

Abstract
Background
The CXCL12/CXCR4 chemokine ligand/receptor axis controls (progenitor) cell homeostasis and trafficking. So far, an atheroprotective role of CXCL12/CXCR4 has only been implied through pharmacological intervention, particularly as the somatic deletion of the CXCR4 gene in mice is embryonically lethal. Moreover, cell-specific effects of CXCR4 in the arterial wall and underlying mechanisms remain elusive, prompting us to investigate the relevance of CXCR4 in vascular cell types for atheroprotection.
Methods
We examined the role of vascular CXCR4 in atherosclerosis and plaque composition by inducing an endothelial cell (EC, BmxCreERT2-driven)-specific or smooth muscle cell (SMC, SmmhcCreERT2- or TaglnCre-driven)-specific-deficiency of CXCR4 in an apolipoprotein E-deficient mouse model. To identify underlying mechanisms for effects of CXCR4, we studied endothelial permeability, intravital leukocyte adhesion, involvement of the Akt/WNT/β-catenin signaling pathway and relevant phosphatases in VE-cadherin expression and function, vascular tone in aortic rings, cholesterol efflux from macrophages, and expression of SMC phenotypic markers. Finally, we analyzed associations of common genetic variants at the CXCR4 locus with the risk for coronary heart disease, along with CXCR4 transcript expression in human atherosclerotic plaques.
Results
The cell-specific deletion of CXCR4 in arterial ECs (n=12-15) or SMCs (n=13-24) markedly increased atherosclerotic lesion formation in hyperlipidemic mice. Endothelial barrier function was promoted by CXCL12/CXCR4, which triggered Akt/WNT/β-catenin-signaling to drive VE-cadherin expression and stabilized junctional VE-cadherin complexes through associated phosphatases. Conversely, endothelial CXCR4-deficiency caused arterial leakage and inflammatory leukocyte recruitment during atherogenesis. In arterial SMCs, CXCR4 sustained normal vascular reactivity and contractile responses, whereas CXCR4-deficiency favored a synthetic phenotype, the occurrence of macrophage-like SMCs in the lesions, and impaired cholesterol efflux. Regression analyses in humans (n=259,796) identified the C-allele at rs2322864 within the CXCR4 locus to be associated with increased risk for coronary heart disease. In line, C/C risk genotype carriers showed reduced CXCR4 expression in carotid artery plaques (n=188), which was furthermore associated with symptomatic disease.
Conclusions
Our data clearly establish that vascular CXCR4 limits atherosclerosis by maintaining arterial integrity, preserving endothelial barrier function, and a normal contractile SMC phenotype. Enhancing these beneficial functions of arterial CXCR4 by selective modulators might open novel therapeutic options in atherosclerosis.
Keywords: atherosclerosis, CXCR4, endothelial cell, permeability, smooth muscle cell phenotype, VE-cadherin, WNT signaling
Introduction
The chemokine receptor CXCR4 and its cognate ligand CXCL12 are best known for their role in the homing of progenitor cells to the bone marrow and their mobilization to the periphery.1 Moreover, the importance of CXCR4 and CXCL12 in cell homeostasis, organ development and vascularization explains why targeted disruption of either gene results in embryonic or perinatal lethality.2–4 Stress-induced progenitor cell mobilization has been linked to a double-edged role of the CXCL12/CXCR4 axis after arterial injury. Whereas recruitment of angiogenic early outgrowth cells mediated by CXCL12/CXCR4 contributes to vascular regeneration, the mobilization of smooth muscle progenitor cells drives injury-induced neointima formation.1,5 However, CXCR4 is not only expressed on progenitor cells, but also on endothelial cells (ECs), vascular smooth muscle cells (SMCs) and on diverse leukocyte subsets, all of which play a role in neointima formation and in native or diet-induced atherosclerosis.1,6 For instance, a protective role of endothelial CXCR4 promoting re-endothelialization after endothelial denudation has been revealed during neointima formation.7 In turn, an atheroprotective role of the CXCL12/CXCR4 axis has been related to the control of neutrophil numbers and activation8,9 as well as microparticle-mediated recruitment of angiogenic cells.10
Given that somatic deletion of the CXCR4 gene in mice is embryonically lethal, the atheroprotective role of CXCR4 has so far been implied by relying on bone marrow chimeras or using pharmacological inhibitors, e.g. systemic treatment with a CXCR4 antagonist.8 Beyond that, vascular cell-specific effects of CXCR4 in the arterial wall and underlying mechanisms have not been elucidated in the context of atherosclerosis. Likewise, genome-wide association studies have validated significant associations between single nucleotide polymorphisms near CXCL12 as a lead gene in the 10q11 locus and the risk of coronary artery disease and early myocardial infarction,11,12 whereas a role of CXCR4 variants, in particular those associated with cardiovascular risk beyond lipid levels,13 remains unknown.
Here, we examined the role of vascular CXCR4 in atherosclerosis by introducing an EC-specific and SMC-specific-deficiency of Cxcr4 in a mouse model of diet-induced atherosclerosis, identified mechanisms underlying atherosclerosis-related effects of CXCR4 in these cell types, and analyzed the association of common CXCR4 variants with the risk for coronary heart disease (CHD) as well as CXCR4 expression in human atherosclerosis.
Methods
Please see online supplemental material for expanded methods.
Animals
All mice were on a C57/Bl6 background (Supplemental Table 1). For atherosclerosis studies, mice were fed a HFD containing 21% fat and 0.15-0.2% cholesterol (Altromin 132010, Sniff TD88137) for 12 weeks. All animal studies were approved by the local ethical committee.
Lesion analysis and intravital microscopy
For analysis of atherosclerotic lesions, the aortic root and thoraco-abdominal aorta were stained for lipid depositions with Oil-Red-O. Lesions in the root were quantified and averaged in 3-5 sections per mouse. Immunohistochemistry was performed to assess cellular composition. Leukocyte adhesion in carotid arteries was investigated using intravital microscopy.
In vivo permeability assay
Endothelial permeability in the aortic arch was studied using Evans blue (1%, 30 min).
Cell culture
Human aortic endothelial cells (hAoECs, Lonza), SV-40-transformed mouse endothelial cells (SVECs) and human aortic vascular smooth muscle cells (hAoSMCs, Lonza) were used.
siRNA silencing and gene expression analysis
Specific knockdown of β-catenin and CXCR4 (Supplemental Table 2) in human aortic endothelial cells (hAoECs) was accomplished by application of RNA interference technology (Qiagen). RNA was isolated using RNeasy kits and Tissuelyser (both Qiagen) and reverse transcribed using MMLV reverse transcriptase (Promega). Quantitative real-time PCR was performed using the Sybrgreen (Supplemental Table 3). Gene expression was normalized to GAPDH using the ΔΔCt method.
Transcription-based reporter assay
WNT/β-catenin signaling activity was quantified in hAoECs using a β-catenin-activated reporter (BAR) system containing multimerized TCF/LEF DNA-binding sites with inserted Gaussia luciferase as a reporter gene. Conditioned media was analyzed using the Gaussia luciferase assay kit (New England Biolabs) and a plate luminometer (Tecan).
In vitro permeability and scratch assay
An in vitro permeability assay (Millipore) was performed with hAoEC monolayers grown in 96-well cell culture inserts with a semi-permeable membrane using FITC-Dextran. A scratch would was introduced by dragging a sterile pipette tip across confluent hAoSMC monolayers to create a cell-free path, which was used for monitoring cell migration.
Vascular reactivity
The thoracic aorta was divided into equal rings. Each aortic ring was connected to a force transducer in organ chambers using a passive tension of 2 g for subsequent manipulation.
Analysis of CXCR4 in humans
We examined the association of 345 common variants at the CXCR4 locus (± 25KB) in association with CHD by using data on altogether 92,516 CHD cases and 167,280 controls (see Supplemental Table 4 for details of data sets included). Human carotid artery endarterectomy specimen (n=218) were collected in a cohort of patients undergoing vascular surgery, and utilization of human vascular tissues was approved by the Ethics Committee of the Medical Faculty at the TU Dresden (EK316122008). Quantification of CXCR4 mRNA expression and genotyping of rs2322864 in human specimens was performed and correlated with the clinical phenotype.
Statistical analysis
All data are expressed as mean±SD or mean±SEM, as indicated. Statistical analyses were performed using GraphPad Prism 6 (GraphPad Software Inc.). After testing for normality, unpaired Student’s t-test (with Welch correction for mouse studies), Mann-Whitney, one-way ANOVA with Tukey’s or Sidak’s multiple comparisons test, Kruskal-Wallis test with Dunn’s post-test, 2-way ANOVA or 2-way repeated measures ANOVA with Bonferroni post-test were used, as appropriate. P-values < 0.05 were considered statistically significant.
Results
CXCR4 on arterial endothelial cells is atheroprotective
To focus on EC-specific effects of CXCR4 in atherosclerosis-prone mice lacking the Apoe gene (Apoe−/−) and subjected to hyperlipidemia, Cxcr4-floxed (Cxcr4fl/fl) Apoe−/− mice were crossed with BmxCreERT2-expressing mice (termed BmxCre), mediating efficient tamoxifen-inducible Cxcr4 deletion in arterial ECs (Figure 1A, Supplemental Figure 1A).7 After 12 weeks of high-fat diet (HFD), BmxCre+Cxcr4fl/flApoe−/− mice displayed larger atherosclerotic lesions in the aortic root (Figure 1B–D), the aortic arch (Figure 1E) and the aorta (Figure 1F), as compared to BmxCre-Cxcr4fl/flApoe−/− controls. Whereas the relative SMC content (Figure 1G) and collagen content (Supplemental Figure 1B) in aortic root lesions were unaltered, macrophage content (Fig. 1H–I, Supplemental Figure 1C) and the necrotic core area (Figure 1J, Supplemental Figure 1D) were increased in BmxCre+Cxcr4fl/flApoe−/− mice. Conversely, the relative presence of ECs in root lesions was decreased in BmxCre+ Cxcr4fl/flApoe−/− mice (Supplemental Figure 1E,F). Whereas the density of ECs lining the plaque cap was changed (Supplemental Figure 1G), the number of intercellular adhesion molecule-1 (ICAM-1)-positive cells lining the plaque cap, likely corresponding to activated ECs, was increased (Figure 1K,L). This correlated with an increased adhesion of inflammatory monocytes and neutrophils to the carotid artery of TNFα-treated BmxCre+Cxcr4fl/flApoe−/− mice on HFD (Figure 1M,N).
Figure 1. CXCR4 on arterial endothelial cells is atheroprotective.
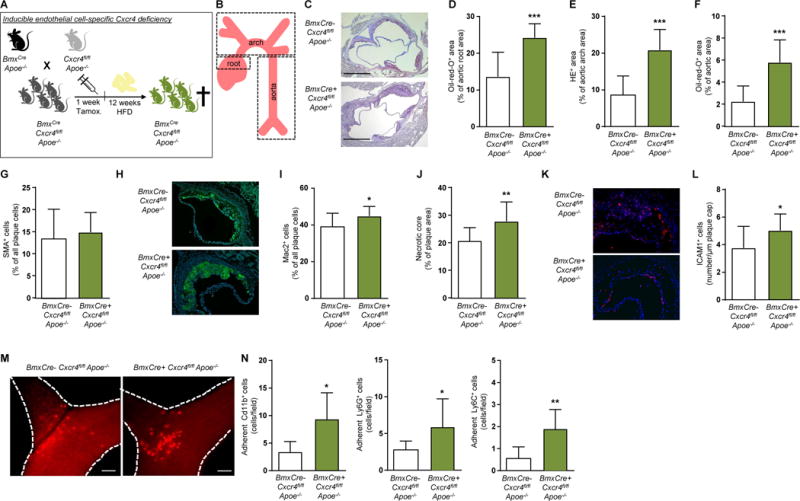
Cxcr4fl/flApoe−/− mice were crossed with BmxCreERT2-expressing mice (termed BmxCre), to allow for tamoxifen-inducible Cxcr4 deletion in arterial endothelial cells. BmxCre+Cxcr4fl/flApoe−/− mice vs BmxCre-Cxcr4fl/flApoe−/− controls were analyzed after 12 weeks of high-fat diet (HFD), unless otherwise indicated. (A,B) Experimental scheme. (C-F) Lesion area measured after Oil-Red-O or HE staining in the aortic root (C, D; n=12-14), aortic arch (E; n=12-15) and aorta (F; n=12-15). Representative images are shown for aortic root (C). Scale bars = 500 µm. (G) SMC content in aortic root lesions, as quantified after staining for SMA (n=12-14). (H,I) Macrophage content in aortic root lesions, as quantified after staining for Mac2 (n=12-14). Representative images are shown in (H). (J) Necrotic core area in aortic root lesions, as quantified by anucleated area measurement (n=12-14). (K, L) The number of ICAM-1+ cells per µm plaque lining in aortic root lesions (n=10-13). Representative images are shown in (K). (M, N) Representative images (M) and quantification (N) of intravital microscopy of leukocyte adhesion to TNFα–stimulated carotid arteries of BmxCre+Cxcr4fl/flApoe−/− vs BmxCre-Cxcr4fl/flApoe−/− mice after 8 weeks of HFD. Leukocyte labeling was performed for CD11b (all myeloid cells), Ly6G (neutrophils) or Ly6C (monocytes) (n=7-10). (D-N) Data represent mean±SD. *P<0.05; **P<0.01; ***P<0.001, as analyzed by Student’s t-test with Welch correction or Mann-Whitney test, as appropriate.
The serum levels of CXCL12 (Supplemental Figure 1H), cholesterol and triglycerides, or body weight did not significantly differ between BmxCre- and BmxCre+Cxcr4fl/flApoe−/− mice after HFD (Supplemental Table 5), nor were numbers of leukocytes, neutrophils, monocytes, lymphocytes and platelets altered in peripheral blood (Supplemental Figure 1I, Supplemental Table 5). In the context of arterial injury, EC-specific Cxcr4 deficiency was found to decrease mobilization of Sca1+CD31+Flk1+ and Lin-Sca1+ progenitor cells,7 often referred to as circulating angiogenic early-outgrowth cells and SMC progenitors,5,14 respectively. As both progenitor subsets have been associated with atheroprotection,15,16 their mobilization was examined by flow cytometry. No differences in these subpopulations or their CXCR4 expression were observed between BmxCre- and BmxCre+ Cxcr4fl/flApoe−/− mice after HFD (Supplemental Figure 1J–Q). This indicates that Bmx is not expressed in these subsets and thus mainly affects resident arterial ECs. Taken together, our data reveal a pro-atherogenic effect of endothelial Cxcr4 deletion, associated with an inflammatory phenotype of the endothelium and increased macrophage content in the atherosclerotic lesions.
CXCR4 on arterial smooth muscle cells is atheroprotective
We next examined the contribution of CXCR4 in resident SMCs to atherosclerosis, crossing Cxcr4fl/flApoe−/− mice with TaglnCre-expressing mice for constitutive Cxcr4 deletion in SMCs17 (Figure 2A, Supplemental Figure 2A). As compared to TaglnCre-Cxcr4fl/flApoe−/− controls, the lesion size in the aortic root (Figure 2B,C) and in the aorta (Figure 2D) was significantly increased in TaglnCre+ Cxcr4fl/flApoe−/− mice after 12 weeks of HFD. No differences in plaque size of the aortic arch (Figure 2E) or in lesional macrophage content or necrotic core area of aortic root lesions (Figure 2F,G, Figure S2B,C) were observed. In contrast, the relative SMC content of aortic root lesions was significantly decreased in TaglnCre+Cxcr4fl/flApoe−/− vs TaglnCre-Cxcr4fl/flApoe−/− mice (Figure 2H). However, absolute SMC numbers collagen content and the percentage of TUNEL+ cells, indicative of apoptosis, were unaltered (Supplemental Figure 2D–F). Whereas leukocyte, neutrophil, monocyte, lymphocyte and platelet counts in peripheral blood, and CXCL12 serum levels were similar in TaglnCre- and TaglCre+ Cxcr4fl/flApoe−/− mice (Supplemental Figure 2G,H, Supplemental Table 6), cholesterol and triglyceride serum levels were lower and body weight was slightly decreased in TaglnCre+Cxcr4fl/flApoe−/− mice (Supplemental Table 6).
Figure 2. CXCR4 on smooth muscle cells is atheroprotective.
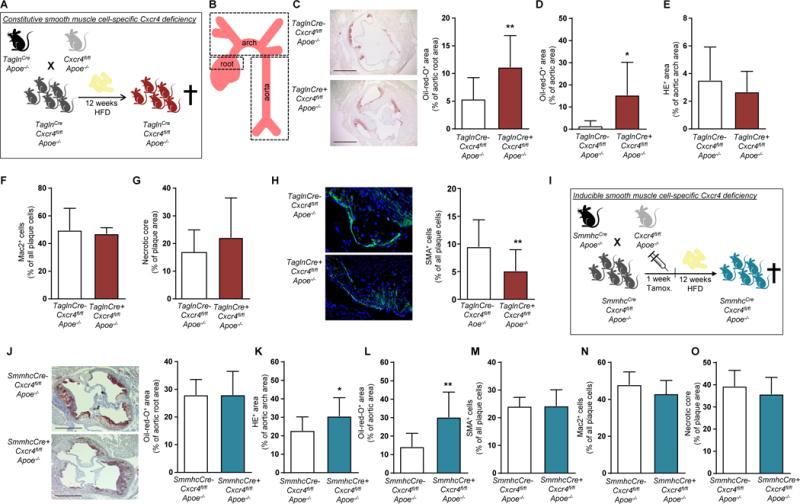
(A–H) Cxcr4fl/flApoe−/− mice were crossed with a TaglnCre-expressing mouse line, triggering constitutive Cxcr4 deletion in SMCs. TaglnCre+Cxcr4fl/flApoe−/− mice vs TaglnCre-Cxcr4fl/flApoe−/− controls were analyzed after 12 weeks of HFD. (A, B) Experimental scheme. (C-E) Lesion area measured after Oil-Red-O or HE staining in the aortic root (C; n=13-24), aorta (D; n=6-16), and aortic arch (D; n=5-11). Representative pictures are shown for aortic root (left panel). Scale bars = 500 µm. (F) Macrophage content in aortic root lesions, quantified after Mac2 staining (n=5-16). (G) Necrotic core area in aortic root lesions, as quantified by anucleated area measurement (n=6-16). (H) SMC content in aortic root lesions was quantified after staining for SMA and expressed relative to the number of plaque cells (n=13-23). Representative images are shown (left panel). (I-O) Cxcr4fl/flApoe−/− mice were crossed with SmmhcCreERT2-expressing mice (termed SmmhcCre) to allow for tamoxifen-inducible Cxcr4 deletion in SMCs. SmmhcCre+Cxcr4fl/fl Apoe−/− mice vs SmmhcCre-Cxcr4fl/flApoe−/− controls were analyzed after 12 weeks of HFD. (I) Experimental scheme. (J-O) Lesional area measured after Oil-Red-O or HE staining in the aortic root (J; n=12-15), aortic arch (K; n=10-13) and aorta (L; n=12-13). Representative pictures are shown for aortic root (J). Scale bars = 500 µm. (M-O) Quantification of SMC and macrophage content expressed relative to the number of plaque cells (M,N, n=12-14) and necrotic core area in aortic root lesions, as quantified by measuring the anucleated area (O, n=12-13). (C-O) Data represent mean±SD. *P<0.05; **P<0.01, as analyzed by Student’s t-test with Welch correction or Mann-Whitney test, as appropriate.
As an alternative to constitutive SMC-specific Cxcr4 deletion, we studied atherosclerosis in SmmhcCre+Cxcr4fl/flApoe−/− mice (Figure 2I), enabling a tamoxifen-inducible Cxcr4 deletion in SMCs.18 Whereas lesion size did not differ in the aortic root (Figure 2J), a significant increase in lesion size was observed in the aortic arch and in the aorta of SmmhcCre+Cxcr4fl/fl Apoe−/− mice vs SmmhcCre-Cxcr4fl/flApoe−/− controls after 12 weeks of HFD (Figure 2K,L). The lesional content of SMCs and macrophages, and the necrotic core area (Figure 2M–O, Supplemental Figure 2I–K) in aortic root lesions were unchanged. No differences in peripheral blood leukocyte and platelet counts, body weight, plasma levels of cholesterol, triglycerides and CXCL12 were observed between SmmhcCre+ and SmmhcCre-Cxcr4fl/flApoe−/− mice after HFD (Supplemental Figure 2L,M, Supplemental Table 7). Furthermore, bone marrow (BM) reconstitution studies revealed that CXCR4 on BM-derived ECs does not affect atherosclerosis, whereas CXCR4 on a subset of BM-derived SMCs is pro-atherogenic (Supplemental Figure 3, Supplemental Table 7 and 8). These effects are apparently unrelated to the function of CXCR4 in resident vascular cells.
Endothelial CXCR4 reduces vascular permeability through WNT/β-catenin
To identify the processes underlying the atheroprotective functions of endothelial CXCR4, we examined its role in vascular permeability, which is crucially regulated by the EC layer,19 also given that increased permeability is associated with enhanced atherosclerosis.20 Histamine-induced vascular permeability was increased in C57/Bl6 mice pre-treated with the CXCR4 antagonist AMD3465 for 16 h (Figure 3A–C), whereas systemic pre-treatment with the CXCR4 ligand CXCL12 for 4 h reduced histamine-induced vascular permeability (Figure 3D). Under conditions of HFD, vascular permeability was increased in Apoe−/− mice with an endothelial-specific Cxcr4 deficiency (Figure 3E–G, PBS-treated group). Similarly, blockade of CXCR4 with AMD increased vascular permeability in Apoe−/− mice but did not affect vascular permeability in BmxCre+ vs BmxCre-Cxcr4fl/flApoe−/− mice (Figure 3F,G). In vitro, the permeability of hAoECs, which express CXCR4 on their surface (Supplemental Figure 4A), to FITC-dextran was increased by activation with the inflammatory cytokine TNFα (Supplemental Figure 4B). In contrast, permeability was reduced by treatment of hAoECs with CXCL12 for 24 h and even by short stimulation with CXCL12 for 15 min (Figure 3H–J). These effects were prevented by the CXCR4 antagonist AMD3100 (Figure 3H–J), indicating that a CXCL12/CXCR4-mediated mechanism protects the EC barrier function. Likewise, blockade of Akt kinase signaling down-stream of CXCR4 using SH5 prevented the reduction in endothelial permeability by CXCL12 (Figure 3K).
Figure 3. Endothelial CXCR4 reduces vascular permeability through WNT signaling.
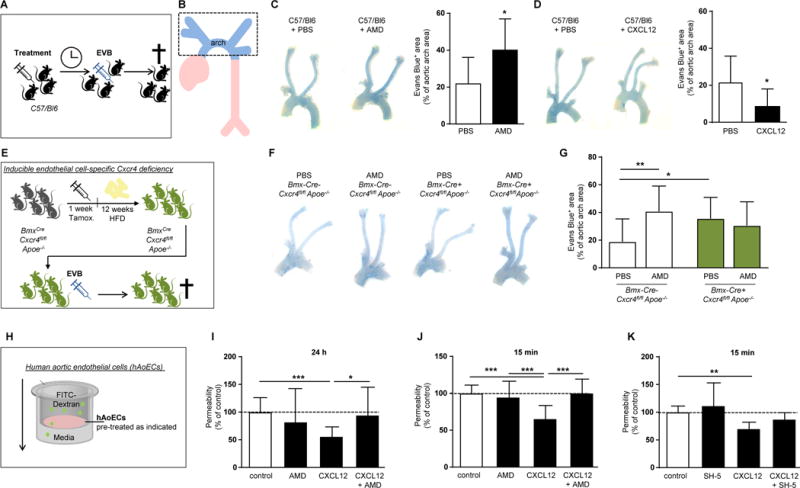
(A, B) Experimental scheme. (C) Histamine-induced (10 µg i.v. for 10 min) vascular permeability to Evans blue in C57/Bl6 mice pretreated with the CXCR4 antagonist AMD3465 (125 µg/mouse) or vehicle (PBS) for 16 hours, as indicated. (n=8-13; Student’s t-test with Welch correction). Representative images are shown (left panel). (D) Histamine-induced (10 µg histamine i.v. for 10 min) vascular permeability to Evans blue in C57/Bl6 mice pretreated for 4 hours with CXCL12 (3 µg) or vehicle (PBS), as indicated (n=8-13; Student’s t-test with Welch correction). Representative images are shown (left panel). (E) Experimental scheme. (F, G) Evans blue extravasation in the aortic arch of BmxCre-Cxcr4fl/flApoe−/− and BmxCre+ Cxcr4fl/flApoe−/− mice after 12 weeks of HFD and treated with the CXCR4 antagonist AMD3465 (125 µg/mouse) or vehicle (PBS) for 12 hours, as indicated. (n=9-21; 2-way ANOVA with Bonferroni post-test). Representative images are shown (left panel). (H) Experimental scheme. (I,J) Permeability of hAoECs to FITC-Dextran after stimulation with CXCL12 (100 ng/ml) for 24 h (I) or 15 min (J). Where indicated, cells were pretreated with the CXCR4 antagonist AMD3100 (I) or AMD3465 (J) at 1 µg/ml for 1 h before CXCL12 stimulation. Data in (I) represent n=17-20 wells from 7 independent experiments (Kruskal-Wallis test with Dunn’s post-test). Data in (J) represent n=11-19 wells from 6 independent experiments (1-way ANOVA with Tukey’s multiple comparison test). (K) Permeability of hAoECs to FITC-Dextran after stimulation for 15 min with CXCL12 (100 ng/ml) and pretreated with the Akt inhibitor SH-5 (20 nM) for 30 min, as indicated (n=6-8 wells from 4 independent experiments; Kruskal-Wallis test with Dunn’s post-test). (C-K) Graphs represent mean mean±SD, *P<0.05; **P<0.01; ***P<0.001.
As WNT signaling has been reported to reduce leakage of the blood-brain barrier21 and β-catenin is present in endothelium covering human atherosclerotic lesions (Supplemental Figure 4C), we examined a potential role for WNT/β-catenin in CXCL12-mediated protection of arterial endothelial barrier function using a luciferase reporter assay (Figure 4A). In hAoECs, CXCL12 induced a marked increase in WNT activity, which was inhibited by a CXCR4 antagonist, by siRNA-mediated CXCR4 knock-down or by endo-IWR1 as a specific WNT inhibitor (Figure 4B, Supplemental Figure 4D). Treatment of hAoECs with CXCL12 up-regulated β-catenin transcript levels (Supplemental Figure 4E). Likewise, CXCL12 increased both cytoplasmic and nuclear β-catenin protein levels, an effect blocked by silencing CXCR4 or by inhibiting WNT signaling with endo-IWR1 (Figure 4C, Supplemental Figure 4F). Accordingly, transcript expression of the WNT target genes MT1, cyclin-D1 and axin-2 was upregulated by CXCL12 (Supplemental Figure 4G). Notably, treatment with the WNT protein WNT3a, which triggers β-catenin stabilization by binding to the Frizzled receptor, or with the GSK3 inhibitor LiCl to stimulate WNT signaling reduced endothelial permeability (Figure 4D). The reduction of endothelial permeability induced by CXCL12 treatment for 24 h or 15 min could be reversed by endo-IWR1 (Figure 4E, Supplemental Figure 4H). Together, our data indicate that CXCL12 improves endothelial barrier function through the WNT pathway.
Figure 4. The CXCL12/CXCR4 axis sustains endothelial barrier function through WNT/β-catenin signaling regulating VE-cadherin.
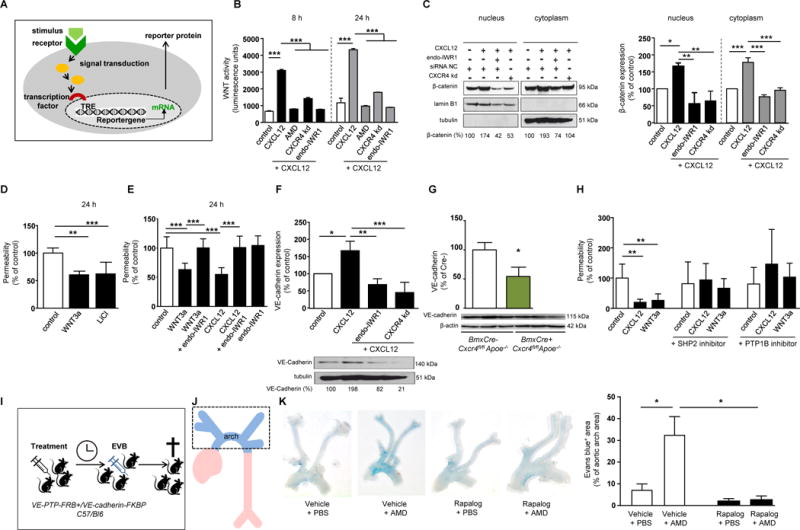
(A) Experimental scheme. (B) WNT activity in hAoECs as determined in a Gaussia luciferase WNT reporter assay, after stimulation with CXCL12 (100 ng/ml) for indicated periods. Cells were pretreated by the CXCR4 antagonist AMD3100 (1 µg/ml) or CXCR4 knock-down was performed using siRNA (CXCR4-kd). WNT activation was blocked by the WNT inhibitor endo-IWR1 (10 µM) (n=3; 1-way ANOVA with Sidak’s multiple comparison test). (C) β-Catenin protein levels in cytoplasmic and nuclear extracts of hAoECs after stimulation with CXCL12 (100 ng/ml) for 24 h. Cells were pretreated with endo-IWR1 (10 µM). Silencing of CXCR4 was performed using siRNA (CXCR4-kd). β-Catenin levels were normalized to cytoplasmic (tubulin) and nuclear (lamin β1) loading controls, respectively, and presented relative to untreated controls, as quantified by densitometry (n=3; 1-way ANOVA with Tukey’s multiple comparison test). A representative western blot and densitometry quantification is shown (left panel). (D) Permeability of hAoECs to FITC-dextran after stimulation with WNT3a (200 ng/ml) or the WNT activator LiCl (30 mM) for 24 h (n=5-10 wells from 4 independent experiments; Kruskal-Wallis test with Dunn’s post-test). (E) Permeability of hAoECs to FITC-dextran after stimulation with WNT3a (200 ng/ml) or CXCL12 (100 ng/ml) for 24 h and pre-treatment with endo-IWR1 (1 µM) for 1 h, as indicated (n=7-9 wells from 3 independent experiments; 1-way ANOVA with Tukey’s multiple comparison test). (F) VE-cadherin protein levels in cytoplasmic extracts of hAoECs after stimulation with CXCL12 (100 ng/ml) for 24 h and treatment with endo-IWR1 (10 µM), as indicated. Knock-down of CXCR4 was performed using siRNA (CXCR4-kd). VE-cadherin expression was normalized to tubulin and presented relative to untreated, as quantified by densitometry (n=3; 1-way ANOVA with Tukey’s multiple comparison test). A representative Western blot and densitometry quantification is shown. Controls received control siRNA (B,C,F). (G) Quantification of VE-cadherin expression in carotid artery lysates of BmxCre+ vs BmxCre-Cxcr4fl/flApoe−/− mice (n=3; Mann-Whitney test). (H) Permeability of hAoECs to FITC-dextran after stimulation with CXCL12 (100 ng/ml) or WNT3a (200 ng/ml) for 24 h and blockade of SHP2 (5 µM) or PTP1B (25 µM) for 1 h, as indicated (n=8-12 wells from 4 independent experiments; Kruskal-Wallis test with Dunn’s post-test). (I-K) Vascular permeability to Evans blue induced by histamine (10 µg i.v., 10 min) in VE-PTP-FRB+/VE-cadherin-FKBP C57/Bl6 knock-in mice pretreated with the CXCR4 antagonist AMD3465 (125 µg) for 12 h and/or Rapalog (250 µg) for 4 h or vehicle, as indicated (n=8-16; 2-way ANOVA with Bonferroni post-test). (A-K) Data present mean±SD (A-J) or mean±SEM (K). *P<0.05; **P<0.01; ***P<0.001.
Endothelial CXCR4 sustains VE-cadherin expression and function
The adherens junctional protein VE-cadherin is an important contributor to endothelial barrier function.19 Because CXCL12 regulates E-cadherin expression and localization in migrating epithelial cells,22 we tested a role of VE-cadherin in the CXCL12/CXCR4-mediated control of endothelial permeability. In hAoECs, CXCL12 increased transcript and protein expression of VE-cadherin (Figure 4F, Supplemental Figure 4I). This was paralleled by reduced protein levels of VE-cadherin in carotid artery lysates of BmxCre+Cxcr4fl/flApoe−/− mice compared to controls (Figure 4G). Of note, inhibiting WNT signaling with endo-IWR1 prevented the up-regulation of VE-cadherin mRNA and protein levels by CXCL12 (Figure 4F, Supplemental Figure 4I), indicating that VE-cadherin expression is increased by the CXCL12/WNT pathway. The VE-cadherin-associated phosphatases SHP2, PTP1B and VE-PTP are known to sustain VE-cadherin function and endothelial barrier integrity.23, 24 Blocking SHP2 and PTPB1 prevented the reduction of endothelial permeability induced by CXCL12 or WNT3a (Figure 4H). Importantly, an increase in histamine-triggered vascular permeability upon CXCR4 blockade could not be observed in engineered VE-PTP-FRB+/VE-cadherin-FKBP C57/Bl6 knock-in mice featuring a non-dissociating, thus stabilized interaction between VE-cadherin and VE-PTP, which is induced by the chemical compound rapalog and has been previously linked to a preservation of the endothelial barrier23 (Figure 4I–K). Together, our data show that the CXCL12/CXCR4 axis maintains endothelial integrity through the activity of VE-cadherin and VE-cadherin-regulating phosphatases.
CXCL12/CXCR4 signaling did not alter the expression of other tight junctional proteins or inflammatory markers in ECs (data not shown). Finally, as vascular tone regulators such as eNOS have been shown to influence atherogenesis25 and the EC layer critically regulates vascular tone, we examined a possible role for endothelial CXCR4 in vascular reactivity. No differences were observed in the vascular contraction or relaxation of aortic rings from BmxCre+ vs BmxCre-Cxcr4fl/flApoe−/− mice after 12 weeks of HFD or chow diet (Supplemental Figure 5A–C). Altogether, these data reveal an important function for endothelial CXCR4 in maintaining endothelial barrier integrity via VE-cadherin expression and function, without directly affecting the inflammatory phenotype of ECs or vascular tone.
CXCR4 on SMCs regulates lesional cholesterol efflux
Cholesterol efflux capacity has been independently and inversely associated with athero-sclerotic cardiovascular disease in patients with familial hypercholesterolemia.26 Analysis of cholesterol efflux revealed a reduction in efflux capacity from lipid-laden macrophages when using serum from TaglnCre+ vs TaglnCre-Cxcr4fl/flApoe−/− mice on HFD as acceptor. Such reduced cholesterol efflux was observed without and with cAMP stimulation, which enhances cellular lipid efflux to the apolipoprotein acceptor ApoA-1 (Figure 5A).27 Similar results were obtained using serum from atherosclerotic SmmhcCre+ vs SmmhcCre-Cxcr4fl/flApoe−/− mice as acceptor (Figure 5B). Accordingly, serum levels of ApoA-1 were significantly reduced in TaglnCre+ vs TaglnCre-Cxcr4fl/flApoe−/− mice (Figure 5C). Thus, reduced cholesterol efflux may contribute to increased lipid retention and associated lesion size, as evidenced in TaglnCre+ and SmmhcCre+Cxcr4fl/flApoe−/− mice (Figure 2).
Figure 5. Effect of CXCR4 deletion on cholesterol efflux, vascular tone and SMC phenotype.
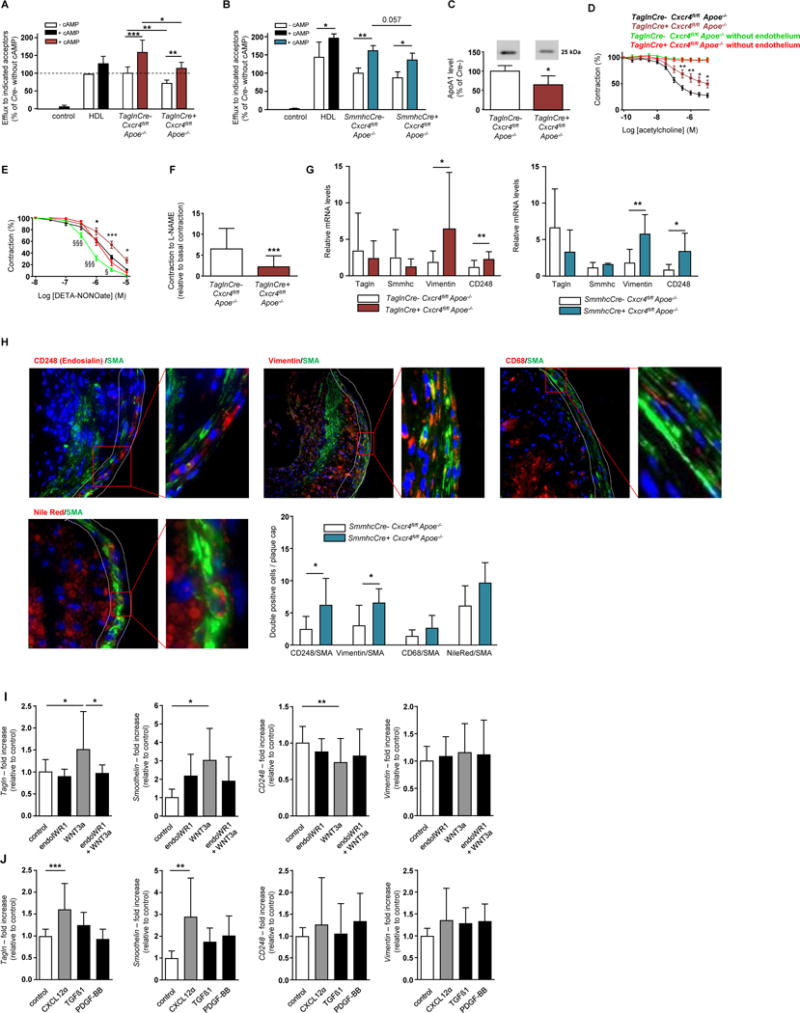
(A, B) Quantification of cholesterol efflux from lipid-laden macrophages using HDL or pooled serum from TaglnCre+ vs TaglnCre-Cxcr4fl/flApoe−/− mice (A, n=6) or from SmmhcCre+ vs SmmhcCre-Cxcr4fl/flApoe−/− mice (B, n=4) after 12 weeks of HFD as acceptor. Data represent mean±SD (Mann-Whitney test for comparing Cre- vs Cre+; 2-way ANOVA with Bonferroni post-test for comparing without vs with cAMP). (C) ApoA1 protein levels in serum of TaglnCre+ vs TaglnCre-Cxcr4fl/flApoe−/− mice after 12 weeks of HFD, as measured by Western blot analysis A representative blot and densitometry quantification is shown (n=6; mean±SD; Mann-Whitney test). (D,E) Acetylcholine-induced endothelium-dependent relaxation (D, n=20 rings with endothelium or n=9-10 rings without endothelium from 8-9 mice) and DETA-NONOate-induced endothelium-independent relaxation (E, n=16-18 rings with endothelium or n=11-16 rings without endothelium from 8-9 mice) of aortic rings from TaglnCre+ vs TaglnCre-Cxcr4fl/flApoe−/− mice after 12 weeks of HFD, with or without removal of the endothelium, as indicated. Data represent mean±SEM (2-way repeated measures ANOVA with Bonferroni post-test for comparing Cre- vs Cre+). (F) Ratio of L-NAME-induced contraction (300 µM L-NAME) to basal pre-contraction without L-NAME in aortic rings from TaglnCre+ vs TaglnCre-Cxcr4fl/flApoe−/− mice after 12 weeks of HFD (n=20-21 rings from 8-9 mice). Data represent mean±SEM (Mann-Whitney test). (G) Relative quantification of transgrelin (Tagln), smooth muscle-myosin heavy chain (Smmhc), vimentin and CD248 (endosialin) mRNA expression in thoracic aortas of TaglnCre+ vs TaglnCre-Cxcr4fl/flApoe−/− and SmmhcCre+ vs SmmhcCre-Cxcr4fl/flApoe−/− mice after normalization to 18S rRNA (n=3-9). Data represent mean±SD, Student’s t-test with Welch correction or Mann-Whitney test, as appropriate. (H) Representative images and quantification of protein expression of CD248 (endosialin), vimentin and CD68 and lipid deposition (Nile red) in atherosclerotic lesions (plaque cap) in mice with SMC-specific Cxcr4 deficiency, as compared to controls (n=6-10). Data represent mean±SD and were analyzed by Student’s t-test with Welch correction or Mann-Whitney test, as appropriate (G,H). (I) Relative quantification of Tagln, smoothelin, CD248 and vimentin mRNA expression in hAoSMCs, pretreated with endoIWR1 (1 µM) and stimulated with WNT3a (200 ng/ml), as indicated. Data are normalized to Gapdh expression (mean±SD, data combined from 3-7 experiments, each performed in triplicate; 1-way ANOVA with Sidak’s multiple comparison test or Kruskal-Wallis test with Dunn’s multiple comparison test, as appropriate). (J) Relative quantification of Tagln, smoothelin, CD248 and vimentin mRNA expression in hAoSMCs stimulated with (protease-resistant) CXCL12 (100-300 ng/ml), TGF-β (2.5 ng/ml) or PDGF-BB (20 ng/ml) as indicated. Data are normalized to Gapdh expression (mean±SD, data combined from 3-5 experiments, each performed in triplicate; 1-way ANOVA with Sidak’s multiple comparison test or Kruskal-Wallis test with Dunn’s multiple comparison test, as appropriate). (A-J) *P<0.05; **P<0.01; ***P<0.001.
To identify mechanisms underlying the reduced SMC content in atherosclerotic lesions of TaglnCre+Cxcr4fl/flApoe−/− mice, we analyzed expression of the proliferation marker PCNA in aortic lysates and found lower levels in atherosclerotic aortas of TaglnCre+Cxcr4fl/flApoe−/− mice as compared to controls after HFD but not a chow diet (Supplemental Figure 5D, E). Using a scratch assay, we observed an increase in the migratory capacity of hAoSMCs after stimulation with CXCL12, which was inhibited using a CXCR4 antagonist (Supplemental Figure 5F). Surface expression of CXCR4 on hAoSMCs could be detected by flow cytometric analysis (Supplemental Figure 5G).
CXCR4 on SMCs supports NO-dependent vascular reactivity
Because SMCs are crucial in regulating vascular tone, we examined the effects of SMC-specific Cxcr4 deficiency on blood pressure, on vascular contraction and relaxation in organ baths. Systolic and diastolic blood pressure did not differ between TaglnCre+ vs TaglnCre- Cxcr4fl/flApoe−/− mice (Supplemental Figure 5H,I). Upon treatment with acetylcholine, the relaxation of aortic rings isolated from TaglnCre+Cxcr4fl/flApoe−/− mice after HFD for 12 weeks was impaired as compared to those from TaglnCre-Cxcr4fl/flApoe−/− controls (Figure 5D). Acetylcholine induces NO production in ECs to trigger SMC relaxation, explaining the unresponsiveness of aortic rings lacking the EC layer (Figure 5D). Reduced relaxation capacity of TaglnCre+Cxcr4fl/flApoe−/− aortic rings was also recorded after treatment with the NO donor DETA-NONOate triggering EC-independent relaxation (Figure 5E). A comparable reduction in DETA-NONOate-induced relaxation was observed in the absence of the EC layer (Figure 5E). Moreover, contraction triggered by the NO synthase inhibitor L-NAME was lower in aortic rings from TaglnCre+Cxcr4fl/fl Apoe−/− mice vs controls after HFD (Figure 5F). Similar results were obtained in rings from mice receiving a chow diet (Supplemental Figure 5J–L). Of note, plasma levels of NO did not differ between genotypes on chow or HFD (Supplemental Figure 5M,N). This indicates that the impairment in vascular reactivity of TaglnCre+Cxcr4fl/flApoe−/− aortic rings was due to reduced responsiveness of SMCs rather than to reduced NO availability. By comparison, tamoxifen-induced SmmhcCre-driven deletion of Cxcr4 resulted in a less pronounced impairment in DETA-NONOate-induced vascular relaxation in SmmhcCre+ vs SmmhcCre-Cxcr4fl/flApoe−/− mice but did not affect acetylcholine-induced relaxation or L-NAME-induced contraction (Supplemental Figure 5O–Q). Collectively, our data indicate that Cxcr4-deficiency in SMCs exerts rather moderate effects on vascular tone per se and that the more marked impairment observed in TaglnCre+Cxcr4fl/flApoe−/− mice is likely attributable to the constitutive Cxcr4-deficiency during vascular development.
CXCR4 maintains SMCs in a contractile phenotype
Vascular SMCs display a significant plasticity and can de-differentiate from a contractile phenotype to a more synthetic phenotype. This SMC phenotype switching has recently been implicated in influencing atherosclerosis.28 We found that mice with SMC-specific Cxcr4 deficiency showed an increased aortic expression of vimentin, a synthetic marker induced by PDGF-BB29, and of CD248 (endosialin), which is involved in PDGF-mediated cellular effects and associated with a synthetic SMC phenotype (Figure 5G, Supplemental Figure 5R,S).30,31 Moreover, staining for vimentin, CD248, CD68 and lipids (Nile red) revealed a higher prevalence of de-differentiated SMCs in plaque caps of these mice (Figure 5H). In vitro, both WNT3a and CXCL12 increased the expression of contractile markers (Tagln, smoothelin, CNN1) in cultured hAoSMCs, whereas the expression of synthetic markers was reduced (for CD248 after WNT3A treatment, Figure 5I, Supplemental Figure 5T) or not altered (after CXCL12 treatment, Figure 5J, Supplemental Figure 5U), respectively. Combined with our finding that the CXCL12/CXCR4 axis drives WNT activation (Figure 4B), these data reveal that the CXCL12/CXCR4/WNT axis supports a contractile phenotype in SMCs.
CXCR4 risk variants are associated with impaired CXCR4 expression
We evaluated the relevance of our findings indicating a protective role of CXCR4 to human atherosclerosis. Immunohistochemistry in carotid endarterectomy specimen detected CXCR4 protein expression in both ECs and SMCs of human atherosclerotic lesions (Figure 6A), as well as the presence of active β-catenin indicating an activation of the WNT pathway (Figure 6B). We next examined the association of all common variants at the CXCR4 locus with coronary heart disease (CHD) using data on altogether 92,516 CHD cases and 167,280 controls (including CARDIoGRAMplusC4D) and fine mapping of data from the 1000 genome project (see Table S9 for details of data sets included). Thus, we identified the C-allele at rs2322864 (minor allele frequency 26%) to be significantly associated with increased CHD risk (OR: 1.04 for every C-allele; P=4.38×10−7) (Figure 6C) and with reduced CXCR4 gene expression in whole-blood upon eQTL analysis (data not shown). Notably, the C/C-genotype at rs2322864 was found to be linked to reduced CXCR4 mRNA expression in atherosclerotic plaques of human carotid arteries (Figure 6D). Moreover, CXCR4 expression was lower in patients with symptomatic vs asymptomatic carotid stenosis, as evident by neurologic events, e.g. transient ischemic attacks (Figure 6E), consistent with the notion that low vascular CXCR4 levels give rise to increased risk for carotid and coronary atherosclerosis.
Figure 6. Relevance of CXCR4 pathway expression to human atherosclerotic disease.
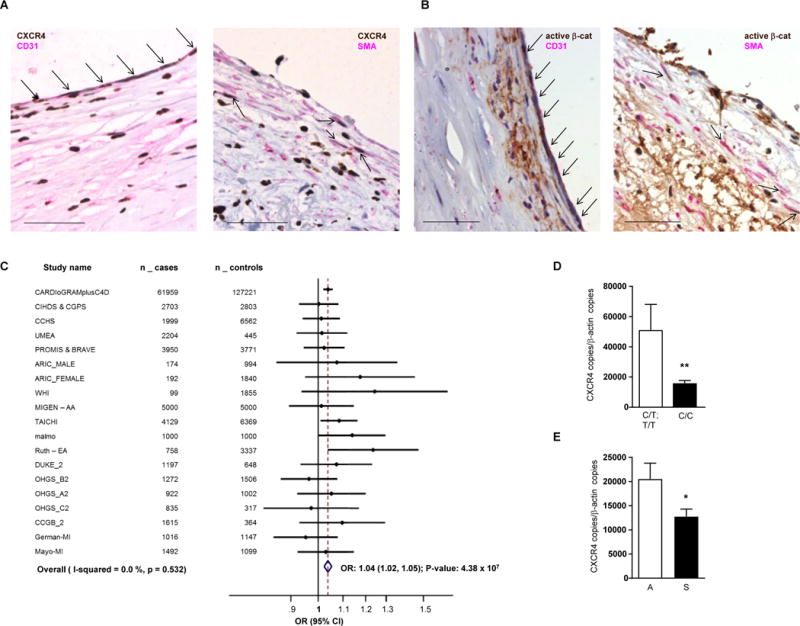
(A) Staining of CXCR4 in human atherosclerotic lesions. Human carotid endarterectomy specimen were stained for CXCR4 (brown) in combination with CD31 (left, pink) or SMA (right, pink). Scale bar = 50 µm. Arrows indicate CXCR4+ ECs (left) or CXCR4+ SMCs (right). (B) Staining for active β-catenin in human atherosclerotic lesions. Human carotid endarterectomy specimen were stained for active β-catenin (ABC; brown) in combination with CD31 (left, pink) or SMA (right, pink). Scale bar = 50 µm. Arrows indicate ABC+ ECs (left) or ABC+ SMCs (right). (C) Forest blot for the associations of CHD risk with rs2322864. We examined the association of 345 common variants at the CXCR4 locus (± 25KB) with CHD by using data on 92,516 CHD cases and 167,280 controls (see Supplemental Table 4 for details of data sets included), most notably interrogating the CARDIoGRAMplusC4D data. We conducted fine-mapping studies in 12,500 myocardial infarction cases and 12,000 controls and genotyped all 512 variants with a minor allele frequency >0.1% identified by the 1000-Genomes project at the CXCR4 locus. A P-value of 5×10−5 was considered as statistically significant based on Bonferroni correction for 857 variants. Odds ratio (OR) with 95% confidence interval (CI) and P-value are given. The C-allele at rs2322864 was found to be associated with increased CHD risk (OR: 1.04; P=4.38×10−7). (D) Association of rs2322864 with CXCR4 expression in carotid endarterectomy specimen (C/T; T/T: n=121 and C/C: n=67, Mann-Whitney test with Bonferroni correction to adjust for multiple comparisons). (E) Correlation of CXCR4 expression with clinical carotid stenosis, as evident by neurologic events, e.g. transient ischemic attacks (A, asymptomatic, n=25; S, symptomatic, n=19, unpaired t-test with Welch correction); CXCR4 expression was normalized to β-actin expression (mean±SEM). *P<0.05.
Discussion
Our data unequivocally reveal that CXCR4 is atheroprotective in vascular cells by sustaining endothelial integrity and promoting the contractile phenotype of SMCs. This conclusion is supported by the increase in atherosclerosis upon arterial EC or SMC-specific deletion of Cxcr4 in mice as well as reduced CXCR4 expression in human carriers of a common CXCR4 risk variant for CHD. Our results corroborate and extend the importance of CXCR4 in atherosclerosis and human disease beyond previous findings using its systemic blockade or disruption in bone marrow as well as its role in stem and cancer cell migration.
Sustained endothelial integrity upon CXCL12/CXCR4 signaling was mediated by i) Akt/WNT signaling, ii) involved the VE-cadherin-associating phosphatases VE-PTP, SHP2 and PTP1B,23,32–34 and was linked with iii) increased VE-cadherin expression and function, as evidenced by reinforced interactions of VE-cadherin and VE-PTP to conserve endothelial barrier function.23 As a result, interference with the CXCL12/CXCR4 axis enhanced Evans blue extravasation into the vascular wall under conditions of acute inflammation and during atherogenesis in vivo. Moreover, endothelial Cxcr4-deficiency was associated with an up-regulation of ICAM-1 expression in arterial endothelial cells covering atherosclerotic lesions, reflecting their activation and promoting atherogenic leukocyte recruitment and increased macrophage content in the lesions. The latter effect was not due to a direct effect of the CXCL12/CXCR4 axis on the expression of inflammatory mediators and adhesion molecules in ECs, as it could not be stimulated or modulated by CXCL12 in vitro. This implies that the marked leukocyte trafficking into early lesions of mice lacking endothelial Cxcr4 was rather related to disturbed endothelial integrity and increased permeability, subsequently enhancing vascular inflammation and adhesion molecule expression on the endothelium.
WNT activation was previously found to decrease leakage of the blood-brain barrier,21 implicating β-catenin transcriptional activity in reduced permeability of brain ECs.35 A link between CXCL12 and WNT has been suggested in peripheral nerve sheath tumors and pancreatic β-cells, triggering cell growth and survival, respectively.36,37 We found that the CXCL12/CXCR4 axis reduces endothelial permeability through Akt/WNT- and VE-cadherin-mediated mechanisms, which may refine and underlie findings that CXCL12 or its analogs can reduce thrombin-induced endothelial permeability and pulmonary vascular leakage in a model of LPS-induced acute respiratory distress syndrome.38,39 Notably extending previous findings23, stabilizing the interaction between VE-cadherin and VE-PTP prevented the impairment of endothelial barrier function upon interference with CXCR4. Surprisingly, atherosclerosis-prone endothelium displays increased β-catenin nuclear localization.40 However, enhancing WNT signaling by blocking the endogenous β-catenin/WNT inhibitor GSK3 reduced atherosclerosis and inflammatory endothelial VCAM-1 expression41, further supporting an atheroprotective role of WNT at least partially at the endothelial level. Findings that endothelial KLF4 is itself a β-catenin target gene42, drives VE-cadherin expression43 and is atheroprotective in ECs44, may provide additional evidence for a down-stream link of CXCL12-induced β-catenin and VE-cadherin expression. Given the involvement of SHP2 in Akt signaling45, our data unveil that the CXCL12-CXCR4 axis can sustain endothelial integrity through i) SHP2/Akt/WNT signaling inducing VE-cadherin expression, and ii) VE-cadherin-associated phosphatases, namely VE-PTP, supporting VE-cadherin stability and function.
In second line of investigation, we show that the CXCL12/CXCR4 axis also favors a contractile over a synthetic phenotype in arterial SMCs to explain increased lesion formation in mice with SMC-specific Cxcr4 deficiency. Phenotype switching of contractile SMCs to a synthetic phenotype has long been considered to contribute to atherogenesis by generating a more inflammatory SMC phenotype.28 A recent study supported this notion by documenting that loss of myocardin, a promotor of the contractile and non-inflammatory SMC phenotype, increased atherosclerosis.46 Here we uncover a role for a chemokine ligand/receptor axis in influencing SMC phenotype in the context of atherosclerosis. In addition to upregulating markers (vimentin and endosialin) associated with the synthetic SMC phenotype29–31, SMC Cxcr4-deficiency increasingly switched SMCs in atherosclerotic lesions to a macrophage-like phenotype featuring co-staining for CD68 and Nile-Red. Such SMC-derived macrophage-like cells have only recently been identified in atherosclerotic lesions, and are suggested to augment atherosclerosis.28,47,48 Likewise, endosialin has been found to promote atherosclerosis through phenotypic remodeling of vascular SMCs.31 In accord with CXCL12 driving WNT signaling, we confirmed that both CXCL12 and WNT3a promoted the contractile phenotype of SMCs in vitro,49 consistent with our observations upon Cxcr4-deficiency in vivo. Combined with a reduced macrophage cholesterol efflux capacity due to lower ApoA1 serum levels, this may underlie atherogenic effects of Cxcr4-deficiency in SMCs. Notably, since an autocrine loop for CXCR4-dependent expression of CXCL12 has been established in ECs,10 a cross-talk of CXCR4-bearing SMCs and ECs via CXCL12 in the arterial wall may also contribute to atheroprotection. Pro-atherogenic effects of CXCR4 in BM-derived SMCs contrasted a protective role in resident SMCs, likely due to differential expression levels or functions depending on the cellular context and origin.
Genome-wide association studies have validated highly significant associations of variants near CXCL12 as a lead gene in the 10q11 locus with the risk for CAD and myocardial infarction.11,12 Based on a more recent analysis, we have now extended the role of CXCL12 to the locus of its receptor CXCR4, showing that a common variant, the C-allele at rs2322864 was associated with increased risk for CHD and the C/C-genotype with reduced CXCR4 expression in whole blood and carotid atherosclerotic plaques. Of note, the odds ratio for CHD risk appeared to be more prominent in predominantly female cohorts. CXCR4 expression was also reduced in patients with symptomatic compared to asymptomatic carotid atherosclerosis, indicative of a more unstable plaque phenotype. Collectively, our data substantiate the protective role of the CXCL12/CXCR4 axis in cells of the arterial wall. As global inducible Cxcr4-deficiency less markedly exacerbate atherosclerosis (Saleheen et al., in preparation), CXCR4 may also confer pro-atherogenic effects in other cell types distinct to neutrophils, ECs or SMCs, however, the identity of this cell type, e.g. in the hematopoietic or other non-vascular cell compartments remains to be elucidated. Our data linking variants in the CXCR4 gene locus with CHD risk support the notion that CXCR4 is a disease-modifying receptor in atherosclerosis, indicating that protective effects may prevail, but cannot explain differences or resolve the directionality in BM-related vs peripheral effects of CXCR4 on atherogenesis.
Our data indicate that specifically enhancing the atheroprotective functions of CXCR4 in different arterial cell types (but not in hematopoietic cells) might open new and exciting therapeutic options. Owing to interference with the role of CXCR4 in progenitor cell mobilization and trafficking, systemic approaches, e.g. using non-cleavable CXCL12 peptides39 or small molecule agonists to boost CXCR4 function, carry a substantial risk of side effects and suboptimal efficacy, e.g. due to pro-atherogenic effects of CXCR4-bearing BM-derived cells. To avoid such obstacles, one could envision a regional application of selectively targeted agonists or modulators through peri- or intravascular routes. This may be accomplished using polymer- or nanoparticle-based delivery, as exemplified for microR-126-3p, which specifically de-represses CXCR4 activity in ECs.10 Likewise, the therapeutic feasibility shown for target site blockers50 could be extended to blocking specific microRNA interactions with CXCR4.
Supplementary Material
Clinical perspective.
What Is New?
We show for the first time that the chemokine receptor CXCR4 in vascular cells limits atherosclerosis.
Mechanistically, the CXCL12/CXCR4 chemokine ligand/receptor axis promotes endothelial barrier function through VE-cadherin expression and the stabilization of junctional VE-cadherin complexes.
In arterial smooth muscle cells (SMCs), CXCR4 sustains vascular reactivity responses and a contractile SMC phenotype, whereas CXCR4-deficiency favors the occurrence of macrophage-like SMCs in atherosclerotic plaques and impairs cholesterol efflux.
In humans we identified a common allele variant within the CXCR4 locus to be associated with reduced CXCR4 expression in carotid artery plaques and increased risk for coronary heart disease.
What Are the Clinical Implications?
Our data reveal that specifically enhancing the atheroprotective functions of arterial CXCR4 by selective modulators may open novel therapeutic options in atherosclerosis.
As systemic approaches to boost CXCR4 function carry a substantial risk of side effects and suboptimal efficacy, e.g. due to pro-atherogenic effects of CXCR4-bearing hematopoietic cells, we envision a regional application of selectively targeted CXCR4 agonists or modulators through peri- or intravascular routes as a novel therapeutic option in atherosclerosis.
Regional boosting of vascular CXCR4 may be accomplished using polymer- or nanoparticle-based delivery, as exemplified for microR-126-3p, which we previously showed to de-repress CXCR4 activity in endothelial cells.
Acknowledgments
We thank Y. Zou (Columbia University, New York), R.H. Adams (University of Münster, Germany), S. Offermanns (University of Heidelberg, Germany) for providing Cxcr4flox, Bmx-CreERT2 and Smmhc-CreERT2 mice, respectively, and J. Jankowski for support with housing. We thank M. Garbe, S. Elbin, R. Soltan, N. Persigehl and B. Zhou for excellent technical assistance.
Funding Sources
This work was funded by Deutsche Forschungsgemeinschaft (SFB1123-A1 to C.W and Y.D. and SFB1123-Z1 to R.M.), NIH (1R01HL122843 to D.S., R.D. and C.W.), the Fondation Leducq Transatlantic Network of Excellence CVGeneF(x) to C.W. and D.R., the German Federal Ministry of Education and Research (01KU1213A to C.W.), the European Research Council (ERC Advanced Grant °692511 to C.W.), the German Centre for Cardiovascular Research (MHA VD1.2, 81Z1600212 and 81X2800151 to C.W., O.S., Y.D., H.N., and R.P.B.), the German Heart Foundation (F/40/12 to H.N.), the START-program (49/13 to H.N.) and the Habilitation program of the Faculty of Medicine, RWTH Aachen (to H.N.).
Footnotes
Author contributions
Y.D. and H.N. designed the study, performed mouse and in vitro experiments, analyzed data and wrote the paper.
E.P.C.V. and R.M. performed and analysed the in vivo experiments.
V.E. performed luciferase assays, immunoblots and qRT-PCR.
M.D. performed in vivo permeability studies.
M.M. and C.N. performed mouse experiments, collected and processed the histological data, performed flow cytometry analysis and in vivo permeability studies.
L.P. performed immunoblots, qRT-PCR and in vitro permeability assays and processed histological data.
Y.J. performed atherosclerotic plaque analysis and immunohistochemistry.
K.S. and R.P.B. performed and analyzed vascular reactivity studies.
K.B. and C.R. contributed to vector design and construction.
W.T. performed flow cytometry analysis and analyzed qRT-PCR data.
B.M.K. and P.B. performed blood pressure measurements.
L.S., R.v.G and T.H. performed in vitro experiments and contributed critical reagents.
P.J.H.K., A.v.d.W. and E.L. performed and analysed human plaque immunohistochemistry.
G.G., D.T. and L.H. performed and analysed human endarterectomy studies.
O.S. performed and analyzed intravital microscopy experiments.
D.V. contributed a critical mouse model.
D.R. was involved in study design and contributed to lipid uptake experiments.
D.S. performed and analysed genome-wide association studies.
C.W. designed and supervised the study, analyzed data and wrote the paper.
All authors discussed the results and commented on the manuscript.
Y.D. and H.N. contributed equally to the study.
Conflict of Interest Disclosures
Besides the research funding and grant support, as detailed for each author under Acknowledgements, there are no conflicts of interest to disclose.
References
- 1.Döring Y, Pawig L, Weber C, Noels H. The CXCL12/CXCR4 chemokine ligand/receptor axis in cardiovascular disease. Front Physiol. 2014;5:212. doi: 10.3389/fphys.2014.00212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zou YR, Kottmann AH, Kuroda M, Taniuchi I, Littman DR. Function of the chemokine receptor CXCR4 in haematopoiesis and in cerebellar development. Nature. 1998;393:595–599. doi: 10.1038/31269. [DOI] [PubMed] [Google Scholar]
- 3.Tachibana K, Hirota S, Iizasa H, Yoshida H, Kawabata K, Kataoka Y, Kitamura Y, Matsushima K, Yoshida N, Nishikawa S, Kishimoto T, Nagasawa T. The chemokine receptor CXCR4 is essential for vascularization of the gastrointestinal tract. Nature. 1998;393:591–594. doi: 10.1038/31261. [DOI] [PubMed] [Google Scholar]
- 4.Nagasawa T, Hirota S, Tachibana K, Takakura N, Nishikawa S, Kitamura Y, Yoshida N, Kikutani H, Kishimoto T. Defects of B-cell lymphopoiesis and bone-marrow myelopoiesis in mice lacking the CXC chemokine PBSF/SDF-1. Nature. 1996;382:635–638. doi: 10.1038/382635a0. [DOI] [PubMed] [Google Scholar]
- 5.Zernecke A, Schober A, Bot I, von Hundelshausen P, Liehn EA, Mopps B, Mericskay M, Gierschik P, Biessen EA, Weber C. SDF1α/CXCR4 axis is instrumental in neointimal hyperplasia and recruitment of smooth muscle progenitor cells. Circ Res. 2005;96:784–791. doi: 10.1161/01.RES.0000162100.52009.38. [DOI] [PubMed] [Google Scholar]
- 6.Weber C, Noels H. Atherosclerosis: Current pathogenesis and therapeutic options. Nat Med. 2011;17:1410–1422. doi: 10.1038/nm.2538. [DOI] [PubMed] [Google Scholar]
- 7.Noels H, Zhou B, Tilstam PV, Theelen W, Li X, Pawig L, Schmitz C, Akhtar S, Simsekyilmaz S, Shagdarsuren E, Schober A, Adams RH, Bernhagen J, Liehn EA, Döring Y, Weber C. Deficiency of endothelial CXCR4 reduces reendothelialization and enhances neointimal hyperplasia after vascular injury in atherosclerosis-prone mice. Arterioscler Thromb Vasc Biol. 2014;34:1209–1220. doi: 10.1161/ATVBAHA.113.302878. [DOI] [PubMed] [Google Scholar]
- 8.Zernecke A, Bot I, Djalali-Talab Y, Shagdarsuren E, Bidzhekov K, Meiler S, Krohn R, Schober A, Sperandio M, Soehnlein O, Bornemann J, Tacke F, Biessen EA, Weber C. Protective role of CXC receptor 4/CXC ligand 12 unveils the importance of neutrophils in atherosclerosis. Circ Res. 2008;102:209–217. doi: 10.1161/CIRCRESAHA.107.160697. [DOI] [PubMed] [Google Scholar]
- 9.Bot I, Daissormont IT, Zernecke A, van Puijvelde GH, Kramp B, de Jager SC, Sluimer JC, Manca M, Herias V, Westra MM, Bot M, van Santbrink PJ, van Berkel TJ, Su L, Skjelland M, Gullestad L, Kuiper J, Halvorsen B, Aukrust P, Koenen RR, Weber C, Biessen EA. CXCR4 blockade induces atherosclerosis by affecting neutrophil function. J Mol Cell Cardiol. 2014;74:44–52. doi: 10.1016/j.yjmcc.2014.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zernecke A, Bidzhekov K, Noels H, Shagdarsuren E, Gan L, Denecke B, Hristov M, Koppel T, Jahantigh MN, Lutgens E, Wang S, Olson EN, Schober A, Weber C. Delivery of microrna-126 by apoptotic bodies induces CXCL12-dependent vascular protection. Sci Signal. 2009;2:ra81. doi: 10.1126/scisignal.2000610. [DOI] [PubMed] [Google Scholar]
- 11.Samani NJ, Erdmann J, Hall AS, Hengstenberg C, Mangino M, Mayer B, Dixon RJ, Meitinger T, Braund P, Wichmann HE, Barrett JH, Konig IR, Stevens SE, Szymczak S, Tregouet DA, Iles MM, Pahlke F, Pollard H, Lieb W, Cambien F, Fischer M, Ouwehand W, Blankenberg S, Balmforth AJ, Baessler A, Ball SG, Strom TM, Braenne I, Gieger C, Deloukas P, Tobin MD, Ziegler A, Thompson JR, Schunkert H. Genome-wide association analysis of coronary artery disease. N Engl J Med. 2007;357:443–453. doi: 10.1056/NEJMoa072366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kathiresan S, Voight BF, Purcell S, Musunuru K, Ardissino D, Mannucci PM, Anand S, Engert JC, Samani NJ, Schunkert H, Erdmann J, Reilly MP, Rader DJ, Morgan T, Spertus JA, Stoll M, Girelli D, McKeown PP, Patterson CC, Siscovick DS, O’Donnell CJ, Elosua R, Peltonen L, Salomaa V, Schwartz SM, Melander O, Altshuler D, Ardissino D, Merlini PA, Berzuini C, Bernardinelli L, Peyvandi F, Tubaro M, Celli P, Ferrario M, Fetiveau R, Marziliano N, Casari G, Galli M, Ribichini F, Rossi M, Bernardi F, Zonzin P, Piazza A, Mannucci PM, Schwartz SM, Siscovick DS, Yee J, Friedlander Y, Elosua R, Marrugat J, Lucas G, Subirana I, Sala J, Ramos R, Kathiresan S, Meigs JB, Williams G, Nathan DM, MacRae CA, O’Donnell CJ, Salomaa V, Havulinna AS, Peltonen L, Melander O, Berglund G, Voight BF, Kathiresan S, Hirschhorn JN, Asselta R, Duga S, Spreafico M, Musunuru K, Daly MJ, Purcell S, Voight BF, Purcell S, Nemesh J, Korn JM, McCarroll SA, Schwartz SM, Yee J, Kathiresan S, Lucas G, Subirana I, Elosua R, Surti A, Guiducci C, Gianniny L, Mirel D, Parkin M, Burtt N, Gabriel SB, Samani NJ, Thompson JR, Braund PS, Wright BJ, Balmforth AJ, Ball SG, Hall AS, Schunkert H, Erdmann J, Linsel-Nitschke P, Lieb W, Ziegler A, Konig I, Hengstenberg C, Fischer M, Stark K, Grosshennig A, Preuss M, Wichmann HE, Schreiber S, Schunkert H, Samani NJ, Erdmann J, Ouwehand W, Hengstenberg C, Deloukas P, Scholz M, Cambien F, Reilly MP, Li M, Chen Z, Wilensky R, Matthai W, Qasim A, Hakonarson HH, Devaney J, Burnett MS, Pichard AD, Kent KM, Satler L, Lindsay JM, Waksman R, Knouff CW, Waterworth DM, Walker MC, Mooser V, Epstein SE, Rader DJ, Scheffold T, Berger K, Stoll M, Huge A, Girelli D, Martinelli N, Olivieri O, Corrocher R, Morgan T, Spertus JA, McKeown P, Patterson CC, Schunkert H, Erdmann E, Linsel-Nitschke P, Lieb W, Ziegler A, Konig IR, Hengstenberg C, Fischer M, Stark K, Grosshennig A, Preuss M, Wichmann HE, Schreiber S, Holm H, Thorleifsson G, Thorsteinsdottir U, Stefansson K, Engert JC, Do R, Xie C, Anand S, Kathiresan S, Ardissino D, Mannucci PM, Siscovick D, O’Donnell CJ, Samani NJ, Melander O, Elosua R, Peltonen L, Salomaa V, Schwartz SM, Altshuler D. Genome-wide association of early-onset myocardial infarction with single nucleotide polymorphisms and copy number variants. Nat Genet. 2009;41:334–341. doi: 10.1038/ng.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Teslovich TM, Musunuru K, Smith AV, Edmondson AC, Stylianou IM, Koseki M, Pirruccello JP, Ripatti S, Chasman DI, Willer CJ, Johansen CT, Fouchier SW, Isaacs A, Peloso GM, Barbalic M, Ricketts SL, Bis JC, Aulchenko YS, Thorleifsson G, Feitosa MF, Chambers J, Orho-Melander M, Melander O, Johnson T, Li X, Guo X, Li M, Shin Cho Y, Jin Go M, Jin Kim Y, Lee JY, Park T, Kim K, Sim X, Twee-Hee Ong R, Croteau-Chonka DC, Lange LA, Smith JD, Song K, Hua Zhao J, Yuan X, Luan J, Lamina C, Ziegler A, Zhang W, Zee RY, Wright AF, Witteman JC, Wilson JF, Willemsen G, Wichmann HE, Whitfield JB, Waterworth DM, Wareham NJ, Waeber G, Vollenweider P, Voight BF, Vitart V, Uitterlinden AG, Uda M, Tuomilehto J, Thompson JR, Tanaka T, Surakka I, Stringham HM, Spector TD, Soranzo N, Smit JH, Sinisalo J, Silander K, Sijbrands EJ, Scuteri A, Scott J, Schlessinger D, Sanna S, Salomaa V, Saharinen J, Sabatti C, Ruokonen A, Rudan I, Rose LM, Roberts R, Rieder M, Psaty BM, Pramstaller PP, Pichler I, Perola M, Penninx BW, Pedersen NL, Pattaro C, Parker AN, Pare G, Oostra BA, O’Donnell CJ, Nieminen MS, Nickerson DA, Montgomery GW, Meitinger T, McPherson R, McCarthy MI, McArdle W, Masson D, Martin NG, Marroni F, Mangino M, Magnusson PK, Lucas G, Luben R, Loos RJ, Lokki ML, Lettre G, Langenberg C, Launer LJ, Lakatta EG, Laaksonen R, Kyvik KO, Kronenberg F, Konig IR, Khaw KT, Kaprio J, Kaplan LM, Johansson A, Jarvelin MR, Janssens AC, Ingelsson E, Igl W, Kees Hovingh G, Hottenga JJ, Hofman A, Hicks AA, Hengstenberg C, Heid IM, Hayward C, Havulinna AS, Hastie ND, Harris TB, Haritunians T, Hall AS, Gyllensten U, Guiducci C, Groop LC, Gonzalez E, Gieger C, Freimer NB, Ferrucci L, Erdmann J, Elliott P, Ejebe KG, Doring A, Dominiczak AF, Demissie S, Deloukas P, de Geus EJ, de Faire U, Crawford G, Collins FS, Chen YD, Caulfield MJ, Campbell H, Burtt NP, Bonnycastle LL, Boomsma DI, Boekholdt SM, Bergman RN, Barroso I, Bandinelli S, Ballantyne CM, Assimes TL, Quertermous T, Altshuler D, Seielstad M, Wong TY, Tai ES, Feranil AB, Kuzawa CW, Adair LS, Taylor HA, Jr, Borecki IB, Gabriel SB, Wilson JG, Holm H, Thorsteinsdottir U, Gudnason V, Krauss RM, Mohlke KL, Ordovas JM, Munroe PB, Kooner JS, Tall AR, Hegele RA, Kastelein JJ, Schadt EE, Rotter JI, Boerwinkle E, Strachan DP, Mooser V, Stefansson K, Reilly MP, Samani NJ, Schunkert H, Cupples LA, Sandhu MS, Ridker PM, Rader DJ, van Duijn CM, Peltonen L, Abecasis GR, Boehnke M, Kathiresan S. Biological, clinical and population relevance of 95 loci for blood lipids. Nature. 2010;466:707–713. doi: 10.1038/nature09270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schober A, Knarren S, Lietz M, Lin EA, Weber C. Crucial role of stromal cell-derived factor-1α in neointima formation after vascular injury in apolipoprotein E-deficient mice. Circulation. 2003;108:2491–2497. doi: 10.1161/01.CIR.0000099508.76665.9A. [DOI] [PubMed] [Google Scholar]
- 15.Zoll J, Fontaine V, Gourdy P, Barateau V, Vilar J, Leroyer A, Lopes-Kam I, Mallat Z, Arnal JF, Henry P, Tobelem G, Tedgui A. Role of human smooth muscle cell progenitors in atherosclerotic plaque development and composition. Cardiovasc Res. 2008;77:471–480. doi: 10.1093/cvr/cvm034. [DOI] [PubMed] [Google Scholar]
- 16.Foteinos G, Hu Y, Xiao Q, Metzler B, Xu Q. Rapid endothelial turnover in atherosclerosis-prone areas coincides with stem cell repair in apolipoprotein e-deficient mice. Circulation. 2008;117:1856–1863. doi: 10.1161/CIRCULATIONAHA.107.746008. [DOI] [PubMed] [Google Scholar]
- 17.Holtwick R, Gotthardt M, Skryabin B, Steinmetz M, Potthast R, Zetsche B, Hammer RE, Herz J, Kuhn M. Smooth muscle-selective deletion of guanylyl cyclase-a prevents the acute but not chronic effects of anp on blood pressure. Proc Natl Acad Sci U S A. 2002;99:7142–7147. doi: 10.1073/pnas.102650499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wirth A, Benyo Z, Lukasova M, Leutgeb B, Wettschureck N, Gorbey S, Orsy P, Horvath B, Maser-Gluth C, Greiner E, Lemmer B, Schutz G, Gutkind JS, Offermanns S. G12-G13-larg-mediated signaling in vascular smooth muscle is required for salt-induced hypertension. Nat Med. 2008;14:64–68. doi: 10.1038/nm1666. [DOI] [PubMed] [Google Scholar]
- 19.Dejana E, Tournier-Lasserve E, Weinstein BM. The control of vascular integrity by endothelial cell junctions: Molecular basis and pathological implications. Dev Cell. 2009;16:209–221. doi: 10.1016/j.devcel.2009.01.004. [DOI] [PubMed] [Google Scholar]
- 20.Davignon J, Ganz P. Role of endothelial dysfunction in atherosclerosis. Circulation. 2004;109:III27–32. doi: 10.1161/01.CIR.0000131515.03336.f8. [DOI] [PubMed] [Google Scholar]
- 21.Reis M, Czupalla CJ, Ziegler N, Devraj K, Zinke J, Seidel S, Heck R, Thom S, Macas J, Bockamp E, Fruttiger M, Taketo MM, Dimmeler S, Plate KH, Liebner S. Endothelial wnt/beta-catenin signaling inhibits glioma angiogenesis and normalizes tumor blood vessels by inducing PDGF-β expression. J Exp Med. 2012;209:1611–1627. doi: 10.1084/jem.20111580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hwang S, Zimmerman NP, Agle KA, Turner JR, Kumar SN, Dwinell MB. E-cadherin is critical for collective sheet migration and is regulated by the chemokine CXCL12 protein during restitution. J Biol Chem. 2012;287:22227–22240. doi: 10.1074/jbc.M112.367979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Broermann A, Winderlich M, Block H, Frye M, Rossaint J, Zarbock A, Cagna G, Linnepe R, Schulte D, Nottebaum AF, Vestweber D. Dissociation of VE-PTP from VE-cadherin is required for leukocyte extravasation and for vegf-induced vascular permeability in vivo. J Exp Med. 2011;208:2393–2401. doi: 10.1084/jem.20110525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Timmerman I, Hoogenboezem M, Bennett AM, Geerts D, Hordijk PL, van Buul JD. The tyrosine phosphatase shp2 regulates recovery of endothelial adherens junctions through control of beta-catenin phosphorylation. Mol Biol Cell. 2012;23:4212–4225. doi: 10.1091/mbc.E12-01-0038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van Haperen R, de Waard M, van Deel E, Mees B, Kutryk M, van Aken T, Hamming J, Grosveld F, Duncker DJ, de Crom R. Reduction of blood pressure, plasma cholesterol, and atherosclerosis by elevated endothelial nitric oxide. J Biol Chem. 2002;277:48803–48807. doi: 10.1074/jbc.M209477200. [DOI] [PubMed] [Google Scholar]
- 26.Ogura M, Hori M, Harada-Shiba M. Association between cholesterol efflux capacity and atherosclerotic cardiovascular disease in patients with familial hyper-cholesterolemia. Arterioscler Thromb Vasc Biol. 2016;36:181–188. doi: 10.1161/ATVBAHA.115.306665. [DOI] [PubMed] [Google Scholar]
- 27.Smith JD, Miyata M, Ginsberg M, Grigaux C, Shmookler E, Plump AS. Cyclic amp induces apolipoprotein E binding activity and promotes cholesterol efflux from a macrophage cell line to apolipoprotein acceptors. J Biol Chem. 1996;271:30647–30655. doi: 10.1074/jbc.271.48.30647. [DOI] [PubMed] [Google Scholar]
- 28.Bennett MR, Sinha S, Owens GK. Vascular smooth muscle cells in atherosclerosis. Circ Res. 2016;118:692–702. doi: 10.1161/CIRCRESAHA.115.306361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Salabei JK, Cummins TD, Singh M, Jones SP, Bhatnagar A, Hill BG. PDGF-mediated autophagy regulates vascular smooth muscle cell phenotype and resistance to oxidative stress. Biochem J. 2013;451:375–388. doi: 10.1042/BJ20121344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Naylor AJ, McGettrick HM, Maynard WD, May P, Barone F, Croft AP, Egginton S, Buckley CD. A differential role for CD248 (endosialin) in pdgf-mediated skeletal muscle angiogenesis. PLoS ONE. 2014;9:e107146. doi: 10.1371/journal.pone.0107146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hasanov Z, Ruckdeschel T, Konig C, Mogler C, Kapel SS, Korn C, Spegg C, Eichwald V, Wieland M, Appak S, Augustin HG. Endosialin promotes atherosclerosis through phenotypic remodeling of vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2017;37:495–505. doi: 10.1161/ATVBAHA.116.308455. [DOI] [PubMed] [Google Scholar]
- 32.Nakamura Y, Patrushev N, Inomata H, Mehta D, Urao N, Kim HW, Razvi M, Kini V, Mahadev K, Goldstein BJ, McKinney R, Fukai T, Ushio-Fukai M. Role of protein tyrosine phosphatase 1β in vascular endothelial growth factor signaling and cell-cell adhesions in endothelial cells. Circ Res. 2008;102:1182–1191. doi: 10.1161/CIRCRESAHA.107.167080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ukropec JA, Hollinger MK, Salva SM, Woolkalis MJ. SHP2 association with VE-cadherin complexes in human endothelial cells is regulated by thrombin. J Biol Chem. 2000;275:5983–5986. doi: 10.1074/jbc.275.8.5983. [DOI] [PubMed] [Google Scholar]
- 34.Gavard J. Endothelial permeability and VE-cadherin: A wacky comradeship. Cell Adh Migr. 2014;8:158–164. doi: 10.4161/cam.29026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Paolinelli R, Corada M, Ferrarini L, Devraj K, Artus C, Czupalla CJ, Rudini N, Maddaluno L, Papa E, Engelhardt B, Couraud PO, Liebner S, Dejana E. Wnt activation of immortalized brain endothelial cells as a tool for generating a standardized model of the blood brain barrier in vitro. PLoS ONE. 2013;8:e70233. doi: 10.1371/journal.pone.0070233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mo W, Chen J, Patel A, Zhang L, Chau V, Li Y, Cho W, Lim K, Xu J, Lazar AJ, Creighton CJ, Bolshakov S, McKay RM, Lev D, Le LQ, Parada LF. CXCR4/CXCL12 mediate autocrine cell-cycle progression in nf1-associated malignant peripheral nerve sheath tumors. Cell. 2013;152:1077–1090. doi: 10.1016/j.cell.2013.01.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu Z, Habener JF. Stromal cell-derived factor-1 promotes survival of pancreatic beta cells by the stabilisation of β-catenin and activation of transcription factor 7-like 2 (tcf7l2) Diabetologia. 2009;52:1589–1598. doi: 10.1007/s00125-009-1384-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kobayashi K, Sato K, Kida T, Omori K, Hori M, Ozaki H, Murata T. Stromal cell-derived factor-1alpha/c-x-c chemokine receptor type 4 axis promotes endothelial cell barrier integrity via phosphoinositide 3-kinase and rac1 activation. Arterioscler Thromb Vasc Biol. 2014;34:1716–1722. doi: 10.1161/ATVBAHA.114.303890. [DOI] [PubMed] [Google Scholar]
- 39.Guo C, Goodwin AJ, Buie JN, Cook JA, Halushka PV, Argraves K, Zingarelli B, Zhang XK, Wang L, Fan H. A stromal cell-derived factor 1 alpha analogue improves endothelial cell function in lipopolysaccharide-induced acute respiratory distress syndrome. Mol Med. 2016;22:115–123. doi: 10.2119/molmed.2015.00240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gelfand BD, Meller J, Pryor AW, Kahn M, Bortz PD, Wamhoff BR, Blackman BR. Hemodynamic activation of β-catenin and T-cell-specific transcription factor signaling in vascular endothelium regulates fibronectin expression. Arterioscler Thromb Vasc Biol. 2011;31:1625–1633. doi: 10.1161/ATVBAHA.111.227827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Choi SE, Jang HJ, Kang Y, Jung JG, Han SJ, Kim HJ, Kim DJ, Lee KW. Atherosclerosis induced by a high-fat diet is alleviated by lithium chloride via reduction of vcam expression in ApoE-deficient mice. Vascul Pharmacol. 2010;53:264–272. doi: 10.1016/j.vph.2010.09.004. [DOI] [PubMed] [Google Scholar]
- 42.Ai Z, Shao J, Wu Y, Yu M, Du J, Shi X, Shi X, Zhang Y, Guo Z. Chir99021 enhances KLF4 expression through β-catenin signaling and miR-7a regulation in J1 mouse embryonic stem cells. PLoS ONE. 2016;11:e0150936. doi: 10.1371/journal.pone.0150936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cowan CE, Kohler EE, Dugan TA, Mirza MK, Malik AB, Wary KK. Kruppel-like factor-4 transcriptionally regulates VE-cadherin expression and endothelial barrier function. Circ Res. 2010;107:959–966. doi: 10.1161/CIRCRESAHA.110.219592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhou G, Hamik A, Nayak L, Tian H, Shi H, Lu Y, Sharma N, Liao X, Hale A, Boerboom L, Feaver RE, Gao H, Desai A, Schmaier A, Gerson SL, Wang Y, Atkins GB, Blackman BR, Simon DI, Jain MK. Endothelial kruppel-like factor 4 protects against atherothrombosis in mice. J Clin Invest. 2012;122:4727–4731. doi: 10.1172/JCI66056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wu CJ, O’Rourke DM, Feng GS, Johnson GR, Wang Q, Greene MI. The tyrosine phosphatase SHP-2 is required for mediating phosphatidylinositol 3-kinase/Akt activation by growth factors. Oncogene. 2001;20:6018–6025. doi: 10.1038/sj.onc.1204699. [DOI] [PubMed] [Google Scholar]
- 46.Ackers-Johnson M, Talasila A, Sage AP, Long X, Bot I, Morrell NW, Bennett MR, Miano JM, Sinha S. Myocardin regulates vascular smooth muscle cell inflammatory activation and disease. Arterioscler Thromb Vasc Biol. 2015;35:817–828. doi: 10.1161/ATVBAHA.114.305218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Feil S, Fehrenbacher B, Lukowski R, Essmann F, Schulze-Osthoff K, Schaller M, Feil R. Transdifferentiation of vascular smooth muscle cells to macrophage-like cells during atherogenesis. Circ Res. 2014;115:662–667. doi: 10.1161/CIRCRESAHA.115.304634. [DOI] [PubMed] [Google Scholar]
- 48.Allahverdian S, Chehroudi AC, McManus BM, Abraham T, Francis GA. Contribution of intimal smooth muscle cells to cholesterol accumulation and macrophage-like cells in human atherosclerosis. Circulation. 2014;129:1551–1559. doi: 10.1161/CIRCULATIONAHA.113.005015. [DOI] [PubMed] [Google Scholar]
- 49.Carthy JM, Luo Z, McManus BM. Wnt3a induces a contractile and secretory phenotype in cultured vascular smooth muscle cells that is associated with increased gap junction communication. Lab Invest. 2012;92:246–255. doi: 10.1038/labinvest.2011.164. [DOI] [PubMed] [Google Scholar]
- 50.Hartmann P, Zhou Z, Natarelli L, Wei Y, Nazari-Jahantigh M, Zhu M, Grommes J, Steffens S, Weber C, Schober A. Endothelial dicer promotes atherosclerosis and vascular inflammation by miRNA-103-mediated suppression of KLF4. Nat Commun. 2016;7:10521. doi: 10.1038/ncomms10521. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.