

Abstract
Aims
Rivaroxaban, a direct inhibitor of activated factor X (FXa), is the only new oral anticoagulant approved for secondary prevention after acute coronary syndrome. Our objective was to identify the possible molecular mechanisms of rivaroxaban that contribute to endothelial function.
Methods
Cell viability and growth of human umbilical vein endothelial cells (HUVEC) were registered. Gene expression studies comparing the effects of rivaroxaban and FXa were conducted by a selective RNA array and confirmed by protein quantification. Wound‐healing experiments on HUVEC, platelet adhesion, enzymatic activity, and cell‐based assays for fibrin formation were performed with rivaroxaban.
Results
Rivaroxaban (50 nM) only altered (>2 fold change) the expression of matrix metallopeptidase 2 and urokinase plasminogen activator (u‐PA), but counteracted the FXa (9 nM)‐induced upregulation of several pro‐inflammatory genes (P < 0.05) and FXa‐enhanced platelet adhesion over HUVEC. Rivaroxaban increased u‐PA protein expression in HUVEC supernatants and enhanced u‐PA activity (up to 4 IU ng−1 of u‐PA). Rivaroxaban (1 nM–1 μM) showed a significant and dose‐dependent positive effect on HUVEC growth that was inhibited by BC‐11‐hydroxibromide, an inhibitor of u‐PA. Healing properties after a wound on HUVEC cultures, and fibrinolytic properties were also shown by rivaroxaban. Both effects were reversed by BC‐11‐hydroxibromide.
Conclusions
Rivaroxaban enhanced viability, growth and migration of HUVEC, mainly by u‐PA activation and upregulation, which also participate in the rivaroxaban‐induced fibrinolytic activity at endothelial level. Rivaroxaban also protected from the pro‐inflammatory effects of FXa on HUVEC. Altogether may improve endothelial functionality and could contribute to the cardiovascular benefits of rivaroxaban.
Keywords: cell‐based fibrinolysis, endothelial functionality, human umbilical vein endothelial cells, rivaroxaban, wound healing
What is Already Known about this Subject
Adjusted indirect comparison suggested that FXa inhibitors (apixaban and rivaroxaban) were associated with lower coronary risk than dabigatran (a direct thrombin inhibitor).
Clinical trials with different oral FXa inhibitors after acute coronary syndrome showed a lack of consistency in the efficacy outcomes: rivaroxaban showed a significant reduction in cardiovascular death and major events compared with placebo (ATLAS‐2 trial), but not apixaban (APPRAISE‐2 trial).
What this Study Adds
Rivaroxaban shows endothelium protective and repairing properties mediated by urokinase plasminogen activator.
Rivaroxaban only modifies the expression of matrix metallopeptidase 2 and urokinase plasminogen activator, but counteracts the pro‐inflammatory effects of FXa.
By increasing urokinase plasminogen activator activity, rivaroxaban would potentiate the fibrinolytic pathway activity in endothelial cells.
Introduction
New oral anticoagulants are novel direct‐acting drugs that are selective for one specific coagulation factor, either thrombin (IIa) or activated coagulation factor X (FXa). In their main indication for prevention of thromboembolic recurrences in patients with venous thromboembolism and atrial fibrillation, they have shown greater advantages and fewer disadvantages compared to vitamin K antagonist drugs 1. Moreover, rivaroxaban was approved as an adjunct antithrombotic therapy in addition to dual antiplatelet therapy in selected patients with acute coronary syndrome 2, 3.
The effects of new oral anticoagulants on the risks of acute coronary events have been revised by meta‐analysis, revealing some discrepancies between them. Adjusted indirect comparison suggested that FXa inhibitors (apixaban and rivaroxaban) were associated with lower coronary risk than dabigatran (a direct thrombin inhibitor) 4. However, two trials have been made with oral FXa inhibitors after acute coronary syndrome, which showed a lack of consistency in the efficacy outcomes, with a significant reduction compared with placebo in the ATLAS‐2 trial (rivaroxaban) 5 but not in the APPRAISE‐2 trial (apixaban) 6, which was prematurely terminated because of an excess of bleeding and no evidence of benefit. This discrepancy is not well explained by a difference in the rates of bleeding, which was increased to a fairly similar extent in both trials.
The impact of an acute coronary syndrome event is not limited to the acute management phase, but also to an elevated risk of residual atherothrombotic events that commonly requires chronic management for months or even years. In order to improve secondary prevention post‐myocardial infarction, the use of rivaroxaban in this setting has been approved in Europe 2.
Direct FXa inhibition is an interesting and useful action mechanism as FXa is the convergence point of the extrinsic and intrinsic components of the coagulation cascade and has kinetics of activation relatively slow compared to the kinetics of thrombin. However, in the control of blood clotting, it is not only the circulating elements of the coagulation cascade that play a role. Vascular endothelial cells are able to produce and express tissue factor and other factors of the coagulation cascade such as factor V, as well as specific surface receptors for FXa 7 or thrombin 8, which facilitate their processing and activity. In the opposite direction, endothelial cells are the main producer of the tissue factor pathway inhibitor, a factor that binds to FXa to later link the tissue factor‐VIIa complex, deactivating it 9, 10. Moreover, heparan sulphate, expressed at the surface of the endothelium, helps to degrade FXa and thrombin 9. Similarly, the expression of t‐PA (plasminogen activator, tissue type) or plasminogen activator inhibitor‐1 11 decanted the equilibrium towards fibrinolysis or thrombus formation, respectively.
In summary, due to its location in the blood‐vessel interface and its functionality, endothelium plays an important role in coagulation and vascular function. Therefore, our objective was to study the effects of rivaroxaban on endothelial cell functionality as a possible way of contribution to the cardiovascular benefits of rivaroxaban.
Methods
Cell culture, growth and viability
Human umbilical vein endothelial cells (HUVEC) were isolated from freshly obtained human umbilical cords donated under informed consent from mothers and following the method previously described 12. All the procedures were approved by the Ethics Committee for Clinical Research at Galicia (Spain), according to the World Medical Association Declaration of Helsinki.
Cell viability of HUVEC was monitored in real time by an impedance‐based technology (RTCA SP Instrument; Roche Applied Science, Mannheim, Germany) under different conditions 13. After seeding in gelatin 0.2% (w/v) pre‐coated E‐plates and 4 h of serum starvation, viability and growth of HUVEC in the presence of rivaroxaban (Bayer), FXa (from bovine plasma, Sigma‐Aldrich), thrombin (Sigma‐Aldrich) or their combinations at different concentrations were registered continuously for 12 h. The system monitored electrical impedance across interdigitated micro‐electrodes integrated on the bottom of cell culture E‐plates. The measurements, called Cell Index (CI) dynamic values, were monitored during all the experiments. All registers were normalized to 1.000 arbitrary units of CI (a.u.) at the beginning of treatments and the ratio of CI between treatments and controls after 12 h of treatment were considered as the measurement of cell growth.
RNA isolation and cDNA synthesis
Post‐confluent HUVEC cultures were treated with rivaroxaban (50 nM), FXa (9 nM) or a combination of rivaroxaban and FXa for 4 h. Control experiments were performed in parallel. Total RNA was obtained with NucleoSpin® kit (Macherey‐Nagel, Düren, Germany) following the manufacturer's instructions and quantified with a spectrophotometer (Nanodrop ND‐1000, Thermo Fischer Scientific, Waltham, MA, USA). Samples of 0.5 μg of total RNA was subjected to genomic DNA elimination before they were reverse‐transcribed using the RT2 first strand cDNA synthesis kit (Qiagen Iberia S.L., Madrid, Spain).
Profiled polymerase chain reaction (PCR) array
cDNA was then amplified with the human Endothelial Cell Biology RT2 profiler PCR array (Qiagen Iberia S.L.). Real‐time PCR was performed on the Mx3005P QPCR System (Agilent Technologies, Santa Clara, CA, USA) and used SYBR green detection with the following thermal profile: 1 cycle: 95°C for 10 min, 40 cycles: 95°C for 15 s followed by 60°C for 1 min, 95°C for 1 min, 65°C for 2 min, and 65– 95°C at 2°C/min (dissociation curve). All data from the PCR was collected by the MXPro QPCR software (Agilent Technologies) and analysed by SA Bioscience's PCR Array Data Analysis template Excel (Qiagen Iberia S.L.).
The PCR array contained five separate housekeeping genes (B2M, beta‐2‐microglobulin; HPRT1, hypoxanthine phosphoribosyl transferase 1; RPL13A, ribosomal protein L13a; GAPDH, glyceraldehyde‐3‐phosphate dehydrogenase and ACTB, β‐actin) that were used for normalization of the sample data. Normalization to the housekeeping genes (HKG) was performed by calculating the ΔCt for each gene of interest (GOI) in the plate (Ct value of GOI‐Ct value of HKG). Any Ct value >35 was considered to be a negative call. Fold change was calculated by the ΔΔCt method: (2^(− ΔΔCt)) is the normalized gene expression (2^(− ΔCt)) in the test sample (treated cells) divided by the normalized gene expression (2^(− ΔCt)) in the control sample (control cells).
Protein expression of urokinase‐plasminogen activator (u‐PA)
HUVEC were cultured in flasks (25 cm2) until confluence. After serum and supplement starvation for at least 3 h, incubation with or without rivaroxaban (10–100 nM) for 4 h were made. After that, cell culture supernatants were removed, and cleaned of particulates by centrifugation and stored at −80°C until use. For u‐PA concentration measurement, supernatants were previously concentrated by ultrafiltration with Amicon® Ultra‐0.5 centrifugal filter devices (Merck, Darmstadt, Germany) of 3000 nominal molecular weight limit cutoff. A concentration factor of 10 was achieved by this method.
After removal of supernatants, cells were gently washed with cold phosphate buffer solution (PBS) and then covered and incubated with cell lysis buffer (Cell Lysis Buffer 1, R&D Systems) at room temperature for 1 h with gentle agitation. Cell lysates were removed from cell debris by centrifugation and stored at −80°C until use. u‐PA concentration was measured in cell lysate concentrated supernatants by quantitative sandwich enzyme immunoassay technique (DUPA00, R&D Systems Europe, Abingdon, UK) following the manufacturer's instructions. Data were expressed as pg ml−1.
Urokinase activity assay
HUVEC cultured until confluence in six‐multiwell plates were serum and supplements starved for at least 3 h prior to incubation with or without rivaroxaban (10–100 nM). After that, cell culture supernatants were removed, cleaned of particulates by centrifugation and stored at −80°C until use. Supernatant concentration was made as explained in the previous section.
Cells were gently washed with PBS and then detached with trypsin 0.25% (Gibco, Gaithersburg, MD, USA) and isolated by centrifugation (250g, 5 min, room temperature). Cell pellets were then lysed with 70 μl of ice‐cold cell lysis buffer provided by the u‐PA activity assay kit (ab174098, Abcam, Cambridge, UK). Cell debris was eliminated by centrifugation and cell lysates were stored at −80°C until use. u‐PA activity in supernatants and cell lysates was measured by a fluorometric kit (ab174098, Abcam) following the manufacturer's instructions. As a negative control of the assay and to test also the inhibitory capability of BC‐11‐hydroxibromide on u‐PA activity, this drug (1–100 μM) was incubated with a series of cell lysates from non‐treated HUVEC for 15 min prior to test the u‐PA activity. Data were expressed as IU ng−1 of u‐PA, being the content in u‐PA measured as described in the previous section.
Wound‐healing test
HUVEC were cultured on 24‐well plates until confluence. After serum and supplements starvation for 3 h, a wound was made in every well with a 1 ml tip and endothelial cells were treated with FXa (9 nM), rivaroxaban (50 nM) or a combination of both in complete medium. BC‐11‐hydroxibromide (10 or 50 μM) was used as u‐PA inhibitor in control experiments, and it was added also to the treatments of FXa, rivaroxaban or their combination. Healing was followed and registered by optical microscopy for 48 h. The percentage of healing over each initial wound size after 36 h was calculated for each treatment and used to compare them. Every treatment was made in duplicate.
Cell‐dependent fibrin formation and fibrinolysis assay
Treatments with rivaroxaban (1 nM–1 μM) for the 4 h were done in EBM‐2 medium. This medium was replaced by 200 μL EBM‐2 medium containing fibrinogen from human plasma (0.17 mg ml−1) and CaCl2 (17 mM). Immediately after the addition of thrombin (0.2 U ml−1), fibrin formation and fibrinolysis were detected as changes in the absorbance at 405 nm (A405) every 5 min for 1 h at 37°C in a microplate multi‐mode spectrophotometer (Synergy 2; BioTek Instruments Inc., Winooski, VT, USA). Thrombin reacts with fibrinogen to form fibrin, which is degraded by plasmin, an enzyme formed by the reaction between plasminogen and t‐ and/or u‐PA. So, total fibrin detected is the result of the equilibrium between its formation and fibrinolysis. In a series of experiments, BC‐11‐hydroxibromide (10 or 50 μM) were added to the medium of reaction. A series of experiments were made in the absence of cells to test the possible direct effects of rivaroxaban on thrombin‐dependent fibrin formation. To this end, the reaction of thrombin (0.2 U ml−1) with fibrinogen (0.17 mg ml−1) in the presence of CaCl2 (17 mM) in EBM‐2 medium was followed by the changes in A405 in the presence of rivaroxaban (1 nM–1 μM). Area under the curves (AUC) of A405 over time for each treatment were referred to their intra‐assay control experiments (considered as 100% of fibrin formation) and expressed as a percentage.
Platelet isolation and adhesion assays to HUVEC monolayers
Human buffy coats from healthy donors were kindly provided by Centro de Transfusiones de Galicia (Santiago de Compostela, Spain). From these samples, platelets were isolated by using the standard methods of Baenziger and Majerus 14. Briefly, samples were centrifuged at 250g for 12 min at room temperature. The upper phase was used as platelet‐rich plasma. Platelets were sedimented by centrifugation at 2200g for 10 min and washed twice with Hepes buffer (10 mM Hepes, 122 mM NaCl, 2.5 mM KCl, 0.5 NaH2PO4, 1.2 mM MgCl2, 10 mM NaHCO3 and 5 mM glucose, pH 7.4), containing 1 μg ml−1 PGE1 (Sigma‐Aldrich). Platelets were stained with 2.5 μM calcein‐acetoxymethyl ester (Invitrogen, Thermo Fischer Scientific) in the dark and 37°C for 15 min. After washing twice with Hepes buffer containing 0.3% bovine serum albumin (Sigma‐Aldrich), platelets were adjusted to a final concentration of 1 × 108 ml−1 in Hepes with 10% foetal bovine serum (Gibco, Gaithersburg, MD, USA). After this isolation and staining, platelets were added to HUVEC monolayers previously treated.
HUVEC for this assay were cultured in black 24‐multiwell plates with clear bottom. After confluence, HUVEC were starved of serum and supplements overnight and treated during 4 h with rivaroxaban (10–100 nM), FXa (9 nM) or a combination of the two. Tumour necrosis factor‐alpha (TNF‐α, 10 ng ml−1) was used as positive control of inflammatory response of HUVEC. After treatment, stained platelets (6 × 106) were incubated over HUVEC during 30 min at 37°C and total calcein fluorescence (Ex/Em: 485/525 nm) was measured in a fluorescence microplate reader (FLUOstar OPTIMA, BMG Labtech, Ortenberg, Germany) in relative fluorescence units. After twice gently washings with endothelial culture medium, the fluorescence of adhered platelets was measured and calculated as the percentage of adhesion for each treatment. Final results of the treatments were expressed as a percentage of the control treatments (considered as 100%).
Drugs and chemicals
Rivaroxaban was generously gifted by Bayer Hispania S.L. FXa, thrombin and fibrinogen were purchased from Sigma‐Aldrich and BC‐11‐hydroxibromide from Tocris Bioscience (R&D Systems, Minneapolis, MN, USA). Specific reagents for each technique were obtained as indicated in each subheading. All other chemicals needed for solutions were of the best quality available.
Data analysis
Data are expressed as mean ± SEM. Two‐group comparisons were performed by the two‐tailed Student's t‐test. Multiple comparisons were evaluated by ANOVA followed by Tukey's test. The RT2 Profiler PCR Array Data Analysis software calculates P‐values using a Student's t‐test (two tail distribution and equal variances between the two samples) based on the replicate 2^(−ΔCt) values for each gene in the treated group compared to the control group. Only individual pairwise comparisons are performed, not any ΔCt comparison across multiple groups at the same time. A P‐value of <0.05 was considered statistically significant. The statistical analyses were performed with SPSS (Statistical Package for the Social Sciences), version 17.0.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY 15, and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 16.
Results
Gene expression modification by rivaroxaban on endothelial cells
A selective analysis of HUVEC transcriptome was made for 84 genes. Changes of mRNA expression were analysed after the treatment with rivaroxaban (50 nM), FXa (9 nM) or their combination during 4 h. The concentrations of these drugs were chosen after a viability/toxicity analysis of rivaroxaban (1 nM–1 μM) and FXa (9–90 nM).
Rivaroxaban did not induce many changes in HUVEC phenotype in our experimental conditions. Major changes were considered for fold changes ≤0.70 or ≥2.00. For the case of rivaroxaban vs. control, major changes are shown in Figure 1A, the most significant being the downregulation of matrix metallopeptidase 2 (MMP2 gene) and the upregulation of u‐PA (PLAU gene). Full results are shown in Supplementary Table S1.
Figure 1.
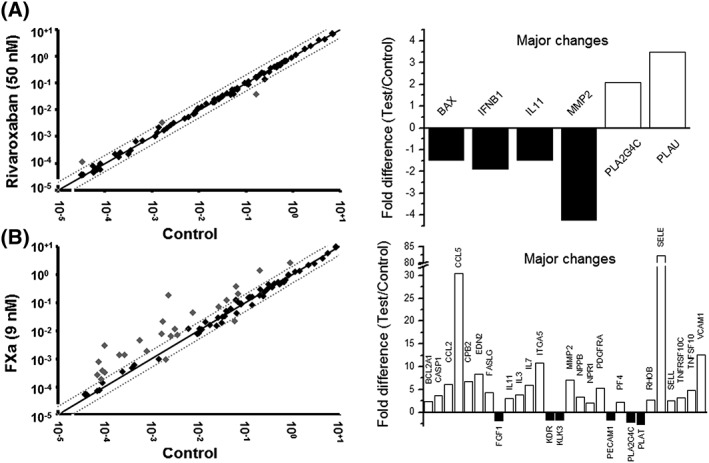
Profiled PCR array. Effects of rivaroxaban (A) or FXa (B) on the transcriptome of selected genes. HUVEC were incubated for 4 h with rivaroxaban, FXa or vehicle (control). Signal intensities of treated vs. control are plotted on left‐hand side, dashed lines and grey points indicate >twofold changes compared with controls. On the right‐hand side, transcripts showing major fold differences are represented in columns: black for downregulations and white for upregulations. Three different samples were analysed for each treatment. Official symbol of the genes as provided by HUGO Gene Nomenclature Committee (HGNC)
Conversely, FXa highly impacted gene expression of HUVEC, the overall result being a marked increase on pro‐inflammatory genes related with cell adhesion to endothelial surface, response to cell injury or extracellular matrix regulation (Figure 1B). Major changes observed were on the following genes: CCL2 (chemokine ligand 2), CCL5 (chemokine ligand 5), ITGA5 (fibronectin receptor), RHOB (ras homologue gene family, member B), SELE (selectin E), SELL (selectin L), TNFRSF10C (tumour necrosis factor receptor superfamily, member 10c), TNFSF10 (tumour necrosis factor superfamily, member 10), VCAM1 (vascular cell adhesion molecule 1), FASLG (fas ligand), IL11 (interleukin 11), IL3 (interleukin 3) and MMP2. Fold changes for all genes analysed are shown in Supplementary Table S2.
Interestingly, the addition of rivaroxaban (50 nM) to the treatment of FXa (9 nM) reverted most of the FXa‐induced gene expression changes, the most pronounced effects being on the genes of endothelin 2, selectin E, vascular cell adhesion molecule‐1 and RANTES (Figure 2). Results for the rest of the genes are shown in Supplementary Table S3.
Figure 2.
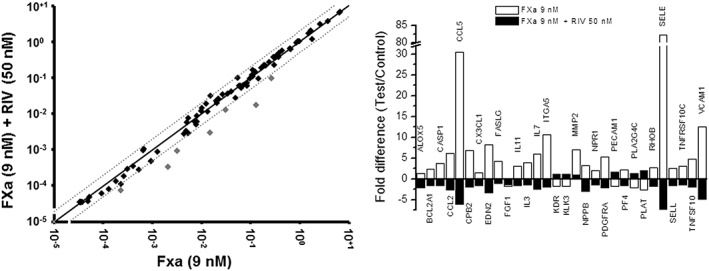
Rivaroxaban vs. FXa. Effects of rivaroxaban (RIV) + FXa vs. effects of FXa alone after 4 h of treatment. Analysis, data presentation on the left‐hand side and symbols for genes as described in Figure 1. On the right‐hand side, representation of the transcripts in columns showing major changes for both treatments: black for FXa + rivaroxaban, white for FXa. Three different samples were analysed for each treatment
Protein expression of u‐PA
Due to the effects shown by rivaroxaban on gene expression, the possible changes in the expression of u‐PA at protein level was checked. Surprisingly, rivaroxaban did not affect u‐PA protein expression in HUVEC lysates in comparison with control experiments (3803.0 ± 566.3 vs. 4059.5 ± 404.5 pg ml−1 for control and rivaroxaban 50 nM, respectively; P > 0.05), after 4 h of treatment (Figure 3A). On the contrary, rivaroxaban increased, in a concentration‐dependent manner, u‐PA protein expression in the HUVEC culture supernatant (36.2 ± 0.8 vs. 53.2 ± 6.2 pg ml−1 for control and rivaroxaban 50 nM, respectively; P < 0.05; Figure 3B). However, the concentrations of the u‐PA found in the supernatants were around two orders of magnitude lower than those found inside the cells.
Figure 3.
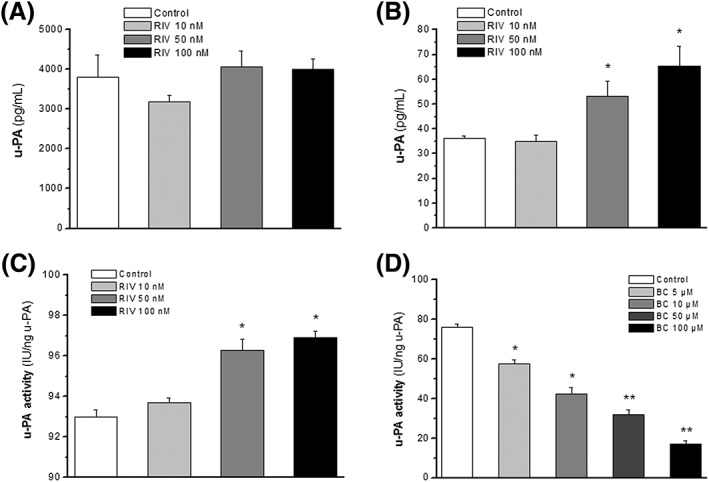
Expression and activity of u‐PA. Effects of rivaroxaban (RIV), at the concentrations indicated, on u‐PA protein expression in HUVEC lysates (A) or in cell culture supernatant (B). Enzyme activity of u‐PA in HUVEC lysates after the treatment with rivaroxaban during 4 h is represented in (C). The effect of direct incubation of BC‐11‐hydroxibromide (BC) in u‐PA activity from HUVEC lysates is shown in (D). Columns represent mean values ± SEM in vertical bars. Results are from at least five independent experiments. *P < 0.05 and **P < 0.01 with respect to their control experiments
Effects of rivaroxaban on u‐PA activity
The activity of u‐PA was measured in HUVEC lysates and in HUVEC culture supernatants after 4 h of treatment with rivaroxaban and compared with control treatments. In culture supernatants u‐PA activity was under the limit of detection of the assay kit, even after concentration of the samples. In HUVEC lysates, rivaroxaban (10–100 nM) increased u‐PA activity in a concentration‐dependent manner (93.0 ± 0.3 vs. 96.3 ± 0.5 IU ng−1 u‐PA for control and rivaroxaban 50 nM, respectively; P < 0.05; Figure 3C) with respect to control experiments.
The specificity of the assay was tested with BC‐11‐hydroxibromide, which was incubated in a series of non‐treated HUVEC lysates (as source of u‐PA) 15 min before the addition of the substrate to begin the enzymatic reaction. In these experiments it was confirmed that BC‐11‐hydroxibromide (1–100 μM) totally blocked the reaction in a concentration‐dependent manner (76.0 ± 1.9 vs. 32.0 ± 2.4 IU ng−1 u‐PA for control and BC‐11‐hydroxibromide 50 μM, respectively; P < 0.05; Figure 3D), demonstrating the specificity of the assay for u‐PA activity.
Endothelial cell viability and rivaroxaban‐enhanced endothelial cell growth
Cell viability was analysed in the presence or absence of rivaroxaban (1 nM–1 μM) for at least 12 h. Endothelial cell growth was linear and continuous in the control conditions during this time, but in the presence of rivaroxaban it was dose‐dependently enhanced, the maximum effect being at 50–100 nM (CI at 12 h were 1.66 ± 0.10 and 1.65 ± 0.23 for rivaroxaban 50 and 100 nM, respectively; P < 0.05 vs. control; Figure 4A). Following this result, 50 nM of rivaroxaban was the concentration used as effective and nontoxic in the following experiments.
Figure 4.
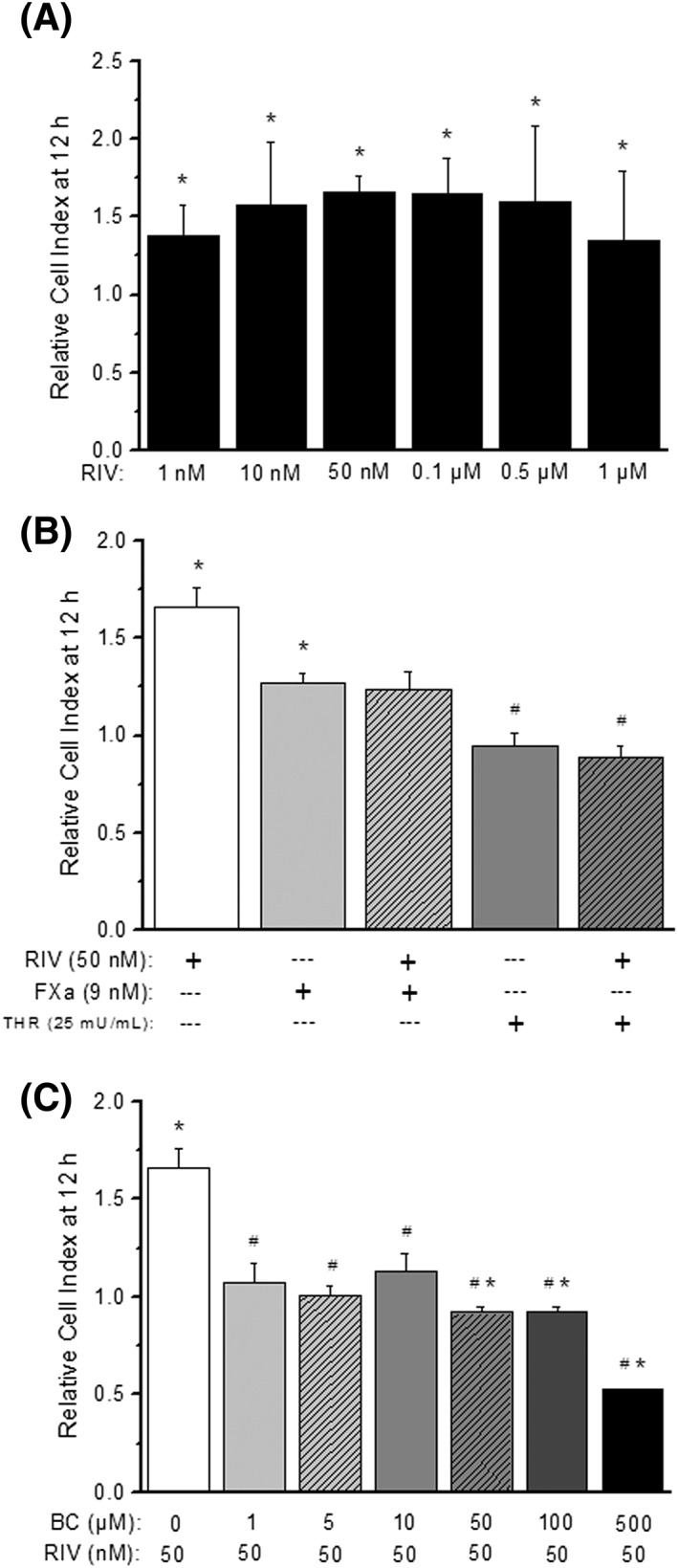
HUVEC growth. HUVEC growth in the presence of the indicated concentrations of rivaroxaban (A), FXa, thrombin or their combinations with rivaroxaban (B) or rivaroxaban in combination with BC‐11‐hydroxibromide (C). Data are expressed as the Cell Index ratio (treatment/control) after 12 h of treatment. Columns represent mean values of eight independent experiments ± SEM shown in vertical bars. *P < 0.05 vs. control. #P < 0.05 vs. rivaroxaban. Abbreviations: BC: BC‐11‐hydroxibromide, FXa: activated factor X, RIV: rivaroxaban, THR: thrombin
The effects of FXa (9–90 nM) or thrombin (25–100 mU ml−1) on HUVEC viability and growth were also tested. The data showed that 9 nM of FXa and 25 mU ml−1 of thrombin were nontoxic and the most growth‐promoting concentrations for these molecules (Supplementary Figure S1). FXa significantly enhanced HUVEC growth in comparison with control experiments (CI at 12 h = 1.27 ± 0.05, P < 0.05 vs. control; Figure 4B), whereas thrombin did not have promoting effects on endothelial cell growth (CI at 12 h = 0.95 ± 0.06, P > 0.05 vs. control; Figure 3B). FXa or thrombin were incubated in the presence of rivaroxaban (50 nM), resulting in no additive effects of the FXa inhibitor over them (Figure 4B). So, rivaroxaban neither modified the growth promoting effect of FXa alone nor stimulated cell growth in the presence of thrombin.
Rivaroxaban‐enhanced HUVEC growth was tested in the presence of different concentrations of BC‐11‐hydroxibromide (1–500 μM), a specific inhibitor of u‐PA. In the presence of this drug, rivaroxaban‐induced HUVEC growth was significantly inhibited in a concentration‐dependent manner, but normal cell viability of HUVEC was not affected by the combination of rivaroxaban 50 nM and BC‐11‐hydroxibromide at concentrations up to 50 μM of the latter (CI at 12 h were 1.66 ± 0.10 and 0.92 ± 0.03 for rivaroxaban 50 nM alone or with BC‐11‐hydroxibromide 50 μM, respectively; P < 0.05; Figure 4C). As a consequence, the experiments performed in the presence of rivaroxaban were made only with concentrations of BC‐11 hydrobromide of 10 or 50 μM at maximum. However, it is important to realize that all the concentrations of BC‐11‐hydroxibromide tested alone (1–500 μM) reduced significantly the normal growth of HUVEC on their own (Supplementary Figure S2).
Endothelial healing properties of rivaroxaban
Rivaroxaban (50 nM) enhanced healing after 36 h of a wound, with respect to control cells (86.4 ± 6.8% vs. 78.2 ± 6.9% of healing for rivaroxaban and control, respectively; P < 0.05; Figure 4), whereas the combination of rivaroxaban and FXa did not induce modification of normal healing. FXa (9 nM) alone did not modify healing significantly with respect to control.
BC‐11‐hydroxibromide significantly reversed the healing effects of rivaroxaban when treated in combination with it and also reduced the effects of FXa (86.4 ± 6.8 vs. 62.9 ± 5.0% for rivaroxaban 50 nM and its combination with BC‐11‐hydroxibromide 50 μM, respectively; P < 0.05; Figure 5). The effects of BC‐11‐hydroxibromide seemed to be dependent on its concentration as a lower concentration (10 μM) also reduced the healing both alone or in combination with rivaroxaban, but to a lesser extent that the concentration of 50 μM (Supplementary Figure S3).
Figure 5.
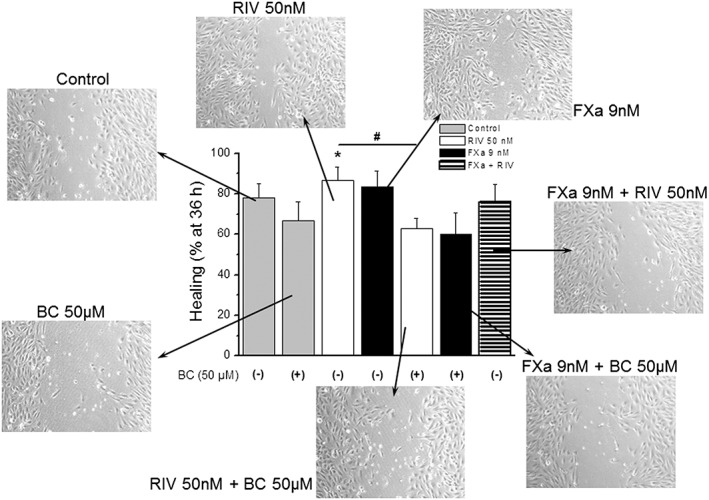
Wound healing. Percentage of healing after 36 h of a wound on HUVEC's cultures w/ or w/o rivaroxaban, FXa, BC‐hydroxibromide or their combinations. Columns represent mean values of five independent experiments ± SEM shown in vertical bars. *P < 0.05 vs. control. #P < 0.05 vs. rivaroxaban. Photographs show typical images of HUVEC cultures (100× magnification) after 36 h of each treatment. Abbreviations as in Figure 4
Endothelial cell‐dependent fibrin formation and fibrinolysis
Fibrin formation was assessed in the presence of HUVEC cultures pretreated or not with rivaroxaban (1 nM–1 μM). Control experiments showed a mean AUC value of 2.54 ± 0.19 after 60 min of reaction (this value was considered the 100% of response). Rivaroxaban, reduced fibrin formation and/or enhanced fibrinolysis significantly in a concentration‐dependent manner from the concentrations of 10 nM up to 1 μM (i.e. 72.2 ± 12.6% of fibrin formation for rivaroxaban 50 nM with respect to control; P < 0.05; Figure 6A).
Figure 6.
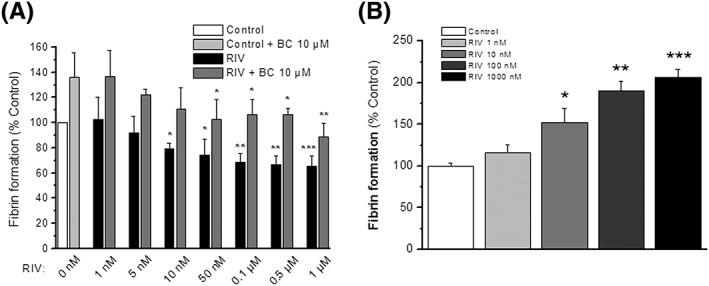
Fibrin formation and fibrinolysis. Percentages of fibrin formation in cell‐based experiments (A) after 4 h of treatment with (black columns) or without (white column) rivaroxaban at the concentrations indicated are shown. In a series of experiments BC‐11‐hydroxibromide (10 μM) was added after the treatment during the time of fibrin formation both to control (light grey column) and to treated cells (grey columns). In a series of experiments thrombin‐induced fibrin formation in the absence of cells was measured (B) in the presence of rivaroxaban (1–1000 nM). Columns represent mean values of the percentage of response ± SEM in vertical bars, referred to control experiments. Results are from at least four independent experiments by sextuplicate. *P < 0.05, **P < 0.01 and ***P < 0.001 with respect to their control experiments. Abbreviations as in Figure 4
In a series of experiments, BC‐11‐hydroxibromide (10 μM) was added during the time of reaction for fibrin production. BC‐11‐hydroxibromide, in comparison with non‐inhibited reactions, significantly reduced fibrinolysis, so it enhanced fibrin detection both in control (136.1 ± 19.3% of fibrin formation in the presence of BC‐11‐hydroxibromide; P < 0.05 with respect to control) and in treated HUVEC cultures with different concentrations of rivaroxaban (Figure 6A). However, the fibrinolytic effect of rivaroxaban treatment persists even in the presence of BC‐11‐hydroxibromide because cells treated with rivaroxaban concentrations of 50 nM or higher showed less fibrin detection than non‐treated cells (136.1 ± 19.3 vs. 102.5 ± 15.6% fibrin formation for BC‐11‐hydroxibromide 10 μM and its combination with rivaroxaban 50 nM, respectively; P < 0.05; Figure 6A).
Moreover, the possible effects of rivaroxaban on thrombin‐dependent fibrin formation were tested in experiments without cells. In these conditions, rivaroxaban (1 nM–1 μM) not only did not inhibit fibrin formation, but it enhances the reaction in a concentration‐dependent manner for the range of 10 nM–1 μM (i.e. 166.2 ± 12.3% of fibrin formation for rivaroxaban 50 nM; P < 0.05 with respect to control; Figure 6B).
Platelet adhesion to HUVEC monolayers
The possible translation of the effects of rivaroxaban to functional adhesion of platelets to endothelium was tested after 4 h of treatment of HUVEC with rivaroxaban (10–100 nM), FXa (9 nM) or their combination. In the conditions of our experiments, the percentage of platelet adhesion was 12.9 ± 0.6% for non‐treated cells (this was considered 100% of control response), and TNF‐α (10 ng ml−1) increased this response to 106.7 ± 1.8% (n = 5; P < 0.05 with respect to control). Rivaroxaban (10–100 nM) alone did not enhance, but even reduced platelet adhesion to endothelial cells. However, FXa (9 nM) significantly increased platelet adhesion to the same extent as TNF‐α (110.1 ± 4.5%; P < 0.05 with respect to control; Figure 7). Importantly, rivaroxaban inhibited the FXa‐induced platelet adhesion to HUVEC in a concentration‐dependent manner (Figure 7), confirming the inhibitory effects of this drug on FXa‐induced pro‐inflammatory effects on endothelial cells.
Figure 7.
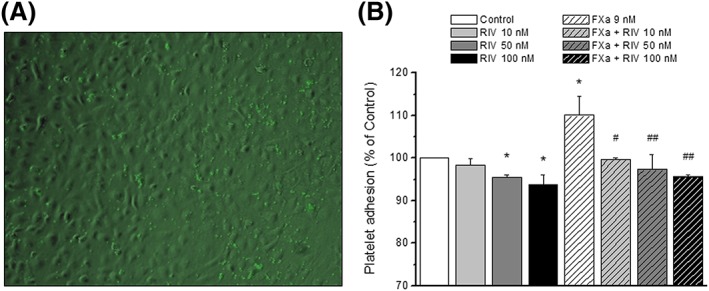
Platelet adhesion to treated HUVEC monolayers. HUVEC were treated during 4 h with rivaroxaban (RIV; 10–100 nM), FXa (9 nM) or their combinations and compared with control (no treatment). (A) Representative merged image of platelet stained with calcein‐AM over a transmitted image of a HUVEC monolayer. (B) mean percentage of response with respect to control (columns) ± SEM (vertical bars) of a minimum of three independent experiments. *P < 0.05 with respect to control; #P < 0.05 and ##P < 0.01 with respect to FXa
Discussion
In this work, for the first time, the effects of rivaroxaban on the functionality of endothelial cells as a possible contributor to the global cardiovascular benefits of the drug have been studied. A significant positive effect on HUVEC's growth and an enhancement of healing after wound was observed by rivaroxaban. A profiled gene expression analysis revealed that rivaroxaban modified the expression of matrix metallopeptidase 2 and u‐PA from up to 84 genes analysed, whereas it counteracted the pro‐inflammatory upregulation induced by FXa on HUVEC, and the effect that could be functionally tested in FXa‐induced platelet adhesion to endothelial cells. Rivaroxaban‐induced upregulation of u‐PA protein was confirmed in HUVEC supernatant and u‐PA activity was also significantly increased by rivaroxaban. This enhanced activity was demonstrated to participate in important endothelial functions. The effects of rivaroxaban on HUVEC growth and healing seem to be mediated by u‐PA activity, as the inhibition of this enzyme with BC‐11‐hydroxibromide almost inhibited its effects. Importantly, the upregulation and enhanced activity of u‐PA by rivaroxaban also determined the fibrinolytic properties showed by the anticoagulant drug in our cell‐based assays. Therefore, rivaroxaban showed endothelium repairing properties, an effect probably mediated by u‐PA upregulation and activation, which also contributes to the rivaroxaban‐enhanced fibrinolytic activity on endothelial cells.
Rivaroxaban showed a concentration‐dependent positive effect on cell viability and growth in comparison with its vehicle. Although to a lesser extent, FXa also showed a positive effect on cell growth, but its combination with rivaroxaban did not show any additive result of both actions. When added to HUVEC in culture, FXa has been shown to be a potent mitogen, stimulating an increase in cell number 17, although other authors reported no direct effect of FXa on endothelial cell proliferation 18. FXa induces endothelial cell activation via a cascade of receptor activation involving docking to effector cell protease receptor‐1 and local proteolytic cleavage of protease‐activated receptor‐2 19. So, the activities induced by FXa on HUVEC are dependent on its catalytic activity and could be inhibited by direct FXa inhibitors 17. Therefore, the combination of FXa and rivaroxaban results in the blockage of the former and what we observed in HUVEC is the residual mitogenic effect of rivaroxaban.
Little is known about the proliferative action of rivaroxaban on endothelial cells. It promoted vessel formation in diabetic mice and improved endothelial progenitor cell function under hyperglycaemic conditions, probably by the upregulation of vascular endothelial growth factor 20. However, our data suggest another mechanism involving the upregulation of u‐PA. An increase in u‐PA protein was found in HUVEC culture supernatants after treatment with rivaroxaban in our experiments. The transient expression of this enzyme in endothelial cells has been demonstrated in different types of physiological angiogenesis and even in wound healing‐associated angiogenesis 21. u‐PA plays a pivotal role in extracellular proteolysis and is thought to be critically involved in the modulation of angiogenesis via the interaction with its u‐PA receptor 22. However, in our experimental conditions, the increase in u‐PA protein in the cell supernatant seems to be insufficient to explain the effects of rivaroxaban on HUVEC. u‐PA concentration in the supernatants was very low in comparison with the u‐PA content in the cells, where we did not find a marked increase in protein expression. Probably, the most relevant effect of rivaroxaban was the increase in u‐PA activity observed in HUVEC, where most of the u‐PA protein was found. The participation of u‐PA activity in the proliferative effect of rivaroxaban was demonstrated by its blockage with BC‐11‐hydroxibromide, a specific inhibitor of u‐PA. This reveals a new mechanistic pathway for rivaroxaban at endothelial level. It has been demonstrated in endothelial cells that u‐PA protects against apoptosis by transcriptional upregulation and partially by mRNA stabilization of inhibition of apoptosis proteins, most prominently the X‐linked inhibitor of apoptosis protein. u‐PA‐induced cell survival involved phosphorylation of p21‐activated kinase 1 and the IkappaB kinase alpha that leads to nuclear factor kappaB p52 activation 23. These mechanisms could be mediating the observed response of rivaroxaban on HUVEC's growth.
Despite its potent mitogenic action, rivaroxaban showed no dramatic alteration of gene expression on HUVEC at the concentration of maximum proliferative effect, which is a desired circumstance for drugs, in order to minimize their possible side effects. Major changes were observed for the downregulation of matrix metallopeptidase 2 and the upregulation of u‐PA and PLA2G4C gene (phospholipase A2, group IVC). Interestingly, u‐PA and matrix metallopeptidase 2 are the main proteins involved in the progression of two‐dimensional co‐culture of bone marrow stromal cells and endothelial cells 24, which can help to explain our positive results in HUVEC proliferation and wound healing. In this study we had the opportunity to show not only the importance of the changes on u‐PA expression but also the relevant functional consequences of enhancing u‐PA activity, as we have discussed for the proliferative action on HUVEC and as we show below for wound‐healing properties and fibrinolysis.
Previous studies had reported the effects of FXa in this regard. In a concentration‐dependent manner, it evokes a pro‐inflammatory response in endothelial cells 25. It also induced the expression of tissue factor and the release of t‐PA and plasminogen activator inhibitor‐1 without affecting u‐PA expression 17. These results agree with our own data. As all these effects have been suggested to be dependent on its catalytic activity 17, 25, it would be expected that rivaroxaban could revert them, as was confirmed by our observations in HUVEC. The experiments of platelet adhesion to HUVEC functionally confirmed the pro‐inflammatory effect of FXa on endothelial cells and the inhibitory effects of rivaroxaban on FXa‐induced platelet adhesion to endothelial cells. The most probable explanation for this result is that rivaroxaban inhibited FXa‐induced upregulation of important adhesion molecules such as αV integrin, vascular cell adhesion molecule‐1, E‐selectin or L‐selectin, some of which are implicated in platelet–endothelial interaction 26, 27. Anti‐inflammatory activity of rivaroxaban has been demonstrated in animal models of ischaemia and reperfusion injury 28. Together with anti‐fibrotic and cardio‐protective activity as pleiotropic effects, it contributes to secondary prevention of cardiovascular events after ischaemia/reperfusion injury.
Specific experiments were designed to test the mitogenic and pro‐migratory ability of rivaroxaban on wound healing and its possible dependence on u‐PA activity. The results clearly showed that rivaroxaban is a potent repairing drug for endothelial wounds in vitro, promoting cell migration to the wound and faster healing than control cultures. Also, the implication of u‐PA on these effects was demonstrated by the inhibition with BC‐11‐hydroxibromide. These results agree with previous suggestions of u‐PA participating in wound healing‐associated processes 21 and in the vascular endothelial growth factor‐induced migration of endothelial cells 29. In fact, u‐PA, but not t‐PA has been involved in neointima formation and neointimal cell accumulation in arteries 30.
It is also important that u‐PA is a serine protease for plasminogen, the inactive form of plasmin. Activation of plasmin triggers a proteolysis cascade that participates in thrombolysis by increasing fibrinolysis. We studied this role in clot degradation in our in vitro model of cell‐based fibrin formation and fibrinolysis. Fibrin formation was induced in the presence of HUVEC cultures previously treated with rivaroxaban. If our hypothesis was correct, rivaroxaban should upregulate u‐PA in cells during the treatment and, most important, enhance the u‐PA protease activity. This should be reflected in an increase in fibrinolysis and, therefore, a reduction in fibrin detection. The experiments probed that, and the incubation with low concentrations of BC‐11‐hydroxibromide during fibrin formation slowed down the reaction, suggesting the implication of u‐PA in this response. Conversely, rivaroxaban did not show direct inhibitory effects on thrombin‐dependent fibrin formation in the experiments in the absence of HUVEC. In fact, it enhances thrombin‐induced fibrin formation, whereas in the presence of HUVEC the effect was the opposite. So, even having a stimulatory effect on direct fibrin formation by thrombin, the net effect of rivaroxaban on HUVEC has a fibrinolytic value. This means that via u‐PA for the activation of plasmin, that degrades fibrin, rivaroxaban could act as a fibrinolytic agent. This finding could have very important clinical outcomes, as a crucial role for t‐PA and u‐PA in the fibrinolytic system has been suggested. In animal models, combined t‐PA and u‐PA deficiency suffer extensive spontaneous fibrin deposition 31. It should also be taken into account that the response of distinct vascular beds to inflammatory stimuli may differentially regulate this fibrinolytic pathway 32. Therefore, our results demonstrate that rivaroxaban enhances u‐PA activity at endothelial cells and, as we have observed, this has important consequences in endothelial functions as growth, healing and fibrinolytic activity. Whether the effect of rivaroxaban on u‐PA is direct or indirect is impossible to say without an exhaustive analysis of the mechanistic ways that rivaroxaban could trigger in endothelial cells and without a molecular study of the possible direct interaction and activation of u‐PA by rivaroxaban. These analyses exceed the objective of the present work. However, altogether, our results suggest that, despite a scarce effect on gene expression modification, rivaroxaban markedly increased u‐PA activity, and the functional changes observed in endothelial cells could be related with this enhanced u‐PA activity. Even more, this activation could also explain the upregulation of u‐PA expression and its increase in secretion/release to the extracellular space, as u‐PA is able to induce its own expression via the receptor of u‐PA in endothelial cells 33.
In conclusion, our results showed endothelium protective and repairing properties of rivaroxaban, as this drug clearly enhanced viability, growth and wound healing on HUVEC. These effects are probably mediated by u‐PA upregulation and its enhanced functional activity. Rivaroxaban also counteracts the pro‐inflammatory effect at endothelial cell level of FXa, probably by its direct inhibition, an effect that showed a functional implication in the inhibition of FXa‐induced platelet adhesion to endothelial cells. Moreover, rivaroxaban via u‐PA activation seems to increase the fibrinolytic pathway activity in endothelial cells, which together with its known anticoagulant activity would help to create a global antithrombotic environment inside the vessels. Altogether, this may contribute to the cardiovascular benefits of rivaroxaban at functional endothelial level.
Competing Interests
The work of E.A. was supported by Bayer Hispania S.L. The other authors have no competing interests to declare.
Thanks to Centro de Transfusiones de Galicia (Santiago de Compostela, Spain) for providing buffy coats. This study was supported by Bayer Hispania S.L.
Contributors
E.A., B.P.D., S.R.R. and J.R.G.J. made substantial contributions to the conception or design of the work; or the acquisition, analysis or interpretation of data for the work. E.A., B.P.D., S.R.R. and J.R.G.J. drafted the work or revised it critically for important intellectual content. All authors gave their final approval of the version to be published. All authors also agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Supporting information
Figure S1 Effects of activated factor X (FXa) and thrombin on HUVEC's growth. HUVEC growth in the presence of the indicated concentrations of FXa or thrombin are shown. Data expressed as the CI ratio (treatment/control) after 12 h of treatment. Columns represent mean values of at least three independent experiments ± SEM shown in vertical bars. *P < 0.05 with respect to control
Figure S2 Effects of BC‐11‐hydrobromide on HUVEC's growth. HUVEC growth in the presence of the indicated concentrations of BC‐11‐hydroxibromide. Data expressed as the CI ratio (treatment/control) after 12 h of treatment. Columns represent mean values of at least three independent experiments ± SEM shown in vertical bars. *P < 0.05 with respect to control
Figure S3 Effects of BC‐11‐hydrobromide on wound and healing assays. Percentage of healing after 36 h of a wound in HUVEC's cultures in the presence or absence of BC‐11‐hydroxibromide or rivaroxaban in the concentrations indicated, or the combination of both. Data expressed as % of healing after 36 h of a wound. Columns represent mean values of at least three independent experiments ± SEM shown in vertical bars. *P < 0.05 with respect to control. #P < 0.05. Abbreviations: BC: BC‐11‐hydroxibromide, RIV: rivaroxaban
Table S1 Endothelial cell biology PCR array. Fold changes are shown as ratios of gene expression in rivaroxaban‐treated vs. control cells
Table S2 Endothelial cell biology PCR array. Fold changes are shown as ratios of gene expression in activated factor X‐treated vs. control cells
Table S3 Endothelial cell biology PCR array. Fold changes are shown as ratios of gene expression in activated factor X + rivaroxaban‐treated vs. activated factor X‐treated cells
Álvarez, E. , Paradela‐Dobarro, B. , Raposeiras‐Roubín, S. , and González‐Juanatey, J. R. (2018) Protective, repairing and fibrinolytic effects of rivaroxaban on vascular endothelium. Br J Clin Pharmacol, 84: 280–291. doi: 10.1111/bcp.13440.
References
- 1. Sardar P, Chatterjee S, Lavie CJ, Giri JS, Ghosh J, Mukherjee D, et al Risk of major bleeding in different indications for new oral anticoagulants: insights from a meta‐analysis of approved dosages from 50 randomized trials. Int J Cardiol 2015; 179: 279–287. [DOI] [PubMed] [Google Scholar]
- 2. Atar D, Bode C, Stuerzenbecher A, Verheugt FW. Anticoagulants for secondary prevention after acute myocardial infarction: lessons from the past decade. Fundam Clin Pharmacol 2014; 28: 353–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cohen M, Iyer D. The "dual‐pathway" strategy after acute coronary syndrome: rivaroxaban and antiplatelet agents in the ATLAS ACS 2‐TIMI 51 trial. Cardiovasc Ther 2014; 32: 224–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Loke YK, Pradhan S, Yeong JK, Kwok CS. Comparative coronary risks of apixaban, rivaroxaban and dabigatran: a meta‐analysis and adjusted indirect comparison. Br J Clin Pharmacol 2014; 78: 707–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mega JL, Braunwald E, Wiviott SD, Bassand JP, Bhatt DL, Bode C, et al Rivaroxaban in patients with a recent acute coronary syndrome. N Engl J Med 2012; 366: 9–19. [DOI] [PubMed] [Google Scholar]
- 6. Alexander JH, Lopes RD, James S, Kilaru R, He Y, Mohan P, et al Apixaban with antiplatelet therapy after acute coronary syndrome. N Engl J Med 2011; 365: 699–708. [DOI] [PubMed] [Google Scholar]
- 7. Bono F, Herault JP, Avril C, Schaeffer P, Lormeau JC, Herbert JM. Human umbilical vein endothelial cells express high affinity receptors for factor Xa. J Cell Physiol 1997; 172: 36–43. [DOI] [PubMed] [Google Scholar]
- 8. Byzova TV, Plow EF. Activation of alphaVbeta3 on vascular cells controls recognition of prothrombin. J Cell Biol 1998; 143: 2081–2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bombeli T, Mueller M, Haeberli A. Anticoagulant properties of the vascular endothelium. Thromb Haemost 1997; 77: 408–423. [PubMed] [Google Scholar]
- 10. Broze GJ Jr. Tissue factor pathway inhibitor. Thromb Haemost 1995; 74: 90–93. [PubMed] [Google Scholar]
- 11. Schleef RR, Loskutoff DJ. Fibrinolytic system of vascular endothelial cells: role of plasminogen activator inhibitors. Haemostasis 1988; 18: 328–341. [DOI] [PubMed] [Google Scholar]
- 12. Paradela‐Dobarro B, Rodiño‐Janeiro BK, Alonso J, Raposeiras‐Roubín S, González‐Peteiro M, González‐Juanatey JR, et al Key structural and functional differences between early and advanced glycation products. J Mol Endocrinol 2016; 56: 23–37. [DOI] [PubMed] [Google Scholar]
- 13. Fandino‐Vaquero R, Fernandez‐Trasancos A, Alvarez E, Ahmad S, Batista‐Oliveira AL, Adrio B, et al Orosomucoid secretion levels by epicardial adipose tissue as possible indicator of endothelial dysfunction in diabetes mellitus or inflammation in coronary artery disease. Atherosclerosis 2014; 235: 281–288. [DOI] [PubMed] [Google Scholar]
- 14. Baenziger NL, Majerus PW. Isolation of human platelets and platelet surface membranes. Methods Enzymol 1974; 31: 149–155. [DOI] [PubMed] [Google Scholar]
- 15. Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH, et al The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 2016; 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE, et al The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 2015; 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Herault JP, Bono F, Avril C, Schaeffer P, Herbert JM. Activation of human vascular endothelial cells by factor Xa: effect of specific inhibitors. Biochem Pharmacol 1999; 57: 603–610. [DOI] [PubMed] [Google Scholar]
- 18. Borensztajn K, Aberson H, Peppelenbosch MP, Spek CA. FXa‐induced intracellular signaling links coagulation to neoangiogenesis: potential implications for fibrosis. Biochim Biophys Acta 2009; 1793: 798–805. [DOI] [PubMed] [Google Scholar]
- 19. Bono F, Schaeffer P, Herault JP, Michaux C, Nestor AL, Guillemot JC, et al Factor Xa activates endothelial cells by a receptor cascade between EPR‐1 and PAR‐2. Arterioscler Thromb Vasc Biol 2000; 20: E107–E112. [DOI] [PubMed] [Google Scholar]
- 20. Wu TC, Chan JS, Lee CY, Leu HB, Huang PH, Chen JS, et al Rivaroxaban, a factor Xa inhibitor, improves neovascularization in the ischemic hindlimb of streptozotocin‐induced diabetic mice. Cardiovasc Diabetol 2015; 14: 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bacharach E, Itin A, Keshet E. In vivo patterns of expression of urokinase and its inhibitor PAI‐1 suggest a concerted role in regulating physiological angiogenesis. Proc Natl Acad Sci U S A 1992; 89: 10686–10690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fibbi G, Caldini R, Chevanne M, Pucci M, Schiavone N, Morbidelli L, et al Urokinase‐dependent angiogenesis in vitro and diacylglycerol production are blocked by antisense oligonucleotides against the urokinase receptor. Lab Invest 1998; 78: 1109–1119. [PubMed] [Google Scholar]
- 23. Prager GW, Mihaly J, Brunner PM, Koshelnick Y, Hoyer‐Hansen G, Binder BR. Urokinase mediates endothelial cell survival via induction of the X‐linked inhibitor of apoptosis protein. Blood 2009; 113: 1383–1390. [DOI] [PubMed] [Google Scholar]
- 24. Li H, Daculsi R, Bareille R, Bourget C, Amedee J. uPA and MMP‐2 were involved in self‐assembled network formation in a two dimensional co‐culture model of bone marrow stromal cells and endothelial cells. J Cell Biochem 2013; 114: 650–657. [DOI] [PubMed] [Google Scholar]
- 25. Senden NH, Jeunhomme TM, Heemskerk JW, Wagenvoord R, van't Veer C, Hemker HC, et al Factor Xa induces cytokine production and expression of adhesion molecules by human umbilical vein endothelial cells. J Immunol 1998; 161: 4318–4324. [PubMed] [Google Scholar]
- 26. Bombeli T, Schwartz BR, Harlan JM. Adhesion of activated platelets to endothelial cells: evidence for a GPIIbIIIa‐dependent bridging mechanism and novel roles for endothelial intercellular adhesion molecule 1 (ICAM‐1), alphavbeta3 integrin, and GPIbalpha. J Exp Med 1998; 187: 329–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gawaz M, Neumann FJ, Dickfeld T, Reininger A, Adelsberger H, Gebhardt A, et al Vitronectin receptor (alpha(v)beta3) mediates platelet adhesion to the luminal aspect of endothelial cells: implications for reperfusion in acute myocardial infarction. Circulation 1997; 96: 1809–1818. [DOI] [PubMed] [Google Scholar]
- 28. Goto M, Miura S, Suematsu Y, Idemoto Y, Takata K, Imaizumi S, et al Rivaroxaban, a factor Xa inhibitor, induces the secondary prevention of cardiovascular events after myocardial ischemia reperfusion injury in mice. Int J Cardiol 2016; 220: 602–607. [DOI] [PubMed] [Google Scholar]
- 29. Prager GW, Breuss JM, Steurer S, Olcaydu D, Mihaly J, Brunner PM, et al Vascular endothelial growth factor receptor‐2‐induced initial endothelial cell migration depends on the presence of the urokinase receptor. Circ Res 2004; 94: 1562–1570. [DOI] [PubMed] [Google Scholar]
- 30. Carmeliet P, Moons L, Herbert JM, Crawley J, Lupu F, Lijnen R, et al Urokinase but not tissue plasminogen activator mediates arterial neointima formation in mice. Circ Res 1997; 81: 829–839. [DOI] [PubMed] [Google Scholar]
- 31. Carmeliet P, Schoonjans L, Kieckens L, Ream B, Degen J, Bronson R, et al Physiological consequences of loss of plasminogen activator gene function in mice. Nature 1994; 368: 419–424. [DOI] [PubMed] [Google Scholar]
- 32. Kumar NG, Clark A, Roztocil E, Caliste X, Gillespie DL, Cullen JP. Fibrinolytic activity of endothelial cells from different venous beds. J Surg Res 2015; 194: 297–303. [DOI] [PubMed] [Google Scholar]
- 33. Li C, Zhang J, Jiang Y, Gurewich V, Chen Y, Liu JN. Urokinase‐type plasminogen activator up‐regulates its own expression by endothelial cells and monocytes via the u‐PAR pathway. Thromb Res 2001; 103: 221–232. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Effects of activated factor X (FXa) and thrombin on HUVEC's growth. HUVEC growth in the presence of the indicated concentrations of FXa or thrombin are shown. Data expressed as the CI ratio (treatment/control) after 12 h of treatment. Columns represent mean values of at least three independent experiments ± SEM shown in vertical bars. *P < 0.05 with respect to control
Figure S2 Effects of BC‐11‐hydrobromide on HUVEC's growth. HUVEC growth in the presence of the indicated concentrations of BC‐11‐hydroxibromide. Data expressed as the CI ratio (treatment/control) after 12 h of treatment. Columns represent mean values of at least three independent experiments ± SEM shown in vertical bars. *P < 0.05 with respect to control
Figure S3 Effects of BC‐11‐hydrobromide on wound and healing assays. Percentage of healing after 36 h of a wound in HUVEC's cultures in the presence or absence of BC‐11‐hydroxibromide or rivaroxaban in the concentrations indicated, or the combination of both. Data expressed as % of healing after 36 h of a wound. Columns represent mean values of at least three independent experiments ± SEM shown in vertical bars. *P < 0.05 with respect to control. #P < 0.05. Abbreviations: BC: BC‐11‐hydroxibromide, RIV: rivaroxaban
Table S1 Endothelial cell biology PCR array. Fold changes are shown as ratios of gene expression in rivaroxaban‐treated vs. control cells
Table S2 Endothelial cell biology PCR array. Fold changes are shown as ratios of gene expression in activated factor X‐treated vs. control cells
Table S3 Endothelial cell biology PCR array. Fold changes are shown as ratios of gene expression in activated factor X + rivaroxaban‐treated vs. activated factor X‐treated cells