

Abstract
Arrhythmogenic Right Ventricular Cardiomyopathy (ARVC) is an inherited cardiomyopathy that is characterized by ventricular arrhythmias, and an increased risk of sudden death (SCD). Although structural abnormalities of the right ventricle predominate, it is well recognized that left ventricular involvement is common particularly in advanced disease, and that left dominant forms occur. The pathologic characteristic of ARVC/D is myocyte loss with fibrofatty replacement. Since the first detailed clinical description of the disorder in 1982, significant advances have been made in understanding this disease. Once the diagnosis of ARVC is established, the single most important clinical decision is whether a particular patient’s SCD risk is sufficient to justify placement of an implantable cardioverter defibrillator (ICD). The importance of this decision reflects the fact that ARVC is a common cause of sudden death in young people, and that sudden cardiac death may be the first manifestation of the disease. This decision is particularly important since these are often young patients who are expected to live for many years. While an ICD can save lives in individuals with this disease, it is also well recognized that ICD therapy is associated with both short and long term complications. Decisions regarding placement of an ICD are based on an estimate of a patient’s risk of SCD, as well as their preferences and values. The primary purpose of this article is to provide a review of literature that concerns risk stratification in patients with ARVC and to place this literature into the framework of the three authors considerable lifetime experiences in caring for patients with ARVC. The most important parameters to consider when determining arrhythmic risk include: 1) electrical instability including frequency of PVC’s and sustained VA, 2) proband status, 3) extent of structural disease, 4) cardiac syncope, 5) male gender, 6) the presence of multiple mutations or a mutation in TMEM 43, and 7) the patient’s willingness to restrict exercise and eliminate participation in competitive or endurance exercise.
Keywords: Arrhythmogenic right ventricular cardiomyopathy, arrhythmogenic right ventricular dysplasia, sudden cardiac death, risk stratification, implantable cardioverter defibrillator, syncope
Introduction
Arrhythmogenic Right Ventricular Cardiomyopathy (ARVC) is an inherited cardiomyopathy that is characterized by ventricular arrhythmias, and an increased risk of sudden death (SCD)1–5. This condition has been referred to with a variety of names including arrhythmogenic right ventricular dysplasia (ARVD), arrhythmogenic right ventricular cardiomyopathy (ARVC), and arrhythmogenic cardiomyopathy (AC). Each of these terms refers to the same condition. For this review article we will use the term arrhythmogenic right ventricular cardiomyopathy (ARVC). Although structural abnormalities of the right ventricle predominate, it is well recognized that left ventricular involvement is common particularly in advanced disease, and that left dominant forms occur2,6. The pathologic characteristic of ARVC/D is myocyte loss with fibrofatty replacement. Since the first detailed clinical description of the disorder in 19821, significant advances have been made in understanding this disease. In the majority of patients, ARVC is a disease of desmosomal dysfunction. A pathogenic mutation can be identified in more than 60% of affected individuals6. Consistent with this, genetic testing has emerged as an important diagnostic tool and is also important for cascade family screening. Once the diagnosis of ARVC is established, the single most important clinical decision is whether a particular patient’s SCD risk is sufficient to justify placement of an implantable cardioverter defibrillator (ICD)2, 7. The importance of this decision reflects the fact that ARVC is a common cause of sudden death in young people8, 9, and that sudden cardiac death may be the first manifestation of the disease10–12. This decision is particularly important since these are often young patients who are expected to live for many years. While an ICD can save lives in individuals with this disease, it is also well recognized that ICD therapy is associated with both short and long term complications13.
The primary purpose of this article is to provide a review of risk stratification in patients with ARVC. This is important since decisions regarding placement of an ICD are based on an estimate of a patient’s risk of SCD, as well as their preferences and values. In order to place the issue of risk stratification in context a brief review of ARVC is provided.
ARVC
ARVC has an estimated prevalence of 1 per 5000 in the population. Patients usually present during the second to fifth decade of life with palpitations, lightheadedness, syncope, or uncommonly with sudden death1, 2, 10–12, 14, 15. It is rare for patients with ARVC to have clinical signs or symptoms of ARVC prior to 12 years of age. It is also uncommon to first develop signs or symptoms of ARVC after the age of 60 years10, 14, 16. Symptoms are largely due to the presence of ventricular arrhythmias. Heart failure (HF) is an uncommon and late manifestation of the disease. Figure 1 summarizes the presenting clinical features, clinical course, arrhythmias and survival outcomes of 439 index-patients diagnosed with ARVC14. The mean age at presentation was 36±14 years. Forty-eight index-patients (11%) presented with cardiac arrest. Of these, 25 were resuscitated and 23 died and the diagnosis was established at autopsy. The median age at cardiac arrest was 25 years. An additional 220 index-patients (50%) presented with a sustained ventricular arrhythmia (VA). An ICD was implanted in 212 (87%) of the 245 index-patients with sustained VA or resuscitated SCD as compared with 139 (81%) of the 171 index-patients presenting without a sustained VA. A sustained VA was seen during follow-up in more than two-thirds of the entire group of patients (301, N=72%). Of the 65 index-patients without an ICD, 31 (48%) experienced a sustained VA during follow-up. Among the index-patients with an ICD, 10 died (3%) 2 of SCD, 3 of HF, 2 of a combination of HF and arrhythmias, and 3 of non-cardiac causes. Among index-patients without an ICD, 11 died (17%) during a median follow- up of 5 years, 10 as a result of a cardiac arrest and 1 of heart failure. The incidence of SCD was 16% in index patients without an ICD as compared to 0.6% among those with an ICD (p<0.001). During long-term follow-up, 54 index-patients (13%) developed symptomatic HF and 18 (4%) had cardiac transplantation. Overall, 391 of the 416 index-patients (94%) who presented alive were living at last follow-up. Figure 2A and 2B show the results of the Kaplan-Meier survival analysis by age for these 416 index-patients for the following outcomes: 1) any ARVD/C related symptoms, 2) sustained VA, 3) cardiac mortality, and 4) cardiac transplantation.
Figure 1.

Schematic representation of the presentation, clinical course and outcome in patients with Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy (ARVD/C) (14). The majority presented with sustained ventricular arrhythmias (VA) and received an implantable cardioverter-defibrillator (ICD) during follow-up. Printed from Groeneweg et al (14)
Figure 2.

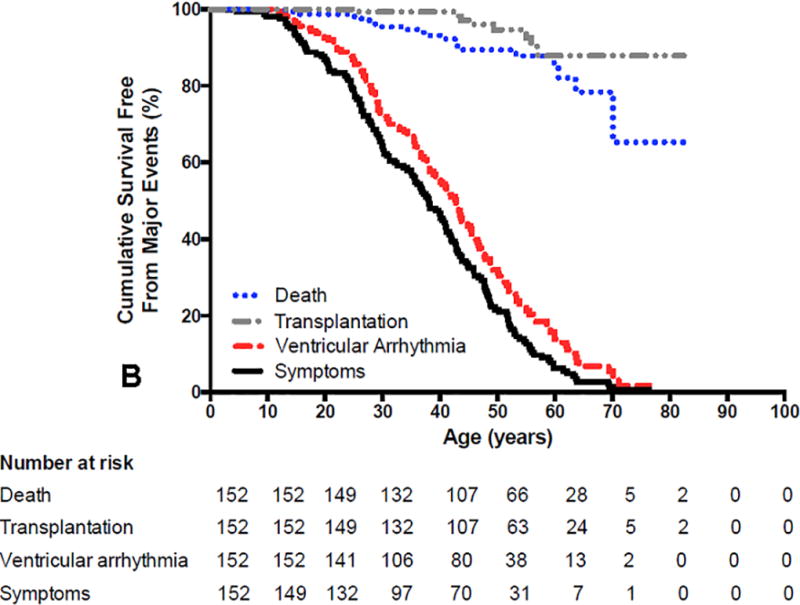
A and B. Survival free from any Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy (ARVD/C) related symptoms, sustained ventricular arrhythmias (VA), cardiac death and cardiac transplantation in ARVD/C index-patients with (2A) pathogenic mutations and (2B) without identified mutations. Symptoms (p=0.005) and sustained VA (p=0.020) occurred more often at younger age in index-patients with mutations. Survival free from cardiac death (p=0.644) and transplantation (p=0.704) was similar in both groups. Reprinted from Groeneweg et al (14)
ARVC is most commonly caused by mutations in desmosomal genes and is inherited in an autosomal dominant pattern with variable penetrance and expressivity. A recent study reported the genetic findings of a large trans-atlantic cohort of 577 patients from 241 unrelated families6. The majority of participants (n=463; 80%) were found to have a single PKP2 mutation with significantly fewer heterozygous carriers of mutations in other ARVD/C-associated genes. Among carriers of single mutations, premature truncating, splice site, and missense mutations were identified in 342 (60%), 130 (23%), and 83 (14%) patients respectively. Patients with SCD or ventricular fibrillation (VF) at presentation (n=36) were younger (median age 23 vs. 36 years; p<0.001) than those presenting with sustained monomorphic ventricular tachycardia (VT). Patients (n=22; 4%) with multiple mutations had significantly earlier occurrence of sustained VT/VF (mean age 28±12 years), lower VT/VF free survival (p=0.037), more frequent left ventricular dysfunction (29%), HF (19%) and cardiac transplantation (9%) as compared to those with only one mutation. DSP mutation carriers had a more than fourfold increased occurrence of left ventricular dysfunction (40%) and HF (13%) than PKP2 carriers. Missense mutation carriers had similar death/transplant free survival and VT/VF penetrance (p=0.137) as compared to those with truncating or splice site mutations.
Management of ARVC
Management of patients with ARVC can be considered to be a five legged stool. The first is to be certain of the correct diagnosis. Misdiagnosis of ARVC is common due to misinterpretation of MR imaging and possible lack of awareness of the 2010 Task Force Criteria17, 18. The diagnosis of ARVC is based on a scoring system with major and minor criteria utilizing a combination of defects in RV morphology and function, characteristic depolarization/ repolarization ECG abnormalities, correct identification of tissue pathology, VA, family history, and the results of genetic testing. The 2010 Task Force Criteria should be used to determine the presence or absence of ARVC17. One approach to this diagnostic classification is to utilize a point classification. Patients are diagnosed as having ARVC if they have 4 points with major criteria equal to two points and minor criteria equal to one point. Patients who have a score of 3 points are classified as having probable ARVC, whereas those with 1 or 2 points are classified as not meeting criteria for ARVC. When considering these guideline recommendations it is important to recognize that there is no single gold-standard diagnostic test. The second leg involves the prediction of the risk of a sustained ventricular arrhythmia to guide decisions regarding ICD implantation. This is the main focus of this review article and will be discussed extensively later in this paper. The third leg is to minimize ICD therapies. This involves a combination of pharmacologic therapy with beta blockers, antiarrhythmic medications, and exercise restriction. These therapies need to be tested in randomized clinical trials. Catheter ablation is another important option for treatment of patients with ARVC who have ventricular tachycardia. This therapy has been markedly improved by the use of epicardial ablation or a combination of endocardial and epicardial ablation for VT19–21. At present this therapy can improve the quality of life by decreasing the frequency of VT episodes. However, it has not been proven to be curative, has not been shown to reduce sudden death risk, and has not been shown to increase survival. It is for this reason that catheter ablation of VT is considered a palliative therapy in patients with ARVC and not a technique that can be used to reduce sudden death risk and need for an ICD. Exercise restriction has been shown to be important to decrease the progression of the disease to reduce the likelihood of sustained ventricular arrhythmias22–27.The fourth leg is to prevent progression of the disease. A recent study compared paired echocardiograms obtained an average of 6.4 years apart in 85 patients with ARVC28. During this time, the RV size increased, and the RV and LV function decreased. This publication, coupled with decades of experience indicates that ARVC is a progressive condition. While clinical studies have not been performed to identify therapies that slow the progression of disease, exercise restriction is likely very important in this regard22–27. James et al have reported that phenotypic expression of ARVC, sustained ventricular arrhythmias, and development of heart failure are related to exercise history23. In this study, HF only developed during follow-up in endurance athletes (18% versus 0%). Similar findings have been reported by Saberniak26. It was notable that the study by Saberniak reported that five ARVC patients, all athletes, underwent cardiac transplantation while no non-athletes were transplanted. The fifth leg is cascade screening of family members. The widespread availability of genetic testing has greatly facilitated this process. Patients diagnosed with ARVC should undergo genetic testing. If a pathogenic mutation is identified, selected testing for this pathogenic mutation can be performed in first degree family members to identify those at risk for development of ARVC. If the same pathogenic mutation is identified in a family member, cardiac evaluation including an ECG, holter, and echocardiogram or MRI should be performed, particularly after puberty. Exercise restriction in mutation carriers is also recommended. Repeat cardiac evaluation should be performed every 2 to 3 years. If no pathogenic mutation is identified in the proband, we generally recommend that cardiac testing, as described above, be performed in all first degree family members every 2 to 3 years depending on the level of physical activity.
Risk Stratification in Patients with ARVC
Since the first description of ARVC by Marcus and Fontaine in 19821, there has been considerable information published about risk stratification in patients with ARVC. This information has largely been the result of single center reports as well as several small multicenter registries. The ultimate approach to risk stratification patients with ARVC can best be determined with a prospective randomized study of ICD implantation. Unfortunately, this type of study has never been performed and in our opinion is unlikely to ever be carried out. Limitations in designing a randomized study include the fact that ARVC is a rare disease with a heterogeneous pattern of clinical presentation. In addition, there are insufficient data to select patients who are at risk for VT but not SCD in order to make a prospective randomized clinical trial acceptable from an ethical perspective.
In general, the risk that a patient with ARVC will experience a sustained VA and/or SCD reflects; 1) whether they have previously experienced a sustained VA, 2) the extent of structural heart disease, 3) the degree of electrical instability including the number of PVCs/24hr and or non-sustained VT 4) cardiac syncope, 5) younger age, 6) male gender, 7) mutation status, and 8) vigorous or sustained exercise. We will review the literature that pertains to each of these risk factors.
Over the past 25 years a number of studies have been performed which have assessed the natural history of ARVC10, 14, 29–33. These studies have focused on the outcomes of populations of ARVC patients without regard to whether a decision had been made to implant an ICD (Table 1) and on defining the outcomes of patients who had an ICD implanted34–42 (Table 2). Interpretation of results of these studies is challenging for many reasons. First, ARVC is an uncommon heterogeneous disease. The number of patients reported in these series is small. Second, the diagnostic criteria for ARVC have evolved over time. Diagnostic criteria for ARVC were initially published in 199443 and were revised in 201017. Advanced MR imaging and genetic testing have only recently become widely available. Third, no uniform standards for phenotyping patients with ARVC and for reporting outcomes have been developed. This makes comparison of published studies difficult. Furthermore, all studies to date phenotyping data are incomplete, which precludes multivariate analysis. Finally, adjunctive therapies in addition to ICD therapy have been different over time. These therapies include antiarrhythmic drugs, VT ablation, and exercise restriction. Virtually all studies which have described the natural history and incidence and predictors of ICD therapy in patients with ARVC have failed to take exercise levels into consideration, reflecting the fact that the link between exercise and the complications of ARVC has only recently been appreciated22–27.
Table 1.
Outcomes of ARVC Patients Regardless of Whether an ICD Was Implanted
| Authors | Publication year |
Study Type | Patient (n) |
Age | Follow- up |
Overall Mortality |
Transplant | Prior VT |
Sudden Death |
Sustained arrhythmia |
Predictors of Mortality / Arrhythmia |
Comments |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Hulot (29) | 2003 | Single Center | 130 | 31±14 | 8.1±7.8 yrs | 18.4 | 79% | 5.4%% | Right heart failure, LV dysfunction, and ventricular tachycardia | No deaths in 10 patients with an ICD, 7 of 14 HF deaths had VT/VF as part of HF exacerbation, no deaths in 28 patients without prior VT | ||
| Pinamonti (30) | 2011 | Single center | 96 | 34±15 | 10.6±7.7 yrs | 13.5 | 7.3 | cardiac dysfunction, LVEF, heart failure symptoms, RVEF, epsilon wave, diuretics, amiodarone | No deaths in the 12 patients implanted with ICD, 3 prim prevention, appropriate therapy in 3/12 | |||
| Brun (31) | 2016 | Multicenter | 88 | not available | 9.1±7.7 | 14 | 4.5 | 59 | 5.6 | VT/NSVT, LV dysfunction | ||
| Kimura (32) | 2016 | Single center | 110 | 48 yrs | 10 yrs | 16 | 1.8 | 67 | 6.3 | 64 | Male sex was a predictor of sustained VT/VF | |
| Groeneweg (14) | 2015 | Multicenter | 416 | 36±14 yrs | 7 yrs | 6 | 4 | 65 | 2.9 | 72 | NA | |
| Mazzanti (33) | 2016 | Single center | 301 | 5.8 | 11.6 | 0 | 8.6 | 8.9 | male gender, younger age, syncope, sustained VT, atrial fibrillation, proband status, exercise level |
Table 2.
Outcomes of ARVC Patients Who Underwent ICD Implantation
| Authors | Publicatio n year |
Study Type |
Patien t (n) |
Age | Follow -up |
Primary preventio n (%) |
Overall Mortalit y (%) |
Transplantatio n (%) |
Appropriat e ICD Interventio n (%) |
ICD Interventio n (VF/Flutter ) (%) |
Predictors of Appropriate Shock |
Predictors of VF |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Corrado (34) | 2003 | multicenter | 132 | 40±15 | 39 ± 27 (3.3) | 28 | 3 | 2.3 | 48 | 24 | NA | younger age, LVEF, cardiac arrest, VT with compromise, unexplained syncope |
| Wichter (35) | 2004 | single center | 60 | 43±16 | 80 ± 43 | 7 | 13 | 3.3 | 68 | 40 | Positive EPS, Extensive RV dysfunction, LV disease | Inducible VF |
| Roguin (36) | 2004 | single center | 42 | 42±26 | 40 | 2 | 2.3 | 78 | NA | Positive EPS, spontaneous VT, male gender, severe RV dysfunction | NA | |
| Piccini (37) | 2005 | single center | 55 | 51 + 11 | 42 | 9 | 3.6 | 73 | 21 | Positive EPS, severe structural disease, NSVT, sustained VT | NA | |
| Corrado (38) | 2010 | multicenter | 106 | 36 ± 18 | 58± 31 | 100 | 0 | 24 | 16 | syncope, NSVT | syncope | |
| Bhonsale (39) | 2011 | single center | 84 | 32 ± 12 | 100 | 2.4 | 8.3 | 48 | 19 | proband status, NSVT, PVC> 1000, pos EPS, | NA | |
| Link (40) | 2014 | multicenter | 108 | 40±14 | 39.6 ± 20.4 | 52 | 0 | 0 | 44 | 20 | Prior VT/VF, inferior T wave inversion | younger age |
| Orgeron (41) | 2017 | single center | 312 | 34 ± 7 | 106 ± 88 | 135 | 2 | 4 | 60 | 19 | spontaneous VT, positive EPS, male, T wave inversion > 3 precordial leads, PVC > 1000 | age < 30, PVC > 1000, syncope, male gender |
Shown in Table 1 is a list of studies which have reported the outcomes of patients with ARVC regardless of ICD implantation14, 29–33. We described in detail the findings of the largest of these studies above14. It can be appreciated that ARVC patients are at significant risk of sudden death, cardiac death, and need for cardiac transplantation over time. Shown in Table 2 is a list of studies which have reported the outcomes of patients with ARVC who have undergone ICD implantation. These studies reveal that patients with ARVC in whom a decision has been made to implant an ICD are at high risk for having VA. During approximately 32 to 106 months of follow-up most studies report the incidence of an appropriate therapy for a sustained ventricular arrhythmia (VT or VF) to be between 40% and 70% and an incidence of an appropriate ICD therapy for VF / V flutter to be approximately 20%. Variables which have been associated with increased risk include a prior sustained VA, NSVT, frequent PVCs, cardiac syncope, proband status, male gender, and the extent of structural disease.
Among these eight studies, the most recent and largest study reporting the outcomes of ICD therapy in patients with ARVC was published in 201741. This study reported the outcomes of 312 ARVC patients (163 men, age at presentation 33.6±13.9) who received an ICD. During a mean follow-up of 8.8±7.3 years, 186 (60%) subjects had an appropriate ICD therapy and 58 (19%) had ICD therapy for ventricular fibrillation/flutter (VF/VFL) (Figure 3). Variables which were identified as predictors of any ICD therapy were ventricular tachycardia (VT) at presentation (HR:1.86, CI:1.38–2.49; p<0.001), inducibility on electrophysiology study (EPS) (3.14, 1.95–5.05; p<0.001), male sex (1.62, 1.20–2.19; p=0.001), inverted T-waves in ≥3 precordial leads (1.66, 1.09–2.52; p=0.018), and premature ventricular contractions (PVC) count ≥1,000/24-hours (2.30, 1.32–4.00; p=0.003) (Figure 4). Among these variables, the only one that remained after multivariable analysis was inducibility at EPS (2.28: 1.10–4.70; p=0.025). Variables which were identified as predictors for an ICD therapy for VF/VFL were: PVC ≥1,000/24 hours (4.39, 1.32–14.61; p=0.016), syncope (1.85, 1.10–3.11; p=0.021), ≤30 years of age at presentation (1.76, 1.04–3.00; p<0.036), and male sex (1.73, 1.01–2.97; p= 0.046) (Figure 5). Younger age at presentation (3.14, 1.32–7.48; p=0.010) and high PVC frequency (4.43, 1.35–14.57; p<0.014) remained as independent predictors of VF/VFL on multivariable analysis. Ninety-eight lead or device related complications occurred in 66 (21%) subjects. Sixty-six of the 98 complications (67%) were lead related and the remainder were generator-related (32, 33%). Six (6%) patients experienced decreased sensing on the right ventricular (RV) lead requiring revision or replacement. Sixty-four of the 312 patients in this series (21%) had inappropriate ICD interventions. The overall mortality was 2%. Cardiac transplantation was required during follow-up in 4% of subjects.
Figure 3.

A–F. Cumulative survival from first appropriate ICD intervention (VT/VF) stratified by inducibility on EPS (A), sex (B), number of T-wave inversions (C), PVCs on Holter monitor (D), history of VT at presentation (E), and primary versus secondary prevention (VT/VF) (F) Reprinted from Orgeron et al (41).
Figure 4.
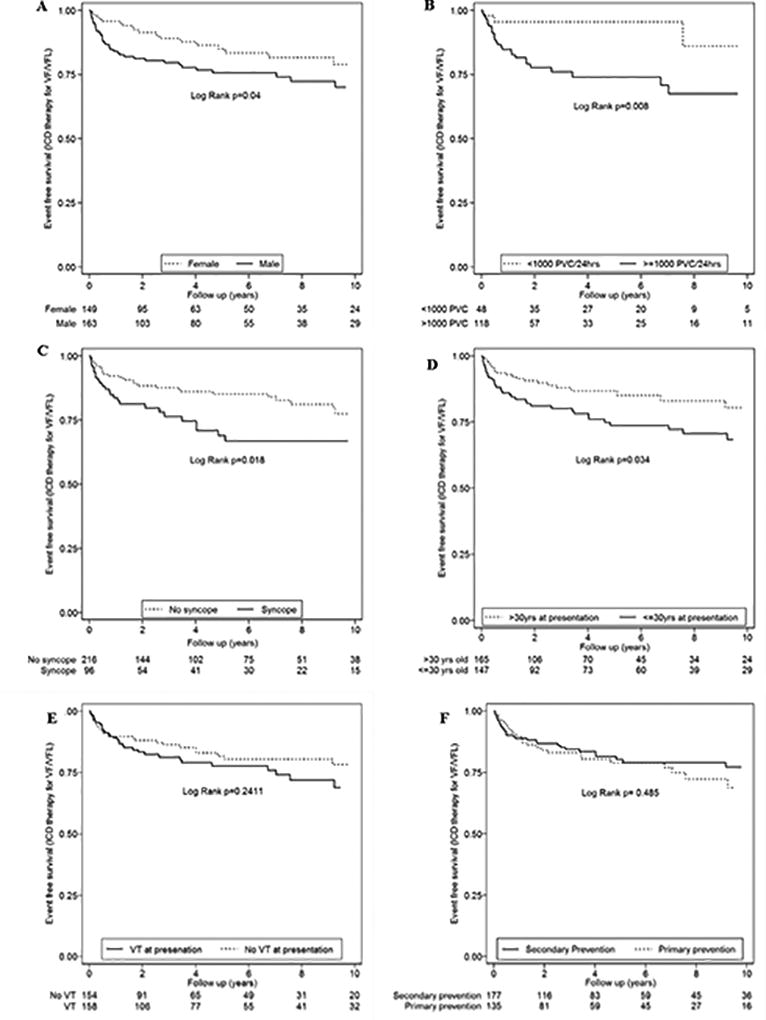
A–F. Cumulative survival from ICD intervention for VF/VFL (CL ≤240ms) stratified by sex (A), PVCs on Holter monitor (B), syncope (C), age at presentation (D), history of VT at presentation (E), and primary versus secondary prevention (VT/VF) (F). Reprinted from Orgeron et al (41).
Figure 5.

A–D. Incidence of sustained ventricular arrhythmias in patients with ARVD/C receiving an ICD for primary prevention based on 4 variables: Inducibility of VT at EPS (A), nonsustained VT (B) PVC frequency (C), and proband status (D). Reprinted from Bhonsale et al (39).
Relatively little is known regarding the outcomes of ARVC patients who have a subcutaneous ICD implanted41, 44. Among the 312 ARVC patients described in the recent large report of the outcomes of ICD therapy in ARVC patients, 7 had a subcutaneous ICD implanted41. The clinical features and outcomes of these patients were not reported separately. Similarly, 15 of the 1637 patients (0.9%) who were included in the recently published post approval study of the subcutaneous ICD had ARVC. The outcomes of this small subset of patients have not been separately reported44.
The objective of the recent study by Brun and colleagues was to examine risk stratification in patients with ARVC who did not have an ICD30. This is important because as it is well recognized that using appropriate ICD therapies for ventricular fibrillation and ventricular flutter overestimates the mortality benefit of ICD therapy. In this study 88 probands from 3 centers were followed for an average of 9.1 + 7.7 years. There were 12 deaths (14%). Among the 36 patients who had never had a sustained VA before enrollment, there were no deaths. There were 5 deaths due to a VA in the 52 patients who had sustained VA before registry enrollment. A relationship was observed between index VT and all-cause mortality (p = 0.052). Among the 5 patients who had an arrhythmic death, the LVEF at baseline was significantly lower (p=0.04) that that of those who did not have an arrhythmic death. No relationship was observed between mortality and syncope, male gender, age, the results of EPS, T wave inversion, QRS duration, and the use of beta blockers. The authors concluded that risk factors for arrhythmic events include prior sustained or nonsustained VT and decreased LV function. These results are in agreement with those of previous outcome studies on ARVC patients with an ICD.
Selected Topics in Risk Stratification of ARVC Patients
Sustained Ventricular Arrhythmias as a Risk Marker
ARVC patients who have experienced a sustained ventricular arrhythmia (sustained VT, ventricular flutter, or ventricular fibrillation) are at a considerable risk of developing recurrent sustained VT as well as ventricular fibrillation (or ventricular flutter as defined as a CL ≤ 240 msec)33, 37, 41. This supports a recommendation for ICD implantation in these patients. Patients with ARVC who have hemodynamically stable VT may be at sufficiently low risk to justify that an ICD not be implanted. For example, in 2003 Corrado reported that this subset of patients had only a 1% subsequent risk of VF34. Whether this finding is reproducible is not known as few other studies have subdivided patients with sustained VT into those with hemodynamically stable versus hemodynamically unstable VT. Some insight in this topic is provided by the recent study by Mazzanti et al33. In this large series, hemodynamically stable VT was a predictor of development of a lethal ventricular arrhythmia. These findings are consistent with the Johns Hopkins clinical experience which supports the concept that patients with ARVC who experience a sustained ventricular arrhythmia, regardless of hemodynamic stability, have a sufficiently high risk of sudden death to warrant placement of an ICD41.
Proband versus Family Member
Probands have a higher arrhythmic risk than family members with ARVC. One of the largest studies to evaluate this was by Bhonsale et al39. (Figure 5). This may be due to the fact that family members are generally diagnosed earlier in the course of the disease. The diagnosis of ARVC in a first degree family member is a major criteria for ARVC17. As a result, evidence of ARVC, such as T wave inversion in leads V1 and V2 above the age of 12 years and > 1000 PVCs/24 hours will confirm the diagnosis of ARVC in a family member. To the degree that the extent of disease predicts arrhythmic risk in ARVC, it is understandable that proband status is an important variable for risk stratification.
Cardiac Syncope is Important for Risk Stratification in ARVC
A recent history of cardiac syncope is an important risk marker in patients with ARVC. This link was recognized first identified by Marcus et al3 and reinforced by studies that described the clinical characteristics of ARVC in patients who died suddenly10–12. It was striking that there was a subset of patients with unexplained syncope occurring within a year or within months of SCD. This link has also been confirmed in reports of ICD therapy in ARVC patients. In a study by Corrado et al, of 106 ARVC patients, syncope as well as a previous episode of sustained VT or VF were important predictors of ICD therapy as well as VT/VF38. These findings have recently been confirmed in a large report examining predictors of ICD therapy in 312 ARVC patients41. Syncope was identified as one of the predictors of ICD therapy for VF/VFL (1.85, 1.10–3.11; p=0.021)41. Because in young individuals syncope is most commonly due to vasovagal causes, careful attention should be paid to defining the circumstances and details of a syncopal episode and to consider this a risk factor for sudden death only in cases of a probable “arrhythmic” nature.
Evidence of Electrical Instability as a Risk Marker
It is now well recognized, that electrical instability is an important risk factor for development of a sustained ventricular arrhythmia in patients with ARVC. Multiple studies have reported that the presence of NSVT as well as frequent PVCs on a 24 hour holter are risk factors for a sustained ventricular arrhythmia37, 38, 41. In a series of 84 ARVC patients who underwent implantation of an ICD for primary prevention, both frequent PVCs and NSVT were identified as significant predictors of appropriate ICD therapy38. Furthermore, these authors reported that the greater the number of PVCs over a 24 hour period, the higher the arrhythmic risk. Another marker of electrical instability is inducibility of sustained VT during an EP study. Several series36, 37,39,41,45 have identified inducibility at EP testing as a risk marker in ARVC patients who have undergone implantation of an ICD. Although others have not found that EP testing is predictive38. Our experience, supports the role of EP testing for risk stratification, especially when a patient has an EP study for diagnostic evaluation. One of the most important differential diagnosis to consider when evaluating a patient with ARVC and VT is idiopathic outflow tract VT. EP testing can not only be helpful in distinguishing these two conditions, but also lead to cure of idiopathic RVOT VT in appropriate patients by mapping and catheter ablation.
Severity of RV and LV Disease
Another variable that is important for risk stratification is the extent of RV and LV disease. Studies have shown that arrhythmic risk is greater in those with more severe disease even if different variables are used to assess the extent of disease. For example, studies have shown that patients with a greater number of T wave inversions on a 12 lead ECG have a higher arrhythmic risk39, 40, 46. There are reports that patients with more extensive abnormalities detected on a SAECG have a higher arrhythmic risk47. Several studies have shown that patients with more extensive RV and/or LV disease have a higher arrhythmic risk35, 48–49. Other studies have reported that the presence of a larger area of scar, as assessed by electroanatomic mapping have more arrhythmias that those with less scar50. This literature supports the view that those patients with more extensive myocardial disease of the heart, as assessed by a number of metrics, are at higher arrhythmic risk than those with little to no structural disease.
Exercise
Another variable for risk stratification is whether the patient with ARVC should participate in sustained and/or high level exercise22–27,51. Over the past five years a number of studies have linked ARVC onset or progression with exercise. These studies have shown that endurance exercise can evoke an earlier presentation of ARVC, and that exercising patients have more severe clinical manifestations of disease such as earlier onset of ventricular arrhythmias, structural dysfunction, heart failure and need for cardiac transplantation
The role of exercise in patients with ARVC as well as the important question of a safe exercise threshold has been reported by the Johns Hopkins ARVC Program24. These studies have shown that patients with ARVC should not engage in high level or endurance exercise, regardless of their genotype. Although it might be potentially safe for asymptomatic mutation carriers to practice the level of exercise as suggested for a healthy adult as recommended by the American Heart Association (AHA), more research is needed to determine the threshold trigger of exercise that produces symptomatic ARVC25.
What about a Role for Gender and Genotype
Men with ARVC are at higher arrhythmia risk than women14, 41, 51–54. While the specific pathophysiologic basis for this observation is unclear it has been suggested that this may be due to a more frequent and intense exercise in men. Others have proposed that this is more closely linked to cellular mechanisms.
Genotype is also of prognostic value. A number of studies have reported that carriers of multiple mutations in the same desmosomal gene, or mutations in two or more genes are at higher arrhythmic risk than those with a single mutation6,55. It is noteworthy that in a multivariable model analysis, multiple desmosomal gene mutations and male gender resulted independent predictors of life-time major arrhythmic events with a Hazard Ratio (HR) of 2.3 and 2.9, respectively55.
It is also recognized that certain ARVC-causing genetic defects may be considered as risk factors for sudden cardiac death. This is especially the case for the “non-desmosomal” TMEM43-p.S358L missense mutation56, which has been shown to be fully penetrant and highly lethal. However, this founder mutation is rare, and is found almost exclusively in Newfoundland.
The presence of phenotypic expression has been shown to be a prerequisite for malignant ventricular arrhythmias and SCD. In a cohort of ARVC desmosomal gene mutation carriers who were prospectively investigated during a long-term follow-up (8.5 years), Zorzi et al.57 found that major arrhythmic events generally occurred in desmosomal gene mutation carriers who fulfilled morphofunctional International Task Force diagnostic criteria and had major risk factors. Desmosomal gene mutation carriers without a definite ARVC phenotypic expression had an uneventful clinical course, with the exception of a 15-year-old desmoplakin gene mutation carrier with previous normal ECG and echocardiographic findings who died suddenly during sleep 2 years later. Postmortem evaluation demonstrated the presence of an epicardial scar in the inferolateral LV region. This finding suggests that lethal ventricular arrhythmias during the concealed phase of ARVC, may be the result of the low sensitivity of routine clinical tests such as ECG and echocardiography for detection of early/minor arrhythmogenic phenotypic substrates, such as an isolated epicardial scar of the LV, for which detection requires more sophisticated imaging technology such as contrast-enhanced cardiac magnetic resonance. Further complicating this issue is that the 2010 Task Force Criteria have shown relatively poor performance for diagnosis (not to mention risk stratification) of isolated LV disease17. These data were recently confirmed by the study of te Riele et al.58 who reported that electrical abnormalities on electrocardiography and holter monitoring precede detectable structural abnormalities in ARVC mutation carriers. Among ARVC mutation carriers, the presence of both electrical and structural abnormalities on CMR identified patients at high risk for arrhythmic events who may benefit from ICD implantation.
Implications for ICD therapy
Recently, an International Task Force of experts from both Europe and USA produced a consensus document on treatment of ARVC7. This International Task Force document provided a summary of existing evidence and a set of recommendations for ICD implantation in patients with ARVC. The document defined three groups of ARVC patients according to their arrhythmic risk (Fig. 6). The high-risk group includes patients with a history of cardiac arrest or sustained VT, as well as those with severe right or left ventricular dysfunction. The indication for ICD implantation in this subset of patients was a class I. The “intermediate risk” category included patients with ≥1 major risk factor (cardiac syncope, non-sustained VT, moderate RV or LV dysfunction, in whom implantation is reasonable (class IIa). A third, lower risk group was defined as those with ≥1 “minor” risk factors (e.g. frequent PVCs, inducible VT at EPS, male gender, complex genotype, younger age, T wave inversion V4 and beyond)). For these patients ICD implantation can be considered, especially if there are multiple risk factors, but should not be systematically performed. And finally, there is a low risk group, in whom the risk of SCD was not sufficiently high to recommend ICD implantation. This includes gene carriers who do not meet diagnostic criteria for ARVC as well as patients who meet these, but do not have high risk markers for SCD. When considering whether to implant an ICD in a patient with ARVC, it is important to note that a family history of SCD, even in a first degree relative is not a risk marker for SCD. Since a family history of ARVC and/or a pathogenic mutation is a major diagnostic criterion for ARVC, these individuals can meet the diagnostic criteria for ARVC in the absence of significant structural or arrhythmic evidence of the disease. This group of individuals is at much lower risk of sudden death, especially if they refrain from vigorous and/or competitive exercise. Finally, patient preferences and values are important and should be respected. Information available concerning risk stratification for sudden death is primarily derived from single center experiences and incomplete phenotyping.
Figure 6.

Flow chart of risk stratification and indications to ICD implantation in ARVC/D. VT, ventricular tachycardia. RV, right ventricle. LV, left ventricle. Major risk factors include syncope, nonsustained VT, and moderate ventricular dysfunction, either RV (RVEF 36% – 46%, or LV (LVEF 36% – 45%). Minor risk factors* include: complex genotype, male gender, proband, positive EP study, T wave inversion in 2 of 3 inferior leads, T wave inversion ≥ 3 precordial leads. Modified from Corrado et al. (7).
Once a decision is made to implant an ICD a final decision concerns which type of ICD to implant. In the absence of a clear indication for atrial pacing, single chamber ICDs are preferred as the risk of early and late complications is reduced. Little data is currently available to guide the decision as whether to implant a subcutaneous ICD or a standard endocardial ICD. The benefits of avoiding an endocardial lead in young people must be balanced against the shorter battery life, the increased risk of inappropriate therapies, and the absence of anti-tachycardia pacing for VT termination. For patients undergoing ICD implantation for primary prevention a strong argument can be made for a subcutaneous ICD whereas for patients who present with sustained monomorphic VT an endocardial device may have clear advantages as the risk of recurrent sustained VT is increased in this subset of patients. Hopefully new studies will be available soon which can provide data to help guide this important decision.
Conclusions
Risk stratification is important for management of patients with ARVC. The most important parameters to consider when determining arrhythmic risk include: 1) electrical instability including frequency of PVC’s and sustained VA, 2) proband status, 3) extent of structural disease, 4) cardiac syncope, 5) male gender, 6) the presence of multiple mutations or a mutation in TMEM43, and 7 agreements to restrict exercise and eliminate participation in competitive or endurance exercise.
Supplementary Material
Acknowledgments
Dr. Calkins is a consultant for Medtronic Inc. and St. Jude Medical. Dr. Calkins receives research support from the St. Jude Medical Foundation and Boston Scientific Corp.
Funding:
Dr Marcus is funded in part by NIH grant #4R01HL116906-04 “Genetics, Mechanisms and Clinical Phenotypes of Arrhythmogenic Cardiomyopathy
Dr Calkins is supported in part by the the Leducq foundation – RHYTHM Network and the Johns Hopkins ARVD Program (www.ARVD.com)
Dr. Corrado is funded by the research grant BIRD (budget for integrated departmental research) 2016, University of Padova, Italy and the TRANSAC (translational arrhythmogenic cardiomyopathy) project, 2013, University of Padova, Italy
Footnotes
Conflict of Interest Disclosure:
Dr Corrado and Dr Marcus have no conflicts of interest to declare.
References
- 1.Marcus FI, Fontaine GH, Guiraudon G, Frank R, Laurenceau JL, Malergue C, Grosgogeat Y. Right ventricular dysplasia: a report of 24 adult cases. Circulation. 1982;65:384–398. doi: 10.1161/01.cir.65.2.384. [DOI] [PubMed] [Google Scholar]
- 2.Corrado D, Link MS, Calkins H. Arrhythmogenic Right Ventricular Cardiomyopathy. N Engl J Med. 2017;376:61–72. doi: 10.1056/NEJMra1509267. [DOI] [PubMed] [Google Scholar]
- 3.Marcus FI, Fontaine GH, Frank R, Gallagher JJ, Reiter MJ. Long-term follow-up in patients with arrhythmogenic right ventricular disease. Eur Heart J. 1989;10(Supplement D):68–73. doi: 10.1093/eurheartj/10.suppl_d.68. [DOI] [PubMed] [Google Scholar]
- 4.Marcus FI. Update of arrhythmogenic right ventricular dysplasia. Card Electrophysiol Rev. 2002;6:54–56. doi: 10.1023/a:1017983004818. [DOI] [PubMed] [Google Scholar]
- 5.Marcus FI, Abidov A. Arrhythmogenic right ventricular cardiomyopathy 2012: diagnostic challenges and treatment. J Cardiovasc Electrophysiol. 2012;23:1149–1153. doi: 10.1111/j.1540-8167.2012.02412.x. [DOI] [PubMed] [Google Scholar]
- 6.Bhonsale A, Groeneweg JA, James CA, Dooijes D, Tichnell C, Jongbloed JD, Murray B, te Riele AS, van den Berg MP, Bikker H, Atsma DE, de Groot NM, Houweling AC, van der Heijden JF, Russell SD, Doevendans PA, van Veen TA, Tandri H, Wilde AA, Judge DP, van Tintelen JP, Calkins H, Hauer RN. Impact of genotype on clinical course in arrhythmogenic right ventricular dysplasia/cardiomyopathy-associated mutation carriers. Eur Heart J. 2015;36:847–855. doi: 10.1093/eurheartj/ehu509. [DOI] [PubMed] [Google Scholar]
- 7.Corrado D, Wichter T, Link MS, Hauer R, Marchlinski F, Anastasakis A, Bauce B, Basso C, Brunckhorst C, Tsatsopoulou A, Tandri H, Paul M, Schmied C, Pelliccia A, Duru F, Protonotarios N, Estes NA, 3rd, McKenna WJ, Thiene G, Marcus FI, Calkins H. Treatment of arrhythmogenic right ventricular cardiomyopathy/dysplasia: an international task force consensus statement. Circulation. 2015;132:441–453. doi: 10.1161/CIRCULATIONAHA.115.017944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bagnall RD, Weintraub RG, Ingles J, Duflou J, Yeates L, Lam L, Davis AM, Thompson T, Connell V, Wallace J, Naylor C, Crawford J, Love DR, Hallam L, White J, Lawrence C, Lynch M, Morgan N, James P, du Sart D, Puranik R, Langlois N, Vohra J, Winship I, Atherton J, McGaughran J, Skinner JR, Semsarian C. A Prospective Study of Sudden Cardiac Death among Children and Young Adults. N Engl J Med. 2016;374:2441–2452. doi: 10.1056/NEJMoa1510687. [DOI] [PubMed] [Google Scholar]
- 9.Finocchiaro G, Papadakis M, Robertus JL, Dhutia H, Steriotis AK, Tome M, Mellor G, Merghani A, Malhotra A, Behr E, Sharma S, Sheppard MN. Etiology of Sudden Death in Sports: Insights From a United Kingdom Regional Registry. J Am Coll Cardiol. 2016;67:2108–2115. doi: 10.1016/j.jacc.2016.02.062. [DOI] [PubMed] [Google Scholar]
- 10.Dalal D, Nasir K, Bomma C, Prakasa K, Tandri H, Piccini J, Roguin A, Tichnell C, James C, Russell SD, Judge DP, Abraham T, Spevak PJ, Bluemke DA, Calkins H. Arrhythmogenic right ventricular dysplasia: a United States experience. Circulation. 2005;112:3823–3832. doi: 10.1161/CIRCULATIONAHA.105.542266. [DOI] [PubMed] [Google Scholar]
- 11.Gupta R, Tichnell C, Murray B, Rizzo S, te Riele A, Tandri H, Judge D, Thiene G, Basso C, Calkins H, James C. Sudden Cardiac Death in Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy (ARVD/C): Clinical and Pathologic Characterization and Comparison with Cases Ascertained Alive. Am J Cardiol. 2017;120:111–117. doi: 10.1016/j.amjcard.2017.03.251. [DOI] [PubMed] [Google Scholar]
- 12.Sadjadieh G, Jabbari R, Risgaard B, Olesen MS, Haunsø S, Tfelt-Hansen J, Winkel BG. Nationwide (Denmark) study of symptoms preceding sudden death due to arrhythmogenic right ventricular cardiomyopathy. Am J Cardiol. 2014;113:1250–1254. doi: 10.1016/j.amjcard.2013.12.038. [DOI] [PubMed] [Google Scholar]
- 13.Olde Nordkamp LR, Postema PG, Knops RE, van Dijk N, Limpens J, Wilde AA, de Groot JR. Implantable cardioverter-defibrillator harm in young patients with inherited arrhythmia syndromes: A systematic review and meta-analysis of inappropriate shocks and complications. Heart Rhythm. 2016;13:443–454. doi: 10.1016/j.hrthm.2015.09.010. [DOI] [PubMed] [Google Scholar]
- 14.Groeneweg J, Bhonsale A, James C, te Riele A, Dooijes D, Tichnell C, Jongbloed JD, Murray B, te Riele AS, van den Berg MP, Bikker H, Atsma DE, de Groot NM, Houweling AC, van der Heijden JF, Russell SD, Doevendans PA, van Veen TA, Tandri H, Wilde AA, Judge DP, van Tintelen JP, Calkins H, Hauer RNl. Clinical Presentation, Long-Term Follow-up, and Outcomes of 1001 Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy Patients and Family Members. Circ Cardiovasc Genet. 2015;8:437–446. doi: 10.1161/CIRCGENETICS.114.001003. In Press. [DOI] [PubMed] [Google Scholar]
- 15.Marcus FI, Zareba W, Calkins H, Towbin JA, Basso C, Bluemke DA, Estes NA, 3rd, Picard MH, Sanborn D, Thiene G, Wichter T, Cannom D, Wilber DJ, Scheinman M, Duff H, Daubert J, Talajic M, Krahn A, Sweeney M, Garan H, Sakaguchi S, Lerman BB, Kerr C, Kron J, Steinberg JS, Sherrill D, Gear K, Brown M, Severski P, Polonsky S, McNitt S. Arrhythmogenic right ventricular cardiomyopathy/dysplasia clinical presentation and diagnostic evaluation: results from the North American Multidisciplinary Study. Heart Rhythm. 2009;6:984–992. doi: 10.1016/j.hrthm.2009.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bhonsale A, te Riele AS, Sawant AC, Groeneweg JA, James CA, Murray B, Tichnell C, Mast TP, van der Pols MJ, Cramer MJ, Dooijes D, van der Heijden JF, Tandri H, van Tintelen JP, Judge DP, Hauer RN, Calkins H. Cardiac Phenotype and Long Term Prognosis of Arrhythmogenic Right Ventricular Cardiomyopathy/Dysplasia Patients with Late Presentation. Heart Rhythm. 2017;14:883–891. doi: 10.1016/j.hrthm.2017.02.013. [DOI] [PubMed] [Google Scholar]
- 17.Marcus FI, McKenna WJ, Sherrill D, Basso C, Bauce B, Bluemke DA, Calkins H, Corrado D, Cox MG, Daubert JP, Fontaine G, Gear K, Hauer R, Nava A, Picard MH, Protonotarios N, Saffitz JE, Sanborn DM, Steinberg JS, Tandri H, Thiene G, Towbin JA, Tsatsopoulou A, Wichter T, Zareba W. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the task force criteria. Circulation. 2010;121:1533–1541. doi: 10.1161/CIRCULATIONAHA.108.840827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rastegar N, Burt JR, Corona-Villalobos CP, te Riele AS, James CA, Murray B, Calkins H, Tandri H, Bluemke DA, Zimmerman SL, Kamel IR. Cardiac MR Findings and potential diagnostic pitfalls in patients evaluated for arrhythmogenic right ventricular cardiomyopathy. Radiographics. 2014;34:1553–1570. doi: 10.1148/rg.346140194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dalal D, Jain R, Tandri H, Dong J, Eid SM, Prakasa K, Tichnell C, James C, Abraham T, Russell SD, Sinha S, Judge DP, Bluemke DA, Marine JE, Calkins H. Long-term efficacy of catheter ablation of ventricular tachycardia in patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy. J Am Coll Cardiol. 2007;50:432–440. doi: 10.1016/j.jacc.2007.03.049. [DOI] [PubMed] [Google Scholar]
- 20.Philips B, te Riele AS, Sawant A, Kareddy V, James CA, Murray B, Tichnell C, Kassamali B, Nazarian S, Judge DP, Calkins H, Tandri H. Outcomes and ventricular tachycardia recurrence characteristics after epicardial ablation of ventricular tachycardia in arrhythmogenic right ventricular dysplasia/cardiomyopathy. Heart Rhythm. 2015;12:716–725. doi: 10.1016/j.hrthm.2014.12.018. [DOI] [PubMed] [Google Scholar]
- 21.Santangeli P, Zado ES, Supple GE, Haqqani HM, Garcia FC, Tschabrunn CM, Callans DJ, Lin D, Dixit S, Hutchinson MD, Riley MP, Marchlinski F. Circ Arrhythm Electropphysiol. 2015;8:1413–1421. doi: 10.1161/CIRCEP.115.003562. [DOI] [PubMed] [Google Scholar]
- 22.Kirchhof P, Fabritz L, Zwiener M, Witt H, Schäfers M, Zellerhoff S, Paul M, Athai T, Hiller KH, Baba HA, Breithardt G, Ruiz P, Wichter T, Levkau B. Age- and training-dependent development of arrhythmogenic right ventricular cardiomyopathy in heterozygous plakoglobin-deficient mice. Circulation. 2006;114:1799–1806. doi: 10.1161/CIRCULATIONAHA.106.624502. [DOI] [PubMed] [Google Scholar]
- 23.James CA, Bhonsale A, Tichnell C, Murray B, Russell SD, Tandri H, Tedford RJ, Judge DP, Calkins H. Exercise Increases Age-Related Penetrance and Arrhythmic Risk in Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy Associated Desmosomal Mutation Carriers. J Am Coll Cardiol. 2013;62:1290–1297. doi: 10.1016/j.jacc.2013.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sawant AC, Bhonsale A, te Riele AS, Tichnell C, Murray B, Russell SD, Tandri H, Tedford RJ, Judge DP, Calkins H, James CA. Exercise has a Disproportionate Role in the Pathogenesis of Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy in Patients Without Desmosomal Mutations. J Am Heart Assoc. 2014;3:e001471, 1–9. doi: 10.1161/JAHA.114.001471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sawant AC, te Riele AS, Tichnell C, Murray B, Bhonsale A, Tandri H, Judge DP, Calkins H, James CA. Safety of American Heart Association-recommended minimum exercise for desmosomal mutation carriers. Heart Rhythm. 2016;13:199–220. doi: 10.1016/j.hrthm.2015.08.035. [DOI] [PubMed] [Google Scholar]
- 26.Saberniak J, Hasselberg NE, Borgquist R, Platonov PG, Sarvari SI, Smith HJ, Ribe M, Holst AG, Edvardsen T, Haugaa KH. Vigorous physical activity impairs myocardial function in patients with arrhythmogenic right ventricular cardiomyopathy and in mutation positive family members. Eur J Heart Fail. 2014;16:1337–1344. doi: 10.1002/ejhf.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ruwald AC, Marcus F, Estes NA, 3rd3, Link M, McNitt S, Polonsky B, Calkins H, Towbin JA, Moss AJ, Zareba W. Association of competitive and recreational sport participation with cardiac events in patients with arrhythmogenic right ventricular cardiomyopathy: results from the North American multidisciplinary study of arrhythmogenic right ventricular cardiomyopathy. Eur Heart J. 2015;36:1735–1743. doi: 10.1093/eurheartj/ehv110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mast TP, James CA, Calkins H, Teske AJ, Tichnell C, Murray B, Loh P, Russell SD, Velthuis BK, Judge DP, Dooijes D, Tedford RJ, van der Heijden JF, Tandri H, Hauer RN, Abraham TP, Doevendans PA, Te Riele AS, Cramer MJ. Evaluation of Structural Progression in Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy. JAMA Cardiol. 2017;2:293–302. doi: 10.1001/jamacardio.2016.5034. [DOI] [PubMed] [Google Scholar]
- 29.Hulot JS, Jouven X, Empana JP, Frank R, Fontaine G. Natural history and risk stratification of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circulation. 2004;110:1879–1884. doi: 10.1161/01.CIR.0000143375.93288.82. [DOI] [PubMed] [Google Scholar]
- 30.Pinamonti B, Dragos AM, Pyxaras SA, Merlo M, Pivetta A, Barbati G, Di Lenarda A, Morgera T, Mestroni L, Sinagra G. Prognostic predictors in arrhythmogenic right ventricular cardiomyopathy: results from a 10-year registry. Eur Heart J. 32:1105–1113. doi: 10.1093/eurheartj/ehr040. 201. [DOI] [PubMed] [Google Scholar]
- 31.Brun F, Groeneweg JA, Gear K, Sinagra G, van der Heijden J, Mestroni L, Hauer RN, Borgstrom M, Marcus FI, Hughes T. Risk Stratification in Arrhythmic Right Ventricular Cardiomyopathy Without Implantable Cardioverter-Defibrillators. JACC Clin Electrophysiol. 2016;2:558–564. doi: 10.1016/j.jacep.2016.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kimura Y, Noda T, Otsuka Y, Wada M, Nakajima I, Ishibashi K, Miyamoto K, Okamura H, Aiba T, Kamakura S, Noguchi T, Anzai T, Satomi K, Ogawa H, Yasuda S, Kusano K. Potentially Lethal Ventricular Arrhythmias and Heart Failure in Arrhythmogenic Right Ventricular Cardiomyopathy What are the Differences Between Men and Women? J Am Coll Cardiol EP. 2016;2:546–555. doi: 10.1016/j.jacep.2016.02.019. [DOI] [PubMed] [Google Scholar]
- 33.Mazzanti A, Ng K, Faragli A, Maragna R, Chiodaroli E, Orphanou N, Monteforte N, Memmi M, Gambelli P, Novelli V, Bloise R, Catalano O, Moro G, Tibollo V, Morini M, Bellazzi R, Napolitano C, Bagnardi V, Priori SG. Arrhythmogenic Right Ventricular Cardiomyopathy: Clinical Course and Predictors of Arrhythmic Risk. J Am Coll Cardiol. 2016;68:2540–2550. doi: 10.1016/j.jacc.2016.09.951. [DOI] [PubMed] [Google Scholar]
- 34.Corrado D, Leoni L, Link MS, Della Bella P, Gaita F, Curnis A, Salerno JU, Igidbashian D, Raviele A, Disertori M, Zanotto G, Verlato R, Vergara G, Delise P, Turrini P, Basso C, Naccarella F, Maddalena F, Estes NA, 3rd, Buja G, Thiene G. Implantable cardioverter-defibrillator therapy for prevention of sudden death in patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia. Circulation. 2003;108:3084–3091. doi: 10.1161/01.CIR.0000103130.33451.D2. [DOI] [PubMed] [Google Scholar]
- 35.Wichter T, Paul M, Wollmann C, Acil T, Gerdes P, Ashraf O, Tjan TD, Soeparwata R, Block M, Borggrefe M, Scheld HH, Breithardt G, Böcker D. Implantable cardioverter/defibrillator therapy in arrhythmogenic right ventricular cardiomyopathy: single-center experience of long-term follow-up and complications in 60 patients. Circulation. 2004;109:1503–1508. doi: 10.1161/01.CIR.0000121738.88273.43. [DOI] [PubMed] [Google Scholar]
- 36.Roguin A, Bomma CS, Nasir K, Tandri H, Tichnell C, James C, Rutberg J, Crosson J, Spevak PJ, Berger RD, Halperin HR, Calkins H. Implantable cardioverter-defibrillators in patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy. J Am Coll Cardiol. 2004;43:1843–1852. doi: 10.1016/j.jacc.2004.01.030. [DOI] [PubMed] [Google Scholar]
- 37.Piccini JP, Dalal D, Roguin A, Bomma C, Cheng A, Prakasa K, Dong J, Tichnell C, James C, Russell S, Crosson J, Berger RD, Marine JE, Tomaselli G, Calkins H. Predictors of appropriate implantable defibrillator therapies in patients with arrhythmogenic right ventricular dysplasia. Heart Rhythm. 2005;2:1188–1194. doi: 10.1016/j.hrthm.2005.08.022. [DOI] [PubMed] [Google Scholar]
- 38.Corrado D, Calkins H, Link MS, Leoni L, Favale S, Bevilacqua M, Basso C, Ward D, Boriani G, Ricci R, Piccini JP, Dalal D, Santini M, Buja G, Iliceto S, Estes NA, 3rd, Wichter T, McKenna WJ, Thiene G, Marcus FI. Prophylactic implantable defibrillator in patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia and no prior ventricular fibrillation or sustained ventricular tachycardia. Circulation. 2010;122:1144–1152. doi: 10.1161/CIRCULATIONAHA.109.913871. [DOI] [PubMed] [Google Scholar]
- 39.Bhonsale A, James CA, Tichnell C, Murray B, Gagarin D, Philips B, Dalal D, Tedford R, Russell SD, Abraham T, Tandri H, Judge DP, Calkins H. Incidence and predictors of implantable cardioverter-defibrillator therapy in patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy undergoing implantable cardioverter-defibrillator implantation for primary prevention. J Am Coll Cardiol. 2011;58:1485–1496. doi: 10.1016/j.jacc.2011.06.043. [DOI] [PubMed] [Google Scholar]
- 40.Link MS, Laidlaw D, Polonsky B, Zareba W, McNitt S, Gear K, Marcus F, Estes NA., 3rd Ventricular arrhythmias in the North American multidisciplinary study of ARVC: predictors, characteristics, and treatment. J Am Coll Cardiol. 2014;64:119–125. doi: 10.1016/j.jacc.2014.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Orgeron GM, James CA, Te Riele A, Tichnell C, Murray B, Bhonsale A, Kamel IR, Zimmerman SL, Judge DP, Crosson J, Tandri H, Calkins H. Implantable Cardioverter-Defibrillator Therapy in Arrhythmogenic Right Ventricular Dysplasia / Cardiomyopathy: predictors of Appropriate Therapy, Outcomes, and Complications. J AM Heart Assoc. 2017:e006242. doi: 10.1161/JAHA.117.006242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Saguner AM, Vecchiati A, Baldinger SH, Rüeger S, Medeiros-Domingo A, Mueller-Burri AS, Haegeli LM, Biaggi P, Manka R, Lüscher TF, Fontaine G, Delacrétaz E, Jenni R, Held L, Brunckhorst C, Duru F, Tanner FC. Different prognostic value of functional right ventricular parameters in arrhythmogenic right ventricular cardiomyopathy/dysplasia. Circ Cardiovasc Imaging. 2014;7:230–239. doi: 10.1161/CIRCIMAGING.113.000210. [DOI] [PubMed] [Google Scholar]
- 43.McKenna WJ, Thiene G, Nava A, Fontaliran F, Blomstrom-Lundqvist C, Fontaine G, Camerini F. Diagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Task Force of the Working Group Myocardial and Pericardial Disease of the European Society of Cardiology and of the Scientific Council on Cardiomyopathies of the International Society and Federation of Cardiology. Br Heart J. 1994;71:215–218. doi: 10.1136/hrt.71.3.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gold MR, Aasbo JD, El-Chami MF, Niebauer M, Herre J, Prutkin JM, Knight B, Kutalek S, Hsu K, Weiss R, Bass E, Husby M, Stivland TM, Burke MC. Subcutaneous implantable cardioverter-defibrillator Post-Approval Study: Clinical characteristics and perioperative results. Heart Rhythm. 2017;14:1456–1463. doi: 10.1016/j.hrthm.2017.05.016. [DOI] [PubMed] [Google Scholar]
- 45.Saguner AM, Medeiros-Domingo A, Schwyzer MA, On CJ, Haegeli LM, Wolber T, Hürlimann D, Steffel J, Krasniqi N, Rüeger S, Held L, Lüscher TF, Brunckhorst C, Duru F. Usefulness of inducible ventricular tachycardia to predict long-term adverse outcomes in arrhythmogenic right ventricular cardiomyopathy. Am J Cardiol. 2013;111:250–257. doi: 10.1016/j.amjcard.2012.09.025. [DOI] [PubMed] [Google Scholar]
- 46.Link MS, Laidlaw D, Polonsky B, Zareba W, McNitt S, Gear K, Marcus F, Estes NA., 3rd Ventricular arrhythmias in the North American multidisciplinary study of ARVC: predictors, characteristics, and treatment. J Am Coll Cardiol. 2014;64:119–125. doi: 10.1016/j.jacc.2014.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Saguner AM, Ganahl S, Baldinger SH, Kraus A, Medeiros-Domingo A, Nordbeck S, Saguner AR, Mueller-Burri AS, Haegeli LM, Wolber T, Steffel J, Krasniqi N, Delacrétaz E, Lüscher TF, Held L, Brunckhorst CB, Duru F. Usefulness of electrocardiographic parameters for risk prediction in arrhythmogenic right ventricular dysplasia. Am J Cardiol. 2014;113:1728–1734. doi: 10.1016/j.amjcard.2014.02.031. [DOI] [PubMed] [Google Scholar]
- 48.Liao YC, Lin YJ, Chung FP, Chang SL, Lo LW, Hu YF, Chao TF, Chung E, Tuan TC, Huang JL, Liao JN, Chen YY, Chen SA. Risk stratification of arrhythmogenic right ventricular cardiomyopathy based on signal averaged electrocardiograms. Int J Cardiol. 2014;174:628–633. doi: 10.1016/j.ijcard.2014.04.169. [DOI] [PubMed] [Google Scholar]
- 49.Saguner AM, Vecchiati A, Baldinger SH, Rüeger S, Medeiros-Domingo A, Mueller-Burri AS, Haegeli LM, Biaggi P, Manka R, Lüscher TF, Fontaine G, Delacrétaz E, Jenni R, Held L, Brunckhorst C, Duru F, Tanner FC. Different prognostic value of functional right ventricular parameters in arrhythmogenic right ventricular cardiomyopathy/dysplasia. Circ Cardiovasc Imaging. 2014;7:230–239. doi: 10.1161/CIRCIMAGING.113.000210. [DOI] [PubMed] [Google Scholar]
- 50.Schuler PK, Haegeli LM, Saguner AM, Wolber T, Tanner FC, Jenni R, Corti N, Lüscher TF, Brunckhorst C, Duru F. Predictors of appropriate ICD therapy in patients with arrhythmogenic right ventricular cardiomyopathy: long term experience of a tertiary care center. PLoS One. 2012;7:e39584. doi: 10.1371/journal.pone.0039584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zorzi A, Migliore F, Elmaghawry M, Silvano M, Marra MP, Niero A, Nguyen K, Rigato I, Bauce B, Basso C, Thiene G, Iliceto S, Corrado D. Electrocardiographic predictors of electroanatomic scar size in arrhythmogenic right ventricular cardiomyopathy: implications for arrhythmic risk stratification. J Cardiovasc Electrophysiol. 2013;24:1321–1327. doi: 10.1111/jce.12246. [DOI] [PubMed] [Google Scholar]
- 52.Chelko SP, Asikamki A, Anderson P, Bedja D, Amat-Alarcon N, DeMuzumder D, Jasti R, MacRae C, Leber R, Kleber AC, Sattiz JE, Judge DP. Central role for GSK3beta in the pathogenesis of arrhythmogenic cardiomyopathy. JCI Insight. 2016;1:e85923. doi: 10.1172/jci.insight.85923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mitchell JH, Haskell W, Snell P, Van Camp SP. Task Force 8: classification of sports. J Am Coll Cardiol. 2005;45:1364–1367. doi: 10.1016/j.jacc.2005.02.015. [DOI] [PubMed] [Google Scholar]
- 54.Choudhary N, Tompkins C, Polonsky B, McNitt S, Calkins H, Mark Estes NA, 3rd, Krahn AD, Link MS, Marcus FI, Towbin JA, Zareba W. Clinical Presentation and Outcomes by Sex in Arrhythmogenic Right Ventricular Cardiomyopathy: Findings from the North American ARVC Registry. J Cardiovasc Electrophysiol. 2016;27:555–562. doi: 10.1111/jce.12947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Akdis D, Saguner Ardan M, Shah Khooshbu, Wei Chuanyu, Medeiros-Domingo Argelia, von Eckardstein Argelia, von Ekardstein Arnold, Luscher Thomas F, Brunckhorst Corinna, Vincent Chen HS, Duru Firat. Sex hormones affect outcome of arrhythmogenic right ventricular cardiomyopathy/dysplasia: from a stem cell derived cardiomyocyte-based model to clinical biomarkers of disease outcome. Eur Heart J. 2017;38:1498–1508. doi: 10.1093/eurheartj/ehx011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rigato I, Bauce B, Rampazzo A, Zorzi A, Pilichou K, Mazzotti E, Migliore F, Marra MP, Lorenzon A, De Bortoli M, Calore M, Nava A, Daliento L, Gregori D, Iliceto S, Thiene G, Basso C, Corrado D. Compound and digenic heterozygosity predicts lifetime arrhythmic outcome and sudden cardiac death in desmosomal gene-related arrhythmogenic right ventricular cardiomyopathy. Circ Cardiovasc Genet. 2013;6:533–542. doi: 10.1161/CIRCGENETICS.113.000288. [DOI] [PubMed] [Google Scholar]
- 57.Merner ND, Hodgkinson KA, Haywood AF, Connors S, French VM, Drenckhahn JD, Kupprion C, Ramadanova K, Thierfelder L, McKenna W, Gallagher B, Morris-Larkin L, Bassett AS, Parfrey PS, Young TL. Arrhythmogenic right ventricular cardiomyopathy type 5 is a fully penetrant, lethal arrhythmic disorder caused by a missense mutation in the TMEM43 gene. Am J Hum Genet. 2008;82:809–821. doi: 10.1016/j.ajhg.2008.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zorzi A, Rigato I, Pilichou K, Perazzolo Marra M, Migliore F, Mazzotti E, Gregori D, Thiene G, Daliento L, Iliceto S, Rampazzo A, Basso C, Bauce B, Corrado D. Phenotypic expression is a prerequisite for malignant arrhythmic events and sudden cardiac death in arrhythmogenic right ventricular cardiomyopathy. Europace. 2016;18:1086–1094. doi: 10.1093/europace/euv205. [DOI] [PubMed] [Google Scholar]
- 59.te Riele AS, Bhonsale A, James CA, Rastegar N, Murray B, Burt JR, Tichnell C, Madhavan S, Judge DP, Bluemke DA, Zimmerman SL, Kamel IR, Calkins H, Tandri H. Incremental value of cardiac magnetic resonance imaging in arrhythmic risk stratification of arrhythmogenic right ventricular dysplasia/cardiomyopathy-associated desmosomal mutation carriers. J Am Coll Cardiol. 2013;62:1761–1769. doi: 10.1016/j.jacc.2012.11.087. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.