

Abstract
Zebrafish is a model organism that affords experimental advantages toward investigating the normal function of genes associated with congenital birth defects. Here we summarize zebrafish studies of genes implicated in orofacial cleft (OFC). The most common use of zebrafish in this context has been to explore the normal function an OFC-associated gene product in craniofacial morphogenesis by inhibiting expression of its zebrafish ortholog. The most frequently deployed method has been to inject embryos with antisense morpholino oligonucleotides targeting the desired transcript. However improvements in targeted mutagenesis strategies have led to widespread adoption of CRISPR/Cas9 technology. A second application of zebrafish has been for functional assays of gene variants found in OFC patients; such in vivo assays are valuable because the success of in silico methods for testing allele severity has been mixed. Finally, zebrafish have been used to test the tissue specificity of enhancers that harbor single nucleotide polymorphisms (SNPs) associated with risk for OFC. We review examples of each of these approaches in the context of genes that are implicated in syndromic and non-syndromic OFC.
Introduction
Orofacial cleft (OFC) malformations – including primarily cleft lip, cleft palate, or a combination of the two – are among the most common structural birth defects (Dixon et al., 2011). About 33% of OFC cases occur in the context of a syndrome, and familial inheritance usually follows a straightforward pattern. Over 100 syndromes include OFC among physical symptoms (Online Inheritance in Man, OMIM). The remaining cases are non-syndromic, meaning that OFC is unaccompanied by obvious malformations in other organs. The incidence of non-syndromic OFC is about 0.1% in the US population overall, but rises to 15–17% in children with an affected family member, and concordance is 50% between identical twins (Grosen et al., 2011; inheritance patterns and risk factors for OFC reviewed in Beaty et al., 2016). These observations point to OFC pathogenesis having both environmental and genetic contributions. The inheritance pattern of non-syndromic OFC is complex because the risk is influenced by alleles of multiple genes.
To determine how mutations in particular genes lead to syndromic OFC or influence risk for non-syndromic OFC, developmental biologists have traditionally favored the mouse model for reverse genetics studies, because of the clear similarity of murine and human craniofacial anatomy. However, palate formation occurs in a stage of mouse development that is inaccessible to the observer. Further, while the majority of murine models of OFC involve the secondary palate, in humans clefts involving the lip and palate are more common than isolated cleft palate (Bush and Jiang, 2012). In mammals the palate is formed from medial outgrowths of maxillary prominences which elevate and fuse above the tongue. By contrast, in zebrafish the roof of the oropharynx is the ethmoid plate which is formed by convergence of frontonasal prominences with the maxillary prominences; posteriorly the ethmoid plate joins the neurocranium, which forms the skull base (Wada et al., 2005; Eberhart et al., 2006; Dougherty et al., 2012). Despite these differences, an array of genes involved in the murine and human palatogenic program is expressed in analogous craniofacial structures in zebrafish embryos between 36–48 hours post fertilization (hpf) suggesting this program is conserved between fish and mammals (Swartz et al., 2011). Hereafter we refer to the zebrafish ethmoid plate as the palate. Experimental advantages of the zebrafish as a model organism include its external development and facility of gene manipulation. Moreover, in transgenic animals cranial neural crest cells can be readily labeled with photo-convertible dyes, which is of particular use in the study craniofacial development as most of the bones in the head are derived from neural crest (McGurk et al., 2014; Rochard et al., 2015). The experimental advantages of zebrafish and the need for higher throughput models have increasingly led researchers to employ this model to: explore the developmental functions of genes implicated in syndromic and non-syndromic forms of OFC; evaluate the functions of rare variants found in patients; and evaluate the tissue specificity of enhancers harboring non-coding variants associated with non-syndromic OFC.
I. Use of zebrafish to explore the developmental functions of genes mutated in patients with OFC
A variety of methods are used for loss-of-function studies in zebrafish. Many large-scale forward mutagenesis screens have been performed in zebrafish (Driever et al., 1996; Haffter and Nusslein-Volhard, 1996; Amsterdam et al., 1999), and in some cases the affected genes are orthologs of OFC-associated genes (e.g., lockjaw is a mutation in the ortholog of TFAP2A, in which mutations cause branchio-occulo-facial syndrome (Knight et al., 2003; Milunsky et al., 2008)). However more frequently reverse genetics methods have been deployed. To induce loss-of-function (LOF) of genes implicated in OFC, injection of antisense morpholino oligonucleotide (MOs) has been the method of choice in the zebrafish community for many years. However, MOs can lead to non-specific phenotypes by two mechanisms. First, MOs may hybridize to unintended transcripts with fortuitous sequence similarity (i.e., off-target effect), although few such events have been carefully documented. Second, many MOs non-specifically induce expression of p53, resulting in widespread cell death in the brain that is partially rescued by concomitant injection of p53 MO (Robu et al., 2007). The p53-dependent phenotype includes characteristic craniofacial features of hypoplastic Meckel’s cartilage, a medially or posteriorly pointing ceratohyal cartilages, and strongly diminished or absent ceratobranchial cartilages (see Table 1 for a schematic of cartilaginous elements in the larval zebrafish head) (Robu et al., 2007). However it is important to note that a grossly similarly phenotype was observed in 25% of 315 insertional mutants systematically screened with Alcian blue; these mutations affected a diverse set of genes (R. Nissen, personal communication)(Amsterdam et al., 1999). This means that instances of this craniofacial phenotype induced by MOs should not be presumed to be non-specific, provided adequate controls for specificity have been performed (reviewed in Eisen and Smith, 2008). Further, in the case of sox9a, the morphant and a LOF mutant – derived from a forward screen in this case – both exhibit this phenotype (Yan et al., 2002). Many of the zebrafish models of OFC discussed below exhibit a phenotype grossly matching this pattern (see Table 1). Because of the unfortunate similarity of this phenotype to one that is a common artifact, for each of the models created with MO, an important goal is to generate a corresponding LOF mutant to confirm that the phenotype is specific.
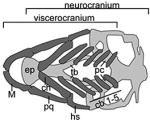
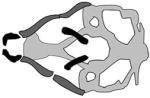
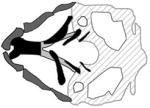
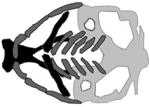
TABLE 1. Zebrafish models of Syndromic and Non-Syndromic Orofacial Cleft.
A table summarizing the phenotypic spectrum of craniofacial cartilage defects in select zebrafish orofacial clefting models. Figures are the author’s interpretations of cartilage structures highlighted from Alcian blue staining performed on specific models between 4–6 dpf and are simplified to exclude smaller cartilages and the notochord. Defective craniofacial cartilages are highlighted in black to distinguish them from otherwise normal viscerocranium (dark grey) and neurocranium (light grey) elements. Craniofacial cartilages with unknown or undocumented structures are represented with translucent placeholders with wild-type structure.
| Human Disease / Gene / Model |
Craniofacial Phenotype | Affected Cartilages | References |
|---|---|---|---|
| N/A |
|
|
|
| Wild-Type | |||
|
| |||
| Bamforth-Lazarus syndrome |
|
|
(Nakada et al., 2009) |
| FOXE1, foxe1 | |||
| morpholino | |||
|
| |||
| Bosma Arhinia Micropthalmia syndrome |
|
|
(Shaw et al., 2017) |
| SMDCH1, smdch1 | |||
| crispr/Cas9; morpholino | |||
|
| |||
| Carnevale, Mingarelli, Malpuech and Michels syndromes (3MC) |
|
|
(Rooryck et al., 2011) |
| COLEC1, colec1 MASP1, masp1 | |||
| morpholino | |||
|
| |||
| Circumferential Skin Creases, Kunz Type |

|
|
(Isrie et al., 2015) |
| MAPRE2, mapre2 | |||
| Morpholino | |||
|
| |||
| Kabuki syndrome Type 1 |

|
|
(Van Laarhoven et al., 2015) |
| KMT2D, kmt2d | |||
| morpholino | |||
|
| |||
| Kabuki syndrome Type 2 |
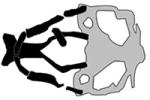
|
|
(Lindgren et al., 2013; Van Laarhoven et al., 2015) |
| KDM6A, kdm6a | |||
| morpholino | |||
|
| |||
| Oblique Cleft |
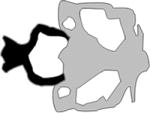
|
|
(Gfrerer et al., 2014) |
| SPECC1L, specc1lb | |||
| morpholino | |||
|
| |||
| Treacher Collins syndrome Type 1 |
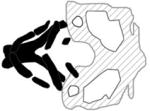
|
|
(Weiner et al., 2012) |
| TCOF1, tcof1 | |||
| morphant | |||
|
| |||
| Treacher Collins syndrome Type 2 |
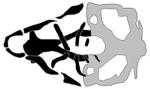
|
|
(Noack Watt et al., 2016) |
| POLR1D, polr1d | |||
| polr1d−/−mutant | |||
|
| |||
| Treacher Collins Syndrome Type 3 |
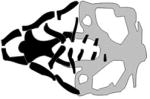
|
|
(Lau et al., 2016; Noack Watt et al., 2016) |
| POLR1C, polr1c | |||
| morpholino, polr1c−/− mutant | |||
|
| |||
| Van der Woude Syndrome Type 1 |
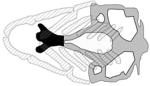
|
|
(Dougherty et al., 2013) |
| IRF6, irf6 | |||
| sox10:dnirf6 | |||
|
| |||
| Van der Woude Syndrome Type 2 |
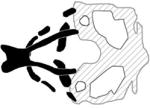
|
|
(Dworkin et al., 2014) |
| GRHL3, grhl3 | |||
| morpholino | |||
|
| |||
| Non-syndromic OFC |
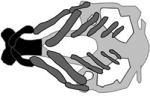
|
|
(Mukherjee et al., 2016) |
| CAPZB, capzb | |||
| capzb−/−mutant | |||
|
|
|||
| CRISPLD2, crispld2 |

|
|
(Yuan et al., 2012) |
| morpholino | |||
|
|
|||
| DLX4, dlx4b |
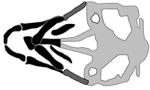
|
|
(Wu et al., 2015) |
| morpholino | |||
|
|
|||
| FAF1, faf1 |

|
|
(Ghassibe-Sabbagh et al., 2011) |
| morpholino | |||
|
|
|||
| FZD6, fzd6 |
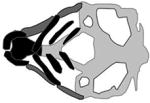
|
|
(Cvjetkovic et al., 2015) |
| morpholino | |||
|
|
|||
| HDAC4, hdac4 |
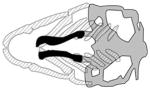
|
|
(DeLaurier et al., 2012) |
| morpholino | |||
|
|
|||
| MIR-140, mir-140 |
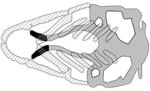
|
|
(Eberhart et al., 2008) |
| duplex | |||
|
|
|||
| PDGFRA, pdgfra |
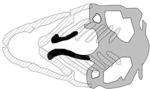
|
|
(Eberhart et al., 2008; McCarthy et al., 2016) |
| mutant | |||
|
|
|||
| WNT9A, wnt9a |

|
|
(Curtin et al., 2011) |
| morpholino | |||
Note, bold font indicates the specific model reviewed in table.
Abbreviations: dn, dominant-negative, N/A, not applicable; N/D, not determined.
As CRISPR/Cas approaches becomes widely available to zebrafish researchers, homozygous LOF mutants can be generated within an acceptable time frame (6 months to a year). Because off-target effects of well-designed CRISPRs are minimal, the confidence that the phenotype present in a LOF mutant is specific to the targeted gene is higher than in the corresponding morphant (Hruscha et al., 2013). However, when there is no phenotype in a LOF mutant generated by CRISPR/Cas9 or other methods, it is important to establish that mRNA, or ideally, protein levels of the targeted gene have been diminished. Second, an important caveat when interpreting the absence of a phenotype in a targeted LOF mutants is that in such mutants there can be compensatory upregulation of expression of paralogous genes, or genes in the same regulatory pathway (Rossi et al., 2015; El-Brolosy and Stainier, 2017). It is not clear how frequent such compensation is, or how frequently it takes each of these two forms. Because such MOs do not induce this compensation, the phenotype of a morphant is often more penetrant than in the corresponding LOF mutant. Careful consideration of all of these issues is essential when interpreting phenotypes in zebrafish LOF models.
Below we summarize some of the published studies that have used zebrafish to examine the roles of genes relevant to human OFC syndromes, using CRISPR/Cas-mediated knockouts, MO-mediated knockdown, and available mutants from large-scale screens. A schematic representation of many of the phenotypes is summarized in Table 1.
A. Syndromic OFC
Bamforth-Lazarus syndrome
Individuals with Bamforth-Lazarus syndrome present with congenital hypothyroidism, spiky hair, and cleft palate (Clifton-Bligh et al., 1998). This syndrome is caused by mutations in the gene that encodes forkhead box E1 (FOXE1) (Clifton-Bligh et al., 1998; Castanet et al., 2002). Members of the FOX family of transcription factors share a highly conserved DNA binding domain, and many have been shown to contribute to embryonic development (Golson and Kaestner, 2016). During embryonic development in zebrafish, the foxe1 transcript is first detected at 12 hours post fertilization (hpf); it is strongly expressed in the oral epithelium by 48 hpf, and in pharyngeal epithelium by 72 hpf (Nakada et al., 2009; Lidral et al., 2015). The injection of MOs targeting the foxe1 start codon (i.e., foxe1 morphants), but not control MOs (control morphants), results in smaller heads, aberrant tail curvature and shortened Meckel’s cartilage, truncated and inverted ceratohyals, and reduced ceratobranchial elements at 5 days post fertilization (dpf) (Nakada et al., 2009). Thus FOXE1 appears to function within oral epithelium, and is necessary for proper development of the underlying cranial neural crest cells (CNCCs).
Bosma arhinia microphthalmia syndrome
Bosma arhinia microphthalmia syndrome (BAMS) is rare congenital disorder whose clinical features include severe hypoplasia or absence of the external nose, often accompanied by eye defects and cleft palate (Bosma et al., 1981; Graham and Lee, 2006). Recently a combination of sequencing approaches (whole-exome, targeted, whole-genome) performed on individuals with BAMS and members of their families revealed a significant mutation burden in patients within a gene called “structural maintenance of chromosomes flexible hinge domain-containing 1” (SMCHD1) (Gordon et al., 2017; Shaw et al., 2017). Mutations in this gene have also been associated with facioscapulohumeral muscular dystrophy-2 (FSHD2) (Lemmers et al., 2012). Interestingly, the SMCHD1 mutations associated with BAMS occur within the portion of the gene that encodes an amino-terminal ATPase domain, whereas those associated with FSHD2 are distributed along the length of the gene (Shaw et al., 2017). Zebrafish smchd1 morphants have aberrations in craniofacial cartilage including a smaller-than-normal palate, a widened angle of the ceratohyal arch, a reduction in the number of ceratobranchial cartilages, and small eyes (Shaw et al., 2017). Each of these phenotypes was also observed in CRISPR/Cas9 smchd1 F0 mutants (Shaw et al., 2017), adding confidence that they result specifically from reduced expression of smchd1 as opposed to being off-target effects of the MOs.
Carnevale, Mingarelli, Malpuech and Michels syndromes
Carnevale, Mingarelli, Malpuech and Michels syndromes were discovered to be allelic and are now called 3MC syndrome (Titomanlio et al., 2005). Among other clinical manifestations, patients diagnosed with these syndromes present with dysmorphic craniofacial features including highly arched eyebrows, ptosis, hypertelorism and cleft lip or palate (Michels et al., 1978; Malpuech et al., 1983; Carnevale et al., 1989; Mingarelli et al., 1996). Sequence analyses of DNA from multiple families of various ethnic backgrounds identified mutations in genes collectin-11 (COLEC11) and mannose-binding lectin-associated serine protease 1 (MASP1), each of which encodes a component of the lectin complement pathway (Rooryck et al., 2011). However, mutations in the COLEC11 and MASP1 genes have not been found in other 3MC families, implying that an additional layer of genetic heterogeneity exists (Urquhart et al., 2016). In zebrafish embryos colec11 is expressed in the pronephric duct, glomeruli and cranial paraxial mesendoderm (Rooryck et al., 2011), and both colec11 morphants and colec11/masp1 double morphants exhibit cleft palate. The angles of the ceratohyals are widened, and both the width and length of the palate are shorter than in control morphants. Moreover, in colec11 and masp1 morphants CNCCs are defective for migration, and in vivo migration experiments involving the implantation of beads soaked with CL-K1 (i.e., the protein encoded by colec11) into zebrafish suggest that CL-K1 is a chemoattractant for CNCCs (Rooryck et al., 2011). These results indicate that the lectin complement pathway is involved in CNCC function, and point to the cellular basis of the phenotypes in 3MC patients.
Circumferential skin creases Kunze type
Multiple, constricting skin creases along the circumference of limbs is seen in patients with circumferential skin creases Kunze type (CSC-KT) syndrome, previously known as “Michelin tire baby” syndrome (Ross, 1969). Associated comorbidities include intellectual disability, cleft palate and other facial dysmorphisms (Kunze and Riehm, 1982; Wouters et al., 2011). The skin creases in CSC-KT generally resolve with age, but the associated craniofacial dysmorphisms persist. CSC-KT syndrome has been linked to mutations in the microtubule-associated protein RP/EB family member 2 (MAPRE2) and tubulin beta class I (TUBB) genes, both of which encode proteins that regulate microtubule turnover (Isrie et al., 2015). Attempts to generate a zebrafish model for CSC-KT have focused on knocking down the mapre2 gene; a definitive TUBB ortholog in the zebrafish genome is yet to be identified (Isrie et al., 2015). Embryos injected with an antisense MO targeting mapre2 exhibit aberrant broadening of the ceratohyal arches by 2 dpf, as well as hypoplastic ceratobranchial arches (Isrie et al., 2015). This work highlights the role of MAPRE2 in craniofacial development and supports the notion that mutations in this gene cause facial dysmorphisms, including cleft palate, in patients with CSC-KT.
Kabuki syndrome
Kabuki syndrome is characterized by multisystem abnormalities including heart defects, skeletal and dental abnormalities, urinary tract defects, hearing loss, structural brain anomalies and craniofacial anomalies (Bogershausen and Wollnik, 2013; Miyake et al., 2013) (Van Laarhoven et al., 2015). A set of patients with Kabuki syndrome were found to have mutation in KDM6A (Lederer et al., 2012), which encodes the demethylase that removes repressive trimethylation of histone H3 at lysine 27 (H3K27me3) thereby establishing transcriptionally permissive state. Zebrafish had two kmd6a paralogs, kmd6a and kmd6al. At 24 hpf kmd6a expression in the head is found in the brain, otic placode and pharyngeal arches. By 96 hpf the kmd6a transcript was restricted to pharyngeal arches and craniofacial cartilages (Lindgren et al., 2013; Van Laarhoven et al., 2015). kmd6a morphants had hypoplastic Meckel’s, palatoquadrate, ceratohyal, and ceratobranchial cartilages, and the ethmoid plate was shorter than normal. These phenotypes were rescued by concomitant injection of human KMD6A mRNA. (Lindgren et al., 2013; Van Laarhoven et al., 2015). Of note the brain was also smaller than normal in kdma6a morphants, so Kdm6a may have indirect roles in craniofacial development. Knockdown of second kdm6a paralog, kdm6al, was reported to yield no phenotype (Van Laarhoven et al., 2015).
A subset of Kabuki patients have mutations in KMT2D encoding a histone methyltransferase that is responsible for tri-methylation of histone H3 at lysine 4 (H3K4me3), an epigenetic mark characteristic of active promoters. Zebrafish kmt2d was reported to be expressed globally, at least up until 24 hpf (Thisse et al., 2001; Van Laarhoven et al., 2015). kmt2d morphants exhibited severe hypoplasia of the viscerocranium, including loss of pharyngeal arches 3–7, Meckel’s cartilage and the ceratohyals. Most kmt2d morphants exhibited a slight shortening of the palate and trabeculae (Van Laarhoven et al., 2015). These genes exemplify how mutations in genes with universal or widespread function can result in craniofacial phenotypes, in humans and in zebrafish.
Oblique facial cleft syndrome
Oblique facial cleft syndrome refers to a rare class of orofacial clefts that extend along either the oro-ocular or naso-ocular axis (Eppley et al., 2005). In 2011 mutations in SPECC1L were identified in two patients with oblique facial clefts (Saadi et al., 2011). They were later also found in patients with Opitz G/BBB syndrome, the letters referring to the initials of patients first diagnosed with it, which is associated with cleft lip and/or palate (Kruszka et al., 2015), and in a family with Teebi hypertelorism syndrome, which is characterized by the presence of frontonasal dysplasia (Bhoj et al., 2015; Wilson et al., 2016). SPECC1L is a cytoskeletal protein that interacts with both microtubules and the actin cytoskeleton. Initial zebrafish studies involving MO-mediated knockdown of specc1, one of the three SPECC1L paralogs (specc1, specc1la, and specc1lb) resulted in a shortening of Meckel’s and ceratohyal cartilage at low MO concentration, and in a complete loss of facial structures at high MO concentration (Saadi et al., 2011). Analysis of the other two paralogs, specc1la and specc1lb, revealed that both are expressed in the oropharyngeal epithelia, and that although specc1la morphants do not have a craniofacial phenotype, specc1lb morphants exhibit late embryonic craniofacial phenotypes including bilateral clefts and an absent mandible (Gfrerer et al., 2014). In specc1lb morphants, CNCCs contributing to the frontonasal prominence failed to integrate with those contributing to the maxillary prominences, and those contributing to lower jaw structures migrated to their correct locations within the pharyngeal segments but failed to converge to form mandibular elements (Gfrerer et al., 2014). These results demonstrate that specc1lb is required for the integration of frontonasal and maxillary prominences, and for the convergence of mandibular prominences. These studies implicate mutations in SPECC1L as the genetic basis of oblique facial clefts, and defects in cell migration as a potential cause of the pathogenesis that leads to oblique facial clefts.
Treacher Collins syndrome
Treacher Collins syndrome (TCS) is a rare congenital disorder that affects derivatives of the first and second branchial arches. Phenotypic presentation varies widely, but TCS can include high or cleft palate, and loss of conductive hearing, among other craniofacial malformations (Dixon, 1996). The genetic etiology of TCS types 1, 2, and 3 can be accounted for by mutations in genes that regulate ribosome biogenesis (Gladwin et al., 1996; Dauwerse et al., 2011). The encoded proteins are for TCS-1, treacle ribosome biogenesis factor 1 (TCOF1), for TCS-2, RNA polymerase I subunit D (POLR1D) and for TCS-3, RNA polymerase I subunit C (POLR1C). Mutations in TCOF1 account for the majority (78%–93%) of TCS cases (Kadakia et al., 2014). The first zebrafish model of TCS was generated by MO-mediated knockdown of zebrafish ortholog tcof1, which is expressed broadly in early embryo stages. tcof1 morphants displayed reduced Meckel’s and ceratobranchial cartilages, widening of the ceratohyal arch angles, and palate retraction resulting in gross frontonasal hypoplasia likely due to an observed marked reduction in pre-migratory and migratory NCC (Weiner et al., 2012). The link between ribosome biogenesis and NCC differentiation remains unclear, but in mouse Tcof1 mutants there is p53-mediated apoptosis in neuroepithelium and loss of NCC (Dixon et al., 2006; Jones et al., 2008). Further evidence for a connection between ribosome biogenesis, the p53 pathway, and craniofacial development comes from the zebrafish fantome mutant which harbors a point mutation leading to truncation of the carboxy terminal domain of a ribosomal biogenesis protein called Wdr43 (Zhao et al., 2014). The phenotype of fantome mutant closely resembles that of tcof1 morphants, and nucleolar localization of Tcof1 is perturbed in the fantome mutant, and inhibition of p53 partially rescues the craniofacial phenotype (Zhao et al., 2014).
A polr1c morphant, and polr1c and polr1d mutant lines model the rarer types of TCS (Lau et al., 2016; Noack Watt et al., 2016). Like tcof1, the polr1c and polr1d genes are expressed broadly during early embryo stages and becomes restricted to pharyngeal arches by 48 hpf. Moreover, polr1c and polr1d mutants both display significant reductions in Meckel’s and ceratobranchial cartilages, with the most severe phenotypes including inversion of the ceratohyals; these phenotypes are largely rescued by concomitant loss of p53 function (Noack Watt et al., 2016). Diminished domains of dlx2, a marker of mature CNCC along with reduced transcription of ribosomal RNA (rRNA) precursors were observed in polr1c and polr1d mutants (Noack Watt et al., 2016). Collectively, these studies highlight an increasingly apparent role of ribosome biogenesis in craniofacial development, expanding the etiologic mechanisms underlying OFC.
Van der Woude syndrome
Van der Woude syndrome (VWS), a congenital disorder with autosomal-dominant inheritance, is characterized by cleft lip, cleft palate, or both, accompanied by pits in the lower lip (Van Der Woude, 1954). Individuals with popliteal pterygium syndrome (PPS) likewise display such craniofacial features, and in addition have popliteal webbing and genital anomalies (Gorlin et al., 1968). Linkage studies of monozygotic twins discordant for VWS, and in multiple families with one or both syndromes, revealed that VWS and PPS are allelic disorders caused by mutations in interferon regulatory factor 6 (IRF6) (Kondo et al., 2002). IRF6 belongs to a family of 9 structurally related transcription factors that are largely involved in governing the transcriptional program of the innate immune response. In zebrafish, a single irf6 ortholog is expressed maternally (Ben et al., 2005; Sabel et al., 2009). Zygotic expression is first detected in embryonic superficial epithelium or enveloping layer (EVL), which is the origin of embryonic periderm, the most superficial layer of epidermis (Kimmel et al., 1990). At later stages, irf6 is expressed in the otic vesicle, olfactory placodes, epithelium of the stomodeum, the epithelial and mesenchymal domains of pharyngeal arches, and the palate (Ben et al., 2005; Sabel et al., 2009; Dougherty et al., 2013).
Because of high levels of maternal mRNA, MO-based methods to decrease irf6 expression yielded no phenotype; however injection of RNA encoding a dominant negative Irf6 (dnIrf6), i.e., the Irf6 DNA binding domain, either alone or fused to the repressor domain of the Engrailed transcription factor, resulted in stalled epiboly, and rupture through the animal pole at high penetrance. Whole mount in situ hybridization revealed strong reduction of expression of keratin genes and other markers of the EVL (Sabel et al., 2009; de la Garza et al., 2013). In rare surviving embryos, which had presumably been injected with a lower dose of the dnIrf6 construct, the pectoral fins were hypoplastic, skin was blistered and the ceratohyal structures were small (Sabel et al., 2009). These results, which are consistent with findings from the mouse Irf6 loss-of-function mutants, and indicate that pathogenesis of OFC in VWS results, at least in part, from defective differentiation of oral epithelium (Ingraham et al., 2006; Richardson et al., 2006; Richardson et al., 2009). Importantly, however in both zebrafish and mouse, there is immunoreactivity for Irf6 in craniofacial mesenchyme as well as oral epithelium. Moreover, forced expression of dominant negative Irf6 in zebrafish neural crest under the control of the sox10 promoter did not cause early death during epiboly but resulted in a shorter palate, without affecting the viscerocranium (Dougherty et al., 2013). These results from zebrafish indicate that lower-than-normal IRF6 function in neural crest may also contribute to the pathogenesis of OFC in VWS patients that with mutations in this gene.
A minority of VWS patients lack mutations in IRF6 but have them in the GRHL3 gene (Peyrard-Janvid et al., 2014). Transcription factors of the Grainy Head Like (i.e., GRHL1–3) family regulate epithelial morphogenesis during development and wound repair (Wang and Samakovlis, 2012). Interestingly, the expression of grhl3, and that of its paralogs grhl1 and grhl2, is strongly reduced in the EVL of dnIrf6-injected embryos (de la Garza et al., 2013). Simultaneous knockdown of grhl1 and grhl3, but not knockdown of either gene singly, prevents the expression of keratin genes and other EVL markers, and results in embryonic death. Forced over-expression of grhl3 in zebrafish embryos injected with dnIrf6 partially restores the expression of EVL markers, implying a transcriptional hierarchy underlying periderm differentiation (de la Garza et al., 2013), However, the regulatory network relevant to the pathogenesis of cleft palate may be more complicated than this, as no genetic interaction between Irf6 and Grhl3 was detected in mice doubly heterozygous for LOF mutations of the two genes. Further, whereas irf6 acts upstream of a Rho GAP (ARHGAP29) (Biggs et al., 2014), GRHL3 lies upstream of a Rho GEF (RhoGEF19) (Boglev et al., 2011). In a separate study, zebrafish grhl3 morphants exhibited reductions in ceratohyal, Meckel’s and palatoquadrate cartilage, and lacked components of the basihyal and ceratobranchial cartilages. In grhl3 morphants, expression of edn1 in the pharyngeal endoderm was reduced, and forced expression of this gene partially rescued lower jaw morphology and the expression of genes that mark ventral cartilage elements (e.g., hand2) (Dworkin et al., 2014).
The fact that both IRF6 and GRHL3 are implicated in OFC points to a role for oral periderm, the tissue where they are both prominently expressed, for normal palatogenesis. Strongly supporting this role, the destruction of periderm with a cell-autonomous toxin leads to cleft palate in mouse (Richardson et al., 2014). Of note, variation near two other genes expressed in periderm, KRT8 and CDH1, is also associated with risk OFC risk genes (Ittiwut et al., 2016; Leslie et al., 2017b; Song et al., 2017). Genes encoding additional members of the gene regulatory network governing periderm differentiation are candidates to harbor mutations that cause or influence risk OFC (see KLF4 below). It is noteworthy however that IRF6 and GRHL3 are expressed in tissues other than periderm, and both are required for neurulation, so the contribution of mutations in these genes to etiology OFC may be complex (reviewed in Kousa et al., 2017).
Robin sequence
Pierre Robin sequence (PRS) comprises mandibular hypoplasia, cleft secondary palate, and downward displacement of the tongue (glossoptosis), which can lead to obstructive apnea and feeding difficulties (Tan et al., 2013). PRS may occur in isolation or as part of a syndrome, such as Stickler syndrome (Printzlau and Andersen, 2004). Haploinsufficiency of the fas-associated factor 1 (FAF1) gene was found to be the cause of cleft palate in a family with PRS (Ghassibe-Sabbagh et al., 2011). In zebrafish faf1 is expressed in the pharyngeal arch primordia between 24–30 hpf. By 56 hpf, the expression domain extends to all cranial arches, with an accumulation of faf1 transcripts prominent in the mandibular and hyoid arches (Ghassibe-Sabbagh et al., 2011). MO-mediated knockdown results in a smaller head and in cartilage defects, including ventrally positioned Meckel’s cartilage, aberrant broadening of angles between the ceratohyals, an incomplete array of pharyngeal arches and overall reduction in neurocranial cartilage structures. Assessment of the expression of sox9a and col2a1 in faf1 morphants revealed altered patterning in the mesenchymal condensation of the pharyngeal arches compared to controls. This observation suggests that defects in cartilage differentiation within the faf1-depleted CNCC population lead to the observed craniofacial defects.
Orofacial cleft 15 (OMIM #616788)
The distal-less (DLX) proteins are homeo-domain transcription factors involved in several morphogenetic programs including, but not limited to, craniofacial development (Robinson and Mahon, 1994; Depew et al., 2002; Gordon et al., 2010). Prompted by the discovery that the DLX6 gene is disrupted in the Nova Scotia duck-tolling retriever dog breed, which has a PRS-like phenotype, DLX5 and DLX6 were sequenced in patients with cleft palate. A missense mutation within the highly conserved DNA binding domain of DLX5 (p.Ile192Met) was identified in 16 patients affected with PRS but was absent from the 1000 Genomes database, indicating it is novel or very rare (Wolf et al., 2014). Moreover, a breakpoint within a regulatory element distal to DLX5 and DLX6 was identified in a multi-generational family presenting with deafness and craniofacial defects including micrognathia and cleft palate (Brown et al., 2010). A novel truncating variant (p.Gln183Argfs*57) of DLX4 was also identified in a mother-son pair that presented with enlarged palpebral fissure, lagophthalmos and bilateral CL/P (Wu et al., 2015). Together these findings support a role for DLX family members in the etiology of CL/P. In zebrafish the two paralogs dlx4a and dlx4b, with respective amino acid sequence similarities of 43% and 37% to human DLX4, are expressed in the anterior and posterior arches of the oral cavity at 72 hpf (Thisse and Thisse, 2004). MO-mediated knockdown of dlx4b results in defective anterior facial protrusion, a decrease in the width of Meckel’s cartilage and an aberrant ceratohyal cartilage angle (Wu et al., 2015). Knockdown of the dlx4a paralog failed to produce a phenotype, implying sub-functionalization of the two paralogs. Similarly, overexpression of either WT or mutant DLX4 mRNA failed to generate a phenotype, ruling out a gain-of-function in the DLX4 truncated product. In summary, this work documents the first findings of DLX4 as OFC relevant gene with support of a craniofacial phenotype in zebrafish dlx4b morphants.
SATB2-associated syndrome
High-resolution FISH mapping of two de novo translocations associated with OFC at the 2q32–q33 locus identified breakpoints – one within an intron of, and the other distal to – the gene encoding special AT-rich sequence binding protein 2 (SATB2)(FitzPatrick et al., 2003). These individuals and others with genetic aberrations in the SATB2 locus exhibit intellectual disability, cleft palate, micrognathia and dental anomalies (FitzPatrick et al., 2003; Leoyklang et al., 2007; Rainger et al., 2014; Zarate et al., 2015). It was recently proposed that all LOF mutations in this gene be classified as SATB2-associated syndrome (SAS)(Docker et al., 2014). SATB2 is a chromatin modifier or transcription factor implicated in osteoblast differentiation (Dobreva et al., 2006). Two studies reported satb2 is highly expressed in the pharyngeal arches 48 hpf (Ahn et al., 2010; Sheehan-Rooney et al., 2010), and one that it can be detected as early as the single cell stage (Ahn et al., 2010). Consistent with this early expression, embryos injected with a satb2 MO stalled during epiboly, adopted an elongated ellipsoidal shape, and died by 13 hpf (Ahn et al., 2010). Epiboly defects in satb2-MO injected larvae were accompanied by an abnormal actin cytoskeleton and aberrant expression of markers of mesoderm (ntl) and neural plate (sox3), interpreted to reflect abnormal cellular migration (Ahn et al., 2010). Like embryos injected with dnIrf6, satb2 morphants died during gastrulation (Ahn et al., 2010). These zebrafish studies demonstrate that SATB2 contributes to cell migration and morphogenesis well before development of the face. An interesting area for research in the future is to see how similar are the cellular underpinnings of the grossly similar phenotypes of embryos depleted of irf6 and those depleted of satb2.
CAPZB-associated syndrome
Patients with mutations in CAPZB have an array of craniofacial defects including cleft palate, micrognathia and muscle defects (Mukherjee et al., 2016). CAPZB regulates the length of actin filaments by capping their growing ends (dos Remedios et al., 2003; Cooper and Sept, 2008). It is highly expressed in pharyngeal arch 1, an embryonic structure through which cells migrate on their way to becoming palate and lower jaw (Cai et al., 2005). In zebrafish, maternal capzb is deposited and zygotic capzb is expressed ubiquitously, with strong expression throughout the craniofacial region and in somites (Mukherjee et al., 2016). Zebrafish capzb LOF mutants exhibit a cleft palate, hypoplastic lower jaw and disorganized muscles, phenotypes that mimic those in humans with LOF mutations in CAPZB. Cell sorting and expression analyses of zebrafish capzb mutants have revealed that the expression of pax3a and pax7a is abnormally upregulated in neural crest cells, and lineage tracing studies have revealed that migration of the frontonasal stream of CNCCs is defective (Mukherjee et al., 2016). These experiments point to cellular underpinnings of OFC in patients with CAPZB mutations.
B. Non-syndromic OFC
CRISPLD2
Several genome-wide scans identified 16q24.1 as a locus that harbors variation associated with risk for non-syndromic OFC (Wyszynski et al., 2003; Field et al., 2004; Marazita et al., 2004a). Targeted sequencing of genes in this region revealed significant association of variants within the gene cysteine-rich secretory protein LCCL domain containing 2 (CRISPLD2, also known as lethal giant larvae homolog 1, LGL1) with this disease in both Caucasian and Hispanic populations (Chiquet et al., 2007). SNPs near CRISPLD2 were also found to be associated with non-syndromic OFC in Chinese populations (Shi et al., 2010; Shen et al., 2011). CRISPLD2 is a glucocorticoid-inducible secreted protein that was originally identified as an important regulator of fetal rat lung development (Kaplan et al., 1999). During zebrafish development, crispld2 is expressed maternally and between 1 dpf and 5 dpf in unidentified tissue or tissues in the craniofacial region (Yuan et al., 2012). A splice-blocking MO, which would be expected to reduce levels of zygotic but not maternal crispld2, induced aberrant widening of ceratobranchials, malformed Meckel’s and palatoquadrate cartilages and cleft in the palate at 5 dpf. A translation blocking MO, which may block translation from maternal and zygotic transcripts, induced complete loss of lower jaw cartilage, and a truncation of the ethmoid plate and trabeculae at 5 dpf (Yuan et al., 2012). Knockdown of crispld2 resulted in elevated cell death in the nervous system and elsewhere that was diminished but not eliminated by concomitant knockdown of p53 (Yuan et al., 2012). It also caused aberrant migration of neural crest cells across the midline (Swindell et al., 2015) and larger-than-normal patches of dlx2a and wnt5a expression in the pharyngeal arches, despite elevated cell death in the neural crest (Yuan et al., 2012). These findings from zebrafish point to a role for crispld2 in migration and possibly survival of NCC; such a role would be harder to study in mouse embryos, where loss of Crispld2 is embryonic lethal (Lan et al., 2009).
FZD6
Meta-analysis of genome-wide scans of families with non-syndromic OFC revealed disease-associated variation in a region of 8q23 encompassing the gene FZD6 (Marazita et al., 2004b). This gene encodes frizzled-6, a planar cell polarity protein that contributes to initiation of non-canonical WNT signaling which is involved in craniofacial development (MacDonald et al., 2009). It also controls the polarity and orientation of migrating CNCCs (Devenport and Fuchs, 2008). In a study by Cvjetkovic et al., a rare intronic variant of FZD6 segregated with affected members in an African-American family (Cvjetkovic et al., 2015). In zebrafish, fzd6 is expressed within the phargyngeal arches, pectoral fin buds and throughout the head between 2–4 dpf. fzd6 morphants exhibited hypoplastic Meckel’s cartilage and ceratohyals, as well as a shorter palate (Sisson and Topczewski, 2009). Further, transient overexpression of fzd6 caused abnormalities in Meckel’s cartilage and the ceratohyals, and loss of the palate. These observations from the zebrafish model support the hypothesis that the FZD6 is the gene in 8q23 that is relevant to etiology of OFC.
HDAC4
Single-marker and haplotype transmission disequilibrium tests performed in 58 non-syndromic OFC case-parent trios revealed preferential transmission to cases of one allele of the SNP rs2121980, near the HDAC4 gene, which encodes histone deacetylase 4 (Park et al., 2006). Loss of HDAC4 function is the cause of brachydactyly mental retardation syndrome (BDMR), which is associated with multiple craniofacial abnormalities (Williams et al., 2010). Although HDAC4 plays a critical role in transcriptional regulation by repressing gene activity (Wang et al., 2014), the mechanism by which loss of HDAC4 causes cleft palate remains unknown. In zebrafish hdac4 is expressed in migrating CNCCs, and later in pharyngeal arches and the palate. Zebrafish hdac4 morphants exhibit distinctive facial shortening at 7 dpf, a short and narrow ethmoid plate and reduced or absent trabeculae (DeLaurier et al., 2012). In these animals, pre-migratory and migratory CNCC populations that normally migrate medial to the eye as palate precursors were lost, either partly or completely, leading to the cleft phenotype. Transient overexpression of hdac4 resulted in midline defects in the craniofacial skeleton, and in cyclopia (DeLaurier et al., 2012). This study reveals that HDAC4 regulates CNCCs and craniofacial morphogenesis and supports HDAC4 as the gene relevant to cleft that is affected, presumably at the level of gene expression, by the allele of rs2121980.
miR-140/PDGFR
Case-control analysis of non-syndromic OFC patients in western China identified a high frequency of the minor allele of the rs7205289 SNP located within the microRNA miR-140 (Li et al., 2010). MicroRNAs (miRNAs) are small, noncoding RNAs that regulate gene expression post-transcriptionally (Lai, 2002; Lewis et al., 2003). Reduction of all miRNAs in neural crest cells (NCCs), through Wnt1-Cre mediated deletion of Dicer, leads to widespread NCC apoptosis and a profound reduction of jaw elements (Huang et al., 2010; Zehir et al., 2010). The minor allele of rs7205289 was shown to adversely affect miR-140 processing in HEK293 cells, and to inhibit the expression of miR-140 in mouse palatal mesenchymal cells (Ghassibe-Sabbagh et al., 2011). In zebrafish mir140 is expressed in the pharyngeal cartilage and fins (Wienholds et al., 2005). Embryos injected with the mir140 duplex exhibit cranial hemorrhage by 2 dpf, shortened ethmoid plate or, in more severe cases, failure of the trabeculae to extend anteriorly (Eberhart et al., 2008). Binding sites for mir140 are present in the 3’ UTR of pdgfra. In mouse, Pdgfra is required during early embryogenesis for the development of NCCs (Soriano, 1997) (Xu et al., 2005). In zebrafish, pdgfra is expressed maternally, and in NCCs, lens placode, head mesenchyme, pharyngeal arches and pectoral fin buds by 48 to 72 hpf (Thisse and Thisse, 2008). Injection of the mir140 duplex reduced levels of the Pdgfra protein, and consequently also Pdgf signaling (Eberhart et al., 2008). Low Pdgf signaling, in turn, inhibits NCC migration, suggesting that mir140 affects palatogenesis by inhibiting Pdgf signaling (Eberhart et al., 2008). pdgfrb is also expressed in the NCCs, and whereas pdgfrb mutants do not exhibit overt craniofacial defects, double pdgfra;pdgfrb mutants have a more severe craniofacial phenotype than pdgfra single mutants (McCarthy et al., 2016).
Based on craniofacial defects in zebrafish and mouse Pdgrfa LOF mutants, targeted PCR-screening of PDGFRA was carried out in 102 unrelated Thai individuals with isolated cleft palate only (CPO) and 500 ethnic matched controls (Rattanasopha et al., 2012). Seven variants appeared to be enriched in cases versus controls. Among these, two variants p.Val544Ala and p. Thr1052Met, were absent from the control group and were predicted to be ‘possibly damaging’ using the PolyPhen algorithm (Rattanasopha et al., 2012). Interestingly, a variant affecting the PDGFRA 3’UTR, which was present in one case and absent from controls, lies within the mir140 binding site and reduces sensitivity of the PDGFRA transcript to regulation by mir140 in reporter assays (comparison was to the reference variant) (Rattanasopha et al., 2012). This is an example of evidence from model systems leading to evaluation of particular genes for the presence of variants associated with OFC risk.
WNT9A and WNT9B
In an analysis of 132 multiplex non-syndromic OFC families and 354 simplex parent-child trios, specific alleles of SNPs near WNT3A, WNT5A, and WNT11, but not those near WNT3, WNT7A, WNT8A, or WNT9B, were associated with non-syndromic OFC (Chiquet et al., 2008). However, a recent large-scale genome-wide association study (GWAS) with replication studies, on a total of 7,404 cases and 16,059 controls from a Chinese population, identified non-syndromic OFC-associated SNPs within WNT9B (Yu et al., 2017). In mouse, Wnt9b is expressed in medial-edge palate epithelium and is required for palate fusion (Juriloff et al., 2006; Lan et al., 2006). In zebrafish embryos, wnt9a, the ortholog of human WNT9A based on shared synteny (Cox et al., 2010), but not wnt9b, the ortholog of WNT9B, is expressed in oral epithelium, implying distinct sub-functionalization of the WNT9 paralogs in the different evolutionary clades (Cox et al., 2010; Curtin et al., 2011). Both paralogs are also expressed in pharyngeal cartilages, with the expression of wnt9a initiating earlier. In zebrafish wnt9a morphants, lower jaw structures fail to form (Curtin et al., 2011). Subsequent evaluation revealed that chondrocytes in wnt9a mutants failed to adopt a normal stacking pattern, and proliferation was reduced (Dougherty et al., 2013); evaluation of wnt9a is required in the extension of chondrocytes in the transverse axis of the palate (Rochard et al., 2016). wnt9 is expressed in the first and second pharyngeal arches at 48 hpf (Jezewski et al., 2008). One group found that the wnt9b MO did not disrupt development (Curtin et al., 2011), whereas a second group observed widespread reduction of craniofacial cartilage, preceded by expanded expression of frzb and pitx2 (Jackson et al., 2015). The discordant morphant phenotypes observed for wnt9b may be due to non-specific effects or genetic background of the particular strain utilized in these different experiments; they emphasize the importance of creating mutants to rule out the possibility of simple technical differences in the use of the MO in different laboratories. These studies support that in zebrafish, wnt9 activity (mediated by wnt9a and possibly wnt9b) is required for jaw formation in fish as in mammals. Thus, the highly accessible zebrafish embryo has provided insight into the specific cellular role for this activity.
II. Use of zebrafish for functional analyses of patient-derived variants
Whole-exome or gene-targeted sequencing of patients with non-syndromic OFC has led to the discovery of rare coding variants of genes that are mutated in syndromic forms of OFC. In silico programs like PolyPhen-2, SIFT, and Mutation Taster, which are based on evolutionary conservation, sometimes combined with molecular modeling, are routinely used to predict whether variants are likely to be deleterious; however the correlation of results from different programs is poor, as is the ability to predict clinically benign variants (Kerr et al., 2017). Thus, assays of protein function in which one can compare the reference variant to a patient-derived counterpart are valuable. Here we list a few examples where zebrafish have been used for functional tests of rare and in one case a common missense variants of genes linked to OFC.
ARHGAP29
Among other candidates in the 1p22 locus, ARHGAP29 emerged as the top candidate for an OFC-risk gene. This conclusion was based on: analysis of gene expression during murine craniofacial development; the discovery that rare coding variants in this gene, but not other candidates, are enriched in cases versus controls (Leslie et al., 2012); and the discovery that non-syndromic OFC-associated, non-coding SNPs affect the expression of ARHGAP29, but not that of other candidate genes tested, in oral epithelium cells (Liu et al., 2017). In a multiplex family in which many members had non-syndromic cleft palate only (NS CPO), exome sequencing revealed rare coding variants in ARHGAP29 in affected individuals; in silico algorithms predicted a p.Ser552Pro patient variant of ARHGAP29 to be deleterious (Liu et al., 2016a). Zebrafish injected with a RNA encoding the reference (wild-type) variant of ARHGAP29 failed to progress through epiboly, possibly because of a disruption in the dynamics of actin polymerization events that are essential for epiboly (Lee, 2014). However, embryos injected with an RNA encoding the p.Ser552Pro variant progressed normally, at the same rate as lacZ-injected controls (Liu et al., 2016a). The inability of the p.Ser552Pro variant to disrupt morphogenesis implies that it has a lower or different activity than the wild-type ARHGAP29, supporting its candidacy as disease-causing.
GRHL3
In two recent GWAS of NS CPO, the lead SNP, rs41268753, was within an exon of GRHL3 (Leslie et al., 2016; Mangold et al., 2016), a gene in which dominant loss-of-function mutations appear to cause Van der Woude syndrome (discussed above) (Peyrard-Janvid et al., 2014). The risk-associated allele of this SNP is a missense variant, p.Thr454Met (Leslie et al., 2016). Overexpression of the missense variant associated with NS CPO in wild-type (WT) zebrafish embryos recapitulates the dnIrf6 phenotype, i.e., rupture during gastrulation (discussed in VWS section), and blocks expression of the periderm marker krt4, similar to the alleles found in VWS patients (de la Garza et al., 2013; Leslie et al., 2016). These findings support rs41268753 as being pathogenic rather than simply being in linkage disequilibrium with a pathogenic variant, and the mechanism of pathogenesis is that it disrupts GRHL3 coding sequence. Because the allele frequency of the risk-associated variant of rs41268753 is much higher than the incidence of NS CPO, it is predicted that the variant of GRHL3 associated with CPO is less penetrant than the ones identified in VWS patients; cell-based reporter assays of GRHL3 transactivation function confirmed this prediction (Leslie et al., 2016).
IRF6
Mutations in IRF6 contribute significantly to syndromic OFC, accounting for up to 70% cases of VWS (Kondo et al., 2002). To test how IRF6 affects zebrafish palatogenesis while avoiding the lethality at epiboly that characterizes the irf6-DBD mutants (Sabel et al., 2009), Dougherty et al. generated strains in which dominant negative irf6 mutants designed from known syndromic OFC variants of IRF6, i.e., IRF6R84C, IRF6 R84H were expressed under the control of the neural-crest-specific sox10 promoter (Dougherty et al., 2013). Remarkably, these lines were viable. The embryos displayed prominent clefts between the median frontonasal process and paired maxillary processes along the seam by which they are joined without affecting ethmoid plate extension. (Dougherty et al., 2013). Meckel’s and palatoquadrate cartilages were smaller than normal in sox10:irf6R84C,H transgenic lines relative to WT. Interestingly extension of the ethmoid plate was not affected in transgenic larvae. These findings suggest that irf6 has essential functions in both neural crest and the oral epithelium, and that disruption of either function can potentially cause OFC.
KLF4
Based on the discovery that KLF4 homologs act downstream of IRF6 in the zebrafish periderm gene regulatory network, Liu et al sequenced the KLF4 gene in a mixed cohort of NSLCP samples from Fillipino, American (USA) and Ethiopian populations, and discovered five novel missense variants (Liu et al., 2016b). Variants p.Arg49Gly and p.His427Tyr are located in the activation and DNA binding domains, respectively, whereas p.Val261Met, p.Ser284Leu and p.Gly239-Tyr242del are located in the repression domain. Of these variants three (p.Arg94Gly, p. Ser284Leu, p. His427Tyr) were predicted to have perturbed function by both SIFT and Polyphen algorithms. However, overexpression of p.Ser26Leu and p.His427Tyr variants, but not the p.Arg94Gly variant, induced lethality in zebrafish embryos during epiboly and caused a reduction in krt4 expression in the EVL, as in embryos injected with dnIrf6 or dnGrhl3 (Liu et al., 2016b). The p.Arg94Gly is an example of a variant predicted to be damaging by the in silico algorithms but that appeared to function normally in a biological assay, emphasizing the importance of functional assays in assessing allele severity.
MAPRE2
Two CSC-KT patient-derived MAPRE2 variants, p.Asn68Ser (inherited, homozygous) and p.Arg143Cys (de novo, heterozygous) were tested for their ability to rescue the phenotype of hypoplastic ceratobranchial cartilages in mapre2 zebrafish morphants. The p.Arg143Cys variant was less effective than WT MAPRE2 at rescuing the morphant phenotype, but the p.Asn68Ser variant more so. This implies that the former is hypoactive and the latter hyperactive, consistent with its hyperactive binding of tubulin (Isrie et al., 2015). These in vivo assays highlight the utility of zebrafish in a LOF-phenotype-rescue-paradigm for testing the disease relevance of candidate syndromic OFC variants.
III. Using zebrafish to test the tissue-specificity of enhancers harboring SNPs that are associated with risk for OFC
As is the case for many complex diseases, SNPs associated with risk for non-syndromic OFC usually lie in non-coding DNA. Presumably most of these variants alter the function of cis-regulatory elements, i.e., enhancers or promoters (an alternative is that they affect a non-coding RNA). Disruption of enhancers can alter craniofacial morphology (Attanasio et al., 2013). Zebrafish are suitable for generating rapid transient reporter assays using fluorescent proteins. Remarkably, human enhancers that are not detectably conserved in zebrafish nonetheless are capable of driving reporter expression in zebrafish in a pattern that reflects expression of the human and zebrafish orthologs (Fisher et al., 2006). This feature has been exploited in several recent studies to test the tissue specificity of enhancers harboring OFC-associated SNPs.
1p22/ARHGAP29
A GWAS can identify the haplotype block that contains a functional SNP, i.e., the SNP at which the risk allele directly affects risk for a disease or trait, but it cannot distinguish the functional SNP from those that are in linkage disequilibrium with it. Towards identifying functional SNPs at the 1p22 locus that are associated with NS CLP, Liu and colleagues hypothesized that functional SNPs would lie within enhancers that are active in a tissue within the face. They amplified chromatin elements containing the 10 most strongly risk-associated SNPs and tested them in transgenic enhancer assays in zebrafish. Two regulatory elements had enhancer activity in oral epithelium and oral mesenchyme (Liu et al., 2017). Chromatin configuration capture assays in a human oral epithelium cell line confirmed that these elements interacted with the transcriptional start site (TSS) of ARHGAP29, and genome engineering confirmed that expression of this gene depends on the allele of the SNPs that is contained in the element, arguing that they are likely to be functional (Liu et al., 2017).
9q22/FOXE1
A SNP at 9p22 was found to reach genome-wide significance in a recent GWAS meta-analysis for orofacial clefts (Leslie et al., 2017a). The SNP lies close to the FOXE1 gene, mutations in which cause Bamforth-Lazarus syndrome, which frequently includes orofacial cleft (Clifton-Bligh et al., 1998; Castanet et al., 2002; Baris et al., 2006). FOXE1 was also the favored candidate risk gene from a family-based genome-wide linkage scan that revealed coding variants in non-syndromic OFC patients (Moreno et al., 2009). Towards identifying functional SNPs at 9q22, chromatin elements were tested for enhancer activity in zebrafish reporter assays, and SNPs were assessed for their effects on enhancer activity of such elements in reporter assays in a human oral epithelium cell line (Lidral et al., 2015). An element −67.7 kb from the FOXE1 TSS was found to be an enhancer that is active in the oral epithelium, pharyngeal arches, thyroid gland and heart tissues. Within this element is a SNP, rs7850258, for which one allele (G) is associated with hypothyroidism and CLP, and another (A) with thyroid cancer. This SNP had allele-dependent effects on enhancer activity in vitro, arguing that it directly influences risk for one or more of these diseases.
FGFR2/10q26 and 17q22/NOG
Towards identifying candidate functional SNPs for non-syndromic OFC, thirteen genomic regions identified by GWAS or other genome-wide approaches were sequenced in more than 1400 case-parent trios (Leslie et al., 2015). In vitro and zebrafish-based evidence indicated that several specific variants directly contribute risk for nonsyndromic clefts. For instance, a de novo mutation within a conserved non-coding element +254 kb downstream of fibroblast growth factor receptor (FGFR2) abrogated the neural crest enhancer activity of the element in transgenic zebrafish. fgfr2 is expressed in the pharyngeal endodermal pouches of this species at 24 hpf, and MO-mediated knockdown affects the craniofacial chondrogenic program (Larbuisson et al., 2013). Another example of a common variant that is strongly associated with NS CLP is rs227727, which lies within a conserved element +105 kb upstream of Noggin (NOG), a gene expressed in superficial and basal layers of palate epithelium. Although this element lacked detectable enhancer activity in zebrafish, an adjacent element at +87 kb had strong enhancer activity in zebrafish periderm. The tandem +87/+105kb element had strong enhancer activity in oral epithelium cells in vitro, and this depended on the allele of rs227727 (Leslie et al., 2015), arguing that it is a functional SNP.
SATB2 locus
Chromosomal breakpoints in patients who present with a syndromic form of OFC (patients also have micrognathia) have been mapped to the large PLCL1-SATB2 gene desert. These breakpoints are located distal (128.5 kb, 749 kb and 896 kb) to the 3’ poly-adenylation signal of SATB2 (Rainger et al., 2014). Comparative genomics approaches assessing orthologous regions of the human and chick genomes identified three cis regulatory elements (CRE 1–3) that overlap with each breakpoint. SATB2 enhancer function was evaluated by testing each of these candidate CREs by transgenic reporter assay and generating stable lines. In a stable reporter line CRE2 recapitulated the endogenous zebrafish satb2 expression domain within the ethmoid plate (Rainger et al., 2014). The activity of CRE 2 was shown to be dependent on the SOX9 transcription factor (Rainger et al., 2014). The results suggest that the translocation breakpoints disrupt the long-range cis- regulation of SATB2 by SOX9.
Concluding thoughts
The rapidly dropping cost of DNA sequencing methods have permitted whole exome sequencing of patients with OFC and other craniofacial malformations, and whole genome sequencing projects are underway. Deep sequencing leads to the discovery of many variants in each patient. Concurrent advances in gene editing have improved our ability to test the function of those variants in animal models. Despite some differences in craniofacial anatomy between fish and mammals, zebrafish bring experimental advantages to these efforts, chiefly external development and relatively low-cost and high-throughput gene editing in comparison to in mouse. Human functional genomics projects are best served by combining the strengths of mouse, zebrafish and in vitro systems. We predict that continued innovations in genetic and embryological methods in zebrafish will mean that the zebrafish model remains a powerful tool in the field of functional genomics.
Acknowledgments
We thank Robert Nissen for permission to share unpublished findings. Research to study zebrafish models of OFC in our groups is supported by grants from the National Institutes of Health NIDCR HD073107 (Cornell) and NIGMS PO1GM061354 (Liao).
Footnotes
The authors have no conflicts of interest to declare.
References
- Ahn HJ, Park Y, Kim S, Park HC, Seo SK, Yeo SY, Geum D. The expression profile and function of Satb2 in zebrafish embryonic development. Mol Cells. 2010;30:377–382. doi: 10.1007/s10059-010-0128-6. [DOI] [PubMed] [Google Scholar]
- Amsterdam A, Burgess S, Golling G, Chen W, Sun Z, Townsend K, Farrington S, Haldi M, Hopkins N. A large-scale insertional mutagenesis screen in zebrafish. Genes Dev. 1999;13:2713–2724. doi: 10.1101/gad.13.20.2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attanasio C, Nord AS, Zhu Y, Blow MJ, Li Z, Liberton DK, Morrison H, Plajzer-Frick I, Holt A, Hosseini R, Phouanenavong S, Akiyama JA, Shoukry M, Afzal V, Rubin EM, FitzPatrick DR, Ren B, Hallgrimsson B, Pennacchio LA, Visel A. Fine tuning of craniofacial morphology by distant-acting enhancers. Science. 2013;342:1241006. doi: 10.1126/science.1241006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baris I, Arisoy AE, Smith A, Agostini M, Mitchell CS, Park SM, Halefoglu AM, Zengin E, Chatterjee VK, Battaloglu E. A novel missense mutation in human TTF-2 (FKHL15) gene associated with congenital hypothyroidism but not athyreosis. J Clin Endocrinol Metab. 2006;91:4183–4187. doi: 10.1210/jc.2006-0405. [DOI] [PubMed] [Google Scholar]
- Beaty TH, Marazita ML, Leslie EJ. Genetic factors influencing risk to orofacial clefts: today's challenges and tomorrow's opportunities. F1000Res. 2016;5:2800. doi: 10.12688/f1000research.9503.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben J, Jabs EW, Chong SS. Genomic, cDNA and embryonic expression analysis of zebrafish IRF6, the gene mutated in the human oral clefting disorders Van der Woude and popliteal pterygium syndromes. Gene Expr Patterns. 2005;5:629–638. doi: 10.1016/j.modgep.2005.03.002. [DOI] [PubMed] [Google Scholar]
- Bhoj EJ, Li D, Harr MH, Tian L, Wang T, Zhao Y, Qiu H, Kim C, Hoffman JD, Hakonarson H, Zackai EH. Expanding the SPECC1L mutation phenotypic spectrum to include Teebi hypertelorism syndrome. Am J Med Genet A. 2015;167A:2497–2502. doi: 10.1002/ajmg.a.37217. [DOI] [PubMed] [Google Scholar]
- Biggs LC, Naridze RL, Demali KA, Lusche DF, Kuhl S, Soll DR, Schutte BC, Dunnwald M. Interferon regulatory factor 6 regulates keratinocyte migration. J Cell Sci. 2014 doi: 10.1242/jcs.139246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogershausen N, Wollnik B. Unmasking Kabuki syndrome. Clin Genet. 2013;83:201–211. doi: 10.1111/cge.12051. [DOI] [PubMed] [Google Scholar]
- Boglev Y, Wilanowski T, Caddy J, Parekh V, Auden A, Darido C, Hislop NR, Cangkrama M, Ting SB, Jane SM. The unique and cooperative roles of the Grainy head-like transcription factors in epidermal development reflect unexpected target gene specificity. Developmental biology. 2011;349:512–522. doi: 10.1016/j.ydbio.2010.11.011. [DOI] [PubMed] [Google Scholar]
- Bosma JF, Henkin RI, Christiansen RL, Herdt JR. Hypoplasia of the nose and eyes, hyposmia, hypogeusia, and hypogonadotrophic hypogonadism in two males. J Craniofac Genet Dev Biol. 1981;1:153–184. [PubMed] [Google Scholar]
- Brown KK, Reiss JA, Crow K, Ferguson HL, Kelly C, Fritzsch B, Morton CC. Deletion of an enhancer near DLX5 and DLX6 in a family with hearing loss, craniofacial defects, and an inv(7)(q21.3q35) Hum Genet. 2010;127:19–31. doi: 10.1007/s00439-009-0736-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush JO, Jiang R. Palatogenesis: morphogenetic and molecular mechanisms of secondary palate development. Development. 2012;139:231–243. doi: 10.1242/dev.067082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai J, Ash D, Kotch LE, Jabs EW, Attie-Bitach T, Auge J, Mattei G, Etchevers H, Vekemans M, Korshunova Y, Tidwell R, Messina DN, Winston JB, Lovett M. Gene expression in pharyngeal arch 1 during human embryonic development. Hum Mol Genet. 2005;14:903–912. doi: 10.1093/hmg/ddi083. [DOI] [PubMed] [Google Scholar]
- Carnevale F, Krajewska G, Fischetto R, Greco MG, Bonvino A. Ptosis of eyelids, strabismus, diastasis recti, hip defect, cryptorchidism, and developmental delay in two sibs. Am J Med Genet. 1989;33:186–189. doi: 10.1002/ajmg.1320330210. [DOI] [PubMed] [Google Scholar]
- Castanet M, Park SM, Smith A, Bost M, Leger J, Lyonnet S, Pelet A, Czernichow P, Chatterjee K, Polak M. A novel loss-of-function mutation in TTF-2 is associated with congenital hypothyroidism, thyroid agenesis and cleft palate. Hum Mol Genet. 2002;11:2051–2059. doi: 10.1093/hmg/11.17.2051. [DOI] [PubMed] [Google Scholar]
- Chiquet BT, Blanton SH, Burt A, Ma D, Stal S, Mulliken JB, Hecht JT. Variation in WNT genes is associated with non-syndromic cleft lip with or without cleft palate. Hum Mol Genet. 2008;17:2212–2218. doi: 10.1093/hmg/ddn121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiquet BT, Lidral AC, Stal S, Mulliken JB, Moreno LM, Arcos-Burgos M, Valencia-Ramirez C, Blanton SH, Hecht JT. CRISPLD2: a novel NSCLP candidate gene. Hum Mol Genet. 2007;16:2241–2248. doi: 10.1093/hmg/ddm176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clifton-Bligh RJ, Wentworth JM, Heinz P, Crisp MS, John R, Lazarus JH, Ludgate M, Chatterjee VK. Mutation of the gene encoding human TTF-2 associated with thyroid agenesis, cleft palate and choanal atresia. Nat Genet. 1998;19:399–401. doi: 10.1038/1294. [DOI] [PubMed] [Google Scholar]
- Cooper JA, Sept D. New insights into mechanism and regulation of actin capping protein. Int Rev Cell Mol Biol. 2008;267:183–206. doi: 10.1016/S1937-6448(08)00604-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox AA, Jezewski PA, Fang PK, Payne-Ferreira TL. Zebrafish Wnt9a,9b paralog comparisons suggest ancestral roles for Wnt9 in neural, oral-pharyngeal ectoderm and mesendoderm. Gene Expr Patterns. 2010;10:251–258. doi: 10.1016/j.gep.2010.05.005. [DOI] [PubMed] [Google Scholar]
- Curtin E, Hickey G, Kamel G, Davidson AJ, Liao EC. Zebrafish wnt9a is expressed in pharyngeal ectoderm and is required for palate and lower jaw development. Mech Dev. 2011;128:104–115. doi: 10.1016/j.mod.2010.11.003. [DOI] [PubMed] [Google Scholar]
- Cvjetkovic N, Maili L, Weymouth KS, Hashmi SS, Mulliken JB, Topczewski J, Letra A, Yuan Q, Blanton SH, Swindell EC, Hecht JT. Regulatory variant in FZD6 gene contributes to nonsyndromic cleft lip and palate in an African-American family. Mol Genet Genomic Med. 2015;3:440–451. doi: 10.1002/mgg3.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dauwerse JG, Dixon J, Seland S, Ruivenkamp CA, van Haeringen A, Hoefsloot LH, Peters DJ, Boers AC, Daumer-Haas C, Maiwald R, Zweier C, Kerr B, Cobo AM, Toral JF, Hoogeboom AJ, Lohmann DR, Hehr U, Dixon MJ, Breuning MH, Wieczorek D. Mutations in genes encoding subunits of RNA polymerases I and III cause Treacher Collins syndrome. Nat Genet. 2011;43:20–22. doi: 10.1038/ng.724. [DOI] [PubMed] [Google Scholar]
- de la Garza G, Schleiffarth JR, Dunnwald M, Mankad A, Weirather JL, Bonde G, Butcher S, Mansour TA, Kousa YA, Fukazawa CF, Houston DW, Manak JR, Schutte BC, Wagner DS, Cornell RA. Interferon regulatory factor 6 promotes differentiation of the periderm by activating expression of Grainyhead-like 3. J Invest Dermatol. 2013;133:68–77. doi: 10.1038/jid.2012.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLaurier A, Nakamura Y, Braasch I, Khanna V, Kato H, Wakitani S, Postlethwait JH, Kimmel CB. Histone deacetylase-4 is required during early cranial neural crest development for generation of the zebrafish palatal skeleton. BMC Dev Biol. 2012;12:16. doi: 10.1186/1471-213X-12-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depew MJ, Lufkin T, Rubenstein JL. Specification of jaw subdivisions by Dlx genes. Science. 2002;298:381–385. doi: 10.1126/science.1075703. [DOI] [PubMed] [Google Scholar]
- Devenport D, Fuchs E. Planar polarization in embryonic epidermis orchestrates global asymmetric morphogenesis of hair follicles. Nat Cell Biol. 2008;10:1257–1268. doi: 10.1038/ncb1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon J, Jones NC, Sandell LL, Jayasinghe SM, Crane J, Rey JP, Dixon MJ, Trainor PA. Tcof1/Treacle is required for neural crest cell formation and proliferation deficiencies that cause craniofacial abnormalities. Proc Natl Acad Sci U S A. 2006;103:13403–13408. doi: 10.1073/pnas.0603730103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon MJ. Treacher Collins syndrome. Hum Mol Genet. 1996;5(Spec No):1391–1396. doi: 10.1093/hmg/5.supplement_1.1391. [DOI] [PubMed] [Google Scholar]
- Dixon MJ, Marazita ML, Beaty TH, Murray JC. Cleft lip and palate: understanding genetic and environmental influences. Nat Rev Genet. 2011;12:167–178. doi: 10.1038/nrg2933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobreva G, Chahrour M, Dautzenberg M, Chirivella L, Kanzler B, Farinas I, Karsenty G, Grosschedl R. SATB2 is a multifunctional determinant of craniofacial patterning and osteoblast differentiation. Cell. 2006;125:971–986. doi: 10.1016/j.cell.2006.05.012. [DOI] [PubMed] [Google Scholar]
- Docker D, Schubach M, Menzel M, Munz M, Spaich C, Biskup S, Bartholdi D. Further delineation of the SATB2 phenotype. Eur J Hum Genet. 2014;22:1034–1039. doi: 10.1038/ejhg.2013.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- dos Remedios CG, Chhabra D, Kekic M, Dedova IV, Tsubakihara M, Berry DA, Nosworthy NJ. Actin binding proteins: regulation of cytoskeletal microfilaments. Physiol Rev. 2003;83:433–473. doi: 10.1152/physrev.00026.2002. [DOI] [PubMed] [Google Scholar]
- Dougherty M, Kamel G, Grimaldi M, Gfrerer L, Shubinets V, Ethier R, Hickey G, Cornell RA, Liao EC. Distinct requirements for wnt9a and irf6 in extension and integration mechanisms during zebrafish palate morphogenesis. Development. 2013;140:76–81. doi: 10.1242/dev.080473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougherty M, Kamel G, Shubinets V, Hickey G, Grimaldi M, Liao EC. Embryonic fate map of first pharyngeal arch structures in the sox10: kaede zebrafish transgenic model. J Craniofac Surg. 2012;23:1333–1337. doi: 10.1097/SCS.0b013e318260f20b. [DOI] [PubMed] [Google Scholar]
- Driever W, Solnica-Krezel L, Schier AF, Neuhauss SC, Malicki J, Stemple DL, Stainier DY, Zwartkruis F, Abdelilah S, Rangini Z, Belak J, Boggs C. A genetic screen for mutations affecting embryogenesis in zebrafish. Development. 1996;123:37–46. doi: 10.1242/dev.123.1.37. [DOI] [PubMed] [Google Scholar]
- Dworkin S, Simkin J, Darido C, Partridge DD, Georgy SR, Caddy J, Wilanowski T, Lieschke GJ, Doggett K, Heath JK, Jane SM. Grainyhead-like 3 regulation of endothelin-1 in the pharyngeal endoderm is critical for growth and development of the craniofacial skeleton. Mech Dev. 2014;133:77–90. doi: 10.1016/j.mod.2014.05.005. [DOI] [PubMed] [Google Scholar]
- Eberhart JK, He X, Swartz ME, Yan YL, Song H, Boling TC, Kunerth AK, Walker MB, Kimmel CB, Postlethwait JH. MicroRNA Mirn140 modulates Pdgf signaling during palatogenesis. Nat Genet. 2008;40:290–298. doi: 10.1038/ng.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberhart JK, Swartz ME, Crump JG, Kimmel CB. Early Hedgehog signaling from neural to oral epithelium organizes anterior craniofacial development. Development. 2006;133:1069–1077. doi: 10.1242/dev.02281. [DOI] [PubMed] [Google Scholar]
- Eisen JS, Smith JC. Controlling morpholino experiments: don't stop making antisense. Development. 2008;135:1735–1743. doi: 10.1242/dev.001115. [DOI] [PubMed] [Google Scholar]
- El-Brolosy MA, Stainier DYR. Genetic compensation: A phenomenon in search of mechanisms. PLoS Genet. 2017;13:e1006780. doi: 10.1371/journal.pgen.1006780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eppley BL, van Aalst JA, Robey A, Havlik RJ, Sadove AM. The spectrum of orofacial clefting. Plast Reconstr Surg. 2005;115:101e–114e. doi: 10.1097/01.prs.0000164494.45986.91. [DOI] [PubMed] [Google Scholar]
- Field LL, Ray AK, Cooper ME, Goldstein T, Shaw DF, Marazita ML. Genome scan for loci involved in nonsyndromic cleft lip with or without cleft palate in families from West Bengal, India. Am J Med Genet A. 2004;130A:265–271. doi: 10.1002/ajmg.a.30252. [DOI] [PubMed] [Google Scholar]
- Fisher S, Grice EA, Vinton RM, Bessling SL, McCallion AS. Conservation of RET regulatory function from human to zebrafish without sequence similarity. Science. 2006;312:276–279. doi: 10.1126/science.1124070. [DOI] [PubMed] [Google Scholar]
- FitzPatrick DR, Carr IM, McLaren L, Leek JP, Wightman P, Williamson K, Gautier P, McGill N, Hayward C, Firth H, Markham AF, Fantes JA, Bonthron DT. Identification of SATB2 as the cleft palate gene on 2q32-q33. Hum Mol Genet. 2003;12:2491–2501. doi: 10.1093/hmg/ddg248. [DOI] [PubMed] [Google Scholar]
- Gfrerer L, Shubinets V, Hoyos T, Kong Y, Nguyen C, Pietschmann P, Morton CC, Maas RL, Liao EC. Functional analysis of SPECC1L in craniofacial development and oblique facial cleft pathogenesis. Plast Reconstr Surg. 2014;134:748–759. doi: 10.1097/PRS.0000000000000517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghassibe-Sabbagh M, Desmyter L, Langenberg T, Claes F, Boute O, Bayet B, Pellerin P, Hermans K, Backx L, Mansilla MA, Imoehl S, Nowak S, Ludwig KU, Baluardo C, Ferrian M, Mossey PA, Noethen M, Dewerchin M, Francois G, Revencu N, Vanwijck R, Hecht J, Mangold E, Murray J, Rubini M, Vermeesch JR, Poirel HA, Carmeliet P, Vikkula M. FAF1, a gene that is disrupted in cleft palate and has conserved function in zebrafish. Am J Hum Genet. 2011;88:150–161. doi: 10.1016/j.ajhg.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gladwin AJ, Dixon J, Loftus SK, Edwards S, Wasmuth JJ, Hennekam RC, Dixon MJ. Treacher Collins syndrome may result from insertions, deletions or splicing mutations, which introduce a termination codon into the gene. Hum Mol Genet. 1996;5:1533–1538. doi: 10.1093/hmg/5.10.1533. [DOI] [PubMed] [Google Scholar]
- Golson ML, Kaestner KH. Fox transcription factors: from development to disease. Development. 2016;143:4558–4570. doi: 10.1242/dev.112672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon CT, Brinas IM, Rodda FA, Bendall AJ, Farlie PG. Role of Dlx genes in craniofacial morphogenesis: Dlx2 influences skeletal patterning by inducing ectomesenchymal aggregation in ovo. Evol Dev. 2010;12:459–473. doi: 10.1111/j.1525-142X.2010.00432.x. [DOI] [PubMed] [Google Scholar]
- Gordon CT, Xue S, Yigit G, Filali H, Chen K, Rosin N, Yoshiura KI, Oufadem M, Beck TJ, McGowan R, Magee AC, Altmuller J, Dion C, Thiele H, Gurzau AD, Nurnberg P, Meschede D, Muhlbauer W, Okamoto N, Varghese V, Irving R, Sigaudy S, Williams D, Ahmed SF, Bonnard C, Kong MK, Ratbi I, Fejjal N, Fikri M, Elalaoui SC, Reigstad H, Bole-Feysot C, Nitschke P, Ragge N, Levy N, Tuncbilek G, Teo AS, Cunningham ML, Sefiani A, Kayserili H, Murphy JM, Chatdokmaiprai C, Hillmer AM, Wattanasirichaigoon D, Lyonnet S, Magdinier F, Javed A, Blewitt ME, Amiel J, Wollnik B, Reversade B. De novo mutations in SMCHD1 cause Bosma arhinia microphthalmia syndrome and abrogate nasal development. Nat Genet. 2017;49:249–255. doi: 10.1038/ng.3765. [DOI] [PubMed] [Google Scholar]
- Gorlin RJ, Sedano HO, Cervenka J. Popliteal pterygium syndrome. A syndrome comprising cleft lip-palate, popliteal and intercrural pterygia, digital and genital anomalies. Pediatrics. 1968;41:503–509. [PubMed] [Google Scholar]
- Graham JM, Jr, Lee J. Bosma arhinia microphthalmia syndrome. Am J Med Genet A. 2006;140:189–193. doi: 10.1002/ajmg.a.31039. [DOI] [PubMed] [Google Scholar]
- Grosen D, Bille C, Petersen I, Skytthe A, Hjelmborg J, Pedersen JK, Murray JC, Christensen K. Risk of oral clefts in twins. Epidemiology. 2011;22:313–319. doi: 10.1097/EDE.0b013e3182125f9c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haffter P, Nusslein-Volhard C. Large scale genetics in a small vertebrate, the zebrafish. Int J Dev Biol. 1996;40:221–227. [PubMed] [Google Scholar]
- Hruscha A, Krawitz P, Rechenberg A, Heinrich V, Hecht J, Haass C, Schmid B. Efficient CRISPR/Cas9 genome editing with low off-target effects in zebrafish. Development. 2013;140:4982–4987. doi: 10.1242/dev.099085. [DOI] [PubMed] [Google Scholar]
- Huang T, Liu Y, Huang M, Zhao X, Cheng L. Wnt1-cre-mediated conditional loss of Dicer results in malformation of the midbrain and cerebellum and failure of neural crest and dopaminergic differentiation in mice. J Mol Cell Biol. 2010;2:152–163. doi: 10.1093/jmcb/mjq008. [DOI] [PubMed] [Google Scholar]
- Ingraham CR, Kinoshita A, Kondo S, Yang B, Sajan S, Trout KJ, Malik MI, Dunnwald M, Goudy SL, Lovett M, Murray JC, Schutte BC. Abnormal skin, limb and craniofacial morphogenesis in mice deficient for interferon regulatory factor 6 (Irf6) Nat Genet. 2006;38:1335–1340. doi: 10.1083/ng1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isrie M, Breuss M, Tian G, Hansen AH, Cristofoli F, Morandell J, Kupchinsky ZA, Sifrim A, Rodriguez-Rodriguez CM, Dapena EP, Doonanco K, Leonard N, Tinsa F, Moortgat S, Ulucan H, Koparir E, Karaca E, Katsanis N, Marton V, Vermeesch JR, Davis EE, Cowan NJ, Keays DA, Van Esch H. Mutations in Either TUBB or MAPRE2 Cause Circumferential Skin Creases Kunze Type. Am J Hum Genet. 2015;97:790–800. doi: 10.1016/j.ajhg.2015.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ittiwut R, Ittiwut C, Siriwan P, Chichareon V, Suphapeetiporn K, Shotelersuk V. Variants of the CDH1 (E-Cadherin) Gene Associated with Oral Clefts in the Thai Population. Genet Test Mol Biomarkers. 2016;20:406–409. doi: 10.1089/gtmb.2015.0325. [DOI] [PubMed] [Google Scholar]
- Jackson H, Prakash D, Litaker M, Ferreira T, Jezewski P. Zebrafish Wnt9b Patterns the First Pharyngeal Arch into D-I-V Domains and Promotes Anterior-Medial Outgrowth. American Journal of Molecular Biology. 2015;5:57–83. [Google Scholar]
- Jezewski PA, Fang PK, Payne-Ferreira TL, Yelick PC. Zebrafish Wnt9b synteny and expression during first and second arch, heart, and pectoral fin bud morphogenesis. Zebrafish. 2008;5:169–177. doi: 10.1089/zeb.2007.0517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones NC, Lynn ML, Gaudenz K, Sakai D, Aoto K, Rey JP, Glynn EF, Ellington L, Du C, Dixon J, Dixon MJ, Trainor PA. Prevention of the neurocristopathy Treacher Collins syndrome through inhibition of p53 function. Nat Med. 2008;14:125–133. doi: 10.1038/nm1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juriloff DM, Harris MJ, McMahon AP, Carroll TJ, Lidral AC. Wnt9b is the mutated gene involved in multifactorial nonsyndromic cleft lip with or without cleft palate in A/WySn mice, as confirmed by a genetic complementation test. Birth Defects Res A Clin Mol Teratol. 2006;76:574–579. doi: 10.1002/bdra.20302. [DOI] [PubMed] [Google Scholar]
- Kadakia S, Helman SN, Badhey AK, Saman M, Ducic Y. Treacher Collins Syndrome: the genetics of a craniofacial disease. Int J Pediatr Otorhinolaryngol. 2014;78:893–898. doi: 10.1016/j.ijporl.2014.03.006. [DOI] [PubMed] [Google Scholar]
- Kaplan F, Ledoux P, Kassamali FQ, Gagnon S, Post M, Koehler D, Deimling J, Sweezey NB. A novel developmentally regulated gene in lung mesenchyme: homology to a tumor-derived trypsin inhibitor. Am J Physiol. 1999;276:L1027–1036. doi: 10.1152/ajplung.1999.276.6.L1027. [DOI] [PubMed] [Google Scholar]
- Kerr ID, Cox HC, Moyes K, Evans B, Burdett BC, van Kan A, McElroy H, Vail PJ, Brown KL, Sumampong DB, Monteferrante NJ, Hardman KL, Theisen A, Mundt E, Wenstrup RJ, Eggington JM. Assessment of in silico protein sequence analysis in the clinical classification of variants in cancer risk genes. J Community Genet. 2017 doi: 10.1007/s12687-016-0289-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimmel CB, Warga RM, Schilling TF. Origin and organization of the zebrafish fate map. Development. 1990;108:581–594. doi: 10.1242/dev.108.4.581. [DOI] [PubMed] [Google Scholar]
- Knight RD, Nair S, Nelson SS, Afshar A, Javidan Y, Geisler R, Rauch GJ, Schilling TF. lockjaw encodes a zebrafish tfap2a required for early neural crest development. Development. 2003;130:5755–5768. doi: 10.1242/dev.00575. [DOI] [PubMed] [Google Scholar]
- Kondo S, Schutte BC, Richardson RJ, Bjork BC, Knight AS, Watanabe Y, Howard E, de Lima RL, Daack-Hirsch S, Sander A, McDonald-McGinn DM, Zackai EH, Lammer EJ, Aylsworth AS, Ardinger HH, Lidral AC, Pober BR, Moreno L, Arcos-Burgos M, Valencia C, Houdayer C, Bahuau M, Moretti-Ferreira D, Richieri-Costa A, Dixon MJ, Murray JC. Mutations in IRF6 cause Van der Woude and popliteal pterygium syndromes. Nat Genet. 2002;32:285–289. doi: 10.1038/ng985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kousa YA, Mansour TA, Seada H, Matoo S, Schutte BC. Shared molecular networks in orofacial and neural tube development. Birth Defects Res. 2017;109:169–179. doi: 10.1002/bdra.23598. [DOI] [PubMed] [Google Scholar]
- Kruszka P, Li D, Harr MH, Wilson NR, Swarr D, McCormick EM, Chiavacci RM, Li M, Martinez AF, Hart RA, McDonald-McGinn DM, Deardorff MA, Falk MJ, Allanson JE, Hudson C, Johnson JP, Saadi I, Hakonarson H, Muenke M, Zackai EH. Mutations in SPECC1L, encoding sperm antigen with calponin homology and coiled-coil domains 1-like, are found in some cases of autosomal dominant Opitz G/BBB syndrome. J Med Genet. 2015;52:104–110. doi: 10.1136/jmedgenet-2014-102677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunze J, Riehm H. A new genetic disorder: autosomal-dominant multiple benign ring-shaped skin creases. Eur J Pediatr. 1982;138:301–303. doi: 10.1007/BF00442501. [DOI] [PubMed] [Google Scholar]
- Lai EC. Micro RNAs are complementary to 3' UTR sequence motifs that mediate negative post-transcriptional regulation. Nat Genet. 2002;30:363–364. doi: 10.1038/ng865. [DOI] [PubMed] [Google Scholar]
- Lan J, Ribeiro L, Mandeville I, Nadeau K, Bao T, Cornejo S, Sweezey NB, Kaplan F. Inflammatory cytokines, goblet cell hyperplasia and altered lung mechanics in Lgl1+/− mice. Respir Res. 2009;10:83. doi: 10.1186/1465-9921-10-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan Y, Ryan RC, Zhang Z, Bullard SA, Bush JO, Maltby KM, Lidral AC, Jiang R. Expression of Wnt9b and activation of canonical Wnt signaling during midfacial morphogenesis in mice. Dev Dyn. 2006;235:1448–1454. doi: 10.1002/dvdy.20723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larbuisson A, Dalcq J, Martial JA, Muller M. Fgf receptors Fgfr1a and Fgfr2 control the function of pharyngeal endoderm in late cranial cartilage development. Differentiation. 2013;86:192–206. doi: 10.1016/j.diff.2013.07.006. [DOI] [PubMed] [Google Scholar]
- Lau MC, Kwong EM, Lai KP, Li JW, Ho JC, Chan TF, Wong CK, Jiang YJ, Tse WK. Pathogenesis of POLR1C-dependent Type 3 Treacher Collins Syndrome revealed by a zebrafish model. Biochim Biophys Acta. 2016;1862:1147–1158. doi: 10.1016/j.bbadis.2016.03.005. [DOI] [PubMed] [Google Scholar]
- Lederer D, Grisart B, Digilio MC, Benoit V, Crespin M, Ghariani SC, Maystadt I, Dallapiccola B, Verellen-Dumoulin C. Deletion of KDM6A, a histone demethylase interacting with MLL2, in three patients with Kabuki syndrome. Am J Hum Genet. 2012;90:119–124. doi: 10.1016/j.ajhg.2011.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SJ. Dynamic regulation of the microtubule and actin cytoskeleton in zebrafish epiboly. Biochem Biophys Res Commun. 2014;452:1–7. doi: 10.1016/j.bbrc.2014.08.005. [DOI] [PubMed] [Google Scholar]
- Lemmers RJ, Tawil R, Petek LM, Balog J, Block GJ, Santen GW, Amell AM, van der Vliet PJ, Almomani R, Straasheijm KR, Krom YD, Klooster R, Sun Y, den Dunnen JT, Helmer Q, Donlin-Smith CM, Padberg GW, van Engelen BG, de Greef JC, Aartsma-Rus AM, Frants RR, de Visser M, Desnuelle C, Sacconi S, Filippova GN, Bakker B, Bamshad MJ, Tapscott SJ, Miller DG, van der Maarel SM. Digenic inheritance of an SMCHD1 mutation and an FSHD-permissive D4Z4 allele causes facioscapulohumeral muscular dystrophy type 2. Nat Genet. 2012;44:1370–1374. doi: 10.1038/ng.2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leoyklang P, Suphapeetiporn K, Siriwan P, Desudchit T, Chaowanapanja P, Gahl WA, Shotelersuk V. Heterozygous nonsense mutation SATB2 associated with cleft palate, osteoporosis, and cognitive defects. Hum Mutat. 2007;28:732–738. doi: 10.1002/humu.20515. [DOI] [PubMed] [Google Scholar]
- Leslie EJ, Carlson JC, Shaffer JR, Butali A, Buxo CJ, Castilla EE, Christensen K, Deleyiannis FW, Leigh Field L, Hecht JT, Moreno L, Orioli IM, Padilla C, Vieira AR, Wehby GL, Feingold E, Weinberg SM, Murray JC, Beaty TH, Marazita ML. Genome-wide meta-analyses of nonsyndromic orofacial clefts identify novel associations between FOXE1 and all orofacial clefts, and TP63 and cleft lip with or without cleft palate. Hum Genet. 2017a doi: 10.1007/s00439-016-1754-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie EJ, Carlson JC, Shaffer JR, Butali A, Buxo CJ, Castilla EE, Christensen K, Deleyiannis FW, Leigh Field L, Hecht JT, Moreno L, Orioli IM, Padilla C, Vieira AR, Wehby GL, Feingold E, Weinberg SM, Murray JC, Beaty TH, Marazita ML. Genome-wide meta-analyses of nonsyndromic orofacial clefts identify novel associations between FOXE1 and all orofacial clefts, and TP63 and cleft lip with or without cleft palate. Hum Genet. 2017b;136:275–286. doi: 10.1007/s00439-016-1754-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie EJ, Liu H, Carlson JC, Shaffer JR, Feingold E, Wehby G, Laurie CA, Jain D, Laurie CC, Doheny KF, McHenry T, Resick J, Sanchez C, Jacobs J, Emanuele B, Vieira AR, Neiswanger K, Standley J, Czeizel AE, Deleyiannis F, Christensen K, Munger RG, Lie RT, Wilcox A, Romitti PA, Field LL, Padilla CD, Cutiongco-de la Paz EM, Lidral AC, Valencia-Ramirez LC, Lopez-Palacio AM, Valencia DR, Arcos-Burgos M, Castilla EE, Mereb JC, Poletta FA, Orioli IM, Carvalho FM, Hecht JT, Blanton SH, Buxo CJ, Butali A, Mossey PA, Adeyemo WL, James O, Braimah RO, Aregbesola BS, Eshete MA, Deribew M, Koruyucu M, Seymen F, Ma L, de Salamanca JE, Weinberg SM, Moreno L, Cornell RA, Murray JC, Marazita ML. A Genome-wide Association Study of Nonsyndromic Cleft Palate Identifies an Etiologic Missense Variant in GRHL3. Am J Hum Genet. 2016;98:744–754. doi: 10.1016/j.ajhg.2016.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie EJ, Mansilla MA, Biggs LC, Schuette K, Bullard S, Cooper M, Dunnwald M, Lidral AC, Marazita ML, Beaty TH, Murray JC. Expression and mutation analyses implicate ARHGAP29 as the etiologic gene for the cleft lip with or without cleft palate locus identified by genome-wide association on chromosome 1p22. Birth Defects Res A Clin Mol Teratol. 2012;94:934–942. doi: 10.1002/bdra.23076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie EJ, Taub MA, Liu H, Steinberg KM, Koboldt DC, Zhang Q, Carlson JC, Hetmanski JB, Wang H, Larson DE, Fulton RS, Kousa YA, Fakhouri WD, Naji A, Ruczinski I, Begum F, Parker MM, Busch T, Standley J, Rigdon J, Hecht JT, Scott AF, Wehby GL, Christensen K, Czeizel AE, Deleyiannis FW, Schutte BC, Wilson RK, Cornell RA, Lidral AC, Weinstock GM, Beaty TH, Marazita ML, Murray JC. Identification of functional variants for cleft lip with or without cleft palate in or near PAX7, FGFR2, and NOG by targeted sequencing of GWAS loci. Am J Hum Genet. 2015;96:397–411. doi: 10.1016/j.ajhg.2015.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis BP, Shih IH, Jones-Rhoades MW, Bartel DP, Burge CB. Prediction of mammalian microRNA targets. Cell. 2003;115:787–798. doi: 10.1016/s0092-8674(03)01018-3. [DOI] [PubMed] [Google Scholar]
- Li L, Meng T, Jia Z, Zhu G, Shi B. Single nucleotide polymorphism associated with nonsyndromic cleft palate influences the processing of miR-140. Am J Med Genet A. 2010;152A:856–862. doi: 10.1002/ajmg.a.33236. [DOI] [PubMed] [Google Scholar]
- Lidral AC, Liu H, Bullard SA, Bonde G, Machida J, Visel A, Uribe LM, Li X, Amendt B, Cornell RA. A single nucleotide polymorphism associated with isolated cleft lip and palate, thyroid cancer and hypothyroidism alters the activity of an oral epithelium and thyroid enhancer near FOXE1. Hum Mol Genet. 2015;24:3895–3907. doi: 10.1093/hmg/ddv047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindgren AM, Hoyos T, Talkowski ME, Hanscom C, Blumenthal I, Chiang C, Ernst C, Pereira S, Ordulu Z, Clericuzio C, Drautz JM, Rosenfeld JA, Shaffer LG, Velsher L, Pynn T, Vermeesch J, Harris DJ, Gusella JF, Liao EC, Morton CC. Haploinsufficiency of KDM6A is associated with severe psychomotor retardation, global growth restriction, seizures and cleft palate. Hum Genet. 2013;132:537–552. doi: 10.1007/s00439-013-1263-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Busch T, Eliason S, Anand D, Bullard S, Gowans LJ, Nidey N, Petrin A, Augustine-Akpan EA, Saadi I, Dunnwald M, Lachke SA, Zhu Y, Adeyemo A, Amendt B, Roscioli T, Cornell R, Murray J, Butali A. Exome sequencing provides additional evidence for the involvement of ARHGAP29 in Mendelian orofacial clefting and extends the phenotypic spectrum to isolated cleft palate. Birth Defects Res A Clin Mol Teratol. 2016a doi: 10.1002/bdra.23596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Leslie EJ, Carlson JC, Beaty TH, Marazita ML, Lidral AC, Cornell RA. Identification of common non-coding variants at 1p22 that are functional for non-syndromic orofacial clefting. Nat. Communications. 2017 doi: 10.1038/ncomms14759. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Leslie EJ, Jia Z, Smith T, Eshete M, Butali A, Dunnwald M, Murray J, Cornell RA. Irf6 directly regulates Klf17 in zebrafish periderm and Klf4 in murine oral epithelium, and dominant-negative KLF4 variants are present in patients with cleft lip and palate. Hum Mol Genet. 2016b;25:766–776. doi: 10.1093/hmg/ddv614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald BT, Tamai K, He X. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev Cell. 2009;17:9–26. doi: 10.1016/j.devcel.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malpuech G, Demeocq F, Palcoux JB, Vanlieferinghen P. A previously undescribed autosomal recessive multiple congenital anomalies/mental retardation (MCA/MR) syndrome with growth failure, lip/palate cleft(s), and urogenital anomalies. Am J Med Genet. 1983;16:475–480. doi: 10.1002/ajmg.1320160405. [DOI] [PubMed] [Google Scholar]
- Mangold E, Bohmer AC, Ishorst N, Hoebel AK, Gultepe P, Schuenke H, Klamt J, Hofmann A, Golz L, Raff R, Tessmann P, Nowak S, Reutter H, Hemprich A, Kreusch T, Kramer FJ, Braumann B, Reich R, Schmidt G, Jager A, Reiter R, Brosch S, Stavusis J, Ishida M, Seselgyte R, Moore GE, Nothen MM, Borck G, Aldhorae KA, Lace B, Stanier P, Knapp M, Ludwig KU. Sequencing the GRHL3 Coding Region Reveals Rare Truncating Mutations and a Common Susceptibility Variant for Nonsyndromic Cleft Palate. Am J Hum Genet. 2016;98:755–762. doi: 10.1016/j.ajhg.2016.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marazita ML, Field LL, Tuncbilek G, Cooper ME, Goldstein T, Gursu KG. Genome-scan for loci involved in cleft lip with or without cleft palate in consanguineous families from Turkey. Am J Med Genet A. 2004a;126A:111–122. doi: 10.1002/ajmg.a.20564. [DOI] [PubMed] [Google Scholar]
- Marazita ML, Murray JC, Lidral AC, Arcos-Burgos M, Cooper ME, Goldstein T, Maher BS, Daack-Hirsch S, Schultz R, Mansilla MA, Field LL, Liu YE, Prescott N, Malcolm S, Winter R, Ray A, Moreno L, Valencia C, Neiswanger K, Wyszynski DF, Bailey-Wilson JE, Albacha-Hejazi H, Beaty TH, McIntosh I, Hetmanski JB, Tuncbilek G, Edwards M, Harkin L, Scott R, Roddick LG. Meta-analysis of 13 genome scans reveals multiple cleft lip/palate genes with novel loci on 9q21 and 2q32-35. Am J Hum Genet. 2004b;75:161–173. doi: 10.1086/422475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy N, Liu JS, Richarte AM, Eskiocak B, Lovely CB, Tallquist MD, Eberhart JK. Pdgfra and Pdgfrb genetically interact during craniofacial development. Dev Dyn. 2016;245:641–652. doi: 10.1002/dvdy.24403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGurk PD, Lovely CB, Eberhart JK. Analyzing craniofacial morphogenesis in zebrafish using 4D confocal microscopy. J Vis Exp. 2014:e51190. doi: 10.3791/51190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michels VV, Hittner HM, Beaudet AL. A clefting syndrome with ocular anterior chamber defect and lid anomalies. J Pediatr. 1978;93:444–446. doi: 10.1016/s0022-3476(78)81154-8. [DOI] [PubMed] [Google Scholar]
- Milunsky JM, Maher TA, Zhao G, Roberts AE, Stalker HJ, Zori RT, Burch MN, Clemens M, Mulliken JB, Smith R, Lin AE. TFAP2A mutations result in branchio-oculo-facial syndrome. Am J Hum Genet. 2008;82:1171–1177. doi: 10.1016/j.ajhg.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mingarelli R, Castriota Scanderbeg A, Dallapiccola B. Two sisters with a syndrome of ocular, skeletal, and abdominal abnormalities (OSA syndrome) J Med Genet. 1996;33:884–886. doi: 10.1136/jmg.33.10.884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyake N, Mizuno S, Okamoto N, Ohashi H, Shiina M, Ogata K, Tsurusaki Y, Nakashima M, Saitsu H, Niikawa N, Matsumoto N. KDM6A point mutations cause Kabuki syndrome. Hum Mutat. 2013;34:108–110. doi: 10.1002/humu.22229. [DOI] [PubMed] [Google Scholar]
- Moreno LM, Mansilla MA, Bullard SA, Cooper ME, Busch TD, Machida J, Johnson MK, Brauer D, Krahn K, Daack-Hirsch S, L'Heureux J, Valencia-Ramirez C, Rivera D, Lopez AM, Moreno MA, Hing A, Lammer EJ, Jones M, Christensen K, Lie RT, Jugessur A, Wilcox AJ, Chines P, Pugh E, Doheny K, Arcos-Burgos M, Marazita ML, Murray JC, Lidral AC. FOXE1 association with both isolated cleft lip with or without cleft palate, and isolated cleft palate. Hum Mol Genet. 2009;18:4879–4896. doi: 10.1093/hmg/ddp444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee K, Ishii K, Pillalamarri V, Kammin T, Atkin JF, Hickey SE, Xi QJ, Zepeda CJ, Gusella JF, Talkowski ME, Morton CC, Maas RL, Liao EC. Actin capping protein CAPZB regulates cell morphology, differentiation, and neural crest migration in craniofacial morphogenesisdagger. Hum Mol Genet. 2016;25:1255–1270. doi: 10.1093/hmg/ddw006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakada C, Iida A, Tabata Y, Watanabe S. Forkhead transcription factor foxe1 regulates chondrogenesis in zebrafish. J Exp Zool B Mol Dev Evol. 2009;312:827–840. doi: 10.1002/jez.b.21298. [DOI] [PubMed] [Google Scholar]
- Noack Watt KE, Achilleos A, Neben CL, Merrill AE, Trainor PA. The Roles of RNA Polymerase I and III Subunits Polr1c and Polr1d in Craniofacial Development and in Zebrafish Models of Treacher Collins Syndrome. PLoS Genet. 2016;12:e1006187. doi: 10.1371/journal.pgen.1006187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JW, Cai J, McIntosh I, Jabs EW, Fallin MD, Ingersoll R, Hetmanski JB, Vekemans M, Attie-Bitach T, Lovett M, Scott AF, Beaty TH. High throughput SNP and expression analyses of candidate genes for non-syndromic oral clefts. J Med Genet. 2006;43:598–608. doi: 10.1136/jmg.2005.040162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peyrard-Janvid M, Leslie EJ, Kousa YA, Smith TL, Dunnwald M, Magnusson M, Lentz BA, Unneberg P, Fransson I, Koillinen HK, Rautio J, Pegelow M, Karsten A, Basel-Vanagaite L, Gordon W, Andersen B, Svensson T, Murray JC, Cornell RA, Kere J, Schutte BC. Dominant mutations in GRHL3 cause Van der Woude Syndrome and disrupt oral periderm development. Am J Hum Genet. 2014;94:23–32. doi: 10.1016/j.ajhg.2013.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Printzlau A, Andersen M. Pierre Robin sequence in Denmark: a retrospective population-based epidemiological study. Cleft Palate Craniofac J. 2004;41:47–52. doi: 10.1597/02-055. [DOI] [PubMed] [Google Scholar]
- Rainger JK, Bhatia S, Bengani H, Gautier P, Rainger J, Pearson M, Ansari M, Crow J, Mehendale F, Palinkasova B, Dixon MJ, Thompson PJ, Matarin M, Sisodiya SM, Kleinjan DA, Fitzpatrick DR. Disruption of SATB2 or its long-range cis-regulation by SOX9 causes a syndromic form of Pierre Robin sequence. Hum Mol Genet. 2014;23:2569–2579. doi: 10.1093/hmg/ddt647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rattanasopha S, Tongkobpetch S, Srichomthong C, Siriwan P, Suphapeetiporn K, Shotelersuk V. PDGFRa mutations in humans with isolated cleft palate. Eur J Hum Genet. 2012;20:1058–1062. doi: 10.1038/ejhg.2012.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson RJ, Dixon J, Jiang R, Dixon MJ. Integration of IRF6 and Jagged2 signalling is essential for controlling palatal adhesion and fusion competence. Hum Mol Genet. 2009;18:2632–2642. doi: 10.1093/hmg/ddp201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson RJ, Dixon J, Malhotra S, Hardman MJ, Knowles L, Boot-Handford RP, Shore P, Whitmarsh A, Dixon MJ. Irf6 is a key determinant of the keratinocyte proliferation-differentiation switch. Nat Genet. 2006;38:1329–1334. doi: 10.1038/ng1894. [DOI] [PubMed] [Google Scholar]
- Richardson RJ, Hammond NL, Coulombe PA, Saloranta C, Nousiainen HO, Salonen R, Berry A, Hanley N, Headon D, Karikoski R, Dixon MJ. Periderm prevents pathological epithelial adhesions during embryogenesis. J Clin Invest. 2014;124:3891–3900. doi: 10.1172/JCI71946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson GW, Mahon KA. Differential and overlapping expression domains of Dlx-2 and Dlx-3 suggest distinct roles for Distal-less homeobox genes in craniofacial development. Mech Dev. 1994;48:199–215. doi: 10.1016/0925-4773(94)90060-4. [DOI] [PubMed] [Google Scholar]
- Robu ME, Larson JD, Nasevicius A, Beiraghi S, Brenner C, Farber SA, Ekker SC. p53 activation by knockdown technologies. PLoS Genet. 2007;3:e78. doi: 10.1371/journal.pgen.0030078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rochard L, Monica SD, Ling IT, Kong Y, Roberson S, Harland R, Halpern M, Liao EC. Roles of Wnt pathway genes wls, wnt9a, wnt5b, frzb and gpc4 in regulating convergent-extension during zebrafish palate morphogenesis. Development. 2016;143:2541–2547. doi: 10.1242/dev.137000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rochard LJ, Ling IT, Kong Y, Liao EC. Visualization of Chondrocyte Intercalation and Directional Proliferation via Zebrabow Clonal Cell Analysis in the Embryonic Meckel's Cartilage. J Vis Exp. 2015:e52935. doi: 10.3791/52935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooryck C, Diaz-Font A, Osborn DP, Chabchoub E, Hernandez-Hernandez V, Shamseldin H, Kenny J, Waters A, Jenkins D, Kaissi AA, Leal GF, Dallapiccola B, Carnevale F, Bitner-Glindzicz M, Lees M, Hennekam R, Stanier P, Burns AJ, Peeters H, Alkuraya FS, Beales PL. Mutations in lectin complement pathway genes COLEC11 and MASP1 cause 3MC syndrome. Nat Genet. 2011;43:197–203. doi: 10.1038/ng.757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross CM. Generalized folded skin with an underlying lipomatous nevus "The Michelin Tire baby". Arch Dermatol. 1969;100:320–323. [PubMed] [Google Scholar]
- Rossi A, Kontarakis Z, Gerri C, Nolte H, Holper S, Kruger M, Stainier DY. Genetic compensation induced by deleterious mutations but not gene knockdowns. Nature. 2015;524:230–233. doi: 10.1038/nature14580. [DOI] [PubMed] [Google Scholar]
- Saadi I, Alkuraya FS, Gisselbrecht SS, Goessling W, Cavallesco R, Turbe-Doan A, Petrin AL, Harris J, Siddiqui U, Grix AW, Jr, Hove HD, Leboulch P, Glover TW, Morton CC, Richieri-Costa A, Murray JC, Erickson RP, Maas RL. Deficiency of the cytoskeletal protein SPECC1L leads to oblique facial clefting. Am J Hum Genet. 2011;89:44–55. doi: 10.1016/j.ajhg.2011.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabel JL, d'Alencon C, O'Brien EK, Otterloo EV, Lutz K, Cuykendall TN, Schutte BC, Houston DW, Cornell RA. Maternal Interferon Regulatory Factor 6 is required for the differentiation of primary superficial epithelia in Danio and Xenopus embryos. Dev Biol. 2009;325:249–262. doi: 10.1016/j.ydbio.2008.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw ND, Brand H, Kupchinsky ZA, Bengani H, Plummer L, Jones TI, Erdin S, Williamson KA, Rainger J, Stortchevoi A, Samocha K, Currall BB, Dunican DS, Collins RL, Willer JR, Lek A, Lek M, Nassan M, Pereira S, Kammin T, Lucente D, Silva A, Seabra CM, Chiang C, An Y, Ansari M, Rainger JK, Joss S, Smith JC, Lippincott MF, Singh SS, Patel N, Jing JW, Law JR, Ferraro N, Verloes A, Rauch A, Steindl K, Zweier M, Scheer I, Sato D, Okamoto N, Jacobsen C, Tryggestad J, Chernausek S, Schimmenti LA, Brasseur B, Cesaretti C, Garcia-Ortiz JE, Buitrago TP, Silva OP, Hoffman JD, Muhlbauer W, Ruprecht KW, Loeys BL, Shino M, Kaindl AM, Cho CH, Morton CC, Meehan RR, van Heyningen V, Liao EC, Balasubramanian R, Hall JE, Seminara SB, Macarthur D, Moore SA, Yoshiura KI, Gusella JF, Marsh JA, Graham JM, Jr, Lin AE, Katsanis N, Jones PL, Crowley WF, Jr, Davis EE, FitzPatrick DR, Talkowski ME. SMCHD1 mutations associated with a rare muscular dystrophy can also cause isolated arhinia and Bosma arhinia microphthalmia syndrome. Nat Genet. 2017;49:238–248. doi: 10.1038/ng.3743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheehan-Rooney K, Palinkasova B, Eberhart JK, Dixon MJ. A cross-species analysis of Satb2 expression suggests deep conservation across vertebrate lineages. Dev Dyn. 2010;239:3481–3491. doi: 10.1002/dvdy.22483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen X, Liu RM, Yang L, Wu H, Li PQ, Liang YL, Xie XD, Yao T, Zhang TT, Yu M. The CRISPLD2 gene is involved in cleft lip and/or cleft palate in a Chinese population. Birth Defects Res A Clin Mol Teratol. 2011;91:918–924. doi: 10.1002/bdra.20840. [DOI] [PubMed] [Google Scholar]
- Shi J, Jiao X, Song T, Zhang B, Qin C, Cao F. CRISPLD2 polymorphisms are associated with non-syndromic cleft lip with or without cleft palate in a northern Chinese population. Eur J Oral Sci. 2010;118:430–433. doi: 10.1111/j.1600-0722.2010.00743.x. [DOI] [PubMed] [Google Scholar]
- Sisson BE, Topczewski J. Expression of five frizzleds during zebrafish craniofacial development. Gene Expr Patterns. 2009;9:520–527. doi: 10.1016/j.gep.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song H, Wang X, Yan J, Mi N, Jiao X, Hao Y, Zhang W, Gao Y. Association of single-nucleotide polymorphisms of CDH1 with nonsyndromic cleft lip with or without cleft palate in a northern Chinese Han population. Medicine (Baltimore) 2017;96:e5574. doi: 10.1097/MD.0000000000005574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soriano P. The PDGF alpha receptor is required for neural crest cell development and for normal patterning of the somites. Development. 1997;124:2691–2700. doi: 10.1242/dev.124.14.2691. [DOI] [PubMed] [Google Scholar]
- Swartz ME, Sheehan-Rooney K, Dixon MJ, Eberhart JK. Examination of a palatogenic gene program in zebrafish. Dev Dyn. 2011;240:2204–2220. doi: 10.1002/dvdy.22713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swindell EC, Yuan Q, Maili LE, Tandon B, Wagner DS, Hecht JT. Crispld2 is required for neural crest cell migration and cell viability during zebrafish craniofacial development. Genesis. 2015;53:660–667. doi: 10.1002/dvg.22897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan TY, Kilpatrick N, Farlie PG. Developmental and genetic perspectives on Pierre Robin sequence. Am J Med Genet C Semin Med Genet. 2013;163C:295–305. doi: 10.1002/ajmg.c.31374. [DOI] [PubMed] [Google Scholar]
- Thisse B, Pflumio S, Furthauer M, Loppin B, Heyer V, Degrave A, Woehl R, Lux A, Steffan T, Charbonnier XQ, Thisse C. Expression of the zebrafish genome during embryogenesis (NIH R01 RR15402).. In 2001 [Google Scholar]
- Thisse B, Thisse C. Fast release clones: a high throughput expression analysis. ZFIN direct data submission. 2004;2 [Google Scholar]
- Thisse C, Thisse B. High-resolution in situ hybridization to whole-mount zebrafish embryos. Nat Protoc. 2008;3:59–69. doi: 10.1038/nprot.2007.514. [DOI] [PubMed] [Google Scholar]
- Titomanlio L, Bennaceur S, Bremond-Gignac D, Baumann C, Dupuy O, Verloes A. Michels syndrome, Carnevale syndrome, OSA syndrome, and Malpuech syndrome: variable expression of a single disorder (3MC syndrome)? Am J Med Genet A. 2005;137A:332–335. doi: 10.1002/ajmg.a.30878. [DOI] [PubMed] [Google Scholar]
- Urquhart J, Roberts R, de Silva D, Shalev S, Chervinsky E, Nampoothiri S, Sznajer Y, Revencu N, Gunasekera R, Suri M, Ellingford J, Williams S, Bhaskar S, Clayton-Smith J. Exploring the genetic basis of 3MC syndrome: Findings in 12 further families. Am J Med Genet A. 2016;170A:1216–1224. doi: 10.1002/ajmg.a.37564. [DOI] [PubMed] [Google Scholar]
- Van Der Woude A. Fistula labii inferioris congenita and its association with cleft lip and palate. Am J Hum Genet. 1954;6:244–256. [PMC free article] [PubMed] [Google Scholar]
- Van Laarhoven PM, Neitzel LR, Quintana AM, Geiger EA, Zackai EH, Clouthier DE, Artinger KB, Ming JE, Shaikh TH. Kabuki syndrome genes KMT2D and KDM6A: functional analyses demonstrate critical roles in craniofacial, heart and brain development. Hum Mol Genet. 2015;24:4443–4453. doi: 10.1093/hmg/ddv180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada N, Javidan Y, Nelson S, Carney TJ, Kelsh RN, Schilling TF. Hedgehog signaling is required for cranial neural crest morphogenesis and chondrogenesis at the midline in the zebrafish skull. Development. 2005;132:3977–3988. doi: 10.1242/dev.01943. [DOI] [PubMed] [Google Scholar]
- Wang S, Samakovlis C. Grainy head and its target genes in epithelial morphogenesis and wound healing. Curr Top Dev Biol. 2012;98:35–63. doi: 10.1016/B978-0-12-386499-4.00002-1. [DOI] [PubMed] [Google Scholar]
- Wang Z, Qin G, Zhao TC. HDAC4: mechanism of regulation and biological functions. Epigenomics. 2014;6:139–150. doi: 10.2217/epi.13.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner AM, Scampoli NL, Calcaterra NB. Fishing the molecular bases of Treacher Collins syndrome. PLoS One. 2012;7:e29574. doi: 10.1371/journal.pone.0029574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wienholds E, Kloosterman WP, Miska E, Alvarez-Saavedra E, Berezikov E, de Bruijn E, Horvitz HR, Kauppinen S, Plasterk RH. MicroRNA expression in zebrafish embryonic development. Science. 2005;309:310–311. doi: 10.1126/science.1114519. [DOI] [PubMed] [Google Scholar]
- Williams SR, Aldred MA, Der Kaloustian VM, Halal F, Gowans G, McLeod DR, Zondag S, Toriello HV, Magenis RE, Elsea SH. Haploinsufficiency of HDAC4 causes brachydactyly mental retardation syndrome, with brachydactyly type E, developmental delays, and behavioral problems. Am J Hum Genet. 2010;87:219–228. doi: 10.1016/j.ajhg.2010.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson NR, Olm-Shipman AJ, Acevedo DS, Palaniyandi K, Hall EG, Kosa E, Stumpff KM, Smith GJ, Pitstick L, Liao EC, Bjork BC, Czirok A, Saadi I. SPECC1L deficiency results in increased adherens junction stability and reduced cranial neural crest cell delamination. Sci Rep. 2016;6:17735. doi: 10.1038/srep17735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf ZT, Leslie EJ, Arzi B, Jayashankar K, Karmi N, Jia Z, Rowland DJ, Young A, Safra N, Sliskovic S, Murray JC, Wade CM, Bannasch DL. A LINE-1 insertion in DLX6 is responsible for cleft palate and mandibular abnormalities in a canine model of Pierre Robin sequence. PLoS Genet. 2014;10:e1004257. doi: 10.1371/journal.pgen.1004257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wouters L, Rodriguez Rodriguez CM, Dapena EP, Poorten VV, Devriendt K, Van Esch H. Circumferential skin creases, cleft palate, typical face, intellectual disability and growth delay: "circumferential skin creases Kunze type". Eur J Med Genet. 2011;54:236–240. doi: 10.1016/j.ejmg.2011.01.003. [DOI] [PubMed] [Google Scholar]
- Wu D, Mandal S, Choi A, Anderson A, Prochazkova M, Perry H, Gil-Da-Silva-Lopes VL, Lao R, Wan E, Tang PL, Kwok PY, Klein O, Zhuan B, Slavotinek AM. DLX4 is associated with orofacial clefting and abnormal jaw development. Hum Mol Genet. 2015;24:4340–4352. doi: 10.1093/hmg/ddv167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyszynski DF, Albacha-Hejazi H, Aldirani M, Hammod M, Shkair H, Karam A, Alashkar J, Holmes TN, Pugh EW, Doheny KF, McIntosh I, Beaty TH, Bailey-Wilson JE. A genome-wide scan for loci predisposing to non-syndromic cleft lip with or without cleft palate in two large Syrian families. Am J Med Genet A. 2003;123A:140–147. doi: 10.1002/ajmg.a.20283. [DOI] [PubMed] [Google Scholar]
- Xu X, Bringas P, Jr, Soriano P, Chai Y. PDGFR-alpha signaling is critical for tooth cusp and palate morphogenesis. Dev Dyn. 2005;232:75–84. doi: 10.1002/dvdy.20197. [DOI] [PubMed] [Google Scholar]
- Yan YL, Miller CT, Nissen RM, Singer A, Liu D, Kirn A, Draper B, Willoughby J, Morcos PA, Amsterdam A, Chung BC, Westerfield M, Haffter P, Hopkins N, Kimmel C, Postlethwait JH. A zebrafish sox9 gene required for cartilage morphogenesis. Development. 2002;129:5065–5079. doi: 10.1242/dev.129.21.5065. [DOI] [PubMed] [Google Scholar]
- Yu Y, Zuo X, He M, Gao J, Fu Y, Qin C, Meng L, Wang W, Song Y, Cheng Y, Zhou F, Chen G, Zheng X, Wang X, Liang B, Zhu Z, Fu X, Sheng Y, Hao J, Liu Z, Yan H, Mangold E, Ruczinski I, Liu J, Marazita ML, Ludwig KU, Beaty TH, Zhang X, Sun L, Bian Z. Genome-wide analyses of non-syndromic cleft lip with palate identify 14 novel loci and genetic heterogeneity. Nat Commun. 2017;8:14364. doi: 10.1038/ncomms14364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Q, Chiquet BT, Devault L, Warman ML, Nakamura Y, Swindell EC, Hecht JT. Craniofacial abnormalities result from knock down of nonsyndromic clefting gene, crispld2, in zebrafish. Genesis. 2012;50:871–881. doi: 10.1002/dvg.22051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarate YA, Perry H, Ben-Omran T, Sellars EA, Stein Q, Almureikhi M, Simmons K, Klein O, Fish J, Feingold M, Douglas J, Kruer MC, Si Y, Mao R, McKnight D, Gibellini F, Retterer K, Slavotinek A. Further supporting evidence for the SATB2-associated syndrome found through whole exome sequencing. Am J Med Genet A. 2015;167A:1026–1032. doi: 10.1002/ajmg.a.36849. [DOI] [PubMed] [Google Scholar]
- Zehir A, Hua LL, Maska EL, Morikawa Y, Cserjesi P. Dicer is required for survival of differentiating neural crest cells. Dev Biol. 2010;340:459–467. doi: 10.1016/j.ydbio.2010.01.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C, Andreeva V, Gibert Y, LaBonty M, Lattanzi V, Prabhudesai S, Zhou Y, Zon L, McCann KL, Baserga S, Yelick PC. Tissue specific roles for the ribosome biogenesis factor Wdr43 in zebrafish development. PLoS Genet. 2014;10:e1004074. doi: 10.1371/journal.pgen.1004074. [DOI] [PMC free article] [PubMed] [Google Scholar]