/well. Cells were incubated at 37 °C in 5% CO2 overnight. Medium was exchanged with fresh growth medium containing 100 nm bafilomycin A1 (Sigma), and incubation continued for another 20 min. TcdB wild-type toxin (1 nm) was incubated with 10 nm of antibody (PA41 mAb or Fab) in serum-free medium at 4 °C for 20 min. Then cells were chilled on ice, and medium was exchanged with ice-cold medium containing TcdB and antibody. Cells were kept on ice for toxin binding for 1 h at 4 °C before they were washed with ice-cold PBS twice to remove unbound toxins. pH-dependent insertion into the plasma membrane was induced by warm, acidified growth medium (37 °C, pH 4.8 or 7.5) for 5 min at 37 °C. After 1 h of further incubation on ice, medium containing released 86Rb+ was removed from the cell plate, and the amount of 86Rb+ released was determined by liquid scintillation counting with TopCount NXT (PerkinElmer Life Sciences).
Statistical analyses
No statistical method was used to predetermine the sample size. The statistical tests used and the number of independent biological replicates for each experiment are indicated in the figure legends. The error bars are also defined in the figure legends. A p value of ≤0.05 was considered significant.
Author contributions
H. K. K., R. C., A. C. N., G. J. R., P. W., and D. B. L. designed the study. H. K. K. designed, performed, and analyzed the EM experiments; performed in vitro autoprocessing and Rac1 glucosylation assays; and purified, crystallized, and determined the X-ray structure of the TcdB-PA41 Fab complex. R. C. designed, developed, performed, and analyzed the functional cell assays, including cell viability, toxin binding and entry, GTD release, and Rac1 modification assays. H. K. K. and R. C. generated expression clones and purified proteins. Z. Z. with assistance from R. A. M. performed and analyzed the rubidium release assay. K. R. and R. W. performed the surface plasmon resonance experiments. X. J. expressed and purified PA41 mAb and Fab. B. W. S. assisted with analysis of the X-ray data. H. K. K., R. C., and D. B. L. wrote the paper. All authors reviewed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We thank Jimmy Ballard (University of Oklahoma Medical Center) for the TcdB sequences from ribotypes 027 and 078 and the B. megaterium pHis1622 expression plasmid containing TcdB holotoxin from C. difficile 027/BI/NAP1 R20291. A portion of the experiments described here used the Vanderbilt robotic crystallization facility, which was supported by National Institutes of Health Grant S10 RR026915, and the Vanderbilt Structural Electron Microscopy Facility, which is supported in part by the Vanderbilt Center for Structural Biology. This research also used resources of the Advanced Photon Source, a United States Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract DE-AC02-06CH11357. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and Michigan Technology Tri-Corridor Grant 085P1000817.
This work was supported in part by United States Department of Veterans Affairs Award BX002943 (to D. B. L.) and Public Health Service, National Institutes of Health Grants AI095755 (to D. B. L.) and AI108778 (to B. W. S.). These studies were partially funded by MedImmune through a collaborative project awarded to D. B. L. MedImmune had the right to review the manuscript but otherwise had no role in the preparation or decision to publish. K. R., R. W., X. J., A. C. N., G. J. R., and P. W. are employees of MedImmune, a unit of AstraZeneca. K. R., R. W., X. J., A. C. N., G. J. R., and P. W. own stock in AstraZeneca. G. J. R. is now employed by MabVax Therapeutics. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S6, Tables S1 and S2, and supporting methods.
The atomic coordinates and structure factors (code 5VQM) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- CDI
- C. difficile infection
- GTD
- glucosyltransferase domain
- CROPs
- combined repetitive oligopeptides
- InsP6
- inositol hexakisphosphate
- CDR
- complementarity-determining region
- HC
- heavy chain
- LC
- light chain
- SPR
- surface plasmon resonance
- Bis-Tris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol.
References
- 1. Smits W. K., Lyras D., Lacy D. B., Wilcox M. H., and Kuijper E. J. (2016) Clostridium difficile infection. Nat. Rev. Dis. Primers 2, 16020 10.1038/nrdp.2016.20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lessa F. C., Mu Y., Bamberg W. M., Beldavs Z. G., Dumyati G. K., Dunn J. R., Farley M. M., Holzbauer S. M., Meek J. I., Phipps E. C., Wilson L. E., Winston L. G., Cohen J. A., Limbago B. M., Fridkin S. K., et al. (2015) Burden of Clostridium difficile infection in the United States. N. Engl. J. Med. 372, 825–834 10.1056/NEJMoa1408913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Carter G. P., Chakravorty A., Pham Nguyen T. A., Mileto S., Schreiber F., Li L., Howarth P., Clare S., Cunningham B., Sambol S. P., Cheknis A., Figueroa I., Johnson S., Gerding D., Rood J. I., et al. (2015) Defining the roles of TcdA and TcdB in localized gastrointestinal disease, systemic organ damage, and the host response during Clostridium difficile infections. MBio 6, e00551. 10.1128/mBio.00551-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Steele J., Mukherjee J., Parry N., and Tzipori S. (2013) Antibody against TcdB, but not TcdA, prevents development of gastrointestinal and systemic Clostridium difficile disease. J. Infect. Dis. 207, 323–330 10.1093/infdis/jis669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Drudy D., Fanning S., and Kyne L. (2007) Toxin A-negative, toxin B-positive Clostridium difficile. Int. J. Infect. Dis. 11, 5–10 10.1016/j.ijid.2006.04.003 [DOI] [PubMed] [Google Scholar]
- 6. Krivan H. C., Clark G. F., Smith D. F., and Wilkins T. D. (1986) Cell surface binding site for Clostridium difficile enterotoxin: evidence for a glycoconjugate containing the sequence Ga1-α1–3Gal-β1–4GlcNAc. Infect. Immun. 53, 573–581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tucker K. D., and Wilkins T. D. (1991) Toxin A of Clostridium difficile binds to the human carbohydrate antigens I, X, and Y. Infect. Immun. 59, 73–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. LaFrance M. E., Farrow M. A., Chandrasekaran R., Sheng J., Rubin D. H., and Lacy D. B. (2015) Identification of an epithelial cell receptor responsible for Clostridium difficile TcdB-induced cytotoxicity. Proc. Natl. Acad. Sci. U.S.A. 112, 7073–7078 10.1073/pnas.1500791112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yuan P., Zhang H., Cai C., Zhu S., Zhou Y., Yang X., He R., Li C., Guo S., Li S., Huang T., Perez-Cordon G., Feng H., and Wei W. (2015) Chondroitin sulfate proteoglycan 4 functions as the cellular receptor for Clostridium difficile toxin B. Cell Res. 25, 157–168 10.1038/cr.2014.169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tao L., Zhang J., Meraner P., Tovaglieri A., Wu X., Gerhard R., Zhang X., Stallcup W. B., Miao J., He X., Hurdle J. G., Breault D. T., Brass A. L., and Dong M. (2016) Frizzled proteins are colonic epithelial receptors for C. difficile toxin B. Nature. 538, 350–355 10.1038/nature19799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Qa'Dan M., Spyres L. M., and Ballard J. D. (2000) pH-induced conformational changes in Clostridium difficile toxin B. Infect. Immun. 68, 2470–2474 10.1128/IAI.68.5.2470-2474.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Genisyuerek S., Papatheodorou P., Guttenberg G., Schubert R., Benz R., and Aktories K. (2011) Structural determinants for membrane insertion, pore formation and translocation of Clostridium difficile toxin B. Mol. Microbiol. 79, 1643–1654 10.1111/j.1365-2958.2011.07549.x [DOI] [PubMed] [Google Scholar]
- 13. Giesemann T., Jank T., Gerhard R., Maier E., Just I., Benz R., and Aktories K. (2006) Cholesterol-dependent pore formation of Clostridium difficile toxin A. J. Biol. Chem. 281, 10808–10815 10.1074/jbc.M512720200 [DOI] [PubMed] [Google Scholar]
- 14. Zhang Z., Park M., Tam J., Auger A., Beilhartz G. L., Lacy D. B., and Melnyk R. A. (2014) Translocation domain mutations affecting cellular toxicity identify the Clostridium difficile toxin B pore. Proc. Natl. Acad. Sci. U.S.A. 111, 3721–3726 10.1073/pnas.1400680111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Reineke J., Tenzer S., Rupnik M., Koschinski A., Hasselmayer O., Schrattenholz A., Schild H., and von Eichel-Streiber C. (2007) Autocatalytic cleavage of Clostridium difficile toxin B. Nature 446, 415–419 10.1038/nature05622 [DOI] [PubMed] [Google Scholar]
- 16. Just I., Selzer J., Wilm M., von Eichel-Streiber C., Mann M., and Aktories K. (1995) Glucosylation of Rho proteins by Clostridium difficile toxin B. Nature 375, 500–503 10.1038/375500a0 [DOI] [PubMed] [Google Scholar]
- 17. Yang Z., Ramsey J., Hamza T., Zhang Y., Li S., Yfantis H. G., Lee D., Hernandez L. D., Seghezzi W., Furneisen J. M., Davis N. M., Therien A. G., and Feng H. (2015) Mechanisms of protection against Clostridium difficile infection by the monoclonal antitoxin antibodies actoxumab and bezlotoxumab. Infect. Immun. 83, 822–831 10.1128/IAI.02897-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yang Z., Schmidt D., Liu W., Li S., Shi L., Sheng J., Chen K., Yu H., Tremblay J. M., Chen X., Piepenbrink K. H., Sundberg E. J., Kelly C. P., Bai G., Shoemaker C. B., et al. (2014) A novel multivalent, single-domain antibody targeting TcdA and TcdB prevents fulminant Clostridium difficile infection in mice. J. Infect. Dis. 210, 964–972 10.1093/infdis/jiu196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yang Z., Shi L., Yu H., Zhang Y., Chen K., Saint Fleur A., Bai G., and Feng H. (2016) Intravenous adenovirus expressing a multi-specific, single-domain antibody neutralizing TcdA and TcdB protects mice from Clostridium difficile infection. Pathog. Dis. 74, ftw078 10.1093/femspd/ftw078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Marozsan A. J., Ma D., Nagashima K. A., Kennedy B. J., Kang Y. K., Arrigale R. R., Donovan G. P., Magargal W. W., Maddon P. J., and Olson W. C. (2012) Protection against Clostridium difficile infection with broadly neutralizing antitoxin monoclonal antibodies. J. Infect. Dis. 206, 706–713 10.1093/infdis/jis416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hernandez L. D., Kroh H. K., Hsieh E., Yang X., Beaumont M., Sheth P. R., DiNunzio E., Rutherford S. A., Ohi M. D., Ermakov G., Xiao L., Secore S., Karczewski J., Racine F., Mayhood T., et al. (2017) Epitopes and mechanism of action of the Clostridium difficile Toxin A-neutralizing antibody actoxumab. J. Mol. Biol. 429, 1030–1044 10.1016/j.jmb.2017.02.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Orth P., Xiao L., Hernandez L. D., Reichert P., Sheth P. R., Beaumont M., Yang X., Murgolo N., Ermakov G., DiNunzio E., Racine F., Karczewski J., Secore S., Ingram R. N., Mayhood T., et al. (2014) Mechanism of action and epitopes of Clostridium difficile toxin B-neutralizing antibody bezlotoxumab revealed by X-ray crystallography. J. Biol. Chem. 289, 18008–18021 10.1074/jbc.M114.560748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wilcox M. H., Gerding D. N., Poxton I. R., Kelly C., Nathan R., Birch T., Cornely O. A., Rahav G., Bouza E., Lee C., Jenkin G., Jensen W., Kim Y.-S., Yoshida J., Gabryelski L., et al. (2017) Bezlotoxumab for prevention of recurrent Clostridium difficile infection. N. Engl. J. Med. 376, 305–317 10.1056/NEJMoa1602615 [DOI] [PubMed] [Google Scholar]
- 24. Mullard A. (2016) FDA approves antitoxin antibody. Nat. Rev. Drug Discov. 15, 811 10.1038/nrd.2016.257 [DOI] [PubMed] [Google Scholar]
- 25. Babcock G. J., Broering T. J., Hernandez H. J., Mandell R. B., Donahue K., Boatright N., Stack A. M., Lowy I., Graziano R., Molrine D., Ambrosino D. M., and Thomas W. D. (2006) Human monoclonal antibodies directed against toxins A and B prevent Clostridium difficile-induced mortality in hamsters. Infect. Immun. 74, 6339–6347 10.1128/IAI.00982-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Anosova N. G., Cole L. E., Li L., Zhang J., Brown A. M., Mundle S., Zhang J., Ray S., Ma F., Garrone P., Bertraminelli N., Kleanthous H., and Anderson S. F. (2015) A combination of three fully human toxin A- and toxin B-specific monoclonal antibodies protects against challenge with highly virulent epidemic strains of Clostridium difficile in the hamster model. Clin. Vaccine Immunol. 22, 711–725 10.1128/CVI.00763-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Qiu H., Cassan R., Johnstone D., Han X., Joyee A. G., McQuoid M., Masi A., Merluza J., Hrehorak B., Reid R., Kennedy K., Tighe B., Rak C., Leonhardt M., Dupas B., et al. (2016) Novel Clostridium difficile anti-toxin (TcdA and TcdB) humanized monoclonal antibodies demonstrate in vitro neutralization across a broad spectrum of clinical strains and in vivo potency in a hamster spore challenge model. PLoS One 11, e0157970 10.1371/journal.pone.0157970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wang H., Sun X., Zhang Y., Li S., Chen K., Shi L., Nie W., Kumar R., Tzipori S., Wang J., Savidge T., and Feng H. (2012) A chimeric toxin vaccine protects against primary and recurrent Clostridium difficile infection. Infect. Immun. 80, 2678–2688 10.1128/IAI.00215-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pruitt R. N., Chambers M. G., Ng K. K., Ohi M. D., and Lacy D. B. (2010) Structural organization of the functional domains of Clostridium difficile toxins A and B. Proc. Natl. Acad. Sci. U.S.A. 107, 13467–13472 10.1073/pnas.1002199107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chumbler N. M., Rutherford S. A., Zhang Z., Farrow M. A., Lisher J. P., Farquhar E., Giedroc D. P., Spiller B. W., Melnyk R. A., and Lacy D. B. (2016) Crystal structure of Clostridium difficile toxin A. Nat. Microbiol. 1, 15002 10.1038/nmicrobiol.2015.2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lanis J. M., Hightower L. D., Shen A., and Ballard J. D. (2012) TcdB from hypervirulent Clostridium difficile exhibits increased efficiency of autoprocessing. Mol. Microbiol. 84, 66–76 10.1111/j.1365-2958.2012.08009.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lanis J. M., Heinlen L. D., James J. A., and Ballard J. D. (2013) Clostridium difficile 027/BI/NAP1 encodes a hypertoxic and antigenically variable form of TcdB. PLoS Pathog. 9, e1003523 10.1371/journal.ppat.1003523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chumbler N. M., Farrow M. A., Lapierre L. A., Franklin J. L., Haslam D. B., Goldenring J. R., and Lacy D. B. (2012) Clostridium difficile toxin B causes epithelial cell necrosis through an autoprocessing-independent mechanism. PLoS Pathog. 8, e1003072 10.1371/journal.ppat.1003072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Papatheodorou P., Zamboglou C., Genisyuerek S., Guttenberg G., and Aktories K. (2010) Clostridial glucosylating toxins enter cells via clathrin-mediated endocytosis. PLoS One 5, e10673 10.1371/journal.pone.0010673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chandrasekaran R., Kenworthy A. K., and Lacy D. B. (2016) Clostridium difficile toxin A undergoes clathrin-independent, PACSIN2-dependent endocytosis. PLoS Pathog. 12, e1006070 10.1371/journal.ppat.1006070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Geny B., and Popoff M. R. (2006) Bacterial protein toxins and lipids: pore formation or toxin entry into cells. Biol. Cell 98, 667–678 10.1042/BC20050082 [DOI] [PubMed] [Google Scholar]
- 37. Popoff M. R. (2014) Clostridial pore-forming toxins: powerful virulence factors. Anaerobe 30, 220–238 10.1016/j.anaerobe.2014.05.014 [DOI] [PubMed] [Google Scholar]
- 38. Chen S., Wang H., Gu H., Sun C., Li S., Feng H., and Wang J. (2016) Identification of an essential region for translocation of Clostridium difficile toxin B. Toxins 8, E241. 10.3390/toxins8080241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhang Y., Shi L., Li S., Yang Z., Standley C., Yang Z., ZhuGe R., Savidge T., Wang X., and Feng H. (2013) A segment of 97 amino acids within the translocation domain of Clostridium difficile toxin B is essential for toxicity. PLoS One 8, e58634 10.1371/journal.pone.0058634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wesche J., Elliott J. L., Falnes P. O., Olsnes S., and Collier R. J. (1998) Characterization of membrane translocation by anthrax protective antigen. Biochemistry 37, 15737–15746 10.1021/bi981436i [DOI] [PubMed] [Google Scholar]
- 41. Fischer A., and Montal M. (2007) Single molecule detection of intermediates during botulinum neurotoxin translocation across membranes. Proc. Natl. Acad. Sci. U.S.A. 104, 10447–10452 10.1073/pnas.0700046104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Klingenberg O., and Olsnes S. (1996) Ability of methotrexate to inhibit translocation to the cytosol of dihydrofolate reductase fused to diphtheria toxin. Biochem. J. 313, 647–653 10.1042/bj3130647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Legler P. M., Compton J. R., Hale M. L., Anderson G. P., Olson M. A., Millard C. B., and Goldman E. R. (2017) Stability of isolated antibody-antigen complexes as a predictive tool for selecting toxin neutralizing antibodies. MAbs 9, 43–57 10.1080/19420862.2016.1236882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Fischer A., and Montal M. (2007) Crucial role of the disulfide bridge between botulinum neurotoxin light and heavy chains in protease translocation across membranes. J. Biol. Chem. 282, 29604–29611 10.1074/jbc.M703619200 [DOI] [PubMed] [Google Scholar]
- 45. Fischer A., Garcia-Rodriguez C., Geren I., Lou J., Marks J. D., Nakagawa T., and Montal M. (2008) Molecular architecture of botulinum neurotoxin E revealed by single particle electron microscopy. J. Biol. Chem. 283, 3997–4003 10.1074/jbc.M707917200 [DOI] [PubMed] [Google Scholar]
- 46. Juris S. J., Melnyk R. A., Bolcome R. E. 3rd, Chan J., and Collier R. J. (2007) Cross-linked forms of the isolated N-terminal domain of the lethal factor are potent inhibitors of anthrax toxin. Infect. Immun. 75, 5052–5058 10.1128/IAI.00490-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Basilio D., Jennings-Antipov L. D., Jakes K. S., and Finkelstein A. (2011) Trapping a translocating protein within the anthrax toxin channel: implications for the secondary structure of permeating proteins. J. Gen. Physiol. 137, 343–356 10.1085/jgp.201010578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Huotari J., and Helenius A. (2011) Endosome maturation. EMBO J. 30, 3481–3500 10.1038/emboj.2011.286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Morrison C. (2015) Antibacterial antibodies gain traction. Nat. Rev. Drug Discov. 14, 737–738 10.1038/nrd4770 [DOI] [PubMed] [Google Scholar]
- 50. Foletti D., Strop P., Shaughnessy L., Hasa-Moreno A., Casas M. G., Russell M., Bee C., Wu S., Pham A., Zeng Z., Pons J., Rajpal A., and Shelton D. (2013) Mechanism of action and in vivo efficacy of a human-derived antibody against Staphylococcus aureus α-hemolysin. J. Mol. Biol. 425, 1641–1654 10.1016/j.jmb.2013.02.008 [DOI] [PubMed] [Google Scholar]
- 51. Lo A. W., Moonens K., De Kerpel M., Brys L., Pardon E., Remaut H., and De Greve H. (2014) The molecular mechanism of Shiga toxin Stx2e neutralization by a single-domain antibody targeting the cell receptor-binding domain. J. Biol. Chem. 289, 25374–25381 10.1074/jbc.M114.566257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Mechaly A., Levy H., Epstein E., Rosenfeld R., Marcus H., Ben-Arie E., Shafferman A., Ordentlich A., and Mazor O. (2012) A novel mechanism for antibody-based anthrax toxin neutralization: inhibition of prepore-to-pore conversion. J. Biol. Chem. 287, 32665–32673 10.1074/jbc.M112.400473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Oganesyan V., Peng L., Damschroder M. M., Cheng L., Sadowska A., Tkaczyk C., Sellman B. R., Wu H., and Dall'Acqua W. F. (2014) Mechanisms of neutralization of a human anti-α-toxin antibody. J. Biol. Chem. 289, 29874–29880 10.1074/jbc.M114.601328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Diep B. A., Hillard J. J., Le V. T. M., Tkaczyk C., Le H. N., Tran V. G., Rao R. L., Dip E. C., Pereira-Franchi E. P., Cha P., Jacobson S., Broome R., Cheng L. I., Weiss W., Prokai L., et al. (2017) Targeting α toxin to mitigate its lethal toxicity in ferret and rabbit models of Staphylococcus aureus necrotizing pneumonia. Antimicrob. Agents Chemother. 10.1128/AAC.02456-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kroh H. K., Chandrasekaran R., Rosenthal K., Woods R., Jin X., Ohi M. D., Nyborg A. C., Rainey G. J., Warrener P., Spiller B. W., and Lacy D. B. (2017) Use of a neutralizing antibody helps identify structural features critical for binding of Clostridium difficile toxin TcdA to the host cell surface. J. Biol. Chem. 10.1074/jbc.M117.781112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ohi M., Li Y., Cheng Y., and Walz T. (2004) Negative staining and image classification: powerful tools in modern electron microscopy. Biol. Proced. Online 6, 23–34 10.1251/bpo70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ludtke S. J., Baldwin P. R., and Chiu W. (1999) EMAN: semiautomated software for high-resolution single-particle reconstructions. J. Struct. Biol. 128, 82–97 10.1006/jsbi.1999.4174 [DOI] [PubMed] [Google Scholar]
- 58. Frank J., Radermacher M., Penczek P., Zhu J., Li Y., Ladjadj M., and Leith A. (1996) SPIDER and WEB: processing and visualization of images in 3D electron microscopy and related fields. J. Struct. Biol. 116, 190–199 10.1006/jsbi.1996.0030 [DOI] [PubMed] [Google Scholar]
- 59. de la Rosa-Trevín J. M., Quintana A., Del Cano L., Zaldívar A., Foche I., Gutiérrez J., Gómez-Blanco J., Burguet-Castell J., Cuenca-Alba J., Abrishami V., Vargas J., Otón J., Sharov G., Vilas J. L., Navas J., et al. (2016) Scipion: a software framework toward integration, reproducibility and validation in 3D electron microscopy. J. Struct. Biol. 195, 93–99 10.1016/j.jsb.2016.04.010 [DOI] [PubMed] [Google Scholar]
- 60. de la Rosa-Trevín J. M., Otón J., Marabini R., Zaldívar A., Vargas J., Carazo J. M., and Sorzano C. O. (2013) Xmipp 3.0: an improved software suite for image processing in electron microscopy. J. Struct. Biol. 184, 321–328 10.1016/j.jsb.2013.09.015 [DOI] [PubMed] [Google Scholar]
- 61. Evans P. (2006) Scaling and assessment of data quality. Acta Crystallogr. D Biol. Crystallogr. 62, 72–82 10.1107/S0907444905036693 [DOI] [PubMed] [Google Scholar]
- 62. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 12 10.1107/S09074449090473375–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., et al. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 10.1107/S0907444910045749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Winter G. (2010) Xia2: an expert system for macromolecular crystallography data reduction. J. Appl. Crystallogr. 43, 186–190 10.1107/S0021889809045701 [DOI] [Google Scholar]
- 65. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 10.1107/S0021889807021206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 10.1107/S0907444909052925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., and Ferrin T. E. (2004) UCSF Chimera: a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 10.1002/jcc.20084 [DOI] [PubMed] [Google Scholar]
- 69. Morin A., Eisenbraun B., Key J., Sanschagrin P. C., Timony M. A., Ottaviano M., and Sliz P. (2013) Collaboration gets the most out of software. Elife 10.7554/eLife.01456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Krissinel E., and Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 10.1016/j.jmb.2007.05.022 [DOI] [PubMed] [Google Scholar]
- 71. de Beer T. A., Berka K., Thornton J. M., and Laskowski R. A. (2014) PDBsum additions. Nucleic Acids Res. 42, D292–D296 10.1093/nar/gkt940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Pruitt R. N., Chumbler N. M., Rutherford S. A., Farrow M. A., Friedman D. B., Spiller B., and Lacy D. B. (2012) Structural determinants of Clostridium difficile toxin A glucosyltransferase activity. J. Biol. Chem. 287, 8013–8020 10.1074/jbc.M111.298414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B., Tinevez J.-Y., White D. J., Hartenstein V., Eliceiri K., Tomancak P., et al. (2012) Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 10.1038/nmeth.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Gupta P., Zhang Z., Sugiman-Marangos S. N., Tam J., Raman S., Julien J. P., Kroh H. K., Lacy D. B., Murgolo N., Bekkari K., Therien A. G., Hernandez L. D., and Melnyk R. A. (2017) Functional defects in Clostridium difficile TcdB toxin uptake identify CSPG4 receptor binding determinants. J. Biol. Chem. 292, 17290–17301 10.1074/jbc.M117.806687 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.