

SUMMARY
Sustained energy starvation leads to activation of AMP-activated protein kinase (AMPK), which coordinates energy status with numerous cellular processes including metabolism, protein synthesis, and autophagy. Here, we report that AMPK phosphorylates the histone methyltransferase EZH2 at T311 to disrupt the interaction between EZH2 and SUZ12, another core component of the polycomb repressive complex 2 (PRC2), leading to attenuated PRC2-dependent methylation of histone H3 at Lys27. As such, PRC2 target genes, many of which are known tumor suppressors, were upregulated upon T311-EZH2 phosphorylation, which suppressed tumor cell growth both in cell culture and mouse xenografts. Pathologically, immunohistochemical analyses uncovered a positive correlation between AMPK activity and pT311-EZH2, and higher pT311-EZH2 correlates with better survival in both ovarian and breast cancer patients. Our finding suggests that AMPK agonists might be promising sensitizers for EZH2 targeting cancer therapies.
Keywords: AMPK, EZH2, Polycomb Repressive Complex 2, phosphorylation, ovarian cancer, metformin
In Brief
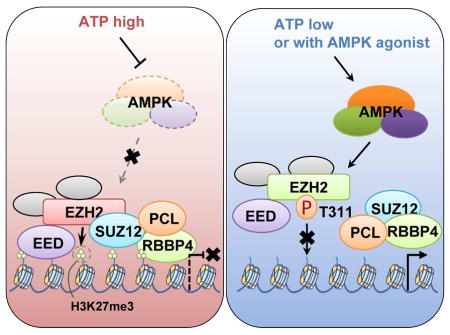
The metabolic state of cell can be connected to gene expression and modification of histones through several mechanisms. Wan et al. find that AMPK-mediated phosphorylation of EZH2 at T311 inhibits PRC2 methyltransferase activity to relieve PRC2-dependent epigenetic silencing and subsequently suppresses tumorigenesis.
INTRODUCTION
Metabolic cues are crucial inputs in dictating acute responses through governing cellular signaling pathways as well as in shaping up long-term transcriptional and epigenetic profiles to control chronic cellular responses (Lu and Thompson, 2012). It has been well documented that metabolites including acetyl-CoA and α-ketoglutarate actively participate in histone acetylation and demethylation, respectively (Cai et al., 2011; Klose and Zhang, 2007). However, given the nature of metabolites that they are small molecules and essential to many different cellular processes, metabolites are not considered as favorable therapeutic targets (Vander Heiden, 2011). Therefore, defining crucial metabolite sensor proteins that are capable of monitoring and coupling metabolic messages directly with chromatin changes to govern cellular responses, would be of great translational value (Vander Heiden, 2011). To this end, mechanistic target of rapamycin (mTOR) and AMP-activated protein kinase (AMPK) have been characterized as the two most important sensors for energy surplus and deprivation state, respectively. Importantly, mTOR is activated by the energy surplus conditions (Zoncu et al., 2011), whereas AMPK is largely activated by elevated cellular ADP or AMP level that is coupled to energy shortage state (Hardie et al., 2012). During the past decade, even though the functions of AMPK in metabolism, protein synthesis, and autophagy have been well characterized (Herzig and Shaw, 2017), it remains less explored about its mechanistic role in global epigenetic alterations that play critical roles in defining cellular chronic reactions to energy state. In addition to its canonical function to govern energy homeostasis, accumulating evidence suggests that AMPK also plays crucial roles in tumorigenesis partly through modulating the expression of genes critical for tumor cell growth and survival (Cheng et al., 2016). Hence, we were interested in revealing the potential role and molecular mechanism(s) of AMPK in regulating epigenetic marks on chromatin to control the fate of cancer cells.
Polycomb repressive complex 2 (PRC2) plays a central role in controlling important cellular processes such as maintaining stem cell pluripotency and promoting cell proliferation (Margueron and Reinberg, 2011). As the primary catalytic subunit of the PRC2 complex, EZH2 methylates histone H3 lysine 27 (H3K27) primarily at promoters of target genes (Melnick, 2012), trimethylated H3K27 (H3K27me3) in turn recruits the polycomb repressive complex 1 (PRC1) to the target gene promoters where PRC1 catalyzes mono-ubiquitination of histone H2A at Lys 119 (Sauvageau and Sauvageau, 2010). These epigenetic modifications in the promoter region act in concert to facilitate epigenetic silencing of target genes (Margueron and Reinberg, 2011).
The core PRC2 complex contains three subunits, the catalytic subunit EZH2 or EZH1, the scaffolding components SUZ12 and EED. In addition to these core subunits, RbAp46/48 (also known as RBBP7/4), zinc-finger protein AEBP2, tudor domain-containing protein PCL isoforms, and Jumonji family protein JARID2 have been shown in complex with the core PRC2 complex to either enhance the enzymatic activity of PRC2 or tether PRC2 to the targeted gene loci (Margueron and Reinberg, 2011). PRC2 is a stable protein complex as depleting any of its core components resulted in a decreased expression of other subunits (Cao et al., 2002; Cao and Zhang, 2004). This finding is supported by the observation that Ezh2−/−, Eed−/− and Suz12−/− ES cells all displayed deficiency in maintaining pluripotency (Margueron and Reinberg, 2011), suggesting that the integrity of the PRC2 core complex is essential to its enzymatic activity.
EZH2 mutations or amplifications have been found in a broad spectrum of human cancers including B-cell lymphoma, ovarian cancer, breast cancer, melanoma, bladder cancer, gastric cancer and other cancers (Kim and Roberts, 2016). Given the evidence of EZH2 as a cancer driver, numerous efforts have been made that led to the development of EZH2 inhibitory compounds including EPZ-6438 (Knutson et al., 2013) and GSK126 (McCabe et al., 2012), both of which are currently used in clinical trials primarily against EZH2-mutated B-cell lymphoma and advanced solid tumors (Kim and Roberts, 2016). However, mixed responses of anti-EZH2 single agent therapies have been reported in both clinical and pre-clinical studies, particularly in the settings of solid tumors, advocating novel combination therapies for EZH2 hyperactive solid tumor patients (Kim and Roberts, 2016).
Here we found that AMPK directly phosphorylates EZH2 at Thr311 to disrupt its interaction with SUZ12 and to inhibit PRC2 enzymatic activity, which is supported by the increased expression of PRC2-repressed genes. Furthermore, the T311E-EZH2 mutant that mimics AMPK-mediated phosphorylation status suppresses tumor cell growth both in vitro and in vivo. Consistent with these results, a positive correlation between AMPK activity and pT311-EZH2 status was observed in ovarian and breast tumor samples, and we also found that higher pT311-EZH2 correlates with better survival in both ovarian and breast cancer patients.
RESULTS
AMPK Suppresses EZH2-mediated Histone H3K27 Trimethylation
AMPK is a hetero-trimeric kinase complex consisting of a catalytic α-subunit, a scaffolding β-subunit and a regulatory γ-subunit (Hardie et al., 2012). Notably, in Ampkα1 and Ampkα2 double knockout (thereafter termed Ampkα DKO) MEFs (Tsou et al., 2011), we observed an upregulation of methylated histone H3K27 and to a lesser extent, elevation in H3K4me3, but not other histone methylation markers we examined (Figure 1A). Re-introducing AMPKα1 largely suppressed Ampkα deletion-induced of H3K27me3 (Figure 1B), and H3K27me3 levels were downregulated after ectopic expression of constitutively active AMPKα1 in breast cancer cells (Figure S1A). These results indicate a direct connection between AMPKα genetic status and the H3K27 methylation levels. Furthermore, activating AMPK by a specific AMPK agonist, A769662 (Cool et al., 2006), attenuated H3K27me3 in WT, but not Ampkα DKO MEFs (Figure 1C). Furthermore, A769662 treatment also led to a decrease of H3K27me3 in various ovarian cancer cell lines (Figure S1B). These findings suggest that the kinase activity of AMPK is required to suppress H3K27me3 in cells.
Figure 1. AMPK Suppresses EZH2-mediated Histone H3K27 Trimethylation.

(A) Immunoblot (IB) analysis of whole cell lysates (WCL) derived from Ampkα WT and Ampkα1−/−; Ampkα2−/− double knock out (DKO) MEFs.
(B) Ampkα1−/−; Ampkα2−/− DKO MEFs were infected with the retroviral construct expressing HA-AMPKα1. Infected cells were selected with 1 μg/ml puromycin for 72 hours to eliminate the non-infected cells before harvesting.
(C) Ampkα WT and DKO MEFs were treated with 100 μM A769662 for the indicated period of time before harvesting.
(D) T98G cells were treated with 2 mM metformin for 2 days before harvesting.
(E) Ampkα WT and DKO MEFs were infected with shGFP control or shEzh2 lentiviral shRNA. The infected cells were selected with 1 μg/ml puromycin for 72 hours to eliminate the non-infected cells before harvesting.
(F) Quantification of the relative H3K27me3 band intensities from three independent experiments. H3K27me3 bands were normalized to TUBULIN, and then normalized to the first lane. Data are represented as mean ± SD, n=3. * P < 0.05, Student’s t test.
(G) shGFP- (as a negative control) and shEZH2-MDA-MB-231 cells were treated with or without 10 μM AMPK inhibitor Compound C for 12 hours before harvesting for IB analysis.
See also Figure S1.
Since AMPK becomes activated when cellular ATP:AMP or ATP:ADP ratio decreases (Hardie et al., 2012), we next sought to examine whether physiological changes of cellular energy status could also result in an alteration of H3K27 methylation level. To this end, we observed that metformin (Figure 1D and S1C) and 2-Deoxy-D-glucose (2DG) treatment (Figure S1D), which have been previously characterized to activate AMPK (Egan et al., 2011; Gwinn et al., 2008; Lamia et al., 2009), both led to a marked decrease in H3K27me3 level in various cell lines we examined. Recently, glucose deprivation has been shown to activate AMPK through aldolase-dependent assembly of a lysosomal complex containing AMPK and its upstream activator LKB1 (Zhang et al., 2017). Interestingly, prolonged glucose deprivation led to a dramatic reduction of H3K27me3 levels (Figure S1E), further supporting a possible negative regulation of H3K27me3 by AMPK. On the other hand, pharmacological inhibition of AMPK kinase activity by the non-specific inhibitor Compound C (Zhou et al., 2001) resulted in an upregulation of H3K27 methylation in multiple breast cancer cell lines (Figure S1F).
Since PRC2 is the primary H3K27 methyltransferase, we next sought to explore the potential role of PRC2 in AMPK-dependent suppression of H3K27me3. In support of a potential role of EZH2 in mediating AMPK-triggered downregulation of H3K27me3, we found that depletion of EZH2 in Ampkα DKO MEFs decreased H3K27me3 levels (Figures 1E–F). Moreover, compared to control cells, inhibiting AMPK by Compound C failed to induce H3K27me3 in EZH2-depleted MDA-MB-231 cells (Figure 1G). These results indicate that AMPK might regulate H3K27me3 in part via modulating the methyltransferase activity of PRC2/EZH2.
To explore potential mechanisms for this regulation, we first looked at the impact of histone phosphorylation. AMPK can directly phosphorylate histone proteins at H3 S10 (Lo et al., 2001) and H2B S36 (Bungard et al., 2010) to promote gene transcription in budding yeast and MEFs, respectively. Consistent with these reports, we found that AMPK phosphorylated histone proteins in vitro (Figure S1G). However, phosphorylated oligonucleosomes could still be efficiently methylated by the PRC2 complex in in vitro methyltransferase experiments (Figure S1G), indicating that phosphorylation of histones by AMPK does not interfere with PRC2-mediated H3K27 trimethylation in vitro. In cells, we observed a context-dependent effect of AMPK-mediated H3 S10 phosphorylation. In MEFs, deletion of Ampkα failed to eliminate H3S10p (Figure 1A). On the other hand, in ovarian cancer cell line OVCAR5, but not OVCAR8, treatment with the specific AMPK agonist A769662 led to a moderate increase of H3S10p (Figure S1B), while H3K27me3 downregulation was observed in both cell lines treated with A769662. These results suggest that although AMPK controls both histone phosphorylation and EZH2-mediated H3K27me3, we failed to demonstrate an obvious correlation between H3S10p and H3K27me3. Numerous reports showed that S10 of H3 is subjected to phosphorylation by a handful of other kinases such as Aurora A (Crosio et al., 2002), Aurora B (Wilkins et al., 2014), and CDK8 (Meyer et al., 2008), thus it is plausible that AMPK-mediated H3 S10 phosphorylation occurs in more specific settings, likely to be in a cellular and/or tissue context-dependent manner.
In skeletal muscle cells, AMPK promotes HDAC5 phosphorylation and abrogates HDAC5-mediated suppression of GLUT4 transcription (McGee et al., 2008). However, compared to WT MEFs, we found that neither HDAC5 expression nor H3 acetylation (Ac-H3) was altered upon the deletion of Ampkα (Figure 1A). Moreover, in keeping with previous reports (McGee et al., 2008), we found that depletion of HDAC5 led to an increase of Ac-H3 in both OVCAR5 and OVCAR8 cells (Figure S1H), and activating AMPK augmented Ac-H3 levels in OVCAR5 cells (Figure S1B). In contrast, H3K27me3 was unaffected by HDAC5 knockdown in these experimental conditions (Figure S1H). Furthermore, A769662 treatment suppresses H3K27me3 regardless of HDAC5 expression status (Figure S1I), supporting the notion that the AMPK-HDAC5 signaling circuit likely has minimal effect on H3K27me3 in our experimental settings. H3K27me3 is deposited onto the nascent chromatin after mitosis, thus a prolonged G1 phase allows cells to accumulate H3K27me3 at the chromatin (Alabert and Groth, 2012). We found that the cell cycle distribution in MEFs and ovarian cancer cells were only slightly affected by Ampkα genetic or activation status (Figure S1J–L), indicating a direct regulation of H3K27me3 by AMPK.
AMPK Abrogates EZH2-mediated Silencing of PRC2 Target Genes
Given the fact that global H3K27me3 is elevated in AMPK inactivated cells (Figure 1A and 1G), we next detected H3K27me3 signals on the chromatin using ChIP-seq assay to assess the physiological consequence of a corresponding enrichment of the repressive epi-mark in genome-wide transcriptional regulation. Notably, compared to wild-type (WT) MEFs, there was an increase of H3K27me3 signals in Ampkα DKO MEFs at and around the transcription start sites (TSS) globally (Figures 2A and S2A). The increased H3K27me3 signals were largely enriched at existing H3K27me3-positive regions (Figures 2B and S2B), which is further supported by the GREAT analysis in Figure S2B that these chromatin regions are indeed highly conserved and H3K27me3-enriched in different cells/tissues. These results indicated that AMPK negatively regulates the global intensity of H3K27me3 at its original chromatin loci, which may suppress the transcriptional repression function of PRC2/EZH2.
Figure 2. AMPK Abrogates EZH2-mediated Epigenetic Silencing of PRC2 Target Genes.

(A) Heat map depicting the H3K27me3 ChIP-Seq signal ±10 kb around its peak summit in both Ampkα WT and DKO MEFs.
(B) Scatter density plot comparing H3K27me3 signals in Ampkα wild type (WT, x-axis) and double knockout (KO, y-axis) MEF cells. Read counts within a 5 kb window around the center of each H3K27me3 peak, which was pooled from both cell conditions, were normalized by 10 million total reads. The color scale indicates the density of peaks.
(C) Ampkα WT and DKO MEFs were subjected to quantitative real-time RT-PCR analyses for relative RNA levels of the indicated genes. Expression levels of these genes were normalized and compared to those of Ampkα WT. Data are means ± SD (n = 3), * P < 0.05, Student’s t test.
(D) Ampkα WT and DKO MEFs were treated with 100 μM A769662 as indicated for 48 hours. Total RNAs were extracted for quantitative real-time RT-PCR analyses for relative RNA levels of the indicated genes. Expression levels of these genes were normalized and compared to those of Ampkα WT without treatment.
(E) Control and 2 mM metformin-treated OVCAR8 cells were subjected to quantitative real-time RT-PCR analyses for relative RNA levels of the indicated genes. Expression levels of these genes were normalized and compared to those of the untreated control cells. Data are means ± SD (n = 3), * P < 0.05; ns, not significant, Student’s t test.
See also Figure S2.
Further RNA-seq analyses of Ampkα DKO cells revealed that genes regulated by EZH2 tend to exhibit an opposite pattern of expression changes upon AMPKα deletion (Figure S2C–D). In particular, target genes that were upregulated by EZH2 silencing were found to be downregulated upon AMPKα deletion in GSEA analysis. This supports the model that AMPK antagonizes EZH2 function, in transcriptional silencing, possibly through negative regulation of its methyltransferase activity towards H3K27. In support of this notion, we found that compared with WT-MEFs, the expression of a cohort of known PRC2 downstream target genes was markedly suppressed in Ampkα DKO MEFs (Figure 2C). Further activation of AMPK by A769662 augmented the expression of these PRC2 target genes in WT, but not Ampkα DKO MEFs (Figure 2D), while depletion of EZH2 in Ampkα DKO MEFs led to increased expression of PRC2 target genes (Figure S2E). Consistently, activating AMPK by metformin led to the elevation of PRC2 target genes in both OVCAR8 (Figure 2E) and T98G (Figure S2F) cells. Taken together, these results suggest that PRC2/EZH2-mediated silencing is globally diminished upon AMPK activation.
AMPK Phosphorylates EZH2 at Threonine 311
To further determine the molecular mechanism by which AMPK inhibits EZH2, we examined the protein sequence of EZH2 and identified a potential AMPK substrate consensus motif (Figures 3A and S3A), which indicates that AMPK might phosphorylate and inactivate EZH2. In support of this hypothesis, we found that EZH2 interacts with AMPKα at endogenous level in cells (Figures 3B and S3B). Under ectopic expression condition, we further demonstrated that EZH2 binds specifically to AMPKα1, but not AMPKα2, in cells (Figure 3C), indicating that different from other AMPK substrates such as ACC1 that can be phosphorylated by both AMPKα1 and α2 (Figure S3C), AMPKα1-containing AMPK kinase complex is likely to be the major isoform to promote EZH2 phosphorylation. To pinpoint the AMPK phosphorylation sites on EZH2, we co-expressed EZH2 and the active AMPKα1 kinase domain in cells followed by immune-purification of EZH2 for subsequent mass spectrometry analysis. Three major phosphorylated serine and threonine sites were mapped (Figure 3A and S3D). Of note, the evolutionally conserved T311 site largely resembles the canonical AMPK substrate motif, whereas other two mapped sites S366 and T367 only minimally match AMPK substrate motifs (Figure 3A).
Figure 3. Identification of EZH2 as a Downstream Phosphorylation Substrate of AMPK.

(A) Sequence alignments of the T311- and the S366/T367-containing regions between EZH2 among various species as well as human EZH1. A schematic representation of EZH2 protein domain structure was shown beneath to illustrate the position of these candidate sites.
(B) Immunoblot (IB) analysis of whole cell lysates (WCL) and anti-EZH2 immunoprecipitates (IP) derived from MDA-MB-231 cells.
(C) IB analysis of WCL and anti-HA IP derived from 293T cells transfected with Flag-EZH2 and the indicated HA-AMPKα constructs.
(D) In vitro kinase assays showing that bacterially purified recombinant WT-EZH2, and to a much lesser extent, S366A/T367A-EZH2, but not the T311A-EZH2 mutant, could be phosphorylated by the AMPK kinase in vitro.
(E) AMPK primarily phosphorylated EZH2 at T311. IB analysis of WCL and anti-Flag IP derived from 293T cells transfected with the indicated constructs.
(F) IB analysis of WCL and anti-HA IP derived from 293T cells transfected with the indicated HA-EZH2, HA-SUZ12 and HA-EED constructs.
See also Figure S3.
Confirming the mass spectrometry analyses, in vitro kinase assays revealed that both the full-length and the N-terminus truncation (aa 1-385) containing the T311 residue, but not the C-terminal SET domain of EZH2 (aa 386-746), could be efficiently phosphorylated by purified AMPK kinase complex (Figure S3E–F). Notably, mutation of T311 to an analine (T311A) displayed a marked reduction in incorporation of 32P-phosphate, while mutation of S366/T367 to alanines only moderately affected AMPK-mediated phosphorylation of EZH2 in vitro (Figure 3D). Consistently, using the AMPK substrate motif antibody, we further showed that T311A mutant was largely impaired in AMPKα1-induced phosphorylation in cells (Figure 3E).
In support of EZH2 as the major target of AMPK among all core subunits of the PRC2 complex, we found that AMPK specifically phosphorylate EZH2, but not EZH1 (Figure S3G–H), other core PRC2 components including SUZ12 and EED (Figure 3F), or H3K27 demethylases UTX and JMJD3 (Figure S3I). Of note, bioinformatics analysis revealed a few cancer patient-derived R308 mutations including R308L, R308Q and R308W (Figure S3J), in which the critical arginine residue within the canonical AMPK substrate motif is mutated. As a result, R308L phenocopied the T311A mutant form of EZH2, both of which are comparably deficient in responding to AMPK-mediated phosphorylation (Figure S3K). This finding suggests a physiological significance of EZH2 phosphorylation at T311 by AMPK. To this end, our results demonstrated mutating the optimal T311 residue or the flanking sequences that match the AMPK canonical phospho-consensus motif largely abolished AMPK-mediated phosphorylation of EZH2. Hence, we mainly focus on elucidating the important role of T311 phosphorylation in modulating PRC2/EZH2 function in the remaining studies.
Activation of AMPK Promotes EZH2 T311 Phosphorylation
To better monitor T311 phosphorylation at the endogenous level, a pT311-EZH2 phosphorylation-specific antibody was generated and validated (Figure S4A–B). Using this specific antibody, we found that activation of AMPK by the specific AMPK agonist A769662 or 2-DG induced EZH2 T311 phosphorylation in WT, but not Ampkα DKO MEFs (Figures 4A and S4C). Similar stimulation of pT311-EZH2 was observed in ovarian cancer cells treated with A769662 (Figures 4B–C), metformin (Figure S4D) or 2DG (Figures S4E–F). These results provide a direct support for our hypothesis that AMPK is a physiological upstream kinase that mediates the phosphorylation of EZH2 at T311, and possibly other sub-optimal sites.
Figure 4. Activation of AMPK Promotes EZH2 T311 Phosphorylation.

(A) Immunoblot (IB) analysis of whole cell lysates (WCL) derived from Ampkα WT and DKO MEFs treated with 100 μM A769662 for the indicated periods of time before harvesting.
(B–C) IB analysis of OVCAR5 (B) and OVCAR8 (C) cells treated with 100 μM A769662 for the indicated time periods before harvesting.
See also Figure S4.
Phosphorylation of EZH2 at T311 by AMPK Disrupts PRC2 Integrity
We next intended to reveal the molecular mechanisms by which AMPK inhibits EZH2 activity via the T311 phosphorylation. To this end, in addition to the phosphorylation-deficient T311A mutant, the phosphorylation mimetic mutant, T311E-EZH2, was generated. We found that compared to the WT, T311E-EZH2 displayed a reduced activity in methylating H3K27 in various cancer cell lines including T98G (Figure 5A), OVCAR5 and OVCAR8 (Figure S5A). Importantly, activating AMPK by A769662 failed to suppress H3K27me3 in T311A-EZH2-expressing OVCAR5 cells (Figure 5B), whereas inhibiting AMPK by Compound C was unable to elevate H3K27me3 in T311E-EZH2-expressing as it did to WT-EZH2 expressing OVCAR5 cells (Figure 5C). Consistently, although ectopic expression of T311A-EZH2 in EZH2-depleted WT-MEFs led to an increase of H3K27me3, it failed to further escalate the level of H3K27 in EZH2-depleted Ampkα DKO MEFs (Figures 5D). Furthermore, similar to the previous identified inhibitory S21D-EZH2 mutant (Cha et al., 2005), T311E-EZH2 was compromised in promoting H3K27 trimethylation in vitro (Figure 5E). These results indicate a negative regulation of EZH2 enzymatic activity by AMPK-mediated phosphorylation at T311.
Figure 5. Phosphorylation of EZH2 at T311 by AMPK Suppresses PRC2 Methyltransferase Activity.

(A) T98G cells stably expressing GFP (as a negative control) or the indicated HA-EZH2 constructs were infected with shGFP or shEZH2 lentiviral constructs. Infected cells were selected with 1 μg/ml puromycin for 72 hours to eliminate the non-infected cells before harvesting for immunoblot (IB) analysis.
(B) shEZH2-OVCAR5 cells stably expressing WT or T311A-EZH2 were treated with 100 μM A769662 for 48 hours before harvesting.
(C) shEZH2-OVCAR5 cells stably expressing WT or T311E-EZH2 were treated with 10 μM Compound C for 48 hours before harvesting.
(D) Ampkα WT and DKO MEFs stably expressing WT or T311A-EZH2 were further depleted with lentiviral shEzh2 construct before harvest for IB analysis.
(E) In vitro methyltransferase assays using immuno-purified PRC2 containing the indicated EZH2 constructs and oligonucleosomes as the substrate.
(F) IB analysis of WCL and anti-Flag immunoprecipitates (IP) derived from 293T cells transfected with HA-SUZ12 and the indicated Flag-EZH2 constructs.
(G) Autoradiography of 35S-labelled SUZ12 bound to bacterially purified His-EZH2 recombinant proteins.
(H) IB analysis of WCL and anti-EZH2 IP derived from OVCAR5 cells infected with EV or pLenti-CMV-HA-AMPKα (1-312) as indicated.
(I) IB analysis of WCL and anti-EZH2 IP derived from OVCAR5 cells treated with 10 mM 2-DG for 1 hour as indicated.
(J) Gel filtration experiments to illustrate that EZH2-containing protein complex migrated differently in T311E-EZH2 expressing OVCAR8 cells than in OVCAR8 cells expressing WT- or T311A-EZH2. OVCAR8 cells stably expressing GFP (as a negative control) or the indicated HA-EZH2 constructs were infected with shGFP or shEZH2 lentiviral constructs. The infected cells were selected with 1 μg/ml puromycin for 72 hours to eliminate the non-infected cells before harvesting for gel filtration analysis using Superose 6 gel filtration chromatography. Prior to running cell lysates, the molecular weight resolution of the column was first estimated by running native molecular weight markers (Thyroglobulin ~669KD, Ferritin ~440KD, Aldolase ~158KD, Conalbumin ~75KD and Ovalbumin ~44KD) and determining their retention times on coomassie-stained SDS-PAGE protein gels.
(K) A schematic illustration of the proposed model for AMPK-mediated phosphorylation of EZH2 disrupts the binding between EZH2 and SUZ12.
See also Figure S5.
Given that T311 resides in the N-terminal regulatory region of the EZH2 protein, which physically interacts with other critical PRC2 components including SUZ12 and EED (Brooun et al., 2016; Justin et al., 2016), we were interested to examine whether phosphorylation of EZH2 affects its binding with other PRC2 components, which has been well demonstrated to play critical roles in optimizing EZH2 methyltransferase function (Cha et al., 2005; Chen et al., 2010; Kaneko et al., 2010; Wei et al., 2011; Xu et al., 2012). To this end, recently resolved crystal structures of the human PRC2 complex show a direct interaction between SUZ12 and the T311 containing region on EZH2 (Brooun et al., 2016; Justin et al., 2016) (Figure S5B). Intriguingly, we found that compared to WT-EZH2, the binding between SUZ12 and T311E-EZH2, but not S21D-EZH2, was significantly reduced both in cells (Figure 5F) and in vitro (Figure 5G). In contrast, the binding between EZH2 and most other PRC2 core components or interacting proteins, including EED (Figure S5C–D), AEBP2 (Figure S5E) and PLU1 (Figure S5F) was not significantly affected by either T311 or S21 phosphorylation mimetic. Notably, the binding between T311E-EZH2 and RBBP4, also named RbAp48, a WD40-repeats domain-containing protein, was also reduced (Figure S5G), likely due to the bridging effect of SUZ12 to direct RBBP4 to PRC2 (Figure S5H) (Ciferri et al., 2012; Ketel et al., 2005; Kloet et al., 2016). Similarly, PCL1, a SUZ12-interacting, PHD-domain containing PRC2 component (Walker et al., 2010), also displayed a reduced interaction with T311E-EZH2 (Figure S5I). In further support of pT311 status as a determining factor for EZH2 interaction with SUZ12, introducing an active AMPKα1 (Figure 5H), activation of AMPK by metformin (Figure 5I) or 2DG (Figures S5J–K) disassociated SUZ12 from EZH2 in cells, while non-phosphorylatable mutant T311A or R308L remained interacting with SUZ12 even after being treated with 2DG (Figure S5K).
Further gel filtration experiment revealed that compared to WT- and T311A-EZH2, chromatin-bound T311E-EZH2 migrated as relatively smaller protein complexes (Figure 5J), supporting a model that upon AMPK-mediated phosphorylation and subsequent disassociation from SUZ12 and SUZ12-bound proteins, pT311-EZH2-containing protein complex is largely catalytically inactive in promoting H3K27 trimethylation (Figure 5K). Notably, in contrast to S21D-EZH2 that displayed a reduced interaction with its primary substrate histone H3 (Cha et al., 2005), we found that T311E-EZH2 still bound to H3 comparably with its WT counterpart (Figure S5L), indicating a different role of AMPK-mediated from AKT-mediated EZH2 phosphorylation. It is worth of mentioning that T311-EZH2 phosphorylation had minimal effect on subcellular localization of EZH2 (Figure S5M), and neither SUZ12 nor EZH2 displayed a significant change in protein stability upon EZH2 T311 phosphorylation (Figures S5N–S). Depletion of core PRC2 components has been shown to disrupt the integrity of the holoenzyme and destabilize PRC2 subunits (Cao and Zhang, 2004; Pasini et al., 2004), our results here indicate that different from the absence of SUZ12, although pT311-EZH2 displayed reduced binding to SUZ12, both EZH2 and SUZ12 might still exist in sub-complexes as evidenced by our gel filtration experiments (Figure 5J). However, it warrants further in-depth mass spectrometry and structural analyses of the exact components within these sub-complexes.
AMPK-mediated Phosphorylation of EZH2 at T311 Attenuates PRC2 Oncogenic Function
EZH2 is frequently overexpressed or mutated in a wide spectrum of human malignancies including prostate, breast and ovarian cancers (Kim and Roberts, 2016). Hyper-activation of PRC2/EZH2 results in epigenetic silencing of various tumor suppressor genes including CDKN2A, CDH1, and DAB2IP to facilitate tumor proliferation, metastasis, and angiogenesis (Albert and Helin, 2010; Margueron and Reinberg, 2011). Therefore, we were interested in further investigating the biological importance of T311 phosphorylating in tumorigenesis. To this end, since activation of AMPK led to a robust induction of T311 phosphorylation on EZH2 in ovarian cancer cell lines OVCAR5 and OVCAR8 (Figure 4B–C and S4C–F), we chose ovarian cancer cell lines to study the functional output of AMPK-mediated EZH2 phosphorylation. Notably, compared to WT-EZH2, stable expression of T311A- and R308L-EZH2 in EZH2-depleted OVCAR8 cells was more potent in suppressing a panel of EZH2 downstream target genes including HOXA11, OLIG2, SOX17 and GATA6 as revealed by real-time RT-PCR experiments (Figure 6A). On the other hand, consistent with the loss-of-function effects of EZH2 phosphorylation at T311 or S21, both T311E and S21D mutants were largely deficient in suppressing these PRC2 target genes, possibly owing to reduced PRC2 integrity or substrate recognition, respectively (Figure 6A).
Figure 6. AMPK-mediated Phosphorylation of EZH2 at T311 Attenuates PRC2 Oncogenic Function.

(A) EZH2-depleted OVCA5 cells stably reintroducing control, WT, T311A, R308L, T311E and S21D EZH2 were quantified for mRNA level expression of EZH2 target genes. mRNA levels of HOXA11, OLIG2, SXO17 and GATA6 were measured by qRT–PCR, and are normalized to GAPDH expression. Data are means ± SEM (n=3).
(B–C) EZH2-depleted OVCAR5 cells stably expressing GFP (as a negative control) or the indicated HA-EZH2 constructs were infected with shGFP or shEZH2 lentiviral constructs. Infected cells were selected with 1 μg/ml puromycin for 72 hours to eliminate the non-infected cells. These cells were subjected to clonogenic survival assays for 10 days. Crystal violet was used to stain the formed colonies (B). The colony numbers were calculated as mean ± SD (n=3), * P < 0.05, Student’s t test (C).
(D–E) OVCAR5 cells generated in (B) were seeded (10,000 cells per well) in 0.5% low-melting-point agarose in DMEM with 10% FBS, layered onto 0.8% agarose in DMEM with 10% FBS. The plates were cultured for 21 days whereupon the colonies >50 μm were counted under a light microscope (D). The colony numbers were plotted as mean ± SD (n=3), * P < 0.05, Student’s t test (E).
(F–G) The tumor size (F) and growth curves (G) for the xenograft experiments using shEZH2-OVCAR5 cells expressing GFP (as a negative control) or the indicated EZH2 construct. In each flank of six nude mice, 1×106 cells were injected subcutaneously. The visible tumors were measured at the indicated days. Error bars represent ±SEM, n=6.
(H–I) EZH2-depleted OVCAR5 cells stably expressing WT or R308L-EZH2 constructs were subjected to clonogenic survival assays for 14 days. 100 μM A769662 was added immediately after plating cells as indicated. Crystal violet was used to stain the formed colonies (H). The colony numbers were calculated as mean ± SD (n=3) (I).
(J–K) EZH2-depleted OVCAR5 cells stably expressing WT or R308L-EZH2 constructs were seeded (10,000 cells per well) in 0.5% low-melting-point agarose in DMEM with 10% FBS, layered onto 0.8% agarose in DMEM with 10% FBS. The plates were cultured for 21 days whereupon the colonies >50 μm were counted under a light microscope. 100 μM A769662 was added immediately after plating cells as indicated (J). The colony numbers were calculated as mean ± SD (n=3) (K).
See also Figure S6.
As a consequence, compared to WT-EZH2, both T311A- and R308L-EZH2 were more potent in promoting proliferation and anchorage-independent growth of OVCAR5 cells as evidenced by colony formation (Figure 6B–C) and soft agar (Figure 6D–E) assays. Notably, OVCAR5 cells harboring cancer patient-derived R308L-EZH2 mutant displayed aggressive tumor growth compared to WT-EZH2-expressing counterparts (Figure 6F–G and S6A–B). On the other hand, compared to WT-EZH2, the T311E mutant was incapable of promoting transformation features when reintroducing into EZH2-depleted cells, indicating that phosphorylation of EZH2 at the T311 residue compromised the oncogenic role of EZH2 (Figure 6B–G and S6A–B). The similar anti-proliferation and anti-migration roles of AMPK were observed in MEFs. We found that compared to their WT counterpart, Ampkα DKO MEFs displayed faster migration in a wound healing experiment (Figure S6C–D). Furthermore, activation of AMPK by A769662 moderately suppressed the proliferation of WT-, but not Ampkα DKO MEFs (Figure S6E–F).
Notably, in contrast to WT-EZH2-expressing cells, A769662-induced activation of AMPK in T311A-EZH2-expressing OVCAR5 cells failed to augment the levels of a group of PRC2 downstream target genes (Figure S6G). Furthermore, compared to WT-EZH2, OVCAR5 cells expressing the R308L-EZH2 mutant were relatively more resistant to the growth inhibitory effect of A769662 (Figure 6H–K). These results further support a crucial role of EZH2 phosphorylation at T311 in mediating AMPK-dependent regulation of PRC2 activity and tumor inhibition (Figure S6H).
EZH2 Phosphorylation at T311 Correlates with Better Survival in Cancer Patients
AMPK serves as a cellular AMP/ATP sensor to maintain energy homeostasis, given its multifaceted functions in fine-tuning multiple metabolic pathways, its role in tumorigenesis is context-dependent (Hardie, 2013). Although recent studies demonstrated that AMPK helps tumor cells survival in part through elevated glucose uptake and redox protection in leukemia-initiating cells (Saito et al., 2015), in other types of cells, AMPK has been well characterized to functions as a tumor suppressor (Zadra et al., 2015). AMPK exerts its tumor suppressive function partly through suppressing mTORC1 activity and lipogenesis via phosphorylating TSC2, RAPTOR and ACC1, respectively (Shackelford and Shaw, 2009). These mechanisms uphold a tumor suppressor role for AMPK, which is further asserted by the accelerated development of lymphoma in Ampkα1−/−;Eμ-Myc mice (Faubert et al., 2013).
AMPK activation is promoted by the phosphorylation at T172 of the AMPKα subunits (Hardie et al., 2012). Therefore, pT172 is widely used as an AMPK activation marker. Using both pT172-AMPKα and pT311-EZH2 antibodies, we observed a statistically significant correlation between pT172-AMPKα and pT311-EZH2 in both ovarian (Figure 7A and 7C) and breast (Figure 7B and 7D) cancer clinical samples. Importantly, high pT311-EZH2 expression correlated with better survival in both types of cancer patients (Figure 7E–F). In contrast, p-T172-AMPKα failed to display a significant correlation with overall survival in both breast cancer and ovarian cancer patients (Figures S7A–B). Thus, these data coherently suggest that although AMPK exerts its multifaceted roles in cancer (Saito et al., 2015; Shaw, 2015), its inhibitory phosphorylation on EZH2 might suppress PRC2 oncogenic function to attenuate tumorigenesis (Figures S6H, S7C).
Figure 7. EZH2 Phosphorylation at T311 Correlates with Better Survival in Cancer Patients.

(A–B) Immunohistochemical staining of p-T172-AMPKα and p-T311-EZH2 in representative ovarian cancer (A) or breast cancer (B) specimens. Brown staining indicates positive immunoreactivity.
(C–D) A positive correlation between p-T172-AMPKα and p-T311-EZH2 levels in human ovarian cancer (C) or breast cancer (D) clinical samples. Statistical significance was determined by a χ2 test.
(E–F) Kaplan–Meier curves showing the overall survival of patients with high or low expression of p-T311-EZH2 in their ovarian cancer (E) or breast cancer (F). Statistical significance was determined by a log-rank test.
See also Figure S7.
DISCUSSION
Both anti- and pro-tumorigenic roles of AMPK have been supported by multiple lines of experimental evidence in distinct tissue/tumor settings (Zadra et al., 2015). The role of AMPK in harnessing unchecked cell growth has been underscored by the findings that AMPK suppresses protein synthesis and de novo fatty acid synthesis (Hardie et al., 2012). Given the pivotal role of PRC2 in promoting cell proliferation, our findings suggest that energy stresses like glucose deprivation and glycolysis blockade activate the AMPK kinase and subsequently relieve EZH2-mediated silencing, which promotes the expression of PRC2 target genes to inhibit cell cycle progression, suppress cell proliferation and promote differentiation. This is another example of AMPK as a determining pivot to connect cellular energy state and pathological development through regulation of epigenetics (Hardie et al., 2012). On the other hand, a recent study showed that AMPK catalyzes H2B phosphorylation, which contributes to AMPK-dependent transcriptional activation of a subset of genes that facilitate cell survival in response to stress (Bungard et al., 2010). In support of a pro-survival role of AMPK in conditions of metabolic stress, it has been shown that AMPK promotes autophagy by phosphorylating ULK1 (Egan et al., 2011) and accelerates fatty acid oxidation to produce ATP (Jeon et al., 2012). Taken together, AMPK governs numerous downstream signaling pathways, depending on different cellular contexts, activation of AMPK leads to either cell proliferation suppression or cell survival under starvation. These findings once again support the complexity of AMPK’s role in dictating cellular fate under stress (Hardie, 2013; Zadra et al., 2015).
Several EZH2 inhibitors that block its methyltransferase activity, such as CPI-1205, EPZ-6438 and GSK126, have been developed and are currently validated in clinical trials for non-Hodgkin’s lymphoma containing gain-of-function mutations of EZH2 or genetically defined solid tumors (Lim et al., 2015). However, acquired resistance to anti-EZH2 therapies have been reported (Kim and Roberts, 2016), suggesting effective combinational therapies are clearly warranted. Our findings that activation of AMPK suppresses PRC2/EZH2 activity suggest that agonists of AMPK might be a promising sensitizer for EZH2-targeting drugs for anti-cancer treatment. Among currently available AMPK agonists, metformin, a first-line drug for treating patients with type 2 diabetes (T2DM) (Foretz et al., 2014), has been widely used in pre-clinical studies to interrogate the effect of energy restriction on tumor cells. Intriguingly, most reports support an anti-cancer function of metformin, which is further evidenced by the observation that in the T2DM population, metformin administration is associated with decreased cancer risks (Foretz et al., 2014). It is noteworthy that metformin directly targets the mitochondrion to inhibit the complex I, resulting in reduced ATP production and subsequently activating AMPK (Pernicova and Korbonits, 2014). This indicates that metformin may exert its anti-cancer functions via other possible mechanisms. Our findings that T311-EZH2 phosphorylation by AMPK impairs PRC2 enzymatic activity (Figures S6H, S7C) advocate for a future comprehensive evaluation of response synergy between direct AMPK agonists and anti-EZH2 inhibitors for treating EZH2-overexpressing solid tumors.
STAR★METHODS
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-EZH2 | Cell Signaling Technology | Cat #5246 |
| Rabbit monoclonal anti-SUZ12 | Cell Signaling Technology | Cat #3737 |
| Rabbit monoclonal anti-AMPKα | Cell Signaling Technology | Cat #5831 |
| Rabbit monoclonal anti-Phospho-AMPK Substrate Motif LXRXX(pS/pT) | Cell Signaling Technology | Cat #5759 |
| Rabbit monoclonal anti-Phospho-T172-AMPKα | Cell Signaling Technology | Cat #2535 |
| Rabbit monoclonal anti-ACC | Cell Signaling Technology | Cat #3676 |
| Rabbit monoclonal anti-Phospho-S79-ACC | Cell Signaling Technology | Cat #11818 |
| Rabbit monoclonal anti-RBBP4 | Cell Signaling Technology | Cat #9067 |
| Rabbit monoclonal anti-histone H3 | Cell Signaling Technology | Cat #4499 |
| Rabbit monoclonal anti-H3K27me2 | Cell Signaling Technology | Cat #9728 |
| Rabbit monoclonal anti-H3K27me3 | Cell Signaling Technology | Cat #9733 |
| Rabbit monoclonal anti-H3K4me3 | Cell Signaling Technology | Cat #9727 |
| Rabbit monoclonal anti-H3K9me2 | Cell Signaling Technology | Cat #4658 |
| Rabbit monoclonal anti-H3K36me2 | Cell Signaling Technology | Cat #2901 |
| Rabbit monoclonal anti-H3K79me2 | Cell Signaling Technology | Cat #5427 |
| Rabbit monoclonal anti-Phospho-S10-H3 | Cell Signaling Technology | Cat #3377 |
| Rabbit polyclonal anti-Phospho-T311-EZH2 | Cell Signaling Technology | Cat #27888 |
| Rabbit polyclonal anti-EED | Abcam | Cat #ab4469 |
| Mouse monoclonal anti-c-Myc | Santa Cruz | Cat #sc-40 |
| Rabbit polyclonal anti-HA | Santa Cruz | Cat #sc-805 |
| Mouse monoclonal anti-HA | Santa Cruz | Cat #sc-7392 |
| Mouse monoclonal anti-HDAC5 | Santa Cruz | Cat #sc-133106 |
| Mouse monoclonal anti-TUBULIN | Sigma-Aldrich | Cat #T5168 |
| Mouse monoclonal anti-VINCULIN | Sigma-Aldrich | Cat #V4505 |
| Mouse monoclonal anti-Flag | Sigma-Aldrich | Cat #F3165 |
| Rabbit polyclonal anti-Flag | Sigma-Aldrich | Cat #F2425 |
| Mouse monoclonal anti-Flag agarose beads | Sigma-Aldrich | Cat #A2220 |
| Mouse monoclonal anti-HA agarose beads | Sigma-Aldrich | Cat #A2095 |
| peroxidase-conjugated anti-mouse secondary antibody | Sigma-Aldrich | Cat #A4416 |
| peroxidase-conjugated anti-rabbit secondary antibody | Sigma-Aldrich | Cat #A4914 |
| IKKα | Cell Signaling Technology | Cat #2682 |
| Rabbit polyclonal anti-acetylated H3 | EMD Millipore | Cat #06-599 |
| Bacterial and Virus Strains | ||
| XL10 Gold Escherichia coli | Agilent | Cat #200314 |
| BL21(DE3) Escherichia coli | Dr. William G. Kaelin, Jr., Dana-Farber Cancer Institute | N/A |
| Biological Samples | ||
| Ovarian tumor samples (n=235) | University of Texas MD Anderson Cancer Center | N/A |
| Breast tumor samples (n=210) | University of Texas MD Anderson Cancer Center or Mackay Memorial Hospital (Taipei, Taiwan) | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| A-769662 | Selleck Chemicals | Cat # S2697; CAS: 844499-71-4 |
| Dorsomorphin (Compound C) 2HCl | Selleck Chemicals | Cat # S7306; CAS: 1219168-18-9 |
| Metformin HCl | Selleck Chemicals | Cat # S1950; CAS: 1115-70-4 |
| 2-Deoxy-D-glucose | Sigma-Aldrich | Cat # D8375; CAS: 154-17-6 |
| Cycloheximide | Sigma-Aldrich | Cat # C7698; CAS: 66-81-9 |
| Chicken polynucleosomes, purified | Epicypher | Cat # 16-0004 |
| AMPK (A1/B1/G2) Kinase | Promega | Cat # V4012 |
| Critical Commercial Assays | ||
| ThruPLEX-FD Prep Kit | Rubicon Genomics | R400427 |
| True-seq RNA sample preparation kit | Illumina | RS-122-2001 |
| Deposited Data | ||
| Raw and analyzed sequencing data | This paper | GEO: GSE97736 |
| Raw data and images | This paper and Mendeley Data | http://dx.doi.org/10.17632/jdf8ddt32n.1 |
| Experimental Models: Cell Lines | ||
| Human: 293T cells | Dr. William G. Kaelin, Jr., Dana-Farber Cancer Institute | N/A |
| Human: T98G cells | Dr. William G. Kaelin, Jr., Dana-Farber Cancer Institute | N/A |
| Human: OVCAR5 cells | Dr. Marc W. Kirschner, Harvard Medical School | N/A |
| Human: OVCAR8 cells | Dr. Marc W. Kirschner, Harvard Medical School | N/A |
| Human: PC3 cells | Dr. Pier Paolo Pandolfi, Beth Israel Deaconess Medical Center | N/A |
| Human: MDA-MB-231 cells | ATCC | CRM-HTB-26 |
| Human: ZR-75-1 cells | ATCC | CRL-1500 |
| Human: MDA-MB-435 cells | ATCC | HTB-129 |
| Human: MCF7 cells | ATCC | HTB-22 |
| Mouse: WT and Ampkα1(−/−);Ampkα2(−/−) Mouse Embryonic Fibroblasts (MEFs) | Dr. Benoit Viollet, Institut Cochin, Paris, France | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: NCRNU-F: CrTac:NCr-Foxn1nu | Taconic Biosciences | NCRNU-F |
| Oligonucleotides | ||
| Primers for real-time RT-qPCR, see Table S1 | This paper | N/A |
| Recombinant DNA | ||
| pCMV-HA-EZH2 | Addgene | Cat # 24230 |
| pGEX-EZH2 | Addgene | Cat # 28060 |
| pET28a-EZH2 | This paper | N/A |
| Mouse EZH1 cDNA | Dr. Stuart H. Orkin, Dana-Farber Cancer Institute | N/A |
| pEBG-AMPKα1(1-312) | Addgene | Cat # 27632 |
| pDONR223-PRKAA1 | Addgene | Cat # 23871 |
| pDONR223-PRKAA2 | Addgene | Cat # 23671 |
| pCMVHA-EED | Addgene | Cat # 24231 |
| pCMVHA-SUZ12 | Addgene | Cat # 24232 |
| pCMV-Flag-EZH2 | This paper | N/A |
| pLenti-CMV-hygro-HA-EZH2 | This paper | N/A |
| Myc-PLU1 | (Klose et al., 2007) | N/A |
| Myc-PCL1 | (Klose et al., 2007) | N/A |
| HA-AEBP2 | (Klose et al., 2007) | N/A |
| HA-UTX | (Klose et al., 2007) | N/A |
| HA-JMJD3 | (Klose et al., 2007) | N/A |
| pLKO-shEZH2 | (Xu et al., 2012) | N/A |
| Software and Algorithms | ||
| ImageJ | Freeware | N/A |
| Prism V7 | Graphpad | N/A |
| Other | ||
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents may be directed to, and will be fulfilled by, the Lead Contact, Dr. Wenyi Wei (wwei2@bidmc.harvard.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Culture
293T, OVCAR5, OVCAR8, T98G, PC3, MDA-MB-231, MDA-MB-435, ZR-75-1, and MCF7 cell lines, were grown in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 2 mM Glutamine, 100 units/ml penicillin and 100 mg/ml streptomycin. Ampkα1, Ampkα2 double knockout MEFs (Tsou et al., 2011) and its WT counterparts were grown in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 2 mM Glutamine, 100 units/ml penicillin and 100 mg/ml streptomycin.
Xenograft Tumor Growth
For assaying tumor growth in the xenograft model, 6-week-old female nude mice (NCr nude from Taconic, CrTac:NCr-Foxn1nu) housed in specific pathogen-free environments were injected subcutaneously with 1.0 × 106 OVCAR5 derivatives (n=6 for each group) mixed with DMEM medium and Matrigel (1:1, vol/vol). Tumor size was measured every other day with a caliper, and the tumor volume was determined by the formula: L × W2 × 0.5, where L is the longest diameter and W is the shortest diameter. 18 days after injection, mice were euthanized and xenografted solid tumors were dissected. All experimental procedures strictly complied with the IACUC guidelines.
Patient Tumor Tissue Samples and Immunohistochemical Staining
Human breast cancer (n=210) and ovarian cancer (n=235) tissue specimens were obtained from patients undergoing surgical resection as primary treatment at The University of Texas MD Anderson Cancer Center or Mackay Memorial Hospital (Taipei, Taiwan) between 1990 and 2009 under the guidelines approved by the Institutional Review Board at MD Anderson, and written informed consent was obtained from patients in all cases at the time of enrollment. Immunohistochemical staining was carried out using a modified ABC method. Briefly, patient tumor samples were deparaffinized and rehydrated. Antigen retrieval was carried out by heating in 0.01 M sodium citrate buffer (pH 6.0) using a microwave oven. To block endogenous peroxidase activity, the sections were treated with 1% hydrogen peroxide in methanol for 30 min. After 1 h preincubation in 10% normal serum to prevent nonspecific staining, the samples were incubated with primary antibodies at 4 °C overnight. The primary antibodies specific to p-T172-AMPKα (Cell Signaling Technology, 1:20) and p-T311-EZH2 (1:100) were used. The sections were then treated with biotinylated secondary antibody, followed by incubations with avidin-biotin peroxidase complex solution for 1 h at room temperature. Color was developed with the 3-amino-9-ethylcarbazole solution. Counterstaining was carried out using Mayer’s hematoxylin. Immunostaining positivity was assessed semiquantitatively using staining intensity and percentage by two independent pathological investigators. H score was determined by multiplying the staining intensity by the percentage of positive tumor cells.
The data were analyzed using statistical software SPSS version 21.0. A P value ≤ 0.05 was defined as statistically significant. The χ2 test was used for statistical analysis of the correlation between p-T172-AMPKα and p-T311-EZH2. Survival curves were estimated by the Kaplan–Meier method, and survival rates in different groups were compared by the log-rank test.
METHOD DETAILS
Site-directed Mutagenesis
Site directed mutagenesis to generate various EZH2 mutants were performed using the QuikChange XL Site-Directed Mutagenesis Kit (Agilent) according to the manufacturer’s instructions.
Lentiviral Packaging and Infection
Lentiviral shRNA virus packaging and subsequent infection of various cell lines were performed according to the protocol described previously (Boehm et al., 2005). Briefly, lentiviral constructs were co-transfected with the pCMV-dR8.91 (Delta 8.9) plasmid containing gag, pol and rev genes and the VSV-G envelope-expressing plasmid into 293T cells. Virus-containing media were collected, filtered before being used for infection.
Immunoblots and Immunoprecipitation
Cells were lysed in EBC buffer (50 mM Tris pH 7.5, 120 mM NaCl, 0.5% NP-40) supplemented with protease inhibitors (Complete Mini, Roche) and phosphatase inhibitors (phosphatase inhibitor cocktail set I and II, Calbiochem). To prepare the Whole Cell Lysates (WCL), 3 × SDS sample buffer was directly added to the cell lysates and sonicate before resolved on SDS-PAGE and immunoblotted with primary antibodies. The protein concentrations of the lysates were measured using the Bio-Rad protein assay reagent on a Beckman Coulter DU-800 spectrophotometer. For immunoprecipitation, cells were lysed in RIPA buffer (50 mM Tris, pH 8.0, 150 mM NaCl, 1.0% NP-40, 0.5% sodium deoxycholate, 0.1% SDS) containing protease and phosphatase inhibitors. The lysates were sonicated before centrifuge at 13,000 rpm and the supernatant was collected for immunoprecipitations. 1 mg lysates were incubated with the appropriate antibody (1–2 μg) for 3–4 h at 4 °C followed by 1 h incubation with Protein A sepharose beads (GE Healthcare). Immuno-complexes were washed five times with NETN buffer (20 mM Tris, pH 8.0, 100 mM NaCl, 1 mM EDTA and 0.5% NP-40) before being resolved by SDS-PAGE and immunoblotted with indicated antibodies. Nuclear/cytoplasmic fractionation was performed as described (Gao et al., 2009b).
In Vitro Binding Assays
Binding to immobilized His-tagged recombinant proteins was performed as described previously (Gao et al., 2009a; Wei et al., 2004). Briefly, the pET28a-EZH2 plasmid with N-terminal 6His tag was transformed into BL21(DE3) competent cells. The recombinant His-EZH2 proteins were expressed by Isopropyl β-D-1-thiogalactopyranoside (IPTG) induction for 18 h at 16 °C. The proteins were purified using Ni-NTA Agarose (Qiagen) according to the manufacturer’s instructions. Agarose-bound His-EZH2 proteins were further incubated with in vitro transcribed and translated 35S-SUZ12 or 35S-EED using TnT Quick Coupled Transcription/Translation System from Promega. The incubation was performed at 4 °C for 3–4 h followed by washing with NETN buffer as described in the Immunoblots and Immunoprecipitation section above. Samples were resolved by SDS-PAGE and subjected to autoradiography.
In Vitro Kinase Assays
AMPK in vitro kinase assays were performed as described previously (Egan et al., 2011). Briefly, bacterially purified GST-EZH2 was incubated with partially purified rat liver active AMPK heterotrimer (Progema) in kinase buffer (25 mM MOPS, pH 7.5, 1 mM EGTA, 0.1 mM Na3VO4, 15 mM MgCl2) supplemented with 300 μM AMP. ATP mix with γ-32P-ATP was added at a 100 μM final concentration. The reaction was initiated by the addition of AMPK in a volume of 30 μl for 60 min at 30 °C followed by the addition of SDS-PAGE sample buffer to stop the reaction before resolved by SDS-PAGE and subsequent autoradiography.
FACS Analysis
Cells were collected and stained with propidium iodide (Roche) as described previously (Wan et al., 2017). Briefly, 1 × 106 cells were trypsinized and washed in PBS twice before being fixed in cold 70% ethanol. The cells were kept on ice for 30 min, then were washed in PBS twice and suspended in 250 μl PBS. 50 μl of a 100 μg/ml sock of RNase and 200 μl propidium iodide (from 50 μg/ml stock solution) were added to remove RNA and stain the DNA. Stained cells were analyzed on a FACSCanto II (BD) at the Moffitt Cancer Center Flow Cytometry core facility.
Mass Spectrometry Analyses
The mass spectrometry procedure was performed as described previously (Liu et al., 2014). Phosphorylated Flag-EZH2 samples were prepared by immunoprecipitation from 293T cells, in the presence of active AMPKα1 kinase domain co-transfected. The EZH2 proteins were resolved by SDS-PAGE. The gel was stained with the Gel-Code Blue reagent, destained and the EZH2 band was excised. Samples were subjected to reduction with 10mM dithiothreitol (DTT) for 30 minutes, alkylation with 55mM iodoacetamide with 45 minutes, and in-gel digestion with trypsin. The digested samples were subjected to reversed phase microcapillary/tandem mass spectrometry (LC/MS/MS) as described previously (Liu et al., 2014),
Gel Filtration Chromatography Analyses
For gel filtration experiment using cell nuclear pellet extracts, cells were washed with phosphate-buffered saline and lysed in 0.5 ml of RIPA buffer (50 mM Tris, pH 8.0, 150 mM NaCl, 1.0% NP-40, 0.5% sodium deoxycholate, 0.1% SDS) containing protease inhibitors (Complete Mini, Roche) and phosphatase inhibitors (phosphatase inhibitor cocktail set I and II, Calbiochem). After centrifuge at 13,000 rpm in a desktop micro-centrifuge, precipitated nuclear pellets were collected and washed twice with Nuclear Buffer (20 mM Tris, pH 8.0, 50 mM NaCl, 1 mM DTT, 5 mM CaCl2), and were then subjected to Micrococcal Nuclease (New England Biolabs) treatment at 37 °C for 15 min followed by centrifuge at 13,000 rpm in a desktop micro-centrifuge. The nuclear pellet extracts were further filtered through a 0.45 μm syringe filter. Total protein concentration was then adjusted to 1 mg/ml with Nuclear Buffer and 500 μl of the nuclear pellet extract was loaded onto a Superose 6 column.
ChIP-sequencing Analyses
MACS2 was employed to call peaks with a broad cutoff set at 0.0001 (Zhang et al., 2008). Peaks that are overlapped with the blacklists from UCSC were filtered. To plot the scatter density figure, all the peaks from both AMPK intact cell line and double knockout cell line were pooled and then merged. Unique reads within a 5 kb window around the center of each peak were counted and normalized to 10 million reads in each cell condition.
GREAT Analysis
GREAT analysis of H3K27me3 peaks in both wild-type (WT) AMPK and double knockout (DKO) MEF cells was performed according to a basal plus extension association rule, which links the peaks with nearby genes (McLean et al., 2010). The rule defines the peak-associated genes as those whose transcription start site (TSS) falls within 5–1000 kb from the peak region. Top annotated gene sets in the Molecular Signatures Database (MSigDB) were used to plot the figure.
RNA-sequencing Analyses
Total RNAs were extracted from either the control MEFs (WT) or AMPKα double knockout (KO) cells. RNA-Seq libraries were constructed using Illumina TruSeq RNA sample preparation kit. The sequencing reads were aligned to mouse genome (mm9) using TopHat 2.0.10 (Bowtie2 2.1.0) (Kim et al., 2013), and read counts for each gene were counted by featureCounts package with default parameters (Liao et al., 2014). DEseq2 1.14.1 was used to call differentially expressed genes between WT and KO cells (Robinson et al., 2010).
GSEA Analysis
Transcriptional profiling upon EZH2 depletion was retrieved from GSE59346 in MEF cells (Wassef et al., 2015) and GSE1566 in T-cells (Su et al., 2005). We defined the EZH2-activated or -repressed targets as those that were significantly down or upregulated after EZH2 knockdown. Limma was used to identify EZH2-regulated genes with a p value less than 0.01 and a fold change of more than 1.6 or 2 for GSE59346 and GSE1566 dataset, respectively. All the AMPK-dependent genes were ranked from the highest to the lowest levels, based on the fold changes after AMPK knockout. A customized script for GSEA analysis was utilized to investigate whether EZH2-activated or -repressed genes were enriched in this ranked gene list (Subramanian et al., 2005).
Real-time RT-PCR Analyses
Total RNA was extracted using the RNeasy mini kit (Qiagen), and the reverse transcription reaction was performed using Power SYBR® Green PCR Master Mix (ABI, 4367659). The real-time RT-PCR was performed with the 7500 Fast Real-time PCR system (ABI). Primers were listed in Table S1.
Clonogenic Survival and Soft Agar Assays
The clonogenic survival and soft agar assays for OVCAR5 ovarian cancer cells were performed as described previously (Wan et al., 2014). Briefly, cells were cultured in 10% FBS containing DMEM before plating into 6-well plate at 1,000 cells per well. 3 weeks later, cells were stained with crystal violet and the colony numbers were counted.
For soft agar assays, cells (10,000 per well) were seeded in 0.5% low-melting-point agarose in DMEM with 10% FBS, layered onto 0.8% agarose in DMEM with 10% FBS. The plates were kept in the cell culture incubator for 80 days after which the colonies >50 μm were counted under a light microscope.
QUANTIFICATION AND STATISTICAL ANALYSIS
Group data are presented as mean ± SD. Comparisons between two groups were made by Student’s paired or unpaired two-tailed t tests as indicated in the figure legends. P values less than 0.05 were considered significant. Analyses were performed using the Microsoft Excel 2010 and GraphPad Prism V7.
DATA AND SOFTWARE AVAILABILITY
The accession number for our RNA-seq and CHIP-seq data at NCBI GEO is GSE97736. Raw gel images and film scans have been deposited to Mendeley Data (http://dx.doi.org/10.17632/jdf8ddt32n.1).
Supplementary Material
Highlights.
AMPK activation attenuates PRC2-mediated epigenetic silencing
AMPK phosphorylates EZH2 at T311 to disrupt EZH2-SUZ12 interaction
EZH2 T311 phosphorylation inhibits PRC2 oncogenic function
EZH2 T311 phosphorylation correlates with better survival in cancer patients
Acknowledgments
We thank Drs. Hiroyuki Inuzuka, Wenjian Gan, Jianping Guo and Qing Yin for critical reading of the manuscript, William Hahn and Stuart Orkin for providing reagents and members of the Wei, Hung, Brown, Xu and Wan labs for useful discussions. W. Wei is a Leukemia and Lymphoma Society Scholar and ACS Research Scholar. This work was supported in part by the NIH grants (W. Wei, R01GM089763, R01GM094777 and R01CA177910; M-C. Hung, R01CA211615; L. Wan, R00CA183914; K. Xu, R00CA178199; J. Zhang, 1K99CA212292; R01GM114142 V. X. Jin), Cancer Prevention Research Institute of Texas Award (M-C. Hung, RP150245; K. Xu, RR140072), National Breast Cancer Foundation, Inc. (M-C. Hung); Breast Cancer Research Foundation (M-C. Hung) and The University of Texas MD Anderson-China Medical University and Hospital Sister Institution Fund (M-C. Hung). V Foundation Translational Award (T2017-010 to K. Xu) and Voelcker Fund Young Investigator Award (K. Xu).
Footnotes
Supplemental Information including Supplemental Figures and one Supplemental Table can be found with this article online.
AUTHOR CONTRIBUTIONS
L.W., K.X., Y.W., and J.Z. performed most of the experiments with assistance from T.H., C.F., Z.Z., Y.V.W., L.H., W.X., W-C.C., W-C.H., C-L.L., Y.C.C., Y.W., J.L., M.Y., V.X.J., J.L., X.D., J.G., S.J. and J.L.. L.W., K.X., Y.W., J.M.S., M.B., M-C.H., and W.W. designed the experiments and supervised the study. L.W. and W.W. wrote the manuscript with help from K.X., Y.W., J.Z., and M-C.H. All authors commented on the manuscript.
DECLARATION OF INTERESTS
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alabert C, Groth A. Chromatin replication and epigenome maintenance. Nature reviews Molecular cell biology. 2012;13:153–167. doi: 10.1038/nrm3288. [DOI] [PubMed] [Google Scholar]
- Albert M, Helin K. Histone methyltransferases in cancer. Seminars in cell & developmental biology. 2010;21:209–220. doi: 10.1016/j.semcdb.2009.10.007. [DOI] [PubMed] [Google Scholar]
- Boehm JS, Hession MT, Bulmer SE, Hahn WC. Transformation of human and murine fibroblasts without viral oncoproteins. Mol Cell Biol. 2005;25:6464–6474. doi: 10.1128/MCB.25.15.6464-6474.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooun A, Gajiwala KS, Deng YL, Liu W, Bolanos B, Bingham P, He YA, Diehl W, Grable N, Kung PP, et al. Polycomb repressive complex 2 structure with inhibitor reveals a mechanism of activation and drug resistance. Nature communications. 2016;7:11384. doi: 10.1038/ncomms11384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bungard D, Fuerth BJ, Zeng PY, Faubert B, Maas NL, Viollet B, Carling D, Thompson CB, Jones RG, Berger SL. Signaling kinase AMPK activates stress-promoted transcription via histone H2B phosphorylation. Science. 2010;329:1201–1205. doi: 10.1126/science.1191241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai L, Sutter BM, Li B, Tu BP. Acetyl-CoA induces cell growth and proliferation by promoting the acetylation of histones at growth genes. Molecular cell. 2011;42:426–437. doi: 10.1016/j.molcel.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P, Jones RS, Zhang Y. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science. 2002;298:1039–1043. doi: 10.1126/science.1076997. [DOI] [PubMed] [Google Scholar]
- Cao R, Zhang Y. SUZ12 is required for both the histone methyltransferase activity and the silencing function of the EED-EZH2 complex. Molecular cell. 2004;15:57–67. doi: 10.1016/j.molcel.2004.06.020. [DOI] [PubMed] [Google Scholar]
- Cha TL, Zhou BP, Xia W, Wu Y, Yang CC, Chen CT, Ping B, Otte AP, Hung MC. Akt-mediated phosphorylation of EZH2 suppresses methylation of lysine 27 in histone H3. Science. 2005;310:306–310. doi: 10.1126/science.1118947. [DOI] [PubMed] [Google Scholar]
- Chen S, Bohrer LR, Rai AN, Pan Y, Gan L, Zhou X, Bagchi A, Simon JA, Huang H. Cyclin-dependent kinases regulate epigenetic gene silencing through phosphorylation of EZH2. Nature cell biology. 2010;12:1108–1114. doi: 10.1038/ncb2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng J, Zhang T, Ji H, Tao K, Guo J, Wei W. Functional characterization of AMP-activated protein kinase signaling in tumorigenesis. Biochim Biophys Acta. 2016;1866:232–251. doi: 10.1016/j.bbcan.2016.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciferri C, Lander GC, Maiolica A, Herzog F, Aebersold R, Nogales E. Molecular architecture of human polycomb repressive complex 2. eLife. 2012;1:e00005. doi: 10.7554/eLife.00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cool B, Zinker B, Chiou W, Kifle L, Cao N, Perham M, Dickinson R, Adler A, Gagne G, Iyengar R, et al. Identification and characterization of a small molecule AMPK activator that treats key components of type 2 diabetes and the metabolic syndrome. Cell metabolism. 2006;3:403–416. doi: 10.1016/j.cmet.2006.05.005. [DOI] [PubMed] [Google Scholar]
- Crosio C, Fimia GM, Loury R, Kimura M, Okano Y, Zhou H, Sen S, Allis CD, Sassone-Corsi P. Mitotic phosphorylation of histone H3: spatio-temporal regulation by mammalian Aurora kinases. Molecular and cellular biology. 2002;22:874–885. doi: 10.1128/MCB.22.3.874-885.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, Vasquez DS, Joshi A, Gwinn DM, Taylor R, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011;331:456–461. doi: 10.1126/science.1196371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faubert B, Boily G, Izreig S, Griss T, Samborska B, Dong Z, Dupuy F, Chambers C, Fuerth BJ, Viollet B, et al. AMPK is a negative regulator of the Warburg effect and suppresses tumor growth in vivo. Cell metabolism. 2013;17:113–124. doi: 10.1016/j.cmet.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foretz M, Guigas B, Bertrand L, Pollak M, Viollet B. Metformin: from mechanisms of action to therapies. Cell metabolism. 2014;20:953–966. doi: 10.1016/j.cmet.2014.09.018. [DOI] [PubMed] [Google Scholar]
- Gao D, Inuzuka H, Korenjak M, Tseng A, Wu T, Wan L, Kirschner M, Dyson N, Wei W. Cdh1 regulates cell cycle through modulating the claspin/Chk1 and the Rb/E2F1 pathways. Mol Biol Cell. 2009a;20:3305–3316. doi: 10.1091/mbc.E09-01-0092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao D, Inuzuka H, Tseng A, Chin RY, Toker A, Wei W. Phosphorylation by Akt1 promotes cytoplasmic localization of Skp2 and impairs APCCdh1-mediated Skp2 destruction. Nature cell biology. 2009b;11:397–408. doi: 10.1038/ncb1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, Shaw RJ. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Molecular cell. 2008;30:214–226. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie DG. The LKB1-AMPK pathway-friend or foe in cancer? Cancer Cell. 2013;23:131–132. doi: 10.1016/j.ccr.2013.01.009. [DOI] [PubMed] [Google Scholar]
- Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nature reviews Molecular cell biology. 2012;13:251–262. doi: 10.1038/nrm3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzig S, Shaw RJ. AMPK: guardian of metabolism and mitochondrial homeostasis. Nature reviews. Molecular cell biology. 2017 doi: 10.1038/nrm.2017.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon SM, Chandel NS, Hay N. AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature. 2012;485:661–665. doi: 10.1038/nature11066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Justin N, Zhang Y, Tarricone C, Martin SR, Chen S, Underwood E, De Marco V, Haire LF, Walker PA, Reinberg D, et al. Structural basis of oncogenic histone H3K27M inhibition of human polycomb repressive complex 2. Nature communications. 2016;7:11316. doi: 10.1038/ncomms11316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko S, Li G, Son J, Xu CF, Margueron R, Neubert TA, Reinberg D. Phosphorylation of the PRC2 component Ezh2 is cell cycle-regulated and up-regulates its binding to ncRNA. Genes & development. 2010;24:2615–2620. doi: 10.1101/gad.1983810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ketel CS, Andersen EF, Vargas ML, Suh J, Strome S, Simon JA. Subunit contributions to histone methyltransferase activities of fly and worm polycomb group complexes. Molecular and cellular biology. 2005;25:6857–6868. doi: 10.1128/MCB.25.16.6857-6868.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biolm. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KH, Roberts CW. Targeting EZH2 in cancer. Nat Med. 2016;22:128–134. doi: 10.1038/nm.4036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloet SL, Makowski MM, Baymaz HI, van Voorthuijsen L, Karemaker ID, Santanach A, Jansen P, Di Croce L, Vermeulen M. The dynamic interactome and genomic targets of Polycomb complexes during stem-cell differentiation. Nature structural & molecular biology. 2016;23:682–690. doi: 10.1038/nsmb.3248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klose RJ, Yan Q, Tothova Z, Yamane K, Erdjument-Bromage H, Tempst P, Gilliland DG, Zhang Y, Kaelin WG., Jr The retinoblastoma binding protein RBP2 is an H3K4 demethylase. Cell. 2007;128:889–900. doi: 10.1016/j.cell.2007.02.013. [DOI] [PubMed] [Google Scholar]
- Klose RJ, Zhang Y. Regulation of histone methylation by demethylimination and demethylation. Nature reviews Molecular cell biology. 2007;8:307–318. doi: 10.1038/nrm2143. [DOI] [PubMed] [Google Scholar]
- Knutson SK, Warholic NM, Wigle TJ, Klaus CR, Allain CJ, Raimondi A, Porter Scott M, Chesworth R, Moyer MP, Copeland RA, et al. Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc Natl Acad Sci U S A. 2013;110:7922–7927. doi: 10.1073/pnas.1303800110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamia KA, Sachdeva UM, DiTacchio L, Williams EC, Alvarez JG, Egan DF, Vasquez DS, Juguilon H, Panda S, Shaw RJ, et al. AMPK regulates the circadian clock by cryptochrome phosphorylation and degradation. Science. 2009;326:437–440. doi: 10.1126/science.1172156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- Lim J, Poulin NM, Nielsen TO. New Strategies in Sarcoma: Linking Genomic and Immunotherapy Approaches to Molecular Subtype. Clinical cancer research : an official journal of the American Association for Cancer Research. 2015 doi: 10.1158/1078-0432.CCR-15-0831. [DOI] [PubMed] [Google Scholar]
- Liu P, Begley M, Michowski W, Inuzuka H, Ginzberg M, Gao D, Tsou P, Gan W, Papa A, Kim BM, et al. Cell-cycle-regulated activation of Akt kinase by phosphorylation at its carboxyl terminus. Nature. 2014;508:541–545. doi: 10.1038/nature13079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo WS, Duggan L, Emre NC, Belotserkovskya R, Lane WS, Shiekhattar R, Berger SL. Snf1--a histone kinase that works in concert with the histone acetyltransferase Gcn5 to regulate transcription. Science. 2001;293:1142–1146. doi: 10.1126/science.1062322. [DOI] [PubMed] [Google Scholar]
- Lu C, Thompson CB. Metabolic regulation of epigenetics. Cell metabolism. 2012;16:9–17. doi: 10.1016/j.cmet.2012.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margueron R, Reinberg D. The Polycomb complex PRC2 and its mark in life. Nature. 2011;469:343–349. doi: 10.1038/nature09784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCabe MT, Ott HM, Ganji G, Korenchuk S, Thompson C, Van Aller GS, Liu Y, Graves AP, Della Pietra A, 3rd, Diaz E, et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature. 2012;492:108–112. doi: 10.1038/nature11606. [DOI] [PubMed] [Google Scholar]
- McGee SL, van Denderen BJ, Howlett KF, Mollica J, Schertzer JD, Kemp BE, Hargreaves M. AMP-activated protein kinase regulates GLUT4 transcription by phosphorylating histone deacetylase 5. Diabetes. 2008;57:860–867. doi: 10.2337/db07-0843. [DOI] [PubMed] [Google Scholar]
- McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, Bejerano G. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol. 2010;28:495–501. doi: 10.1038/nbt.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melnick A. Epigenetic therapy leaps ahead with specific targeting of EZH2. Cancer Cell. 2012;22:569–570. doi: 10.1016/j.ccr.2012.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer KD, Donner AJ, Knuesel MT, York AG, Espinosa JM, Taatjes DJ. Cooperative activity of cdk8 and GCN5L within Mediator directs tandem phosphoacetylation of histone H3. The EMBO journal. 2008;27:1447–1457. doi: 10.1038/emboj.2008.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasini D, Bracken AP, Jensen MR, Lazzerini Denchi E, Helin K. Suz12 is essential for mouse development and for EZH2 histone methyltransferase activity. The EMBO journal. 2004;23:4061–4071. doi: 10.1038/sj.emboj.7600402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pernicova I, Korbonits M. Metformin--mode of action and clinical implications for diabetes and cancer. Nat Rev Endocrinol. 2014;10:143–156. doi: 10.1038/nrendo.2013.256. [DOI] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito Y, Chapple RH, Lin A, Kitano A, Nakada D. AMPK Protects Leukemia-Initiating Cells in Myeloid Leukemias from Metabolic Stress in the Bone Marrow. Cell stem cell. 2015;17:585–596. doi: 10.1016/j.stem.2015.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauvageau M, Sauvageau G. Polycomb group proteins: multi-faceted regulators of somatic stem cells and cancer. Cell stem cell. 2010;7:299–313. doi: 10.1016/j.stem.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shackelford DB, Shaw RJ. The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat Rev Cancer. 2009;9:563–575. doi: 10.1038/nrc2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw RJ. AMPK Keeps Tumor Cells from Starving to Death. Cell stem cell. 2015;17:503–504. doi: 10.1016/j.stem.2015.10.007. [DOI] [PubMed] [Google Scholar]
- Su IH, Dobenecker MW, Dickinson E, Oser M, Basavaraj A, Marqueron R, Viale A, Reinberg D, Wulfing C, Tarakhovsky A. Polycomb group protein ezh2 controls actin polymerization and cell signaling. Cell. 2005;121:425–436. doi: 10.1016/j.cell.2005.02.029. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsou P, Zheng B, Hsu CH, Sasaki AT, Cantley LC. A fluorescent reporter of AMPK activity and cellular energy stress. Cell metabolism. 2011;13:476–486. doi: 10.1016/j.cmet.2011.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden MG. Targeting cancer metabolism: a therapeutic window opens. Nature reviews Drug discovery. 2011;10:671–684. doi: 10.1038/nrd3504. [DOI] [PubMed] [Google Scholar]
- Walker E, Chang WY, Hunkapiller J, Cagney G, Garcha K, Torchia J, Krogan NJ, Reiter JF, Stanford WL. Polycomb-like 2 associates with PRC2 and regulates transcriptional networks during mouse embryonic stem cell self-renewal and differentiation. Cell stem cell. 2010;6:153–166. doi: 10.1016/j.stem.2009.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan L, Chen M, Cao J, Dai X, Yin Q, Zhang J, Song SJ, Lu Y, Liu J, Inuzuka H, et al. The APC/C E3 Ligase Complex Activator FZR1 Restricts BRAF Oncogenic Function. Cancer Discov. 2017;7:424–441. doi: 10.1158/2159-8290.CD-16-0647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan L, Tan M, Yang J, Inuzuka H, Dai X, Wu T, Liu J, Shaik S, Chen G, Deng J, et al. APC(Cdc20) suppresses apoptosis through targeting Bim for ubiquitination and destruction. Developmental cell. 2014;29:377–391. doi: 10.1016/j.devcel.2014.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wassef M, Rodilla V, Teissandier A, Zeitouni B, Gruel N, Sadacca B, Irondelle M, Charruel M, Ducos B, Michaud A, et al. Impaired PRC2 activity promotes transcriptional instability and favors breast tumorigenesis. Genes Dev. 2015;29:2547–2562. doi: 10.1101/gad.269522.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei W, Ayad NG, Wan Y, Zhang GJ, Kirschner MW, Kaelin WG., Jr Degradation of the SCF component Skp2 in cell-cycle phase G1 by the anaphase-promoting complex. Nature. 2004;428:194–198. doi: 10.1038/nature02381. [DOI] [PubMed] [Google Scholar]
- Wei Y, Chen YH, Li LY, Lang J, Yeh SP, Shi B, Yang CC, Yang JY, Lin CY, Lai CC, et al. CDK1-dependent phosphorylation of EZH2 suppresses methylation of H3K27 and promotes osteogenic differentiation of human mesenchymal stem cells. Nature cell biology. 2011;13:87–94. doi: 10.1038/ncb2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkins BJ, Rall NA, Ostwal Y, Kruitwagen T, Hiragami-Hamada K, Winkler M, Barral Y, Fischle W, Neumann H. A cascade of histone modifications induces chromatin condensation in mitosis. Science. 2014;343:77–80. doi: 10.1126/science.1244508. [DOI] [PubMed] [Google Scholar]
- Xu K, Wu ZJ, Groner AC, He HH, Cai C, Lis RT, Wu X, Stack EC, Loda M, Liu T, et al. EZH2 oncogenic activity in castration-resistant prostate cancer cells is Polycomb-independent. Science. 2012;338:1465–1469. doi: 10.1126/science.1227604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zadra G, Batista JL, Loda M. Dissecting the Dual Role of AMPK in Cancer: From Experimental to Human Studies. Molecular cancer research : MCR. 2015;13:1059–1072. doi: 10.1158/1541-7786.MCR-15-0068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang CS, Hawley SA, Zong Y, Li M, Wang Z, Gray A, Ma T, Cui J, Feng JW, Zhu M, et al. Fructose-1,6-bisphosphate and aldolase mediate glucose sensing by AMPK. Nature. 2017;548:112–116. doi: 10.1038/nature23275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108:1167–1174. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nature reviews Molecular cell biology. 2011;12:21–35. doi: 10.1038/nrm3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.