

Abstract
Microglia, the resident immune cells of the CNS, are primary regulators of the neuroimmune response to injury. Type I interferons (IFNs), including the IFNαs and IFNβ, are key cytokines in the innate immune system. Their activity is implicated in the regulation of microglial function both during development and in response to neuroinflammation, ischemia, and neurodegeneration. Data from numerous studies in multiple sclerosis (MS) and stroke suggest that type I IFNs can modulate the microglial phenotype, influence the overall neuroimmune milieu, regulate phagocytosis, and affect blood–brain barrier integrity. All of these IFN-induced effects result in numerous downstream consequences on white matter pathology and microglial reactivity. Dysregulation of IFN signaling in mouse models with genetic deficiency in ubiquitin specific protease 18 (USP18) leads to a severe neurological phenotype and neuropathological changes that include white matter microgliosis and pro-inflammatory gene expression in dystrophic microglia. A class of genetic disorders in humans, referred to as pseudo-TORCH syndrome (PTS) for the clinical resemblance to infection-induced TORCH syndrome, also show dysregulation of IFN signaling, which leads to severe neurological developmental disease. In these disorders, the excessive activation of IFN signaling during CNS development results in a destructive interferonopathy with similar induction of microglial dysfunction as seen in USP18 deficient mice. Other recent studies implicate “microgliopathies” more broadly in neurological disorders including Alzheimer’s disease (AD) and MS, suggesting that microglia are a potential therapeutic target for disease prevention and/or treatment, with interferon signaling playing a key role in regulating the microglial phenotype.
Keywords: Microglia, Interferons, White matter, Usp18, Pseudo-TORCH syndrome
Introduction
Microglia are key components in the innate immune system of the central nervous system (CNS) [1, 2], however recent studies also suggest that microglia have vital functions in development [3, 4] and homeostasis of the CNS [5–7]. Microglia are produced from a subset of yolk sac cells originating early during embryonic development that migrate into the brain [8–10], and display a unique transcriptome that distinguishes them from both CNS and peripheral immune cells [11]. Developmental functions of microglia include refinement of synaptic networks, promotion of developmental apoptosis, removal of apoptotic cell corpses, positioning of neurons within the developing cortex, and precise secretion of growth factors for neuronal survival (reviewed by Ransohoff [12]). Microglial contributions to adult brain function include adjusting neuronal firing rates to optimize neural networks and producing neuromodulatory factors that support synaptic plasticity [12]. Under normal physiologic conditions, microglia are characterized by a small cell body with fine, ramified processes and low expression of surface antigens [13]. In the unperturbed brain, microglial processes are dynamic structures with constant changes in process length and position [14]. Microglia express an armamentarium of genes (microglia sensome) encoding surface receptor proteins of various types to sense their local microenvironment. These genes include pattern recognition, chemokine/cytokine, purinergic, and Fc receptors among others [15]. Upon encountering signals from invading pathogens or damaged cells, microglia rapidly undergo marked phenotypic, metabolic and gene expression changes [16, 17]. In many cases these encounters induce a transient classical ‘activation’ state in microglia with release of specific pro-inflammatory chemokines and cytokines [18–20] as well as marked morphological [13] and functional changes [16, 17]. Over time, the microglial transcriptomic and phenotypic responses to a stimulus may evolve toward a more immunomodulatory, anti-inflammatory or even neuroprotective state [21, 22]. Aging, neurological disease, and neurodegeneration also trigger microglial reactivity and induce distinctive changes in the microglial transcriptome [23]. Microarray analysis of ex vivo sorted microglia have identified a common microglial gene expression signature in mouse models of neurodegeneration and aging [24, 25], which prominently features expression of type 1 interferon stimulated genes (ISGs) [24].
Type 1 interferons (IFNs), key cytokines in the innate immune system, include 13 IFNα subtypes and IFNβ, all of which signal through the IFNAR receptor complex [26, 27]. The type I IFN surface receptor is a heterodimer consisting of IFNAR1 and IFNAR2, that are associated with tyrosine kinases Janus kinase 1 (Jak1) and Tyk2 (Fig. 1). Receptor binding by type I IFNs brings the IFNAR1 and IFNAR2 subunits together and results in cross-phosphorylation and activation of Jak1 and Tyk2, and subsequent phosphorylation of STAT2, which then associates with STAT1 through an SH2-p-Tyr interaction, leading to the phosphorylation of STAT1. Phosphorylated STAT1 and STAT2 form a complex with IFN regulatory factor 9 (IRF9) called ISGF3. The ISGF3 complex then binds to IFN-stimulated response elements (ISRE) in promoter regions of multiple genes and stimulates type I IFN-dependent gene transcription. Proteins encoded by interferon-stimulated genes (ISGs) include ISG15, MX1, IRF8, IFIT proteins, MX2, USP18, IFI27, CXCL10, CCL8, and CCL5 [28–30]. Type I IFNs have numerous immunomodulatory functions in both the innate and adaptive immune systems [29]. Although type 1 IFNs are classically up-regulated in response to viral infection, recent studies have implicated them as key regulators of the neuroimmune response triggered by non-infectious causes of CNS injury [29, 31, 32] and type I IFNs are also important in experimental neuroprotective phenomena such as LPS preconditioning [33].
Fig. 1.
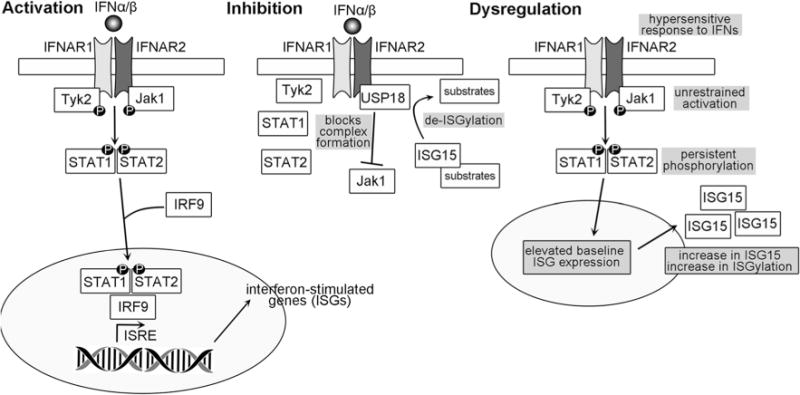
Type I Interferon signaling pathway. Activation—IFNα or IFNβ binding to the IFNAR receptor complex, a heterodimer consisting of IFNAR1 and IFNAR2, induces cross-phosphorylation of the proteins Tyk2 and Jak1, which are associated with IFNAR1 and IFNAR2, respectively. Subsequently, STAT1 and STAT2 are phosphorylated and associate with IRF9 to form a transcriptional complex that translocates into the nucleus and interacts with interferon-stimulated response elements (ISRE) to induce transcription of interferon-stimulated genes (ISGs) including isg15, mx1, usp18 and ifit1. Inhibition—Interferon signaling is restrained or inhibited by USP18, which associates with IFNAR2 and competes for binding with Jak1, thus blocking the association between IFNAR and Jak1 and preventing phosphorylation and the downstream cascades. Dysregulation—In the mouse USP18 KO and in human patients with deficient USP18, the interferon signaling pathway is dysregulated at several levels. INFAR complex formation and activation is unrestrained in the absence of signal, and hypersensitized to inflammatory events that produce low levels of IFNs. This results in persistent phosphorylation of STAT1 and STAT2 and elevated ISG expression. ISG15 accumulates as a consequence of increased transcription and is not cleaved from ISGylated proteins. Consequently, microglia are activated and secrete pro-inflammatory cytokines to induce CNS inflammation. Clinical outcomes of this observed in humans and mice are hydrocephalus, microcephaly, white matter microgliopathy, and early death
Microglia-related genes have now been associated with a number of specific neurological and neuropsychiatric disorders [34]. In particular, genetic variations in cd33 [35, 36], trem2 [37, 38], and apoE [39], all three of which encode proteins expressed in microglia and involved in Aβ clearance, are risk factors for Alzheimer’s disease (AD) [40, 41]. For example, TREM2 is a microglial specific protein, which binds to apolipoproteins, including APOE, and facilitates Aβ clearance [41]. However, a large meta-analysis of many AD patients also revealed several other susceptibility loci that impact other genes related to the innate immune functions of microglia including cr1, hla-drb5, clu, abca7 [42]. These genes have multiple and varied effects on the ability of microglia to phagocytose, metabolize lipids, and engage in critical cell signaling processes [43]. Loss-of-function mutations in the microglia-specific genes dap12 or trem2 cause polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy (Nasu-Hakola disease) [44]. This disorder is characterized by psychotic symptoms and dementia and is associated with diffuse abnormalities in CNS white matter [44]. Another neurological disorder with marked involvement of CNS white matter is hereditary diffuse leukoencephalopathy with spheroids (HDLS). HDLS is caused by mutations in the csf1r gene that encodes the tyrosine protein kinase that acts as cellular receptor for both colony stimulating factor-1 and IL-34 [45, 46]. CSF1R plays an essential role in hematopoetic precursor cell development, particularly cells of myeloid lineage, and inhibition of CSF1R signaling results in rapid and dramatic depletion of CNS microglia [47]. Prolonged treatment with a CSF1R antagonist prevented neuronal loss in a mouse model of AD [48] and improved functional outcome following extensive neuronal loss in the hippocampus [49]. Interestingly, CSF1R antagonist pre-treatment exacerbated post-stroke inflammation and brain infarction [50]. These seemingly divergent effects suggest that microglia play variable roles in the CNS injury response, and treatments must be carefully tailored to the specific disease or injury context. Other genes encoding microglial gene products, including TNFRSF1A and IRF8, have been identified at susceptibility loci in multiple sclerosis (MS) [51]. The latter of these molecules is an interferon regulatory factor (IRF), and there is growing evidence for a strong connection between interferon signaling, microglial dysfunction, and neurological disease, which will be discussed within this review.
Type I Interferon Signaling in Microglia Modulates Neuroimmune Response to CNS Injury
Microglia may play a role in the clearance of myelin debris through their phagocytic activity, and may also provide support to oligodendrocytes and axons. Disruptions in microglial activation may result in deficits within these roles, leading to detrimental effects within white matter. Activated microglia are characteristic in many neuroinflammatory disorders and have been observed early in the process of CNS inflammation, often before the onset of disease symptoms and infiltration of peripheral immune cells into the CNS [32, 52]. Once activated, microglia can exhibit phagocytosis activity, which can be modulated by type I IFNs in differing ways. Type I IFNs increase phagocytosis, including the phagocytosis of T cells [53]. Clearance of infiltrating immune cells is necessary for resolution of an inflammatory episode. Additionally, type I IFNs mediate the pruning of degenerating axons [54], which may be beneficial for maintenance of homeostasis. Clearance of degraded myelin, apoptotic cells, and cellular debris is necessary for the reduction of inflammation and facilitates axonal outgrowth, but may also result in worsening disease progression in some scenarios due to overpruning of healthy synapses [55, 56], suggesting that tight regulation of phagocytosis is necessary for a balance between beneficial clearance and overactive phagocytosis resulting in pathology. The modulatory effects of type I IFNs on microglial phagocytosis suggest potential therapeutic benefit in targeting this pathway in diseases where aberrant microglial phagocytosis of healthy synapses [55] can lead to the progression of neurodegenerative diseases.
Interestingly, Toll-like receptor (TLR)-4 signaling appears to also be required for type I IFN mediated clearance of axonal debris [57], which suggests that multiple innate immune pathways are critical in the normal functioning of microglia and in regulating their responses to injury and inflammation. The molecular basis for glial activation typically involves TLR signaling, which can induce type 1 IFNs under the appropriate stimulus [19, 20, 31]. TLRs are capable of recognizing a number of structural motifs in viral and microbial RNA and DNA, pathogen-associated molecular patterns (PAMPs), as well as endogenous damage-associated molecular patterns (DAMPs). The latter group includes factors such as heat shock protein-70 (HSP70) and fibronectin [58]. In microglia, TLR4 is highly expressed, as is its co-receptor, CD14 [58, 59]. CD14 is required for TLR4-agonist induced production of IFNβ, and CD14-dependent IFNβ induction in turn regulates TLR4 activity and curbs excessive pro-inflammatory reactions in microglia [58]. Recent studies suggest that under certain conditions, HSP70 can stimulate IFNβ production by microglia via TLR2 and/or TLR4 signaling [60]. Innate immune pathways, including TLRs [59, 61, 62] and type 1 IFN signaling [33, 61, 63], are furthermore implicated in neuroprotective responses to ischemia and stroke. Of special note, TLR3 activation strongly induces production of type 1 IFNs by glial cells [19, 20] and we show here that TLR3 and TLR4 agonists can act synergistically in vitro to induce robust release of IFNα from microglia (Fig. 2). These data suggest that the outcome of activating certain innate immune pathways, for example TLR4 alone by specific DAMPs released due to injury, may be modulated by treatments that activate synergistic TLRs, TLR3 in this example, to change the inflammatory milieu to a neuroprotective phenotype. Furthermore, such in vitro studies shed light on the complexities of an in vivo system, where multiple innate immune pathways may be engaged, to various outcomes that may be unexpected based on simple in vitro studies examining the activation or inhibition of a sole TLR. A recent in vivo study on the rat spinal cord suggests that a TLR4 agonist promotes macrophage activation and phagocytosis of debris after a demyelinating insult, oligodendrocyte precursor proliferation, and remyelination of damaged axons in the spinal cord white matter [64]. The implications of these studies are that microglia could be engaged through TLR-signaling, perhaps targeting TLR3 and TLR4, in the brain to perform similar functions to promote the remyelination of white matter lesions.
Fig. 2.
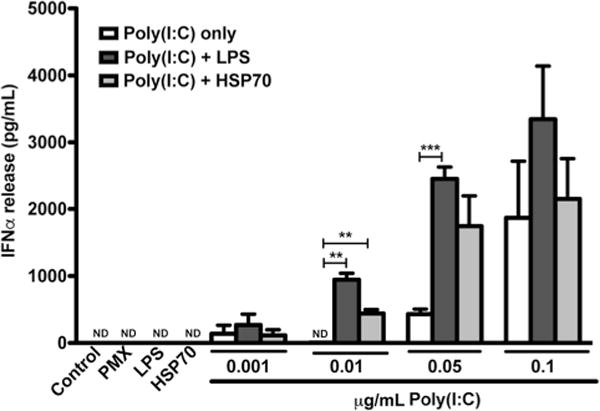
The TLR3 agonist, poly I:C, induces IFNα release and exhibits synergy with TLR4 agonists lipopolysaccharide (LPS) and heat shock protein 70 (HSP). No IFNα release could be detected from primary cultured microglia under control conditions or after treatment with LPS inhibitor polymyxin b (PMX). Nor could IFNα release be detected upon stimulation with TLR4 agonists alone (LPS, HSP70). Primary microglia stimulated with poly I:C alone did release IFNα. Stimulation with poly(I:C) and LPS (TLR3 and TLR4 co-stimulation) synergistically enhanced the release of IFNα in a dose dependent manner that reached a plateau effect, as did treatment with both poly I:C and HSP70 at specific doses. Methods—Primary microglia were harvested and cultured as previously described [65, 66]. Microglia were treated with indicated concentrations (μg/mL) of poly I:C (InvivoGen, San Diego, CA), 100 EU/mL LPS (Control Standard Endo-toxin; Associates of Cape Cod, E. Falmouth, MA), 10 μg/mL HSP70 (BioVision, Milpitas, CA), and/or 10 μg/mL polymyxin b (Sigma, St. Louis, MO). Media from cell culture was collected and IFNα levels determined by ELISA (R&D Systems, Minneapolis, MN)
In experimental autoimmune encephalomyelitis (EAE), a mouse model widely used to gain insights into the pathophysiology of MS and other demyelinating diseases with white matter effects, interferon dysregulation has been reported. Both EAE and MS result in inflammation and demyelination of axons in the CNS. Multiple clinical studies of MS patients have demonstrated conclusively that IFNβ reduces the frequency of clinical events and delays the progression of disability in relapsing-remitting MS, however the treatment is not without side effects [67]. In vivo mouse studies in EAE aiming to elucidate the mechanisms behind IFNβ treatment and response in MS patients have revealed early onset of local IFNβ production, and ISG expression, in the CNS shortly after inoculation of animals with a MOG-peptide to induce EAE [32]. IFNAR1 deficient animals, immunized with MOG to induce EAE, exhibited earlier onset of symptoms, increased infiltration of macrophages into the CNS, exacerbated symptoms and increased mortality compared to immunized WT animals [32]. Microglial cells were strongly implicated as the primary responders to IFNβ signaling in this study, with type I IFNs heavily modulating the activation state of microglia [32]. The timing of these effects suggests that IFNβ is an early response to neuroinflammation, with the data from the IFNAR1 deficient animals implying that it is neuroprotective and may delay onset of symptoms. The contribution of IFNβ facilitates a transition from a pro-inflammatory to a regulatory immune environment, thus ameliorating symptoms of MS. Additional mouse studies on the suppressive effect that administration of the TLR3 agonist poly I:C has on EAE suggest that this effect is mediated by induction of type I IFNs [68]. In this study, myeloid cells in the meninges and choroid plexus, parenchymal microglia, and CD45high leukocytes produced type I IFNs in response to poly I:C exposure, and consequently microglia and astrocytes up-regulated the ISG products IRF7, IRF9, and CXCL10 [68]. Interestingly, microglia expressed higher levels of IRF7 than astrocytes [68], suggesting a greater response and engagement of the interferon signaling machinery in microglia. Our results showing a synergistic effect of TLR3/TLR4 on type I IFN production (Fig. 2) suggest that TLR3-type 1 IFN mediated effects, such as the ones described above in EAE, could be enhanced by addition of other TLR agonists in a form of combination therapy.
As noted above, IFNAR1 deficient animals that had been inoculated with MOG-peptide to induce EAE had increased infiltration of macrophages into the CNS [32]. While this may be due, in part, to regulation of cytokine/chemokine production, which may enhance chemotaxis of peripheral immune cells into the CNS, there is also a growing body of evidence that suggests IFNβ is capable of affecting the integrity of the blood brain barrier (BBB). Studies on the effects of type I IFN treatment in MS suggest that IFNβ stabilizes the BBB, which results in reduced infiltration of peripheral immune cells into the CNS [69–71]. Similarly, in a model of ischemic stroke, IFNβ was found to promote integrity of the BBB, block infiltration of immune cells, and reduce infarct volume [72]. IFNβ knock-out (KO) mice showed a higher accumulation of leukocytes in the brain after middle cerebral artery occlusion (MCAO) [73], as did IFNAR1 KO mice [31], suggesting increased permeability of the BBB to cellular infiltrates in the absence of intact type 1 IFN signaling. These effects of type I IFNs on the BBB integrity may have several consequences for diseases or injury with a marked white matter component. A growing body of data suggests that peripheral immune cells may be the primary effectors of axonal damage in MS and EAE [74], and therapies that reduce the infiltration of peripheral immune cells after ischemic stroke show improved post-stroke outcomes and recovery in rats [75, 76]. Thus treatment of disease or injury of white matter pathology with type I IFNs may have multiple positive effects on disease progression or inflammatory resolution, not only through microglial-dependent mechanisms, but also by preserving the BBB integrity to prevent peripheral immune cell infiltration. The peripheral immune cells produce large quantities of cytokines, many regarded as “neurotoxic” [77–79], which affect not only oligodendrocytes and axons, but also influence microglial activity, which can have further negative effects on oligodendrocytes and axonal integrity.
Recent publications suggest that microglia could play a protective role in stroke [80–83], with interferon pathways strongly implicated in microglial responses to ischemia. In a study comparing WT and IFNβ KO mice, there were no significant differences in infarct volume after MCAO (stroke) [33], suggesting that IFNβ is not an outcome-determining factor in the acute stroke response. However, when IFNβ was administered immediately prior to MCAO, but not immediately after MCAO, there was a 35% reduction in infarct volume [33], suggesting that type 1 IFNs are neuroprotective against ischemia. The critical difference that these studies highlight is that timing of interferon administration or expression is important. However, the lack of improvement following administration of IFNβ to wild-type mice after stroke as well as the finding that IFNβ deficient mice show similar outcomes following stroke, suggests that the role of IFNβ in the acute phase of stroke is not an outcome-determining parameter. Instead, we posit that type I IFNs may play a role in the subacute and chronic phases of stroke recovery, promoting resolution of inflammation and a quiescence of microglial and peripheral immune cells. Studies by our lab and others also suggest that type I IFNs play a role prior to stroke by inducing neuro- or axonal-protection, and thus type I IFNs may be used to induce a preconditioning effect.
Ischemic preconditioning (IPC) is a brief period of ischemia that confers robust neuroprotection against subsequent prolonged ischemic events, including stroke [84–86]. It is important to note that preconditioning does not prevent stroke, but ameliorates the response and results in a smaller infarct volume and improved post-stroke recovery [33, 87, 88]. IPC induces a reprogramming of the transcriptional response to stroke, which results in a neuroprotective phenotype, rather than the destructive inflammatory phenotype observed after stroke alone [87–89]. The mechanisms behind IPC are complex and many, and have been reviewed extensively elsewhere [90]. Of note, however, is that TLRs and type I IFNs are specifically and strongly implicated in the establishment of IPC in many studies [33, 61–63, 91], which implies that immune cells, and microglia by extension, may be important in this phenomenon. Using a model of white matter preconditioning, our lab has been able to demonstrate that interferon signaling in microglia is required for IPC. Following an IPC stimulus, optic nerves, a pure white matter tract, were collected and subjected to oxygen-glucose deprivation (“ischemic stroke-like conditions”) [61]. As parameters for evaluating the preconditioning effect in this ex vivo model, compound action potentials (CAPs) and axonal integrity were measured and found to be improved in preconditioned mouse optic nerves, and neurofilament staining measures of axonal integrity more closely resembled sham controls than non-preconditioned optic nerves [61]. These preconditioning effects were abolished entirely in ex vivo optic nerve preparations from TLR4 KO and IFNAR1 KO mice. When IFNAR1fl/fl mice were crossed with LysMCre mice to create a myeloid cell-targeted knockdown of IFNAR1, the preconditioning effect was also abolished [61]. These data strongly suggest that IPC-mediated neuroprotection is dependent on interferon signaling specifically in microglia in a white matter model of ischemia. Another recent study also strongly suggests that microglia are neuroprotective in the post-stroke brain. When microglia were depleted by a drug targeting CSF1R, post-ischemic inflammation and infarction were exacerbated [50]. Taken together, these data suggest that context is also important. While microglial phagocytosis may become dysregulated and lead to the pruning of healthy synapses resulting in pathology [55, 56], in the post-ischemic brain, microglial functions including regulation of astrocyte activity [50] are necessary for restraining inflammatory processes and preventing tissue damage and neuronal death. As the era of personalized medicine evolves, multiple factors including the timing of drug administration, disease-specific context of treatment, and an individual’s genetic profile will all heavily influence treatment plans for diseases and disorders in which microglia are implicated, either as primary or secondary mechanism of injury or neuroprotection.
Studies on in vivo, endogenous effects of type I interferons and related genes have shed some light on the role these play in the post-stroke brain, and suggest therapeutic targeting of type I interferons and related signaling pathways. Using IFNβ KO mice, Inacio et al. [73] identified a role for endogenous IFNβ signaling in exerting anti-inflammatory actions in experimentally induced focal cerebral ischemia, another model of ischemic stroke. Consistent with these findings, genetic deficiency of CD14, required for microglial production of type I IFNs, results in marked increases in post-MCAO infarct volume [58]. IFNβ modulates inflammatory processes in stroke, in part by suppressing inflammatory cytokine production and reducing microglial activation, resulting in reduced infarct volume and improved neurobehavioral outcomes [92]. Both of these outcomes are important in experimental stroke research. Infarct volume is a measure of cerebral infarction, tissue in which neuronal death has occurred, which is an undesirable consequence of ischemic stroke. Neurobehavioral outcomes are considered a reliable metric of functional outcomes of an experimental therapy. However, neurobehavior and infarct volume may not correlate [93], and a therapy that results in improved neurobehavioral function but no change in infarct volume, may still be considered successful and desirable for patients, for whom post-stroke immobility and disability, as well as cognition and neuropsychiatric status, are leading concerns [94].
Dysregulation of Type I IFN Signaling Leads to White Matter Specific Microgliopathy
Although the activation of microglia is necessary to respond to pathogens and damage signals from injured or apoptotic cells, excessive microglial activation is implicated in the pathogenesis of various neurological disorders [34]. The ability of microglia to restrain inflammation, for example, via the secretion of dampening immunomodulators such as IL-10 and TGF-β [95], is also critical in a variety of neurological disorders. Although, as stated above, the type I IFN response is protective in a variety of models of neurological disease, excessive induction or over-activation of type I IFN signaling may be detrimental as well—and the potency of this effect may be greatest in white matter [96]. In particular, Goldmann et al., recently demonstrated that the ubiquitin-specific protease (USP) 18 (also named UBP43 [97]) is a key molecule imposing microglial quiescence in the white matter. Mice with genetic deficiency in usp18 exhibit a severe neurological phenotype including convulsions, tremor, and loss of balance with early mortality [97]. Gross neuropathological assessment shows marked hydrocephalus secondary to aqueductal stenosis and ependymal cell necrosis associated with early mortality [97]. Goldmann et al. demonstrated that usp18 is abundantly and specifically expressed in white matter microglia [98]. While there were no obvious histopathological abnormalities in the gray matter, a significant increase of Iba1+ microglia was detectable in several white matter regions in USP18 KO mice, and microglia had a white matter selective pro-inflammatory phenotype [98]. Additionally, USP18 KO brains exhibited clusters of microglia in the white matter that strongly resembled the neuropathological state in several human microgliopathies (Table 1) [98]. Furthermore, these mice exhibited constitutive activation of type I interferon signaling pathways resulting in markedly elevated expression of multiple ISGs [98]. The neuropathological state in the USP18 KO mice could be accurately described as a microglia-targeted, white matter specific, type I interferonopathy [98].
Table 1.
Primary and secondary human microgliopathies
| Disease | Clinical hallmarks | Gene(s) | Cited studies |
|---|---|---|---|
| Human diseases in which microgliopathy plays a primary role | |||
| Nasu-Hakola disease | Progressive dementia, multifocal bone cysts | DAP12 or TREM2 | Paloneva et al. [44] |
| Hereditary diffuse leukoencephalopathy with spheroids (HDLS) | White matter disease, personality and behavioral changes, dementia, depression, Parkinsonism, seizures | CSF1R | Nicholson et al. [45], Rademakers et al. [46] |
| Pseudo-TORCH syndrome (PTS), including Aicardi–Goutières syndrome | Microcephaly, white matter disease, cerebral atrophy, CNS calcifications, neonatal seizures | USP18, ISG15, TREX1, RNASEH2(A, B, or C isoforms), SAMHD1, ADAR1, IFIH1 | Goldmann et al. [98], Meuwissen et al. [99], Zhang et al. [100], Crow and Rehwinkel [101], Rice et al. [102] |
| Human diseases exhibiting white matter microgliopathy as a pathological feature | |||
| Multiple sclerosis | Demyelination, muscle weakness, ataxia, vision problems, chronic pain and/or exhaustion | Demyelinating lesions with microglial clusters | Prinz et al. [32], Marziniak and Meuth [67] |
| Alzheimer’s disease | Progressive loss of neurons and synapses leading to cognitive decline | Microglial clustering around plaques | Michailidou et al. [103], Hong et al. [55] |
| Stroke | Brain infarction and neuronal death leading to impaired motor function | Late-stage infarct border clustering of microglia and peripheral immune cells | Michailidou et al. [103], Becker et al. [75], Becker et al. [76] |
| Traumatic brain injury | Highly variable cognitive, emotional, and motor impairments | Complement-enriched microglia clusters | Michailidou et al. [103] |
The mechanisms connecting USP18 to interferon signaling, and how USP18 may affect this pathway, are numerous (Fig. 1). One function of USP18 protein is to proteolytically cleave and remove the conjugated ubiquitin-like modifier ISG15 from multiple cellular target proteins [104]. ISG15 is one of many interferon stimulated gene products [105] and is key in the process of ISGylation, which is implicated in a range of biological activities, including antiviral defense and innate immune system function [97, 105, 106]. As noted above, mice lacking usp18 exhibit dysregulation of ISG15 and injury due to necrosis of ependymal cells concurrent with hydrocephalus and early death [107]. However, these phenotypic alterations were not reversed or improved in isg15/usp18 double KO mice and thus were suggested to be ISG15-independent [97]. USP18 deficiency results in prolonged activation of STAT1, a key regulatory factor in the type 1 IFN signaling pathway (Fig. 1), specifically in white matter microglia and particularly within the context of a challenge to the immune system [96–98]. Goldmann et al., elegantly confirmed the ISG15-independent nature of the prolonged microglial STAT1 activation in white matter by demonstrating the absence of these findings in mice with a single point mutation in usp18 that replaces a key cysteine residue and renders the protein proteolytically inactive [98]. They further demonstrated that white matter microglia from mice with a different point mutation disrupting the interaction between USP18 and IFNAR showed prolonged STAT1 activation mirroring the interferon signaling findings in the USP18 KO mice [98]. These findings indicated that USP18 restrains type I IFN signaling, and defects in USP18 results in hypersensitized microglia with persistent activation after a stimulus [96, 98].
In humans, maternal exposure to microbial pathogens can cause severe damage and result in TORCH syndrome. Consequences of TORCH syndrome include microcephaly, white matter disease, cerebral atrophy, and calcifications in the CNS. Pseudo-TORCH syndrome (PTS) refers to infants born with a clinical phenotype that matches TORCH, but for which there is a non-infectious cause [99]. The genetic cause in most PTS cases is unknown. However, one known rare Mendelian mimic of congenital infection, overlapping with and falling under the umbrella of PTS, is Aicardi-Goutières syndrome (AGS). AGS is genetically heterogeneous, caused by mutations in any of the following genes: three prime repair exonuclease (trex1), ribonuclease H2 subunits a, b or c (rnaseh2a, b or c), SMA and HD domain containing deoxynucleoside triphosphate triphosphohydrolase 1 (samhd1), adenosine deaminase RNA specific (adar1) or interferon induced with helicase C domain 1 (ifih1) [101, 102]. AGS results from an aberrant accumulation and/or sensing of IFN-stimulatory nucleic acids, which is then misrepresented as viral/non-self by the innate immune machinery, leading to a persistent induction of type 1 IFN-mediated inflammation. Similar increases in type 1 IFN production have also been documented in genetic disorders in which other components of the IFN signaling pathway are constitutively activated [100, 108–110]. This group of disorders includes cases of ISG15 deficiency in humans, which exhibit abnormally strong type I IFN responses and elevated levels of type I IFNs, intracranial calcification, a predisposition towards epileptic seizures, and low levels of USP18 [100]. In another recent study, Meuwissen et al. [99] made clinical observations of two unrelated families affected with PTS demonstrating that patients with a genetic mutation in usp18 displayed a neuropathological and neuroimmune phenotype matching mouse models of usp18 deficiency, including a strong induction of the innate immune system as evidenced by robust induction of activated astrocytes and microglia in patient brains, and high constitutive activation of phosphorylated STAT1 [99]. Fibroblasts derived from patient tissues also demonstrated high levels of ISG transcripts as a consequence of unrestrained IFN signaling [99]. The similarities between USP18 deficient human tissue from PTS brains and fetal brains infected with diseases resulting in TORCH syndrome suggest that timely regulation of IFN signaling is crucial for non-pathological development of the brain in utero. Microglia function as the primary immune cells of the CNS, and they are the primary targets of innate immune signaling molecules, including interferons, in the brain. This concept is supported by recent studies in mouse described above demonstrating the importance of USP18 for modulating IFN signaling in microglia [98].
Neurotherapeutic Targets
Targeting white matter microglia may have translational applications to diseases in which white matter microglia display pathologies. In MS, a frequently reported observation in the brain of donors are clusters of microglial cells within regions of normal appearing white matter at the borders of lesions [103]. These microglial clusters result in the deposition of Complement C3 in chronic MS, and also in late stage stroke and traumatic brain injury (TBI) lesions [103], suggesting that these microglial clusters might be a feature of chronic neurodegeneration following white matter predominant CNS injury. However, further studies are needed to show if these are also present in other neurodegenerative disorders such as Parkinson’s, Amyotrophic Lateral Sclerosis (ALS) and AD. Although the contribution of these clusters to disease or injury outcome is unknown, a recent study demonstrated that complement signaling resulted in microglial phagocytosis of synapses early in AD onset [55]. A combination of aberrant microglial accumulation (clustering), combined with increased reactivity (including complement signaling) may all be factors that contribute to microglial dysregulation, resulting in degeneration of synapses in pre-disease states, such as early pre-symptomatic AD. Thus, targeting white matter microglia may enable the manipulation of these clusters, and/or other non-cluster associated white matter microglia, in a desirable and clinically beneficial manner to promote resolution of disease and effect clearance of harmful debris. Regulation of axonal and cellular debris clearance is necessary for the maintenance of a healthy CNS, however a balance must be maintained between clearance of debris and clearance of healthy synapses [53, 54, 56, 57]. Regulation of microglial phagocytosis is attributable to pathways such as TLRs and type I interferon signaling, which have numerous components optimal for pharmacological targeting. For example, USP18 as discussed above, specific components of the Jak/Stat pathway within the type I IFN signaling cascade or TLR signaling associated components such as CD14, TRIF, and Myd88. Additionally, ISGylation is understood to be important for modulation of the interferon signaling pathway [111], but the exact mechanisms and consequences have not been fully elucidated and further research into ISGylation may reveal novel therapeutic targets for modulating this potent and molecularly unique pathway.
Several drugs have shown promise in modulating microglial phenotype under some conditions, including minocycline [112], IL-1 receptor agonists [113], small molecules targeting the IL-1 pathway [114], and other pharmacological agents. Similar strategies could be developed for targeting components of interferon signaling, for example USP18 and IRFs. The identification of ifit1, an ISG, as an immunological bottleneck [115] also suggests that downstream components like ISGs themselves, may be potential targets for modulating microglial response and phenotype. Modulating endogenous production of type 1 IFNs in the CNS, and/or regulating IFN receptors are potentially viable therapeutic strategies. Type 1 IFNs induce an entire program of ISG expression and wide ranging post-translational modifications that have not been studied in microglia, or any CNS cell type. Some ISG products may prove to be important in mediating the IPC response and could become novel targets for therapeutic intervention in stroke, as suggested by the neuroprotective effects that IFNβ exerts on ischemic stroke [63, 73].
There have been a number of recent and exciting developments in our ability to specifically target microglia experimentally (reviewed by Wieghofer et al. [116]). These advances have included microglia selective/specific genetic conditional knockdown and knock-in approaches as well as pharmacologic agents that show varying degrees of microglial selectivity/specificity [116]. Engraftment of bone marrow derived cells, with attempts to drive these cells down a differentiation pathway to resemble mature microglia, have had mixed results with respect to inducing bone marrow derived cells to infiltrate and populate the brain and in directly transplanting cells [34]. Combination therapies involving the pharmacological depletion of the endogenous microglia to facilitate engraftment of donor-derived infiltrates show some encouraging results [117]. However, this study highlights the need for further research into achieving long-term or permanent eradication of host microglia, which is complicated by the existence of a pool of endogenous progenitor cells in the CNS capable of repopulating the brain with microglia after withdrawal of a depleting drug [49, 117]. Achieving complete and permanent depletion of native microglia may be essential to successful microglial transplant therapies. However, with future research, immune cell transplantation strategies may become viable therapeutic options in combination with gene therapy and pharmacologics, or other novel treatments. This combinatory approach may be especially useful in cases of microgliopathies due to inborn genetic errors, such as those observed in AGS and other PTS disorders. Furthermore, as genetic components of neurodegeneration are being identified in microglia, gene therapies combined with transplantation may serve to repopulate the brain with reprogrammed microglia.
Future Directions
Innate immune signaling pathways such as TLRs and type I IFNs are critical to regulating multiple aspects of microglial pathology, ranging from phagocytosis to stimulation of peripheral immune cells. Furthermore, these components are implicated in other processes, which regulate the CNS microenvironment and have more systemic effects, such as maintaining integrity of the BBB. Further characterization of these innate immune pathways in microglia will allow for more comprehensive identification and understanding of the components involved in microglial reactivity and return to quiescence in neurological disorders characterized by ‘sterile’ inflammation. For example, the studies on the role of USP18 in white matter microglia suggest that USP18 is critical in the regulation of interferon signaling and lead to the identification of important molecules that could be targeted by pharmacological compounds to modulate interferon signaling, and by extension microglial function in a number of neurological disorders. The white matter specificity of the USP18 effect on microglia is of particular interest and further developments in this area may have implications for an entire range of neurological disorders in which there is a preponderance of white matter pathology including demyelinating disease, multiple cerebro-vascular disorders associated with leukoencephalopathy and vascular cognitive impairment, infectious or post-infectious leukoencephalopathies and, as we have discussed above, genetic leukoencephalopathies, including ones associated with a destructive interferonopathy. In order to most efficiently and specifically target microglia in these disparate clinical settings, a greater understanding of microglial heterogeneity, particularly differences in microglial phenotype and function in white versus grey matter or in one specific brain region versus another, may be necessary.
Acknowledgments
We thank Dorender Dankwa for her assistance with microglial cultures and ELISA analysis. This manuscript was funded by NIH F32NS100245 (A.M.) and NIH R01NS076620 (J.R.W.).
References
- 1.Benarroch EE. Microglia: multiple roles in surveillance, circuit shaping, and response to injury. Neurology. 2013;81(12):1079–1088. doi: 10.1212/WNL.0b013e3182a4a577. [DOI] [PubMed] [Google Scholar]
- 2.Michell-Robinson MA, Touil H, Healy LM, Owen DR, Durafourt BA, Bar-Or A, Antel JP, Moore CS. Roles of microglia in brain development, tissue maintenance and repair. Brain. 2015;138(Pt 5):1138–1159. doi: 10.1093/brain/awv066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bilimoria PM, Stevens B. Microglia function during brain development: new insights from animal models. Brain Res. 2015;1617:7–17. doi: 10.1016/j.brainres.2014.11.032. [DOI] [PubMed] [Google Scholar]
- 4.Cunningham CL, Martinez-Cerdeno V, Noctor SC. Microglia regulate the number of neural precursor cells in the developing cerebral cortex. J Neurosci. 2013;33(10):4216–4233. doi: 10.1523/JNEUROSCI.3441-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ji K, Akgul G, Wollmuth LP, Tsirka SE. Microglia actively regulate the number of functional synapses. PloS ONE. 2013;8(2):e56293. doi: 10.1371/journal.pone.0056293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, Micheva KD, Mehalow AK, Huberman AD, Stafford B, Sher A, Litke AM, Lambris JD, Smith SJ, John SW, Barres BA. The classical complement cascade mediates CNS synapse elimination. Cell. 2007;131(6):1164–1178. doi: 10.1016/j.cell.2007.10.036. [DOI] [PubMed] [Google Scholar]
- 7.Checchin D, Sennlaub F, Levavasseur E, Leduc M, Chemtob S. Potential role of microglia in retinal blood vessel formation. Invest Ophthalmol Vis Sci. 2006;47(8):3595–3602. doi: 10.1167/iovs.05-1522. [DOI] [PubMed] [Google Scholar]
- 8.Alliot F, Godin I, Pessac B. Microglia derive from progenitors, originating from the yolk sac, and which proliferate in the brain. Brain Res Dev Brain Res. 1999;117(2):145–152. doi: 10.1016/s0165-3806(99)00113-3. [DOI] [PubMed] [Google Scholar]
- 9.Alliot F, Lecain E, Grima B, Pessac B. Microglial progenitors with a high proliferative potential in the embryonic and adult mouse brain. Proc Natl Acad Sci USA. 1991;88(4):1541–1545. doi: 10.1073/pnas.88.4.1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, Mehler MF, Conway SJ, Ng LG, Stanley ER, Samokhvalov IM, Merad M. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. 2010;330(6005):841–845. doi: 10.1126/science.1194637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chiu IM, Morimoto ET, Goodarzi H, Liao JT, O’Keeffe S, Phatnani HP, Muratet M, Carroll MC, Levy S, Tavazoie S, Myers RM, Maniatis T. A neurodegeneration-specific gene-expression signature of acutely isolated microglia from an amyotrophic lateral sclerosis mouse model. Cell Rep. 2013;4(2):385–401. doi: 10.1016/j.celrep.2013.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ransohoff RM. A polarizing question: do M1 and M2 microglia exist? Nat Neurosci. 2016;19(8):987–991. doi: 10.1038/nn.4338. [DOI] [PubMed] [Google Scholar]
- 13.Ransohoff RM. How neuroinflammation contributes to neurodegeneration. Science. 2016;353(6301):777–783. doi: 10.1126/science.aag2590. [DOI] [PubMed] [Google Scholar]
- 14.Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308(5726):1314–1318. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- 15.Hickman SE, Kingery ND, Ohsumi TK, Borowsky ML, Wang LC, Means TK, El Khoury J. The microglial sensome revealed by direct RNA sequencing. Nat Neurosci. 2013;16(12):1896–1905. doi: 10.1038/nn.3554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kettenmann H, Hanisch UK, Noda M, Verkhratsky A. Physiology of microglia. Physiol Rev. 2011;91(2):461–553. doi: 10.1152/physrev.00011.2010. [DOI] [PubMed] [Google Scholar]
- 17.Hanisch UK, Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci. 2007;10(11):1387–1394. doi: 10.1038/nn1997. [DOI] [PubMed] [Google Scholar]
- 18.Gertig U, Hanisch UK. Microglial diversity by responses and responders. Front Cell Neurosci. 2014;8:101. doi: 10.3389/fncel.2014.00101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Town T, Jeng D, Alexopoulou L, Tan J, Flavell RA. Microglia recognize double-stranded RNA via TLR3. J Immunol. 2006;176(6):3804–3812. doi: 10.4049/jimmunol.176.6.3804. [DOI] [PubMed] [Google Scholar]
- 20.Costello DA, Lynch MA. Toll-like receptor 3 activation modulates hippocampal network excitability, via glial production of interferon-beta. Hippocampus. 2013;23(8):696–707. doi: 10.1002/hipo.22129. [DOI] [PubMed] [Google Scholar]
- 21.Hayakawa K, Qiu J, Lo EH. Biphasic actions of HMGB1 signaling in inflammation and recovery after stroke. Ann N Y Acad Sci. 2010;1207:50–57. doi: 10.1111/j.1749-6632.2010.05728.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stevens SL, Vartanian KB, Stenzel-Poore MP. Reprogramming the response to stroke by preconditioning. Stroke. 2014;45(8):2527–2531. doi: 10.1161/STROKEAHA.114.002879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Crotti A, Ransohoff RM. Microglial physiology and pathophysiology: insights from genome-wide transcriptional profiling. Immunity. 2016;44(3):505–515. doi: 10.1016/j.immuni.2016.02.013. [DOI] [PubMed] [Google Scholar]
- 24.Holtman IR, Raj DD, Miller JA, Schaafsma W, Yin Z, Brouwer N, Wes PD, Moller T, Orre M, Kamphuis W, Hol EM, Boddeke EW, Eggen BJ. Induction of a common microglia gene expression signature by aging and neurodegenerative conditions: a co-expression meta-analysis. Acta Neuropathol Commun. 2015;3:31. doi: 10.1186/s40478-015-0203-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Raj DD, Jaarsma D, Holtman IR, Olah M, Ferreira FM, Schaafsma W, Brouwer N, Meijer MM, de Waard MC, van der Pluijm I, Brandt R, Kreft KL, Laman JD, de Haan G, Biber KP, Hoeijmakers JH, Eggen BJ, Boddeke HW. Priming of microglia in a DNA-repair deficient model of accelerated aging. Neurobiol Aging. 2014;35(9):2147–2160. doi: 10.1016/j.neurobiolaging.2014.03.025. [DOI] [PubMed] [Google Scholar]
- 26.Stark GR. How cells respond to interferons revisited: from early history to current complexity. Cytokine Growth Factor Rev. 2007;18(5–6):419–423. doi: 10.1016/j.cytogfr.2007.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stark GR, Kerr IM, Williams BR, Silverman RH, Schreiber RD. How cells respond to interferons. Annu Rev Biochem. 1998;67:227–264. doi: 10.1146/annurev.biochem.67.1.227. [DOI] [PubMed] [Google Scholar]
- 28.Borden EC, Williams BR. Interferon-stimulated genes and their protein products: what and how? J Interferon Cytokine Res. 2011;31(1):1–4. doi: 10.1089/jir.2010.0129. [DOI] [PubMed] [Google Scholar]
- 29.Brendecke SM, Prinz M. How type I interferons shape myeloid cell function in CNS autoimmunity. J Leukoc Biol. 2012;92(3):479–488. doi: 10.1189/jlb.0112043. [DOI] [PubMed] [Google Scholar]
- 30.Owens T, Khorooshi R, Wlodarczyk A, Asgari N. Interferons in the central nervous system: a few instruments play many tunes. Glia. 2014;62(3):339–355. doi: 10.1002/glia.22608. [DOI] [PubMed] [Google Scholar]
- 31.Khorooshi R, Owens T. Injury-induced type I IFN signaling regulates inflammatory responses in the central nervous system. J Immunol. 2010;185(2):1258–1264. doi: 10.4049/jimmunol.0901753. [DOI] [PubMed] [Google Scholar]
- 32.Prinz M, Schmidt H, Mildner A, Knobeloch KP, Hanisch UK, Raasch J, Merkler D, Detje C, Gutcher I, Mages J, Lang R, Martin R, Gold R, Becher B, Bruck W, Kalinke U. Distinct and nonredundant in vivo functions of IFNAR on myeloid cells limit autoimmunity in the central nervous system. Immunity. 2008;28(5):675–686. doi: 10.1016/j.immuni.2008.03.011. [DOI] [PubMed] [Google Scholar]
- 33.Marsh B, Stevens SL, Packard AE, Gopalan B, Hunter B, Leung PY, Harrington CA, Stenzel-Poore MP. Systemic lipopolysaccharide protects the brain from ischemic injury by reprogramming the response of the brain to stroke: a critical role for IRF3. J Neurosci. 2009;29(31):9839–9849. doi: 10.1523/JNEUROSCI.2496-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Prinz M, Priller J. Microglia and brain macrophages in the molecular age: from origin to neuropsychiatric disease. Nat Rev Neurosci. 2014;15(5):300–312. doi: 10.1038/nrn3722. [DOI] [PubMed] [Google Scholar]
- 35.Hollingworth P, Harold D, Sims R, Gerrish A, Lambert JC, Carrasquillo MM, Abraham R, Hamshere ML, Pahwa JS, Moskvina V, Dowzell K, Jones N, Stretton A, Thomas C, Richards A, Ivanov D, Widdowson C, Chapman J, Lovestone S, Powell J, Proitsi P, Lupton MK, Brayne C, Rubinsztein DC, Gill M, Lawlor B, Lynch A, Brown KS, Passmore PA, Craig D, McGuinness B, Todd S, Holmes C, Mann D, Smith AD, Beaumont H, Warden D, Wilcock G, Love S, Kehoe PG, Hooper NM, Vardy ER, Hardy J, Mead S, Fox NC, Rossor M, Collinge J, Maier W, Jessen F, Ruther E, Schurmann B, Heun R, Kolsch H, van den Bussche H, Heuser I, Kornhuber J, Wiltfang J, Dichgans M, Frolich L, Hampel H, Gallacher J, Hull M, Rujescu D, Giegling I, Goate AM, Kauwe JS, Cruchaga C, Nowotny P, Morris JC, Mayo K, Sleegers K, Bettens K, Engelborghs S, De Deyn PP, Van Broeckhoven C, Livingston G, Bass NJ, Gurling H, McQuillin A, Gwilliam R, Deloukas P, Al-Chalabi A, Shaw CE, Tsolaki M, Singleton AB, Guerreiro R, Muhleisen TW, Nothen MM, Moebus S, Jockel KH, Klopp N, Wichmann HE, Pankratz VS, Sando SB, Aasly JO, Barcikowska M, Wszolek ZK, Dickson DW, Graff-Radford NR, Petersen RC, Alzheimer’s Disease Neuroimaging Initiative. van Duijn CM, Breteler MM, Ikram MA, DeStefano AL, Fitzpatrick AL, Lopez O, Launer LJ, Seshadri S, CHARGE consortium. Berr C, Campion D, Epelbaum J, Dartigues JF, Tzourio C, Alperovitch A, Lathrop M, EADI consortium. Feulner TM, Friedrich P, Riehle C, Krawczak M, Schreiber S, Mayhaus M, Nicolhaus S, Wagenpfeil S, Steinberg S, Stefansson H, Stefansson K, Snaedal J, Bjornsson S, Jonsson PV, Chouraki V, Genier-Boley B, Hiltunen M, Soininen H, Combarros O, Zelenika D, Delepine M, Bullido MJ, Pasquier F, Mateo I, Frank-Garcia A, Porcellini E, Hanon O, Coto E, Alvarez V, Bosco P, Siciliano G, Mancuso M, Panza F, Solfrizzi V, Nacmias B, Sorbi S, Bossu P, Piccardi P, Arosio B, Annoni G, Seripa D, Pilotto A, Scarpini E, Galimberti D, Brice A, Hannequin D, Licastro F, Jones L, Holmans PA, Jonsson T, Riemenschneider M, Morgan K, Younkin SG, Owen MJ, O’Donovan M, Amouyel P, Williams J. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat Genet. 2011;43(5):429–435. doi: 10.1038/ng.803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Naj AC, Jun G, Beecham GW, Wang LS, Vardarajan BN, Buros J, Gallins PJ, Buxbaum JD, Jarvik GP, Crane PK, Larson EB, Bird TD, Boeve BF, Graff-Radford NR, De Jager PL, Evans D, Schneider JA, Carrasquillo MM, Ertekin-Taner N, Younkin SG, Cruchaga C, Kauwe JS, Nowotny P, Kramer P, Hardy J, Huentelman MJ, Myers AJ, Barmada MM, Demirci FY, Baldwin CT, Green RC, Rogaeva E, St George-Hyslop P, Arnold SE, Barber R, Beach T, Bigio EH, Bowen JD, Boxer A, Burke JR, Cairns NJ, Carlson CS, Carney RM, Carroll SL, Chui HC, Clark DG, Corneveaux J, Cotman CW, Cummings JL, DeCarli C, DeKosky ST, Diaz-Arrastia R, Dick M, Dickson DW, Ellis WG, Faber KM, Fallon KB, Farlow MR, Ferris S, Frosch MP, Galasko DR, Ganguli M, Gearing M, Geschwind DH, Ghetti B, Gilbert JR, Gilman S, Giordani B, Glass JD, Growdon JH, Hamilton RL, Harrell LE, Head E, Honig LS, Hulette CM, Hyman BT, Jicha GA, Jin LW, Johnson N, Karlawish J, Karydas A, Kaye JA, Kim R, Koo EH, Kowall NW, Lah JJ, Levey AI, Lieberman AP, Lopez OL, Mack WJ, Marson DC, Martiniuk F, Mash DC, Masliah E, McCormick WC, McCurry SM, McDavid AN, McKee AC, Mesulam M, Miller BL, Miller CA, Miller JW, Parisi JE, Perl DP, Peskind E, Petersen RC, Poon WW, Quinn JF, Rajbhandary RA, Raskind M, Reisberg B, Ringman JM, Roberson ED, Rosenberg RN, Sano M, Schneider LS, Seeley W, Shelanski ML, Slifer MA, Smith CD, Sonnen JA, Spina S, Stern RA, Tanzi RE, Trojanowski JQ, Troncoso JC, Van Deerlin VM, Vinters HV, Vonsattel JP, Weintraub S, Welsh-Bohmer KA, Williamson J, Woltjer RL, Cantwell LB, Dombroski BA, Beekly D, Lunetta KL, Martin ER, Kamboh MI, Saykin AJ, Reiman EM, Bennett DA, Morris JC, Montine TJ, Goate AM, Blacker D, Tsuang DW, Hakonarson H, Kukull WA, Foroud TM, Haines JL, Mayeux R, Pericak-Vance MA, Farrer LA, Schellenberg GD. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat Genet. 2011;43(5):436–441. doi: 10.1038/ng.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Benitez BA, Jin SC, Guerreiro R, Graham R, Lord J, Harold D, Sims R, Lambert JC, Gibbs JR, Bras J, Sassi C, Harari O, Bertelsen S, Lupton MK, Powell J, Bellenguez C, Brown K, Medway C, Haddick PC, van der Brug MP, Bhangale T, Ortmann W, Behrens T, Mayeux R, Pericak-Vance MA, Farrer LA, Schellenberg GD, Haines JL, Turton J, Braae A, Barber I, Fagan AM, Holtzman DM, Morris JC, 3C Study Group, EADI consortium, Alzheimer’s Disease Genetic consortium, Alzheimer’s Disease Neuroimaging Initiative, GERAD Consortium. Williams J, Kauwe JS, Amouyel P, Morgan K, Singleton A, Hardy J, Goate AM, Cruchaga C. Missense variant in TREML2 protects against Alzheimer’s disease. Neurobiol Aging. 2014;35(6):1510.e1519–1526. doi: 10.1016/j.neurobiolaging.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, Cruchaga C, Sassi C, Kauwe JS, Younkin S, Hazrati L, Collinge J, Pocock J, Lashley T, Williams J, Lambert JC, Amouyel P, Goate A, Rademakers R, Morgan K, Powell J, St George-Hyslop P, Singleton A, Hardy J, Alzheimer Genetic Analysis Group TREM2 variants in Alzheimer’s disease. N Engl J Med. 2013;368(2):117–127. doi: 10.1056/NEJMoa1211851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Saunders AM, Strittmatter WJ, Schmechel D, George-Hyslop PH, Pericak-Vance MA, Joo SH, Rosi BL, Gusella JF, Crapper-MacLachlan DR, Alberts MJ, et al. Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer’s disease. Neurology. 1993;43(8):1467–1472. doi: 10.1212/wnl.43.8.1467. [DOI] [PubMed] [Google Scholar]
- 40.Mhatre SD, Tsai CA, Rubin AJ, James ML, Andreasson KI. Microglial malfunction: the third rail in the development of Alzheimer’s disease. Trends Neurosci. 2015;38(10):621–636. doi: 10.1016/j.tins.2015.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yeh FL, Wang Y, Tom I, Gonzalez LC, Sheng M. TREM2 Binds to apolipoproteins, including APOE and CLU/APOJ, and thereby facilitates uptake of amyloid-beta by Microglia. Neuron. 2016;91(2):328–340. doi: 10.1016/j.neuron.2016.06.015. [DOI] [PubMed] [Google Scholar]
- 42.Lambert JC, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C, DeStafano AL, Bis JC, Beecham GW, Grenier-Boley B, Russo G, Thorton-Wells TA, Jones N, Smith AV, Chouraki V, Thomas C, Ikram MA, Zelenika D, Vardarajan BN, Kamatani Y, Lin CF, Gerrish A, Schmidt H, Kunkle B, Dunstan ML, Ruiz A, Bihoreau MT, Choi SH, Reitz C, Pasquier F, Cruchaga C, Craig D, Amin N, Berr C, Lopez OL, De Jager PL, Deramecourt V, Johnston JA, Evans D, Lovestone S, Leten-neur L, Moron FJ, Rubinsztein DC, Eiriksdottir G, Sleegers K, Goate AM, Fievet N, Huentelman MW, Gill M, Brown K, Kamboh MI, Keller L, Barberger-Gateau P, McGuiness B, Larson EB, Green R, Myers AJ, Dufouil C, Todd S, Wallon D, Love S, Rogaeva E, Gallacher J, St George-Hyslop P, Clarimon J, Lleo A, Bayer A, Tsuang DW, Yu L, Tsolaki M, Bossu P, Spalletta G, Proitsi P, Collinge J, Sorbi S, Sanchez-Garcia F, Fox NC, Hardy J, Deniz Naranjo MC, Bosco P, Clarke R, Brayne C, Galimberti D, Mancuso M, Matthews F, European Alzheimer’s Disease Initiative, Genetic and Environmental Risk in Alzheimer’s Disease, Alzheimer’s Disease Genetic Consortium, Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) Moebus S, Mecocci P, Del Zompo M, Maier W, Hampel H, Pilotto A, Bullido M, Panza F, Caffarra P, Nacmias B, Gilbert JR, Mayhaus M, Lannefelt L, Hakonarson H, Pichler S, Carrasquillo MM, Ingelsson M, Beekly D, Alvarez V, Zou F, Valladares O, Younkin SG, Coto E, Hamilton-Nelson KL, Gu W, Razquin C, Pastor P, Mateo I, Owen MJ, Faber KM, Jonsson PV, Combarros O, O’Donovan MC, Cantwell LB, Soininen H, Blacker D, Mead S, Mosley TH, Jr, Bennett DA, Harris TB, Fratiglioni L, Holmes C, de Bruijn RF, Passmore P, Montine TJ, Bettens K, Rotter JI, Brice A, Morgan K, Foroud TM, Kukull WA, Hannequin D, Powell JF, Nalls MA, Ritchie K, Lunetta KL, Kauwe JS, Boerwinkle E, Riemenschneider M, Boada M, Hiltuenen M, Martin ER, Schmidt R, Rujescu D, Wang LS, Dartigues JF, Mayeux R, Tzourio C, Hofman A, Nothen MM, Graff C, Psaty BM, Jones L, Haines JL, Holmans PA, Lathrop M, Pericak-Vance MA, Launer LJ, Farrer LA, van Duijn CM, Van Broeckhoven C, Moskvina V, Seshadri S, Williams J, Schellenberg GD, Amouyel P. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet. 2013;45(12):1452–1458. doi: 10.1038/ng.2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Villegas-Llerena C, Phillips A, Garcia-Reitboeck P, Hardy J, Pocock JM. Microglial genes regulating neuroinflammation in the progression of Alzheimer’s disease. Curr Opin Neurobiol. 2016;36:74–81. doi: 10.1016/j.conb.2015.10.004. [DOI] [PubMed] [Google Scholar]
- 44.Paloneva J, Manninen T, Christman G, Hovanes K, Mandelin J, Adolfsson R, Bianchin M, Bird T, Miranda R, Salmaggi A, Tranebjaerg L, Konttinen Y, Peltonen L. Mutations in two genes encoding different subunits of a receptor signaling complex result in an identical disease phenotype. Am J Hum Genet. 2002;71(3):656–662. doi: 10.1086/342259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nicholson AM, Baker MC, Finch NA, Rutherford NJ, Wider C, Graff-Radford NR, Nelson PT, Clark HB, Wszolek ZK, Dickson DW, Knopman DS, Rademakers R. CSF1R mutations link POLD and HDLS as a single disease entity. Neurology. 2013;80(11):1033–1040. doi: 10.1212/WNL.0b013e31828726a7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rademakers R, Baker M, Nicholson AM, Rutherford NJ, Finch N, Soto-Ortolaza A, Lash J, Wider C, Wojtas A, DeJesus-Hernandez M, Adamson J, Kouri N, Sundal C, Shuster EA, Aasly J, MacKenzie J, Roeber S, Kretzschmar HA, Boeve BF, Knopman DS, Petersen RC, Cairns NJ, Ghetti B, Spina S, Garbern J, Tselis AC, Uitti R, Das P, Van Gerpen JA, Meschia JF, Levy S, Broderick DF, Graff-Radford N, Ross OA, Miller BB, Swerd-low RH, Dickson DW, Wszolek ZK. Mutations in the colony stimulating factor 1 receptor (CSF1R) gene cause hereditary diffuse leukoencephalopathy with spheroids. Nat Genet. 2011;44(2):200–205. doi: 10.1038/ng.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Elmore MR, Najafi AR, Koike MA, Dagher NN, Spangenberg EE, Rice RA, Kitazawa M, Matusow B, Nguyen H, West BL, Green KN. Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron. 2014;82(2):380–397. doi: 10.1016/j.neuron.2014.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Spangenberg EE, Lee RJ, Najafi AR, Rice RA, Elmore MR, Blurton-Jones M, West BL, Green KN. Eliminating microglia in Alzheimer’s mice prevents neuronal loss without modulating amyloid-beta pathology. Brain. 2016;139(Pt 4):1265–1281. doi: 10.1093/brain/aww016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rice RA, Spangenberg EE, Yamate-Morgan H, Lee RJ, Arora RP, Hernandez MX, Tenner AJ, West BL, Green KN. Elimination of Microglia improves functional outcomes following extensive neuronal loss in the hippocampus. J Neurosci. 2015;35(27):9977–9989. doi: 10.1523/JNEUROSCI.0336-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jin WN, Shi SX, Li Z, Li M, Wood K, Gonzales RJ, Liu Q. Depletion of microglia exacerbates postischemic inflammation and brain injury. J Cereb Blood Flow Metab. 2017 doi: 10.1177/0271678X17694185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.De Jager PL, Jia X, Wang J, de Bakker PI, Ottoboni L, Aggarwal NT, Piccio L, Raychaudhuri S, Tran D, Aubin C, Briskin R, Romano S, International MSGC. Baranzini SE, McCauley JL, Pericak-Vance MA, Haines JL, Gibson RA, Naeglin Y, Uitdehaag B, Matthews PM, Kappos L, Polman C, McArdle WL, Strachan DP, Evans D, Cross AH, Daly MJ, Compston A, Sawcer SJ, Weiner HL, Hauser SL, Hafler DA, Oksenberg JR. Meta-analysis of genome scans and replication identify CD6, IRF8 and TNFRSF1A as new multiple sclerosis susceptibility loci. Nat Genet. 2009;41(7):776–782. doi: 10.1038/ng.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ponomarev ED, Shriver LP, Maresz K, Dittel BN. Microglial cell activation and proliferation precedes the onset of CNS autoimmunity. J Neurosci Res. 2005;81(3):374–389. doi: 10.1002/jnr.20488. [DOI] [PubMed] [Google Scholar]
- 53.Chan A, Seguin R, Magnus T, Papadimitriou C, Toyka KV, Antel JP, Gold R. Phagocytosis of apoptotic inflammatory cells by microglia and its therapeutic implications: termination of CNS autoimmune inflammation and modulation by interferon-beta. Glia. 2003;43(3):231–242. doi: 10.1002/glia.10258. [DOI] [PubMed] [Google Scholar]
- 54.Hosmane S, Tegenge MA, Rajbhandari L, Uapinyoying P, Kumar NG, Thakor N, Venkatesan A. Toll/interleukin-1 receptor domain-containing adapter inducing interferon-beta mediates microglial phagocytosis of degenerating axons. Journal Neurosci. 2012;32(22):7745–7757. doi: 10.1523/JNEUROSCI.0203-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, Merry KM, Shi Q, Rosenthal A, Barres BA, Lemere CA, Selkoe DJ, Stevens B. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science. 2016 doi: 10.1126/science.aad8373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Huizinga R, van der Star BJ, Kipp M, Jong R, Gerritsen W, Clarner T, Puentes F, Dijkstra CD, van der Valk P, Amor S. Phagocytosis of neuronal debris by microglia is associated with neuronal damage in multiple sclerosis. Glia. 2012;60(3):422–431. doi: 10.1002/glia.22276. [DOI] [PubMed] [Google Scholar]
- 57.Rajbhandari L, Tegenge MA, Shrestha S, Ganesh Kumar N, Malik A, Mithal A, Hosmane S, Venkatesan A. Toll-like receptor 4 deficiency impairs microglial phagocytosis of degenerating axons. Glia. 2014;62(12):1982–1991. doi: 10.1002/glia.22719. [DOI] [PubMed] [Google Scholar]
- 58.Janova H, Bottcher C, Holtman IR, Regen T, van Rossum D, Gotz A, Ernst AS, Fritsche C, Gertig U, Saiepour N, Gronke K, Wrzos C, Ribes S, Rolfes S, Weinstein J, Ehrenreich H, Pukrop T, Kopatz J, Stadelmann C, Salinas-Riester G, Weber MS, Prinz M, Bruck W, Eggen BJ, Boddeke HW, Priller J, Hanisch UK. CD14 is a key organizer of microglial responses to CNS infection and injury. Glia. 2016;64(4):635–649. doi: 10.1002/glia.22955. [DOI] [PubMed] [Google Scholar]
- 59.Olson JK, Miller SD. Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. J Immunol. 2004;173(6):3916–3924. doi: 10.4049/jimmunol.173.6.3916. [DOI] [PubMed] [Google Scholar]
- 60.Kim MY, Shu Y, Carsillo T, Zhang J, Yu L, Peterson C, Longhi S, Girod S, Niewiesk S, Oglesbee M. hsp70 and a novel axis of type I interferon-dependent antiviral immunity in the measles virus-infected brain. J Virol. 2013;87(2):998–1009. doi: 10.1128/JVI.02710-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hamner MA, Ye Z, Lee RV, Colman JR, Le T, Gong DC, Ransom BR, Weinstein JR. Ischemic preconditioning in white matter: magnitude and mechanism. J Neurosci. 2015;35(47):15599–15611. doi: 10.1523/JNEUROSCI.2544-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pradillo JM, Fernandez-Lopez D, Garcia-Yebenes I, Sobrado M, Hurtado O, Moro MA, Lizasoain I. Toll-like receptor 4 is involved in neuroprotection afforded by ischemic preconditioning. J Neurochem. 2009;109(1):287–294. doi: 10.1111/j.1471-4159.2009.05972.x. [DOI] [PubMed] [Google Scholar]
- 63.Stevens SL, Leung PY, Vartanian KB, Gopalan B, Yang T, Simon RP, Stenzel-Poore MP. Multiple preconditioning paradigms converge on interferon regulatory factor-dependent signaling to promote tolerance to ischemic brain injury. J Neurosci. 2011;31(23):8456–8463. doi: 10.1523/JNEUROSCI.0821-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Church JS, Milich LM, Lerch JK, Popovich PG, McTigue DM. E6020, a synthetic TLR4 agonist, accelerates myelin debris clearance, Schwann cell infiltration, and remyelination in the rat spinal cord. Glia. 2017;65(6):883–899. doi: 10.1002/glia.23132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Weinstein JR, Quan Y, Hanson JF, Colonna L, Iorga M, Honda S, Shibuya K, Shibuya A, Elkon KB, Moller T. IgM-dependent phagocytosis in microglia is mediated by complement receptor 3, not Fcalpha/mu receptor. J Immunol. 2015;195(11):5309–5317. doi: 10.4049/jimmunol.1401195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Moller T, Hanisch UK, Ransom BR. Thrombin-induced activation of cultured rodent microglia. J Neurochem. 2000;75(4):1539–1547. doi: 10.1046/j.1471-4159.2000.0751539.x. [DOI] [PubMed] [Google Scholar]
- 67.Marziniak M, Meuth S. Current perspectives on interferon Beta-1b for the treatment of multiple sclerosis. Adv Ther. 2014;31(9):915–931. doi: 10.1007/s12325-014-0149-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Khorooshi R, Morch MT, Holm TH, Berg CT, Dieu RT, Draeby D, Issazadeh-Navikas S, Weiss S, Lienenklaus S, Owens T. Induction of endogenous Type I interferon within the central nervous system plays a protective role in experimental autoimmune encephalomyelitis. Acta Neuropathol (Berl) 2015;130(1):107–118. doi: 10.1007/s00401-015-1418-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kraus J, Ling AK, Hamm S, Voigt K, Oschmann P, Engel-hardt B. Interferon-beta stabilizes barrier characteristics of brain endothelial cells in vitro. Ann Neurol. 2004;56(2):192–205. doi: 10.1002/ana.20161. [DOI] [PubMed] [Google Scholar]
- 70.Stone LA, Frank JA, Albert PS, Bash CN, Calabresi PA, Maloni H, McFarland HF. Characterization of MRI response to treatment with interferon beta-1b: contrast-enhancing MRI lesion frequency as a primary outcome measure. Neurology. 1997;49(3):862–869. doi: 10.1212/wnl.49.3.862. [DOI] [PubMed] [Google Scholar]
- 71.Gesuete R, Packard AE, Vartanian KB, Conrad VK, Stevens SL, Bahjat FR, Yang T, Stenzel-Poore MP. Poly-ICLC preconditioning protects the blood-brain barrier against ischemic injury in vitro through type I interferon signaling. J Neurochem. 2012;123(Suppl 2):75–85. doi: 10.1111/j.1471-4159.2012.07946.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Veldhuis WB, Derksen JW, Floris S, Van Der Meide PH, De Vries HE, Schepers J, Vos IM, Dijkstra CD, Kappelle LJ, Nicolay K, Bar PR. Interferon-beta blocks infiltration of inflammatory cells and reduces infarct volume after ischemic stroke in the rat. J Cereb Blood Flow Metab. 2003;23(9):1029–1039. doi: 10.1097/01.WCB.0000080703.47016.B6. [DOI] [PubMed] [Google Scholar]
- 73.Inacio AR, Liu Y, Clausen BH, Svensson M, Kucharz K, Yang Y, Stankovich T, Khorooshi R, Lambertsen KL, Issazadeh-Navikas S, Deierborg T. Endogenous IFN-beta signaling exerts anti-inflammatory actions in experimentally induced focal cerebral ischemia. J Neuroinflammation. 2015;12:211. doi: 10.1186/s12974-015-0427-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yamasaki R, Lu H, Butovsky O, Ohno N, Rietsch AM, Cialic R, Wu PM, Doykan CE, Lin J, Cotleur AC, Kidd G, Zorlu MM, Sun N, Hu W, Liu L, Lee JC, Taylor SE, Uehlein L, Dixon D, Gu J, Floruta CM, Zhu M, Charo IF, Weiner HL, Ransohoff RM. Differential roles of microglia and monocytes in the inflamed central nervous system. J Exp Med. 2014;211(8):1533–1549. doi: 10.1084/jem.20132477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Becker K, Kindrick D, Relton J, Harlan J, Winn R. Antibody to the alpha4 integrin decreases infarct size in transient focal cerebral ischemia in rats. Stroke. 2001;32(1):206–211. doi: 10.1161/01.str.32.1.206. [DOI] [PubMed] [Google Scholar]
- 76.Becker KJ, McCarron RM, Ruetzler C, Laban O, Sternberg E, Flanders KC, Hallenbeck JM. Immunologic tolerance to myelin basic protein decreases stroke size after transient focal cerebral ischemia. Proc Natl Acad Sci USA. 1997;94(20):10873–10878. doi: 10.1073/pnas.94.20.10873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Huang J, Upadhyay UM, Tamargo RJ. Inflammation in stroke and focal cerebral ischemia. Surg Neurol. 2006;66(3):232–245. doi: 10.1016/j.surneu.2005.12.028. [DOI] [PubMed] [Google Scholar]
- 78.Shichita T, Ago T, Kamouchi M, Kitazono T, Yoshimura A, Ooboshi H. Novel therapeutic strategies targeting innate immune responses and early inflammation after stroke. J Neurochem. 2012;123(Suppl 2):29–38. doi: 10.1111/j.1471-4159.2012.07941.x. [DOI] [PubMed] [Google Scholar]
- 79.Shichita T, Sugiyama Y, Ooboshi H, Sugimori H, Nakagawa R, Takada I, Iwaki T, Okada Y, Iida M, Cua DJ, Iwakura Y, Yoshimura A. Pivotal role of cerebral interleukin-17-producing gammadeltaT cells in the delayed phase of ischemic brain injury. Nat Med. 2009;15(8):946–950. doi: 10.1038/nm.1999. [DOI] [PubMed] [Google Scholar]
- 80.Garden GA, Moller T. Microglia biology in health and disease. J Neuroimmune Pharmacol. 2006;1(2):127–137. doi: 10.1007/s11481-006-9015-5. [DOI] [PubMed] [Google Scholar]
- 81.Lalancette-Hebert M, Gowing G, Simard A, Weng YC, Kriz J. Selective ablation of proliferating microglial cells exacerbates ischemic injury in the brain. J Neurosci. 2007;27(10):2596–2605. doi: 10.1523/JNEUROSCI.5360-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Nedergaard M, Dirnagl U. Role of glial cells in cerebral ischemia. Glia. 2005;50(4):281–286. doi: 10.1002/glia.20205. [DOI] [PubMed] [Google Scholar]
- 83.Weinstein JR, Koerner IP, Moller T. Microglia in ischemic brain injury. Futur Neurol. 2010;5(2):227–246. doi: 10.2217/fnl.10.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gidday JM. Cerebral preconditioning and ischaemic tolerance. Nat Rev Neurosci. 2006;7(6):437–448. doi: 10.1038/nrn1927. [DOI] [PubMed] [Google Scholar]
- 85.Kariko K, Weissman D, Welsh FA. Inhibition of toll-like receptor and cytokine signaling—A unifying theme in ischemic tolerance. J Cereb Blood Flow Metab. 2004;24(11):1288–1304. doi: 10.1097/01.WCB.0000145666.68576.71. [DOI] [PubMed] [Google Scholar]
- 86.Zhang J, Yang ZJ, Klaus JA, Koehler RC, Huang J. Delayed tolerance with repetitive transient focal ischemic preconditioning in the mouse. Stroke. 2008;39(3):967–974. doi: 10.1161/STROKEAHA.107.497412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Dirnagl U, Becker K, Meisel A. Preconditioning and tolerance against cerebral ischaemia: from experimental strategies to clinical use. Lancet Neurol. 2009;8(4):398–412. doi: 10.1016/S1474-4422(09)70054-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Stenzel-Poore MP, Stevens SL, King JS, Simon RP. Preconditioning reprograms the response to ischemic injury and primes the emergence of unique endogenous neuroprotective phenotypes: a speculative synthesis. Stroke. 2007;38(2 Suppl):680–685. doi: 10.1161/01.STR.0000251444.56487.4c. [DOI] [PubMed] [Google Scholar]
- 89.Stenzel-Poore MP, Stevens SL, Simon RP. Genomics of preconditioning. Stroke. 2004;35(11 Suppl 1):2683–2686. doi: 10.1161/01.STR.0000143735.89281.bb. [DOI] [PubMed] [Google Scholar]
- 90.McDonough A, Weinstein JR. Neuroimmune response in ischemic preconditioning. Neurotherapeutics. 2016;13(4):748–761. doi: 10.1007/s13311-016-0465-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Stevens SL, Ciesielski TM, Marsh BJ, Yang T, Homen DS, Boule JL, Lessov NS, Simon RP, Stenzel-Poore MP. Toll-like receptor 9: a new target of ischemic preconditioning in the brain. J Cereb Blood Flow Metab. 2008;28(5):1040–1047. doi: 10.1038/sj.jcbfm.9600606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kuo PC, Scofield BA, Yu IC, Chang FL, Ganea D, Yen JH. Interferon-beta Modulates Inflammatory Response in Cerebral Ischemia. J Am Heart Assoc. 2016 doi: 10.1161/JAHA.115.002610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Fisher M, Hanley DF, Howard G, Jauch EC, Warach S, Group S Recommendations from the STAIR V meeting on acute stroke trials, technology and outcomes. Stroke. 2007;38(2):245–248. doi: 10.1161/01.STR.0000255951.37434.aa. [DOI] [PubMed] [Google Scholar]
- 94.Dirnagl U, Endres M. Found in translation: preclinical stroke research predicts human pathophysiology, clinical phenotypes, and therapeutic outcomes. Stroke. 2014;45(5):1510–1518. doi: 10.1161/STROKEAHA.113.004075. [DOI] [PubMed] [Google Scholar]
- 95.Colton CA. Heterogeneity of microglial activation in the innate immune response in the brain. J Neuroimmune Pharmacol. 2009;4(4):399–418. doi: 10.1007/s11481-009-9164-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Takata K, Ginhoux F. Poised for action: USP18 restrains microglial activation in the white matter. EMBO J. 2015;34(12):1603–1605. doi: 10.15252/embj.201591899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Knobeloch KP, Utermohlen O, Kisser A, Prinz M, Horak I. Reexamination of the role of ubiquitin-like modifier ISG15 in the phenotype of UBP43-deficient mice. Mol Cell Biol. 2005;25(24):11030–11034. doi: 10.1128/MCB.25.24.11030-11034.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Goldmann T, Zeller N, Raasch J, Kierdorf K, Frenzel K, Ketscher L, Basters A, Staszewski O, Brendecke SM, Spiess A, Tay TL, Kreutz C, Timmer J, Mancini GM, Blank T, Fritz G, Biber K, Lang R, Malo D, Merkler D, Heikenwalder M, Knobeloch KP, Prinz M. USP18 lack in microglia causes destructive interferonopathy of the mouse brain. EMBO J. 2015;34(12):1612–1629. doi: 10.15252/embj.201490791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Meuwissen ME, Schot R, Buta S, Oudesluijs G, Tinschert S, Speer SD, Li Z, van Unen L, Heijsman D, Goldmann T, Lequin MH, Kros JM, Stam W, Hermann M, Willemsen R, Brouwer RW, Van IWF, Martin-Fernandez M, de Coo I, Dudink J, de Vries FA, Bertoli Avella A, Prinz M, Crow YJ, Verheijen FW, Pellegrini S, Bogunovic D, Mancini GM. Human USP18 deficiency underlies type 1 interferonopathy leading to severe pseudo-TORCH syndrome. J Exp Med. 2016;213(7):1163–1174. doi: 10.1084/jem.20151529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zhang X, Bogunovic D, Payelle-Brogard B, Francois-Newton V, Speer SD, Yuan C, Volpi S, Li Z, Sanal O, Mansouri D, Tezcan I, Rice GI, Chen C, Mansouri N, Mahdaviani SA, Itan Y, Boisson B, Okada S, Zeng L, Wang X, Jiang H, Liu W, Han T, Liu D, Ma T, Wang B, Liu M, Liu JY, Wang QK, Yalnizoglu D, Radoshevich L, Uze G, Gros P, Rozenberg F, Zhang SY, Jouanguy E, Bustamante J, Garcia-Sastre A, Abel L, Lebon P, Notarangelo LD, Crow YJ, Boisson-Dupuis S, Casanova JL, Pellegrini S. Human intracellular ISG15 prevents interferon-alpha/beta over-amplification and auto-inflammation. Nature. 2015;517(7532):89–93. doi: 10.1038/nature13801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Crow YJ, Rehwinkel J. Aicardi-Goutieres syndrome and related phenotypes: linking nucleic acid metabolism with autoimmunity. Hum Mol Genet. 2009;18(R2):R130–136. doi: 10.1093/hmg/ddp293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Rice GI, Kasher PR, Forte GM, Mannion NM, Greenwood SM, Szynkiewicz M, Dickerson JE, Bhaskar SS, Zampini M, Briggs TA, Jenkinson EM, Bacino CA, Battini R, Bertini E, Brogan PA, Brueton LA, Carpanelli M, De Laet C, de Lonlay P, del Toro M, Desguerre I, Fazzi E, Garcia-Cazorla A, Heiberg A, Kawaguchi M, Kumar R, Lin JP, Lourenco CM, Male AM, Marques W, Jr, Mignot C, Olivieri I, Orcesi S, Prabhakar P, Rasmussen M, Robinson RA, Rozenberg F, Schmidt JL, Steindl K, Tan TY, van der Merwe WG, Vanderver A, Vassallo G, Wakeling EL, Wassmer E, Whittaker E, Livingston JH, Lebon P, Suzuki T, McLaughlin PJ, Keegan LP, O’Connell MA, Lovell SC, Crow YJ. Mutations in ADAR1 cause Aicardi-Goutieres syndrome associated with a type I interferon signature. Nat Genet. 2012;44(11):1243–1248. doi: 10.1038/ng.2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Michailidou I, Naessens DM, Hametner S, Guldenaar W, Kooi EJ, Geurts JJ, Baas F, Lassmann H, Ramaglia V. Complement C3 on microglial clusters in multiple sclerosis occur in chronic but not acute disease: Implication for disease pathogenesis. Glia. 2016 doi: 10.1002/glia.23090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Malakhov MP, Malakhova OA, Kim KI, Ritchie KJ, Zhang DE. UBP43 (USP18) specifically removes ISG15 from conjugated proteins. J Biol Chem. 2002;277(12):9976–9981. doi: 10.1074/jbc.M109078200. [DOI] [PubMed] [Google Scholar]
- 105.Zhang D, Zhang DE. Interferon-stimulated gene 15 and the protein ISGylation system. J Interferon cytokine Res. 2011;31(1):119–130. doi: 10.1089/jir.2010.0110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ritchie KJ, Hahn CS, Kim KI, Yan M, Rosario D, Li L, de la Torre JC, Zhang DE. Role of ISG15 protease UBP43 (USP18) in innate immunity to viral infection. Nat Med. 2004;10(12):1374–1378. doi: 10.1038/nm1133. [DOI] [PubMed] [Google Scholar]
- 107.Ritchie KJ, Malakhov MP, Hetherington CJ, Zhou L, Little MT, Malakhova OA, Sipe JC, Orkin SH, Zhang DE. Dysregulation of protein modification by ISG15 results in brain cell injury. Genes Dev. 2002;16(17):2207–2212. doi: 10.1101/gad.1010202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Crow YJ, Casanova JL. STING-associated vasculopathy with onset in infancy–a new interferonopathy. N Engl J Med. 2014;371(6):568–571. doi: 10.1056/NEJMe1407246. [DOI] [PubMed] [Google Scholar]
- 109.Crow YJ, Manel N. Aicardi-Goutieres syndrome and the type I interferonopathies. Nat Rev Immunol. 2015;15(7):429–440. doi: 10.1038/nri3850. [DOI] [PubMed] [Google Scholar]
- 110.Liu Y, Jesus AA, Marrero B, Yang D, Ramsey SE, Montealegre Sanchez GA, Tenbrock K, Wittkowski H, Jones OY, Kuehn HS, Lee CC, DiMattia MA, Cowen EW, Gonzalez B, Palmer I, DiGiovanna JJ, Biancotto A, Kim H, Tsai WL, Trier AM, Huang Y, Stone DL, Hill S, Kim HJ, St Hilaire C, Gurprasad S, Plass N, Chapelle D, Horkayne-Szakaly I, Foell D, Barysenka A, Candotti F, Holland SM, Hughes JD, Mehmet H, Issekutz AC, Raffeld M, McElwee J, Fontana JR, Minniti CP, Moir S, Kastner DL, Gadina M, Steven AC, Wingfield PT, Brooks SR, Rosenzweig SD, Fleisher TA, Deng Z, Boehm M, Paller AS, Goldbach-Mansky R. Activated STING in a vascular and pulmonary syndrome. N Engl J Med. 2014;371(6):507–518. doi: 10.1056/NEJMoa1312625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Malakhova OA, Yan M, Malakhov MP, Yuan Y, Ritchie KJ, Kim KI, Peterson LF, Shuai K, Zhang DE. Protein ISGylation modulates the JAK-STAT signaling pathway. Genes Dev. 2003;17(4):455–460. doi: 10.1101/gad.1056303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Filipovic R, Zecevic N. Neuroprotective role of minocycline in co-cultures of human fetal neurons and microglia. Exp Neurol. 2008;211(1):41–51. doi: 10.1016/j.expneurol.2007.12.024. [DOI] [PubMed] [Google Scholar]
- 113.Favrais G, van de Looij Y, Fleiss B, Ramanantsoa N, Bonnin P, Stoltenburg-Didinger G, Lacaud A, Saliba E, Dammann O, Gallego J, Sizonenko S, Hagberg H, Lelievre V, Gressens P. Systemic inflammation disrupts the developmental program of white matter. Ann Neurol. 2011;70(4):550–565. doi: 10.1002/ana.22489. [DOI] [PubMed] [Google Scholar]
- 114.Rivera JC, Sitaras N, Noueihed B, Hamel D, Madaan A, Zhou T, Honore JC, Quiniou C, Joyal JS, Hardy P, Sennlaub F, Lubell W, Chemtob S. Microglia and interleukin-1beta in ischemic retinopathy elicit microvascular degeneration through neuronal semaphorin-3A. Arterioscler Thromb Vasc Biol. 2013;33(8):1881–1891. doi: 10.1161/ATVBAHA.113.301331. [DOI] [PubMed] [Google Scholar]
- 115.McDermott JE, Vartanian KB, Mitchell H, Stevens SL, Sanfilippo A, Stenzel-Poore MP. Identification and validation of Ifit1 as an important innate immune bottleneck. PloS ONE. 2012;7(6):e36465. doi: 10.1371/journal.pone.0036465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Wieghofer P, Knobeloch KP, Prinz M. Genetic targeting of microglia. Glia. 2015;63(1):1–22. doi: 10.1002/glia.22727. [DOI] [PubMed] [Google Scholar]
- 117.Capotondo A, Milazzo R, Politi LS, Quattrini A, Palini A, Plati T, Merella S, Nonis A, di Serio C, Montini E, Naldini L, Biffi A. Brain conditioning is instrumental for successful microglia reconstitution following hematopoietic stem cell transplantation. Proc Natl Acad Sci USA. 2012;109(37):15018–15023. doi: 10.1073/pnas.1205858109. [DOI] [PMC free article] [PubMed] [Google Scholar]