

Abstract
We describe a novel approach to separate two ribosome populations from the same cells and use this method, and RNA-seq, to identify the mRNAs bound to S. cerevisiae ribosomes with and without Rps26, a protein linked to the pathogenesis of Diamond Blackfan Anemia (DBA). These analyses reveal that Rps26 contributes to mRNA-specific translation by recognition of the Kozak sequence in well-translated mRNAs, and that Rps26-deficient ribosomes preferentially translate mRNA from select stress response pathways. Surprisingly, exposure of yeast to these stresses leads to the formation of Rps26-deficient ribosomes and to the increased translation of their target mRNAs. These results describe a novel paradigm, the production of specialized ribosomes, which play physiological roles in augmenting the well-characterized transcriptional stress response with a heretofore unknown translational response, thereby creating a feed forward loop in gene-expression. Moreover, the simultaneous gain-of-function and loss-of-function phenotypes from Rps26-deficient ribosomes can explain the pathogenesis of DBA.
Translational control of gene expression is integral to the maintenance of protein homeostasis1–4. Recent findings from ribosomal profiling studies show that different mRNAs are recruited to ribosomes with vastly differing efficiencies5,6. Furthermore, translational efficiency for any given mRNA can vary under different cellular conditions5,7–9. While the molecular mechanisms underlying these differences remain poorly understood, one element that is known to affect an mRNA’s translation efficiency is the sequence immediately upstream of the start codon. Originally described by Kozak, nucleotide changes in this sequence affect protein production by an order of magnitude10–12. Recent studies have confirmed the importance of a 10 nucleotide window upstream of the start-codon13–15, and structural studies have identified a contact between eIF2α and the highly conserved nucleotide at the −3 position (with the AUG being in positions 1-3)16. How the other upstream bases are recognized is not well understood, partly because the mRNA in these studies did not conform to the Kozak consensus, and partly because a 21 amino acid C-terminal extension of Rps26, which is located nearby, was not resolved in the structure.
Haploinsufficiency of ribosomal proteins underlies a number of diseases, such as Diamond Blackfan Anemia (DBA), congenital asplenia, and 5q- syndrome17–20. These diseases exhibit a range of tissue specific symptoms, but share two seemingly paradoxical phenotypes: proliferative and growth deficiencies (often coupled with developmental defects), paired with a greatly increased risk of cancer20–22. While the defects in rapidly proliferating tissues are perhaps expected from insufficiency in ribosomal proteins, lack of ribosomes would predict a resistance and not a susceptibility to cancer. Rps26 is the third most commonly mutated protein in DBA23, and its location in the mRNA exit channel16, as well as crosslinking data24, predict contacts with the mRNA upstream of the start codon during translation initiation.
Here we report a purification method that enables separation of ribosomes lacking (ΔRps26) and containing (+Rps26) Rps26, and then use RNA-seq to identify the mRNAs bound to both ribosome pools. The data show that both types of ribosomes bind to a specific subset of mRNAs, and that mRNAs bound to ΔRps26 ribosomes are translated more poorly in wild type cells than those enriched in +Rps26 ribosomes. Furthermore, mRNAs enriched in ΔRps26 ribosomes lack the sequence conservation upstream of the Kozak sequence. Luciferase reporter assays confirm that Rps26 is required for preferential translation of mRNAs with an adenosine at position −4, and furthermore demonstrate that mRNAs with a guanosine at position −4 are preferentially translated by ΔRps26 ribosomes. Pathway analysis shows clustering of mRNAs bound to ΔRps26 ribosomes in the Hog1 and Rim101 pathways, which regulate the response to high salt and pH stress, respectively. Correspondingly, Rps26 depletion leads to constitutive activation of these pathways and therefore increased resistance to high salt and pH. Finally, we show that upon exposure to high salt or pH, cells generate ΔRps26 ribosomes, thereby allowing for preferential translation of mRNAs with mutations in the −4 position of the Kozak sequence. These data reveal the molecular basis for recognition of the Kozak sequence, and suggest that perturbed protein homeostasis could play a role in the pathogenesis of DBA. Moreover, they demonstrate that the well-characterized transcriptional response to high salt and pH stress is augmented by a hitherto unknown translational response, which involves changes in the composition of ribosomes to enable preferential translation of a subset of stress-related mRNAs. Thus, Rps26-deficient ribosomes have physiological roles.
RESULTS
ΔRps26 and +Rps26 ribosomes bind different mRNAs
Structural and crosslinking data demonstrate that Rps26 is located in the mRNA exit channel, adjacent to the Kozak sequence16,24. To assess if Rps26 influences the repertoire of mRNAs translated by the ribosome, we purified Rps26-replete (+Rps26) and -deficient (ΔRps26) ribosomes from a yeast strain in which ΔRps26 ribosomes are selectively TAP-tagged (Figure 1a). In this strain Rps26 and Rps3, a distal protein, are under galactose-inducible control. Furthermore, Rps3-TAP is under the control of the doxycycline (dox)-repressible TET promoter, allowing for separate induction of Rps3-TAP when Rps26 and Rps3 are repressed. Importantly, Rps3-TAP fully complements the absence of Rps325, and ribosomes containing Rps3-TAP are recruited into polysomes akin to those with untagged Rps3 (Figure S1a-d). After 4h in glucose, cells were harvested and lysed in the presence of cycloheximide to maintain mRNA-ribosome interactions, and ΔRps26 ribosomes separated from +Rps26 ribosomes by affinity purification on IgG beads (Figure 1b). To exclude free mRNAs, the flow-through fraction was loaded onto a sucrose gradient and only the ribosome bound fractions were collected. SDS-PAGE analysis and immunoblotting of the eluate (ΔRps26 ribosomes) and the flow-through (+Rps26 ribosomes) show that this method efficiently separates the two ribosome populations (Figure 1b). Furthermore, Northern blotting and Bioanalyzer results demonstrate that the Rps26-deficient ribosomes contain mature 18S rRNA (Figure 1c & Figure S1f-g). RNAs were isolated from these ribosome pools, sequenced on an Illumina NexSeq 500, and analyzed with the DESeq2 algorithm. Notably, because the two ribosome pools were isolated from the same cells, and therefore encountered the same mRNAs, the RNAseq data from both pools can be directly compared.
Figure 1. Isolation of ΔRps26 ribosomes and characterization of bound mRNAs.
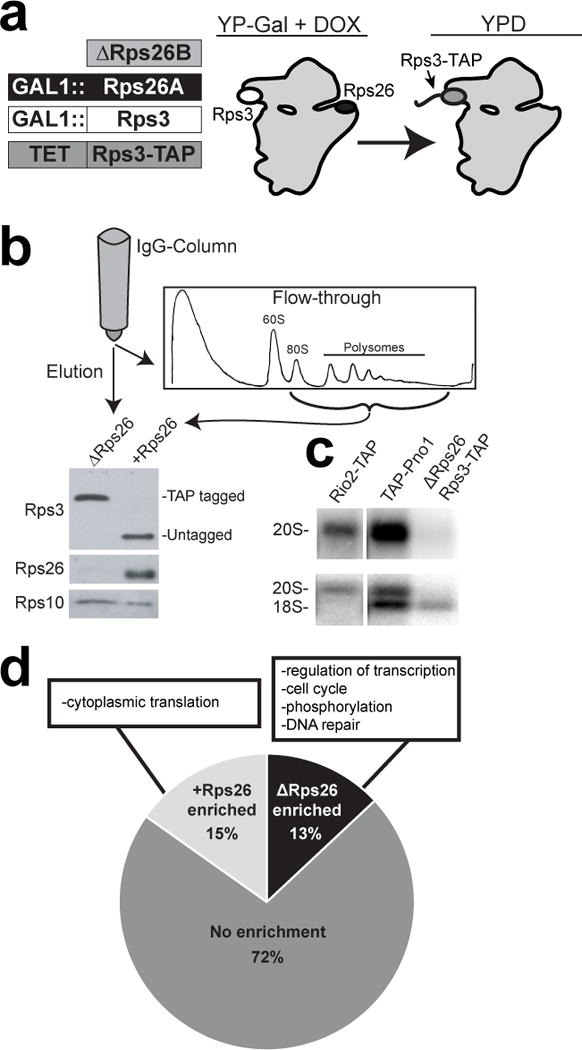
a, Strain used to purify ribosomes lacking Rps26 (ΔRps26). b, Separation of ΔRps26 and +Rps26 ribosomes by affinity purification with Rps3-TAP. The flow-through was loaded on a sucrose gradient and only the fractions containing mRNA-ribosome complexes were used for sequencing analysis. Western blotting of the elution and flow-through for (tagged and untagged) Rps3, and Rps26. Rps10 was used as a loading control. This experiment was repeated at least five times and representative data are shown. c, Northern blot analysis of ΔRps26 ribosomes compared to immature ribosomes captured by TAP-tagging the assembly factors Rio2 or Pno1. Samples were probed against ITS1 (20S rRNA) (top panel) and 18S rRNA (bottom panel). This experiment was repeated three times and representative data are shown. d, Distribution of mRNAs enriched (Padj<0.05 by DESeq2) in +Rps26 and ΔRps26 ribosomes. mRNAs in each pool: +: 865, Δ: 741, no enrichment: 4084. The GO-terms with the most genes represented in each pool are also listed. See also Supplementary Figure 1. Original blot images are in Supplementary Data Set 3.
RNA-seq analysis showed that 88-95% of reads from the +Rps26 sample and 89-95% of the reads from the ΔRps26 sample mapped to the yeast genome. Further, 90% of the ORFs (5219 out of a total of 5784) had 128 or more reads per experiment, a threshold experimentally determined to reduce the false appearance of gene regulation5. Of the remaining 565 ORFs, 274 (49%) are annotated as ‘uncharacterized’ or ‘dubious’ and may not encode proteins (Figure S2c). Furthermore, data from three independently grown and purified samples show high correlation between experiments (Figure S2d). Thus, RNA-seq identifies the overwhelming majority of ORFs with high confidence and reproducibility.
Using a conservative cutoff of padj < 0.05, more than one quarter of all mRNAs are significantly enriched in one of the ribosome pools. Of these, 13% are enriched in the ΔRps26 pool, while 15% are preferentially bound to +Rps26 ribosomes (Figure 1c, Supplementary Data Set 1), and all are enriched more than 1.5 fold. Importantly, control experiments where Rps3-TAP-bound mRNAs are sequenced demonstrate that the differences between these samples do not arise from the TAP-tag on Rps26-deficient ribosomes (Figure S1h).
To validate these data, the effects from Rps26 depletion on polysome recruitment of mRNAs enriched in the two ribosome pools were determined. Rps26 was depleted in a dox-repressible Rps26 strain that accumulates ΔRps26 ribosomes in dox, but is similar to the parent strain in its absence (Figure 2a and S3a-d). If ribosome pulldowns reflect functional interactions with translating ribosomes, Rps26 depletion should selectively decrease the polysome recruitment of mRNAs enriched in the +Rps26 pool. As previously described26, sucrose gradient fractionation was coupled with RT-qPCR to track the distribution and number of ribosomes bound to mRNAs enriched in the +Rps26 versus ΔRps26 pools when Rps26 was replete or depleted (Figure S3e-h). Quantifying these analyses shows that Rps26 depletion has a smaller effect on polysome recruitment of mRNAs preferentially bound by ΔRps26 ribosomes compared to mRNAs enriched on +Rps26 ribosomes (Figure 2b-c), validating the sequencing data in vivo.
Figure 2. ΔRps26 ribosomes bind a distinct set of poorly translated mRNAs.
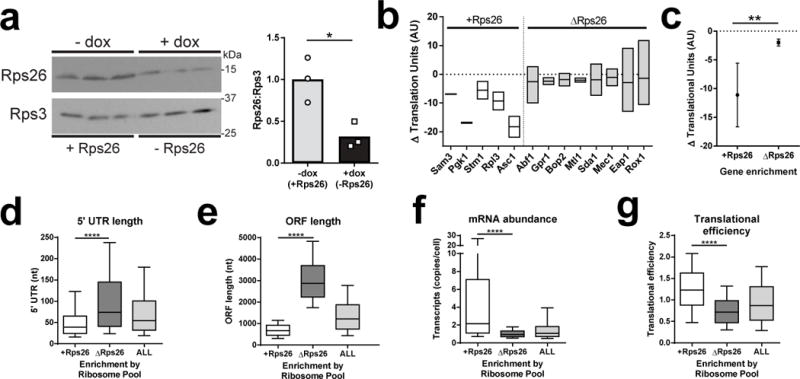
a, Ribosomes isolated from TET:Rps26 cells exposed to dox are probed for Rps26 occupancy relative to Rps3. Western blot of ribosomes purified in parallel from three separate cultures (left) and its quantitation (right). By t-test, (*) p<0.05 t=3.795, DF=4. This experiment was repeated four times and representative data are shown. b, Translational downregulation (measured by bound ribosomes) of mRNAs enriched in +Rps26 or ΔRps26 ribosomes after Rps26 depletion. Three independent experiments were run for Abf1, Eap1 and Rox1, duplicates for all others. Box plots represent the range, midline is the mean. c, Analysis of all mRNAs in b; error bars= SD, (**) p=0.0016 by the Kolmogorov-Smirnov test. d-g, Metagene analysis using existing datasets60,63–65, by the Kolmogorov-Smirnov test (****) p<0.0001. Number of mRNAs analyzed: 5′ UTR length; all=4363, +=763, Δ=575. ORF length; all=5690, +=763, Δ=575; abundance; all=5459, +=804, Δ=739. TE: all=5476, +=838, Δ=736; Boxplot whiskers represent the 10-90th percentile. Boxes represent the interquartile range, midline is the median. See also Supplementary Figures 2 & 3. Original blot images are in Supplementary Data Set 3.
Further analysis revealed that the mRNAs enriched on +Rps26 and ΔRps26 ribosomes differed in their physical characteristics. Specifically, mRNAs enriched on ΔRps26 ribosomes have longer 5′ untranslated regions, longer ORFs, and are less abundant than +Rps26-enriched mRNAs, or the transcriptome as a whole (Figure 2d-f). This holds true even if mRNAs encoding ribosomal proteins, which tend to be short, are excluded (Figure S2e). In contrast, there were no significant differences in the average length of the 3′-UTR or transcript half-lives (Figure S2f). While the molecular basis for these observations is unclear, long 5′-UTRs and ORFs are associated with less efficient translation5,27,28. Importantly, mRNAs enriched on ΔRps26 ribosomes are also much less-efficiently translated than mRNAs enriched on +Rps26 ribosomes, and the entire transcriptome. In contrast, +Rps26 ribosomes enrich highly translated mRNAs (Figure 2g), consistent with a role for Rps26 in recognition of the Kozak sequence, which imparts highly efficient translation.
Rps26-based transcript preference is mediated by nucleotides upstream of the start codon
Weblogo analysis was used to interrogate the sequence enrichment of mRNAs bound to ΔRps26 and +Rps26 ribosomes29. Crosslinking24 and structural16 data indicate that Rps26 is bound in the mRNA exit channel, potentially contacting residues −4, and −7-10 (relative to the start-codon). Thus, our analysis of sequence enrichment focused on the sequence immediately upstream of the start codon. This analysis shows mRNAs enriched on +Rps26 ribosomes are likely to contain an A at residue −3, as discovered by Kozak30. In addition, adenosines are moderately enriched at residues −1, −2, −4, −7, −8 and −10 (Figure 3a), broadly recapitulating the Kozak consensus for yeast13,31,32. This finding is consistent with the notion that +Rps26 ribosomes preferentially translate mRNAs containing adenosines at these positions. In contrast, ΔRps26-enriched mRNAs lack conservation at any position other than −3, and reflect the entire yeast transcriptome (Figure 3b-c). Thus, the selection of mRNAs containing adenosines at positions −1, −2, −4, −7, −8 and −10 relies on Rps26.
Figure 3. Rps26 promotes translation by recognizing specific residues in the Kozak sequence.
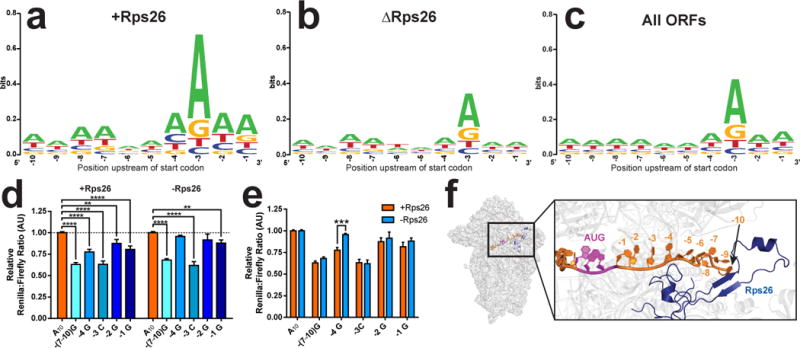
a-c, Weblogo conservation analysis of mRNAs enriched in +Rps26 (a, n=865) and ΔRps26 (b, n=741) ribosomes versus all mRNAs in our dataset (c, n=5696). d, Luciferase reporter assays from TET:Rps26 cells, grown in the absence or presence of dox, to test effects from Kozak sequence mutations with replete (+) or depleted (-) Rps26. For +Rps26 from left to right, by two-way ANOVA: (****) p<0.0001 t=10.24, p<0.0001 t=6.229, p<0.0001 t=7.594, (**) p=0.0017 t=3.193, p<0.0001 t=4.84. For -Rps26 from left to right: (****) p<0.0001 t=8.869, p=0.2228 t=1.223, p<0.0001 t=8.328, p=0.0603 t=2.179, (**) p=0.0064 t=3.116. DF=184. e, Effect from Rps26 depletion on recognition of individual point mutations. From left to right: p=0.6562 t=1.193, (***) p=0.0002 t=4.235, p=0.9712 t=0.2146, p=0.7879 t=0.8371, p=0.5621 t=1.438 by two-way ANOVA, DF=184. For d and e, number of independent cultures: −(7→10)G and −4G, n=15; −3C, n=7; −2G, n=12; −1G, n=13. Bars represent mean values, error bars = SEM. See also Supplementary Figure 4. f, Rps26 (blue) binds mRNA near the −4 position when the start codon (purple) is in the P-site (adapted from 3J8116). The C-terminal 21 amino acids of Rps26 are not resolved in this structure.
To test if translation of mRNAs with a Kozak consensus sequence upstream of the start codon requires Rps26, we developed a dual luciferase reporter assay. Yeast were transformed with plasmids encoding firefly luciferase preceded by the adenosine sequence enriched in +Rps26 ribosomes (A10) and Renilla luciferase preceded by various upstream sequences, each containing mutations at different positions of the Kozak sequence (Figure S4a); thus, the ratio of Renilla/firefly luciferase quantifies the effects of a given upstream sequence on translation relative to the A10 sequence.
In Rps26-replete yeast, mutation of individual adenosines at the −1, −2, −3 and −4 positions, as well as −7 to −10 together, all reduced translation (Figure 3d, left). These data demonstrate that residues upstream of the start codon impact translation, which is consistent with Kozak’s studies in mammalian cells10–12,33, and with more recent work in yeast13. In contrast, in Rps26-depleted cells translation of the −4G and the −2G mutants is indistinguishable from the A10 mRNA (Figure 3d, right). Thus, Rps26 is required for preferential translation of mRNAs with an A at the −4 and −2 positions. Further, translation of mRNAs containing −4G is increased in Rps26-deficient versus +Rps26 yeast (Figure 3e). This latter conclusion is consistent with the sequencing data, which show that, relative to the transcriptome as a whole, ΔRps26 ribosomes specifically enrich mRNAs with a −4G position. This comes at the loss of mRNAs with an A or C at that position (Figure S4b). In contrast, Rps26-containing ribosomes enrich A or C and deplete G from the −4 position (Figure S4b). This effect is specific to Rps26, as depletion of Rps3 or Rps17 has no such effect on translation of the −4G reporter (Figure S4d). Because Rps26 depletion leads to moderate 20S rRNA accumulation34, we wondered if the accumulation of 20S rRNA and not the depletion of Rps26 was responsible for the observed defect in recognition of the −4 residue. We therefore analyzed translation of the luciferase reporter in wild type yeast and yeast lacking the 40S assembly factor Ltv1 (ΔLtv1). Similar to the Rps26 depletion strain, moderate 20S rRNA accumulation is observed in this strain35,36. Importantly, translation of the luciferase reporter is sensitive to the residue at the −4 position in ΔLtv1 yeast (Figure S4e), demonstrating that it is depletion of Rps26 and not accumulation of 20S rRNA that is responsible for the observed defect in recognition of the −4 residue.
Importantly, Rps26 directly contacts the −4 position, as well as an rRNA residue directly neighboring an interactor of the −2 residue (Figure 3f, and reference16). Thus, the sequencing data, reporter assays and structural studies all demonstrate that Rps26 recognizes the −4 and −2 position of the Kozak sequence, and that this is necessary for preferential translation of mRNAs with a Kozak consensus of A or C at that position. ΔRps26 ribosomes have lost this preference and instead show a slight preference for mRNAs containing a −4G.
Accumulation of ΔRps26 ribosomes activates the Hog1 and Rim101 pathways
To assess if mRNAs enriched in ΔRps26 ribosomes clustered in specific biological pathways, where they could produce specific biological outcomes, mRNAs enriched in each ribosome pool were used as a basis for GO-term enrichment analysis on GeneCodis37 (Figure 1c & Supplementary Data Set 2). +Rps26 ribosome-enriched mRNAs encode mainly ribosomal proteins and translation factors. This is consistent with previous findings that mRNAs encoding ribosomal proteins are among the best-translated5,26. In contrast, ΔRps26 ribosome-enriched transcripts were associated with highly regulated processes, including transcriptional control, phosphorylation, cell cycle, DNA repair, and, notably, well-characterized stress-responsive signaling pathways including the Hog1 pathway38, a MAP-kinase cascade that responds to high salt stress, and the Rim101 pathway39, which responds to pH stress (Figure 4a-b).
Figure 4. Accumulation of ΔRps26 ribosomes activates specific biological pathways.
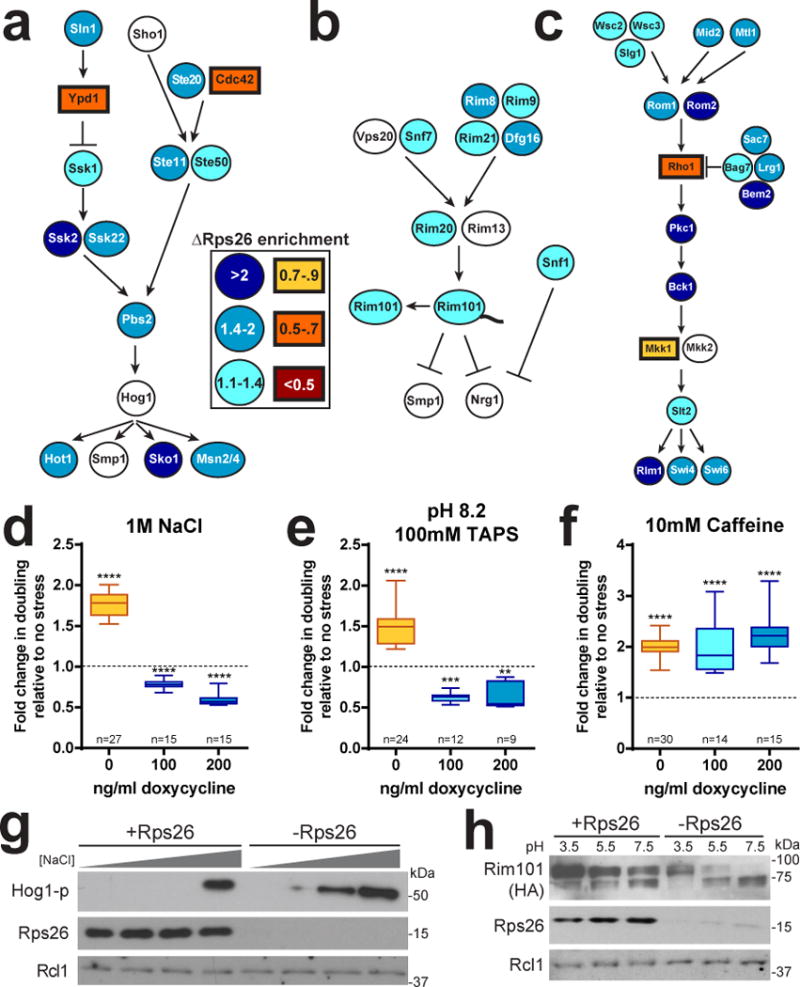
Enrichment of mRNAs encoding components of the Hog MAPK (a), the Rim101 (b), or the cell wall integrity (CWI) (c) pathways in ΔRps26 or +Rps26 ribosomes. mRNAs enriched in ΔRps26 ribosomes are blue-tinted ovals, mRNAs enriched in +Rps26 ribosomes are orange-tinted rectangles and those not enriched in either are white. Changes in doubling time upon addition salt (d), high pH (e), or caffeine (f), in cells containing (-dox) or deficient (+doc) of Rps26. Boxes indicate interquartile range, the midline is the median and whiskers represent the range. Values were compared to no stress (fold change = 1) with the Wilcoxon Signed Rank test, (****) p<0.0001 and (**) p=0.0039. From left to right, the sum of signed ranks (W) for d (378, −120, −120), e (300, −78, −45) and f (465, 105, 120) for cells exposed to 0, 100, and 200 ng/ml of dox, respectively. Data is from n independently grown cultures. g, Hog1 phosphorylation in Rps26-containing and deficient cells after exposure to increasing concentrations of NaCl (0-500mM). h, Proteolytic activation of Rim101 at different pH values in Rps26-containing and deficient cells. Original blot images are in Supplementary Data Set 3. Western blots were repeated four times from independent experiments and representative data are shown.
Since Rps26 depletion increases the relative translation of mRNAs enriched in ΔRps26 ribosomes (Figures 2b-c), we hypothesized that cells depleted of Rps26 are more resistant to these stresses. To test this prediction, we measured growth rates for Rps26-replete and -depleted yeast in rich media with and without high salt, or high pH. Surprisingly, Rps26-deficient cells grow faster in high salt and high pH than in rich media, while Rps26-replete cells are salt and pH sensitive (Figure 4d-e). Additionally, Rps26-deficient yeast displayed increased phosphorylation of the Hog1 MAP-kinase at lower concentrations of NaCl (Figure 4g) and C-terminal cleavage of the Rim101 protein, a hallmark of the high pH response pathway, even in mildly acidic conditions (Figure 4h). Thus, both the Hog1 MAPK and the Rim101 pathway are induced when Rps26 is depleted. This phenotype was pathway selective, as Rps26-deficient yeast are not resistant to caffeine, which activates a distinct MAP kinase cascade to produce cell wall stress40 (Figure 4c, f). Further, high salt and pH produces a growth defect in yeast strains depleted of two other late-binding ribosomal proteins, Rps3 and Rps17 (Figure S5a-g), demonstrating the specific role of Rps26 deficiency in salt and pH resistance. Finally, ΔLtv1 cells also do not show stress resistance, demonstrating that stress resistance does not arise from accumulation of 20S pre-rRNA in these cells (Figure S5h-i).
Yeast form ΔRps26 ribosomes in response to osmotic and high-pH stress
Given the high salt and high pH stress phenotypes manifest in Rps26-deficient yeast, we reasoned that the formation of ΔRps26 ribosomes might be part of the cellular response to these stresses. To test this, wild-type yeast were grown in rich media containing high salt, high pH or caffeine. Ribosomes were then purified from these stressed cells and from non-stressed control cells, and Rps26 protein levels were analyzed by western blot. Levels of 5 additional proteins from the small ribosomal subunit (Rps 0, 3, 5, 8, and Asc1) were assessed as controls. Notably, when normalized to either of these proteins, ribosomes from cells grown under high-salt or high-pH conditions contained less Rps26 than ribosomes from unstressed cells, whereas the levels of Rps26 in cells exposed to caffeine were comparable to non-stressed control cells (Figure 5a-c). These data indicate that formation of Rps26-deficient ribosomes is part of the response to high salt and high pH values.
Figure 5. Cells generate ΔRps26 ribosomes in response to high salt and high pH.
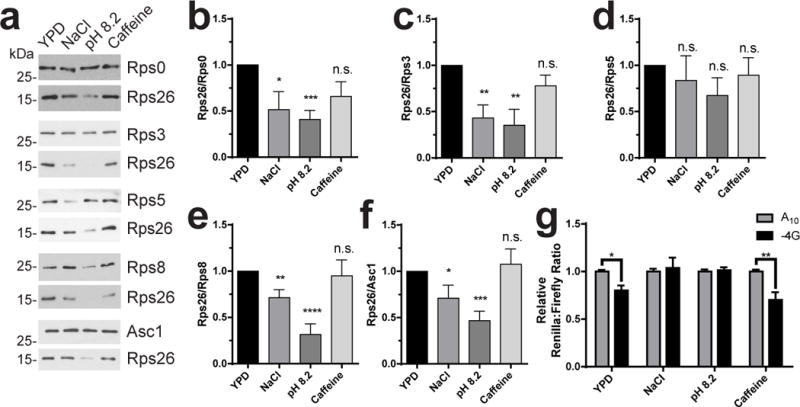
a, Levels of Rps0, Rps3, Rps5, Rps8, Rps26 and Asc1 in ribosomes purified from wild type yeast grown in YPD alone, or YPD with 1M NaCl, pH 8.2 or 10 mM caffeine were analyzed by western blotting. Because all ribosomal proteins but Rps26 are nearly identical in size, separate gels were run to analyze each of these proteins relative to Rps26, and the Rps26 and RpsX bands from each of these are shown. b-f, Quantification of data in panel A. Data were from ribosomes grown and purified in three or four (caffeine) independent replicates for each condition, each run on two different sets of gels and analyzed by t-test. For b: NaCl (*) p=0.0388 t=2.469 DF=8; pH (***) p=0.0003 t=5.967 DF=8; Caff. (ns) p=0.1018 t=1.802 DF=10. For c: NaCl (**) p=0.0025 t=4.015 df=10; pH (**) p=0.0036 t=3.773 DF=10; Caff. (ns) p=0.1343 t=1.606 DF=12. For d: NaCl (ns) p=0.5534 t=0.6132 DF=10; pH (ns) p=0.1184 t=1.708 DF=10; Caff. (ns) p=0.5866 t=0.5618 DF=10. For e: NaCl (**) p=0.008 t=3.299 DF=10; pH (****) p<0.0001 t=7.599 DF=8; Caff. (ns) p=0.8089 t=0.2472 DF=12. For f: NaCl (*) p=0.048 t=2.287 DF=9; pH (***) p=0.0004 t=5.132 DF=10; Caff. (ns) p=0.6934 t=0.4039 DF=12. Bars represent mean values, error bars = SEM. g, Luciferase assays of WT cells after a 4 hour exposure to different stress conditions. For YPD n=7, and for all others n=6, independently grown cultures. By two-way ANOVA: YPD (*) p=0.0219 t=2.817, NaCl (ns) p=0.8563 t=0.4983, pH (ns) p=0.8673 t=0.1681, and Caff. (**) p=0.0014 t=3.884 with DF=42. Bars represent mean values, error bars = SEM. Original blot images are in Supplementary Data Set 3.
We next validated the in vivo translational effects of this physiological depletion of Rps26 using the dual luciferase assay. Above, we have shown that translation of the −4G Renilla reporter is increased in Rps26-deficient yeast, where it is indistinguishable from that of the A10 Renilla reporter (Figure 3d, right). We used this functional signature of Rps26-deficient ribosomes as a read-out for the formation and functionality of Rps26-deficient ribosomes under stress. WT cells transformed with the A10 or the −4G Renilla reporters were exposed to salt, high pH or caffeine stress as above. As expected from the accumulation of ΔRps26 ribosomes in the presence of high salt or high pH, translation of the −4G reporter recovered to the level of the A10 construct when WT cells were exposed to salt or high pH stress, but not caffeine (Figure 5d). These data strongly suggest that the ΔRps26 ribosomes form when yeast are exposed to high salt or high pH translate −4G containing mRNAs. Thus, in response to high salt and high pH stress, cells produce ΔRps26 ribosomes, thereby augmenting the well-characterized transcriptional response to these stresses with preferential translation of mRNAs from these stress response pathways by ΔRps26 ribosomes.
DISCUSSION
Rps26 enforces the translational program encoded by the Kozak sequence
The data herein demonstrate key roles for Rps26 in recognizing adenosines at positions −2 and −4 of the Kozak sequence, leading to preferential translation of mRNAs containing these adenosines. Thus, these data provide a molecular basis for the effects from mutations in the Kozak sequence uncovered 30 years ago10–12,33 (Figure 6a). Because of the critical role for Rps26 in recognition of the Kozak sequence, which enables efficient translation, Rps26 depletion decreases the translation of normally highly-translated mRNAs, including those encoding ribosomal proteins, explaining the overall reduced ribosome numbers in Rps26 depleted cells (Figure S3e-f). Notably, ΔRps26 ribosomes are also revealed to selectively augment the translation of mRNAs containing a −4G. This might arise simply from increased competitiveness of such mRNAs, as strong Kozak mRNAs are no longer favored. Additionally, or alternatively, Rps26-deficiency might ameliorate a steric hindrance that arises from the −4G in wild type ribosomes, or −4G-containing mRNAs might adopt a different structure. Regardless, Rps26 enforces the translational program encoded by the Kozak sequence, while Rps26-deficient ribosomes decode a separate translational program.
Figure 6. Disruption of protein homeostasis by Rps26 insufficiency.
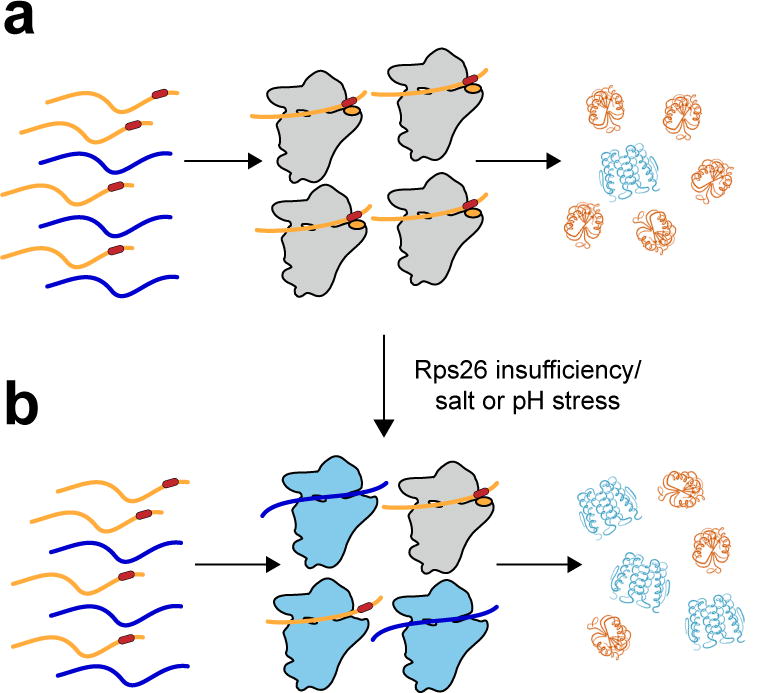
a, Rps26 (orange ovals) recognizes positions −2 and −4 in the Kozak sequence (dark red rectangles), thereby enhancing translation of mRNAs with canonical Kozak sequences. b, When Rps26 is insufficient, or at high salt or pH, ribosomes lacking Rps26 (light blue) accumulate, increasing the translation of mRNAs with Kozak sequence deviations, thereby leading to changes in protein homeostasis.
Remarkably, mRNAs upregulated by Rps26 depletion are not randomly distributed throughout the yeast transcriptome, but instead cluster in specific pathways, including the well-characterized and highly conserved Hog1 MAPK and the high pH Rim101 pathways. Growth assays and immunoblotting reveals that these pathways are constitutively activated in Rps26-deficient yeast, indicating that the decreased translation of otherwise well-translated mRNAs, and the increased translation of mRNAs with mutations in the Kozak sequence, lead to a perturbation in cellular protein homeostasis upon Rps26 depletion. Collectively, the data support a model whereby ΔRps26 ribosomes, such as found in DBA, play pathogenic roles by disabling the translation of essential mRNAs, including those encoding for ribosomal proteins, leading to reduced ribosome production, and by selectively enhancing the translation of otherwise poorly translated mRNAs characterized by long 5′-UTRs and weak Kozak sequences.
Interestingly, while the large majority of mRNAs encoding ribosome assembly factors either show no enrichment, or are enriched in ΔRps26 ribosomes, the mRNA encoding Fap7 is enriched in +Rps26 ribosomes (Figure S3i). Consequently, Fap7 levels are decreased in Rps26 depleted cells (Figure S3j). This finding explains why 20S rRNA is moderately accumulated in Rps26-depleted cells34, even though Rps26-deficient ribosomes contain 18S rRNA (Figure 1c & Figure S1f-g). Furthermore, depletion of Fap7 in Rps26 depleted cells explains why overexpression of Fap7 rescues growth of ΔTsr2 cells41: because Tsr2 stabilizes Rps2642,43, ΔTsr2 cells are expected to be Rps26 deficient, which leads to Fap7 depletion.
How can ribosomal protein haploinsufficiency lead to cancer?
In mammalian cells, activation of pro-growth pathways by translational upregulation of specific mRNAs might account for the cancer predisposition observed in DBA patients, while the overall decrease in translation of ribosomal proteins (Supplementary Data Sets 1&2), and the resulting loss of ribosomes (Figure S3e-f), accounts for growth and developmental defects. Of note, other ribosomal proteins have also been linked to DBA23, and several from the small subunit are also located near the mRNA channel. These might be similarly involved in recognition of specific mRNA features, or binding of translation initiation factors, which can have mRNA-specific effects44–46, with net effects on altering the translational repertoire. Importantly, the method described herein for purifying ribosome subpopulations will allow assessment of mRNA-specific effects from depletion of other ribosomal proteins, or rRNA modifications, which have also been linked to cancer47,48.
Physiological roles for ribosomes lacking ribosomal proteins
In their groundbreaking work, Barna and colleagues have demonstrated that deficiency of the large ribosomal subunit protein Rpl38 leads to specific developmental pathologies49,50. Conceptually similar to Rps26, Rpl38 is required for translation of a specific subset of mRNAs from the Hox family. In contrast, Rps26 is required for ribosome recruitment of highly translated mRNAs, including those encoding ribosomal proteins.
Nevertheless, the data herein not only demonstrate that Rps26-deficient ribosomes are deficient in translation of mRNAs containing a strong Kozak sequence, they also demonstrate that Rps26-deficient ribosomes selectively increase the translation of mRNAs with mutations at the −4 position in the Kozak sequence, including those encoding proteins from the Hog1 and Rim101 stress response pathways. Most importantly, the data show that Rps26-deficient ribosomes are produced by wild type yeast upon exposure to high salt and pH stress (Figure 6b). Thus, Rps26-deficient ribosomes also play physiological roles during stress, creating a feed forward loop that coordinates translational and transcriptional programs that allow for cell survival in the face of changes in the extracellular milieu. Importantly, the physiological relevance of Rps26-deficient ribosomes may explain how these can escape ribosome quality control mechanisms during assembly51,52 and function of ribosomes53,54.
How are Rps26-deficient ribosomes formed?
The observation that Rps26-deficient ribosomes are formed during high salt and high pH stress leads us to ask if these are formed from mature Rps26-containing ribosomes, or, whether instead, newly made 40S subunits are produced lacking Rps26. To start addressing these questions we used RT-qPCR to investigate if the Rps26 mRNA levels decrease more under stress than those for other r-proteins, which could allow for the production of Rps26-deficient ribosomes. The data in Figure S6a-c demonstrate that Rps26 levels decrease similar to those of other ribosomal proteins, perhaps less. Furthermore, forced expression of Rps26 via the galactose-promoter does not have an effect on the stress sensitivity of yeast, even though the levels of Rps26 mRNA are increased about 11-fold (Figure S6d-e). Together, these data indicate that it is not likely that down-regulation of Rps26 levels leads to the de novo formation of Rps26-deficient ribosomes, consistent with the observation that the stress conditions, which lead to formation of Rps26-deficient ribosomes, do not support active ribosome assembly55–57. We thus speculate that Rps26-deficient ribosomes are formed from pre-existing ribosomes. In that regard, it is interesting to note that a specific chaperone for Rps26, Tsr2, has been identified42,43. In addition to delivering Rps26 to nascent ribosomes, Tsr2 might store Rps26 and allow for the fully reversible loss and reincorporation of the protein.
ONLINE METHODS
Yeast strains and plasmids
Saccharomyces cerevisiae
Saccharomyces cerevisiae strains used in this study are listed in Table S1 and were obtained from the GE Dharmacon Yeast Knock-Out Collection or were created by standard recombination techniques58. Identity of generated strains was verified by PCR and western blotting. Plasmids used in this work are listed in Table S2.
Isolation of mRNAs bound to ΔRps26 and wild type ribosomes
Cells for this experiment were generated by transforming strain yKK636 (GAL::Rp26; GAL::Rps3) with pKK3566 (TET:Rps3-TAP). An overnight culture in YPGal supplemented with 0.2 μg/ml dox was grown to mid-log phase. Cells were washed three times with prewarmed YPGal media and inoculated into YPD media at an OD600 of 0.15. After 4h, cells were harvested after addition of 100μg/ml cycloheximide (Sigma Aldrich). TAP purification was performed as described51,59, except that 100μg/ml cycloheximide was included in all steps and only the IgG binding and elution steps were performed. The IgG eluate (containing ΔRps26 ribosomes), as well as the flow-through (containing +Rps26 ribosomes), were collected and fractionated on a 10-50% sucrose gradient as described51. Fractions containing the 80S and polysomes were pooled. Protein was precipitated from these fractions using the TCA-DOC method and analyzed by western blotting. RNA was isolated by phenol-chloroform-isoamyl alcohol extraction.
Illumina Sequencing
Purified RNA was quantified in a Qubit 2.0 Fluorometer (Invitrogen) and run on an Agilent 2100 Bioanalyzer (Agilent Technologies) for quality assessment and then treated with DNase I (New England Biolabs). The DNase-treated total RNA was depleted of ribosomal RNA using the Ribo-Zero™ Gold Yeast Kit (Illumina), and then processed with the TruSeq Stranded Total RNA sample prep kit (Illumina). Briefly, chemically fragmented RNA was random hexamer primed and reverse transcribed to generate the first strand cDNA. The second strand was synthesized incorporating dUTP in place of dTTP, preserving strand information. The double-stranded cDNA was then end-repaired, 3′-adenylated and ligated to PCR adaptors. The purified adaptor-ligated DNA is PCR amplified to generate the final libraries. The final size of fragments is 200-600bp with insert sizes ranging from 80-450bp. The validated libraries were pooled at equimolar ratios, and loaded onto the NextSeq 500 flow cell at a final concentration of 1.8 pM.
Bioinformatic processing
Demultiplexed and quality filtered raw reads (fastq) generated from the NextSeq 500 were trimmed to remove adaptor sequences using Flexbar 2.4 and aligned to the S. cerevisiae genome (S288C, SacCer3) from the Saccharomyces Genome Database60 using TopHat version 2.0.961. HT seq-count version 0.6.1 was used to generate gene counts and differential gene expression analysis on three biological replicates was performed using DESeq2 using standard settings62.
Identification and characterization of enriched transcripts
Transcripts considered enriched in each pool were analyzed using existing datasets of mRNA characteristics. Translational efficiency, 5′ and 3′ UTR length and mRNA abundance and half-life measurements were derived from existing datasets63–65. GO-term enrichment was determined using GENECODIS37. Enrichment of nucleotides upstream of the start codon in transcripts that were associated with Rps26-mediated selection was determined using Weblogo analysis29. ORF-length data were taken from the SGD_features file listed above. Upstream bases were acquired from the S. cerevisiae reference genome60 using the custom-built program STARTSEQ, written in Python.
Purification of ribosomes from TET:Rps26 cells
Cells were grown to mid-log phase and flash frozen in Ribosome buffer (20 mM Hepes/KOH (pH 7.4), 100 mM KOAc, 2.5 mM Mg(OAc)2) supplemented with 1 mg/ml heparin, protease inhibitor cocktail (Sigma) and 2 mM DTT. 400 μL of each clarified yeast lysate was layered over 100 μL of sucrose cushion (Ribo Buffer, 500 mM KCl, 1 M sucrose, 2 mM DTT) and spun in a Beckman TLA 100.1 rotor at 100,000 RPM for 2.5 h. The resulting pellets were resuspended in high salt buffer (Ribo Buffer, 500 mM KCl, 1 mg/ml heparin, 2 mM DTT) and analyzed on SDS-PAGE followed by Western blotting.
qPCR mRNA shift assay
A yeast strain with dox-inducible Rps26 deficiency (TET:Rps26) was generated by transforming yKK491 with pKK3792. Depletion of Rps26 was induced by inoculating YPD media (with or without 0.2 μg/ml dox) with TET:Rps26 yeast at an OD600 of 0.05. Cultures were grown until they reached mid-log phase and prepared for sucrose gradient fractionation as above. 5000 OD units of clarified lysate (~50μl of lysate) were loaded onto a 25-45% sucrose gradient and spun in an SW-41Ti rotor (Beckman) at 40,000rpm for 165 min. Gradients were fractionated and the following samples were collected: (i) unbound RNAs, (ii) free 40S and 60S ribosomal subunits, (iii) 80S monosomes, as well as one each (iv-xiii) containing two, three, four ribosomes, etc. We were able to clearly resolve peaks for up to 9 bound ribosomes. The final two samples contained 10-11, or 12-13 ribosomes, respectively. After addition of 0.65ng of in vitro transcribed RNase P RNA from B. subtilis to each sample, which was used for normalization, RNA was isolated by phenol-chloroform extraction from a fixed percentage of each sample’s total volume, and reverse transcription was performed using Protoscript II (New England Biolabs) per manufacturer’s instructions. qPCR was performed with Excella 2X SYBR master mix per manufacturer’s instructions on a BioRad IQ2 using primers listed in Table S3. The percentage of mRNA in each sample was calculated by comparing each sample’s Ct value to the smallest Ct value obtained for that gradient. This ΔCt was then transformed into arbitrary reference units of mRNA content, normalized to RNaseP levels, and then divided by total mRNA content to represent the fraction of mRNA in that sample. Translational units (TU) were calculated by multiplying the mRNA content in each gradient sample with the number of ribosomes bound in that sample, and summing these over all gradient fractions. The Δ Translational Units value was obtained by subtracting the TU of each gene in Rps26 replete cells from the TU in Rps26 depleted cells. For each gradient fraction:
qPCR from total cellular RNA
Total RNA was isolated from cells growing in mid-log phase by hot-phenol extraction. After ethanol precipitation, 1ug of purified RNA was reverse transcribed and analyzed by qPCR as described above.
Depletion of Rps3 and Rps15
TET:Rps3 cells were generated by transforming yKK493 with pKK4015 and TET:Rps17 cells by transforming yKK489 with pKK3968. Both strains were grown as described for TET:Rps26.
Luciferase Assay
TET:Rps26 cells were transformed with plasmids encoding firefly luciferase preceded by an 10-adenosine upstream sequence and Renilla luciferase preceded by one of six sequences (listed in Table S1 and Figure S4a). Cells were then grown in selective media, depleted of Rps26 as described above and harvested in mid-log phase. Control experiments demonstrate that the copy numbers of these plasmids do not change upon dox addition (Figure S4c), ensuring that dox-dependent differences we see arise from differential translation, although we cannot exclude effects on mRNA transcription. For luciferase assays under stress, WT cells were grown for 4h under stress conditions (or in minimal media as controls) as detailed below and harvested in mid-log phase. Cells were lysed and luciferase activity was measured using the Promega Dual-Luciferase Reporter Assay System on a Perkin Elmer EnVision 2104 Multilabel Reader according the manufacturer’s protocol with assay volumes scaled down to 15%.
Stress response growth curves
For stress-tolerance tests, TET:Rps26 cells were grown and depleted of Rps26 as described above with either 0.2 or 0.1 μg/ml dox as indicated. Cells in mid-log phase were transferred to stress media (or control cultures) at OD 0.1 to test stress tolerance. Composition of stress-media was as follows: YPD + 1M NaCl, YPD + 10 mM caffeine. For high-pH stress, cells were grown in YPD + 100mM TAPS buffered to pH 7 (no stress) or pH 8.2 with NaOH. Cells were grown at 30°C with rapid shaking and doubling times were measured in the Bioscreen C Automated Microbiology Growth Curve Analysis System (Growth Curves USA) or with a Synergy 2 multi-mode microplate reader (BioTek).
Stress pathway activation
HOG pathway activation was tested by growing TET:Rps26 cells to mid log phase at 30°C in YPD with or without dox as above. Cells were then transferred to YPD containing 0, 100mM, 300mM or 500mM NaCl for 5 min, collected and analyzed by western blotting. Rim101 pathway activation was tested as described39 with TET:Rps26 cells transformed with pKK3678 (3HA-Rim101).
Ribosome purification from stressed cells
BY4741 yeast cells were grown to mid-log phase before seeding into different media. Cells were inoculated into stress media at a starting O.D. 0.7 and harvested after 4h. Ribosomes were purified as previously described66.
Antibodies
The phospho-p38 (D3F9) antibody from Cell Signaling Technologies was used to detect Hog1-P67. HA-tagged Rim101 was detected using the anti-HA antibody [HA.C5] from Abcam (ab18181). The anti-TAP antibody (CAB1001) was from ThermoFisher Scientific. Anti-Rps5 is from ProteinTech (16964-1-AP). Polyclonal antibodies were gifts from M. Seedorf (Rps3), G. Dieci (Rps8)68, L. Valášek (Rps0) and A. Link (Asc1). The monoclonal antibody against Tub1, developed by J. Frankel, was obtained from the Developmental Studies Hybridoma Bank, created by the NICHD of the NIH and maintained at The University of Iowa. The Fap7 and Rcl1 antibodies were raised against purified recombinant proteins by Josman, Llc. Rps10 and Rps26 antibodies were raised against peptides from each protein by New England Peptide. All four antibodies were tested against yeast lysates and either recombinant protein or purified 40S ribosomal subunits.
Statistics
Various statistical tests were used as appropriate, and as indicated in the respective figures. Unpaired, two-tailed, Student’s t-tests were used on small datasets. For larger datasets, or where there was not an assumption or normality, the nonparametric Kolmogorov-Smirnov test was used. For testing changes in growth curve rates from a hypothetical value of 1 (no change), the nonparametric two-tailed Wilcoxon Signed Rank test was used. Finally, a two-way ANOVA with Holm-Sidak correction for multiple comparisons was used for datasets in which two factors (ie, cell type and reporter construct) were tested.
Data Availability Statement
Sequencing data have been at NCBI-GEO under accession code GSE86203. Source data for Figures 1–5 and Figures S1-S6 are available with the paper online. Other data and custom scripts will be made available upon request. The methods of data collection can be found in the Life Sciences Reporting Summary.
Supplementary Material
Acknowledgments
We thank J. Cleveland, J. Joyce and members of the Karbstein lab for comments. This work was supported by National Institutes of Health awards R01-GM086451 (to K.K.) and F31-GM116406 (to MBF). HG was supported in part by the PGA National Women’s Cancer Awareness Fellowship. MBF also acknowledges support by the Richard & Helen DeVos Graduate Fellowship. The research of KK was supported in part by a Faculty Scholar grant from the Howard Hughes Medical Institute (55108536). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
AUTHOR CONTRIBUTIONS
Experiments were designed by MBF, HG and KK. MBF, HG and ELP performed and analyzed the experiments. EAW wrote software scripts to analyze sequencing data. The manuscript was written and edited by MBF, HG and KK, and all authors support the conclusions.
There are no competing financial interests.
References
- 1.Holcik M, Sonenberg N. Translational control in stress and apoptosis. Nat Rev Mol Cell Biol. 2005;6:318–27. doi: 10.1038/nrm1618. [DOI] [PubMed] [Google Scholar]
- 2.Silvera D, Formenti SC, Schneider RJ. Translational control in cancer. Nat Rev Cancer. 2010;10:254–66. doi: 10.1038/nrc2824. [DOI] [PubMed] [Google Scholar]
- 3.Van Der Kelen K, Beyaert R, Inzé D, De Veylder L. Translational control of eukaryotic gene expression. Crit Rev Biochem Mol Biol. 2009;44:143–68. doi: 10.1080/10409230902882090. [DOI] [PubMed] [Google Scholar]
- 4.Kong J, Lasko P. Translational control in cellular and developmental processes. Nat Rev Genet. 2012;13:383–394. doi: 10.1038/nrg3184. [DOI] [PubMed] [Google Scholar]
- 5.Ingolia NT, Ghaemmaghami S, Newman JRS, Weissman JS. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 2009;324:218–23. doi: 10.1126/science.1168978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ingolia NT, Lareau LF, Weissman JS. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell. 2011;147:789–802. doi: 10.1016/j.cell.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brar G, et al. High-resolution view of the yeast meiotic program revealed by ribosome profiling. Science. 2012;335:552–7. doi: 10.1126/science.1215110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zid BM, O’Shea EK. Promoter sequences direct cytoplasmic localization and translation of mRNAs during starvation in yeast. Nature. 2014;514:117–121. doi: 10.1038/nature13578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stumpf CR, Moreno MV, Olshen AB, Taylor BS, Ruggero D. The translational landscape of the mammalian cell cycle. Mol Cell. 2013;52:574–582. doi: 10.1016/j.molcel.2013.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kozak M. Point mutations close to the AUG initiator codon affect the efficiency of translation of rat preproinsulin in vivo. Nature. 1984;308:241–246. doi: 10.1038/308241a0. [DOI] [PubMed] [Google Scholar]
- 11.Kozak M. Point mutations define a sequence flanking the AUG initiator codon that modulates translation by eukaryotic ribosomes. Cell. 1986;44:283–92. doi: 10.1016/0092-8674(86)90762-2. [DOI] [PubMed] [Google Scholar]
- 12.Kozak M. At least six nucleotides preceding the AUG initiator codon enhance translation in mammalian cells. J Mol Biol. 1987;196:947–950. doi: 10.1016/0022-2836(87)90418-9. [DOI] [PubMed] [Google Scholar]
- 13.Dvir S, et al. Deciphering the rules by which 5′-UTR sequences affect protein expression in yeast. PNAS. 2013;110:E2792–801. doi: 10.1073/pnas.1222534110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim Y, et al. The immediate upstream region of the 5′-UTR from the AUG start codon has a pronounced effect on the translational efficiency in Arabidopsis thaliana. Nucleic Acids Res. 2014;42:485–498. doi: 10.1093/nar/gkt864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen SJ, Lin G, Chang KJ, Yeh LS, Wang CC. Translational efficiency of a non-AUG initiation codon is significantly affected by its sequence context in yeast. J Biol Chem. 2008;283:3173–80. doi: 10.1074/jbc.M706968200. [DOI] [PubMed] [Google Scholar]
- 16.Hussain T, et al. Structural Changes Enable Start Codon Recognition by the Eukaryotic Translation Initiation Complex. Cell. 2014;159:597–607. doi: 10.1016/j.cell.2014.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Farrar JE, et al. Ribosomal protein gene deletions in Diamond-Blackfan anemia. Blood. 2011;118:6943–6951. doi: 10.1182/blood-2011-08-375170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bolze A, et al. Ribosomal Protein SA Haploinsufficiency in Humans with Isolated Congenital Asplenia. Science. 2013;976 doi: 10.1126/science.1234864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ebert BL, et al. Identification of RPS14 as a 5q- syndrome gene by RNA interference screen. Nature. 2008;451:335–9. doi: 10.1038/nature06494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Burwick N, Shimamura A, Liu JM. Non-Diamond Blackfan anemia disorders of ribosome function: Shwachman Diamond syndrome and 5q- syndrome. Semin Hematol. 2011;48:136–43. doi: 10.1053/j.seminhematol.2011.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stumpf CR, Ruggero D. The cancerous translation apparatus. Curr Opin Genet Dev. 2011;21:474–83. doi: 10.1016/j.gde.2011.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Armistead J, Triggs-Raine B. Diverse diseases from a ubiquitous process: the ribosomopathy paradox. FEBS Lett. 2014;588:1491–500. doi: 10.1016/j.febslet.2014.03.024. [DOI] [PubMed] [Google Scholar]
- 23.Boria I, et al. The ribosomal basis of Diamond-Blackfan Anemia: mutation and database update. Hum Mutat. 2010;31:1269–79. doi: 10.1002/humu.21383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pisarev AV, Kolupaeva VG, Yusupov MM, Hellen CUT, Pestova TV. Ribosomal position and contacts of mRNA in eukaryotic translation initiation complexes. EMBO J. 2008;27:1609–21. doi: 10.1038/emboj.2008.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Koch B, et al. Yar1 protects the ribosomal protein Rps3 from aggregation. J Biol Chem. 2012;287:21806–15. doi: 10.1074/jbc.M112.365791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Arava Y, et al. Genome-wide analysis of mRNA translation profiles in Saccharomyces cerevisiae. Proc Natl Acad Sci U S A. 2003;100:3889–94. doi: 10.1073/pnas.0635171100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hurowitz EH, Brown PO. Genome-wide analysis of mRNA lengths in Saccharomyces cerevisiae. Genome Biol. 2003;5:R2. doi: 10.1186/gb-2003-5-1-r2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu H, et al. Characterization and evolution of 5′ and 3′ untranslated regions in eukaryotes. Gene. 2012;507:106–11. doi: 10.1016/j.gene.2012.07.034. [DOI] [PubMed] [Google Scholar]
- 29.Crooks G, Hon G, Chandonia J, Brenner S. WebLogo: a sequence logo generator. Genome Res. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kozak M. Compilation and analysis of sequences upstream from the translational start site in eukaryotic mRNAs. Nucleic Acids Res. 1984;12:857–72. doi: 10.1093/nar/12.2.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hamilton R, Watanabe CK, de Boer H, de Boer HA. Compilation and comparison of the sequence context around the AUG startcodons in Saccharomyces cerevisiae mRNAs. Nucleic Acids Res. 1987;15:3581–3593. doi: 10.1093/nar/15.8.3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nakagawa S, Niimura Y, Gojobori T, Tanaka H, Miura K. Diversity of preferred nucleotide sequences around the translation initiation codon in eukaryote genomes. Nucleic Acids Res. 2008;36:861–871. doi: 10.1093/nar/gkm1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kozak M. Possible role of flanking nucleotides in recognition of the AUG initiator codon by eukaryotic ribosomes. Nucleic Acids Res. 1981;9:5233–52. doi: 10.1093/nar/9.20.5233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ferreira-Cerca S, Pöll G, Gleizes PE, Tschochner H, Milkereit P. Roles of eukaryotic ribosomal proteins in maturation and transport of pre-18S rRNA and ribosome function. Mol Cell. 2005;20:263–75. doi: 10.1016/j.molcel.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 35.Ghalei H, et al. Hrr25/CK1δ-directed release of Ltv1 from pre-40S ribosomes is necessary for ribosome assembly and cell growth. J Cell Biol. 2015;208:745–759. doi: 10.1083/jcb.201409056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pertschy B, et al. RNA helicase Prp43 and its co-factor Pfa1 promote 20 to 18 S rRNA processing catalyzed by the endonuclease Nob1. J Biol Chem. 2009;284:35079–35091. doi: 10.1074/jbc.M109.040774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carmona-Saez P, Chagoyen M, Tirado F, Carazo JM, Pascual-Montano A. GENECODIS: a web-based tool for finding significant concurrent annotations in gene lists. Genome Biol. 2007;8:R3. doi: 10.1186/gb-2007-8-1-r3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hohmann S. Control of high osmolarity signalling in the yeast Saccharomyces cerevisiae. FEBS Lett. 2009;583:4025–4029. doi: 10.1016/j.febslet.2009.10.069. [DOI] [PubMed] [Google Scholar]
- 39.Hayashi M, Fukuzawa T, Sorimachi H, Maeda T. Constitutive Activation of the pH-Responsive Rim101 Pathway in Yeast Mutants Defective in Late Steps of the MVB/ESCRT Pathway. Mol Cell Biol. 2005;25:9478–9490. doi: 10.1128/MCB.25.21.9478-9490.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Levin D. Cell wall integrity signaling in Saccharomyces cerevisiae. Microbiol Mol Biol Rev. 2005;69:262–91. doi: 10.1128/MMBR.69.2.262-291.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Peña C, Schütz S, Fischer U, Chang Y, Panse VG. Prefabrication of a ribosomal protein subcomplex essential for eukaryotic ribosome formation. Elife. 2016;5 doi: 10.7554/eLife.21755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Peng WT, et al. A panoramic view of yeast noncoding RNA processing. Cell. 2003;113:919–933. doi: 10.1016/s0092-8674(03)00466-5. [DOI] [PubMed] [Google Scholar]
- 43.Schütz S, et al. A RanGTP-independent mechanism allows ribosomal protein nuclear import for ribosome assembly. Elife. 2014;3:e03473. doi: 10.7554/eLife.03473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee ASY, Kranzusch PJ, Cate JHD. eIF3 targets cell-proliferation messenger RNAs for translational activation or repression. Nature. 2015;522:111–114. doi: 10.1038/nature14267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Truitt ML, et al. Differential Requirements for eIF4E Dose in Normal Development and Cancer Article Differential Requirements for eIF4E Dose in Normal Development and Cancer. Cell. 2015;162:59–71. doi: 10.1016/j.cell.2015.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wolfe AL, et al. RNA G-quadruplexes cause eIF4A-dependent oncogene translation in cancer. Nature. 2014;513:65–70. doi: 10.1038/nature13485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dong XY, et al. SnoRNA U50 is a candidate tumor suppressor gene at 6q14.3 with a mutation associated with clinically significant prostate cancer. Hum Mol Genet. 2008;17:1031–1042. doi: 10.1093/hmg/ddm375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dong XY, et al. Implication of snoRNA U50 in human breast cancer. J Genet Genomics. 2009;36:447–454. doi: 10.1016/S1673-8527(08)60134-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kondrashov N, et al. Ribosome-mediated specificity in Hox mRNA translation and vertebrate tissue patterning. Cell. 2011;145:383–97. doi: 10.1016/j.cell.2011.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xue S, et al. RNA regulons in Hox 5′ UTRs confer ribosome specificity to gene regulation. Nature. 2014;517:33–38. doi: 10.1038/nature14010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Strunk BS, Novak MN, Young CL, Karbstein K. A Translation-Like Cycle Is a Quality Control Checkpoint for Maturing 40S Ribosome Subunits. Cell. 2012;150:111–21. doi: 10.1016/j.cell.2012.04.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lebaron S, et al. Proofreading of pre-40S ribosome maturation by a translation initiation factor and 60S subunits. Nat Struct Mol Biol. 2012;19:744–53. doi: 10.1038/nsmb.2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.LaRiviere FJ, Cole SE, Ferullo DJ, Moore MJ. A late-acting quality control process for mature eukaryotic rRNAs. Mol Cell. 2006;24:619–26. doi: 10.1016/j.molcel.2006.10.008. [DOI] [PubMed] [Google Scholar]
- 54.Cole S, LaRiviere F, Merrikh C, Moore M. A convergence of rRNA and mRNA quality control pathways revealed by mechanistic analysis of nonfunctional rRNA decay. Mol Cell. 2009;34:440–450. doi: 10.1016/j.molcel.2009.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gasch AP, et al. Genomic expression programs in the response of yeast cells to environmental changes. Mol Biol Cell. 2000;11:4241–4257. doi: 10.1091/mbc.11.12.4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Warner JR. The economics of ribosome biosynthesis in yeast. Trends Biochem Sci. 1999;24:437–440. doi: 10.1016/s0968-0004(99)01460-7. [DOI] [PubMed] [Google Scholar]
- 57.Viladevall L, et al. Characterization of the calcium-mediated response to alkaline stress in Saccharomyces cerevisiae. J Biol Chem. 2004;279:43614–43624. doi: 10.1074/jbc.M403606200. [DOI] [PubMed] [Google Scholar]
METHODS-ONLY REFERENCES
- 58.Longtine MS, et al. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast. 1998;14:953–61. doi: 10.1002/(SICI)1097-0061(199807)14:10<953::AID-YEA293>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 59.Strunk B, et al. Ribosome Assembly Factors Prevent Premature Translation Initiation by 40S Assembly Intermediates. Science. 2011;333:1449–1453. doi: 10.1126/science.1208245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Engel SR, et al. The reference genome sequence of Saccharomyces cerevisiae: then and now. G3 (Bethesda) 2014;4:389–98. doi: 10.1534/g3.113.008995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–11. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-Seq data with DESeq2. Genome Biol. 2014;15:1–21. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Csárdi G, Franks A, Choi DS, Airoldi EM, Drummond DA. Accounting for Experimental Noise Reveals That mRNA Levels, Amplified by Post-Transcriptional Processes, Largely Determine Steady-State Protein Levels in Yeast. PLOS Genet. 2015;11:e1005206. doi: 10.1371/journal.pgen.1005206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nagalakshmi U, et al. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science. 2008;320:1344–9. doi: 10.1126/science.1158441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang Y, et al. Precision and functional specificity in mRNA decay. Proc Natl Acad Sci U S A. 2002;99:5860–5865. doi: 10.1073/pnas.092538799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Acker MG, Kolitz SE, Mitchell SF, Nanda JS, Lorsch JR. Reconstitution of yeast translation initiation. Methods Enzymol. 2007;430:111–45. doi: 10.1016/S0076-6879(07)30006-2. [DOI] [PubMed] [Google Scholar]
- 67.Tanigawa M, Kihara A, Terashima M, Takahara T, Maeda T. Sphingolipids regulate the yeast high-osmolarity glycerol response pathway. Mol Cell Biol. 2012;32:2861–70. doi: 10.1128/MCB.06111-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dieci G, Bottarelli L, Ottonello S. A general procedure for the production of antibody reagents against eukaryotic ribosomal proteins. Protein Pept Lett. 2005;12:555–60. doi: 10.2174/0929866054395860. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequencing data have been at NCBI-GEO under accession code GSE86203. Source data for Figures 1–5 and Figures S1-S6 are available with the paper online. Other data and custom scripts will be made available upon request. The methods of data collection can be found in the Life Sciences Reporting Summary.