

Abstract
Background
Combined immunotherapy approaches are promising cancer treatments. We evaluated anti–programmed cell death protein 1 (PD-1) treatment combined with gene-mediated cytotoxic immunotherapy (GMCI) performed by intratumoral injection of a prodrug metabolizing nonreplicating adenovirus (AdV-tk), providing in situ chemotherapy and immune stimulation.
Methods
The effects of GMCI on PD ligand 1 (PD-L1) expression in glioblastoma were investigated in vitro and in vivo. The efficacy of the combination was investigated in 2 syngeneic mouse glioblastoma models (GL261 and CT-2A). Immune infiltrates were analyzed by flow cytometry.
Results
GMCI upregulated PD-L1 expression in vitro and in vivo. Both GMCI and anti–PD-1 increased intratumoral T-cell infiltration. A higher percentage of long-term survivors was observed in mice treated with combined GMCI/anti–PD-1 relative to single treatments. Long-term survivors were protected from tumor rechallenge, demonstrating durable memory antitumor immunity. GMCI led to elevated interferon gamma positive T cells and a lower proportion of exhausted double positive PD1+TIM+CD8+ T cells. GMCI also increased PD-L1 levels on tumor cells and infiltrating macrophages/microglia. Our data suggest that anti–PD-1 treatment improves the effectiveness of GMCI by overcoming interferon-induced PD-L1–mediated inhibitory signals, and GMCI improves anti–PD-1 efficacy by increasing tumor-infiltrating T-cell activation.
Conclusions
Our data show that the GMCI/anti–PD-1 combination is well tolerated and effective in glioblastoma mouse models. These results support evaluation of this combination in glioblastoma patients.
Keywords: AdV-tk, anti-PD1, glioblastoma, immunotherapy
Importance of the study
Here, we used murine glioblastoma models to investigate the combination of anti–PD-1 checkpoint blockade with GMCI, an immunostimulatory approach using a nonreplicating adenovirus vector to deliver the herpes simplex virus thymidine kinase gene to tumor cells, which metabolizes ganciclovir to a toxic metabolite. Our data show the combination results in high numbers of long-term survivors in mouse models of glioblastoma and provide strong support for clinical trials.
T-cell activation is tightly controlled by ligand/receptor interactions that restrain the adaptive immune response, and which cancer cells hijack to evade antitumor immunity.1 Immune checkpoint inhibitors (ICIs), which allow T cells to overcome these inhibitory signals, have emerged as an important approach for cancer treatment.2 Several ICIs, including antibodies targeting programmed cell death protein 1 (PD-1), PD ligand 1 (PD-L1), and cytotoxic T-lymphocyte associated antigen 4 (CTLA-4) have demonstrated durable long-term clinical responses in various tumor types, leading to regulatory approvals.3–5 However, typically less than 30% of patients exhibit a durable response and outcomes are dependent on tumor neoantigen load, preexisting inflammatory infiltrates, intratumoral PD-L1 expression, and intact interferon gamma (IFN-γ) signaling, as well as yet unknown factors.6,7 Efforts to improve the proportion of responders include combining multiple ICIs, combining ICIs with immunostimulatory therapies including chemotherapy, and specifically targeting inhibitory cells.8–11
Gene-mediated cytotoxic immunotherapy (GMCI) is a gene transfer approach that has been evaluated in clinical trials for recurrent and primary glioblastoma.12–14 GMCI involves intratumoral injection of aglatimagene besadenovec (AdV-tk), a nonreplicating adenovirus, expressing the herpes simplex virus thymidine kinase (HSV-tk) gene, followed by administration of the prodrug ganciclovir (GCV). Thymidine kinase (TK) converts GCV into a toxic nucleotide analog, acting as an in situ chemotherapeutic. GMCI results in activation of adaptive and innate immunity, with type I immune responses having been observed,15 creating a microenvironment that can produce acute and memory responses to protect against tumor growth.15–18 GMCI has shown efficacy in the treatment of murine cancer models and encouraging results in human clinical trials, including a phase II clinical trial in newly diagnosed glioblastoma.19–21
Glioblastoma is the most common malignant primary brain tumor and has a median survival of less than 15 months.22,23 Glioblastoma is generally not considered to be immunogenic, possessing a relatively moderate mutational burden and being highly immunosuppressive.24,25 So far, the only reported responses to immune checkpoint blockade in glioblastoma have been in case studies of individuals with somatic mutations that cause a hypermutated phenotype, which are rare in glioblastoma.26 Nonetheless, a range of immunotherapies are being examined for glioblastoma.27
In a recently published clinical study of GMCI in newly diagnosed glioblastoma, median overall survival was 17.1 months for GMCI with standard of care (SOC) versus 13.5 months for SOC alone.14 Analysis of glioblastoma tissue in patients treated with GMCI showed increased tumor-infiltrating CD8+ T cells, supporting an immunostimulatory role for GMCI.14,21 Notably, patients who had gross total surgical tumor resection had significantly increased median overall survival of 25.1 months with GMCI compared with 16.3 months in patients with SOC alone.
We hypothesized that GMCI treatment of glioblastoma would be enhanced if combined with immune checkpoint blockade to increase antitumor T-cell responses. Here we report that in murine glioblastoma models, GMCI resulted in upregulation of PD-L1 in the tumor microenvironment and that addition of an anti–PD-1 blocking monoclonal antibody resulted in higher numbers of long-term survivors, with durable long-term antitumor immunity. Analysis of infiltrating T cells indicated that GMCI improves anti–PD-1 efficacy by increasing the activation of tumor-infiltrating CD8+ T cells, suggesting that the effectiveness of this combination is due to a mutually beneficial reciprocal enhancement of the activity of the 2 modalities. Building on existing evidence of single-agent clinical safety and efficacy, the data reported here support clinical trials to evaluate the combination of GMCI and anti–PD-1 therapy for glioblastoma.
Materials and Methods
Cell Culture and Reagents
GL261Luc2 glioma cells were purchased from Perkin-Elmer. CT-2A glioma cells were from Thomas Seyfried and transduced with luciferase-expressing lentivirus (Genecopeia). Cells were grown in Dulbecco’s modified Eagle’s medium (Life Technologies) containing 100 μg/mL G418 or 4 μg/mL puromycin, respectively, supplemented with 10% fetal bovine serum (Sigma-Aldrich) and 1% penicillin-streptomycin. Human glioblastoma stemlike cells (GSCs) were maintained in neurobasal medium supplemented with B27 (Invitrogen), 100 μg/mL penicillin/streptomycin, GlutaMAX (Invitrogen), and 20 ng/mL each of human epidermal growth factor and fibroblast growth factor 2 (PeproTech). Cells were mycoplasma free and their identities confirmed by short tandem repeat profiling (IDEXX Laboratories). Good Manufacturing Practices–grade nonreplicating serotype 5 adenovirus containing the HSV TK gene driven by a Rous sarcoma virus long terminal repeat promoter in the region of the deleted E1 wild-type adenoviral genes (AdV-tk) was produced by Advantagene. The vector has been characterized and approved for clinical use.
Vector Transduction Assay
All cell lines were plated at 200000 cells per well in 6-well plates. For human cells, plates were precoated with 2.5 μg/mL fibronectin (EMD Millipore). The next day, 500 μL of media containing diluted vector stock was added. After 3 hours, media containing 5 μg/mL GCV was added. Treated cells were stained with sulforhodamine B. In some experiments, cells were treated with IFN-β (1000 U/mL) (PBL Assay Science) and/or MAR1-5A3 monoclonal antibody against IFNAR1 (BioXcel) (10 μg/mL).
Fluorescence Microscopy
Anti-mouse PD-L1 (124301, BioLegend) and anti-H2AX Ser139 (9718, CST) were used for immunofluorescence microscopy. Cells were seeded on 24-well plates containing a circular glass coverslip (#1 thickness) the day before transduction. Four days posttransduction, cells were fixed with 4% paraformaldehyde (Sigma-Aldrich) in phosphate buffered saline (PBS) (pH 7.4). After blocking with 5% donkey serum/0.5% Tween 20, 0.02% TX100/ PBS for 1hour at room temperature, cells were washed 3 times with washing buffer (0.5% Tween 20, 0.02% TX100/ PBS) and stained with PD-L1 (1:100) or H2AX Ser139 (1:100). The next day the samples were washed and incubated for 2 hours with secondary antibody (1:500 Donkey anti-rat Alexa488 or Alexa594, Jackson Laboratories) plus Hoechst 33342 (1:3000, H3570, Life Technology). Images were captured with a Zeiss LSM710 confocal microscope.
Enzyme-Linked Immunosorbent Assay and Flow Cytometry In Vitro
A murine IFN-β ELISA (BioLegend) was used to detect IFN-β after vector transduction. Flow cytometry was performed using anti–hPD-L1, anti–mPD-L1, and 7-aminoactinomycin D (BD Bioscience).
In Vivo Studies
Six-week-old female C57BL/6 mice were purchased from Envigo. GL261Luc2 (100000 cells) or CT-2ALuc (500000 cells) in 5 μL Hanks’ Balanced Salt Solution (HBSS) was injected intracranially (2mm right lateral, 1mm frontal to the bregma, and 3mm deep). After 7 days, 3 μL of AdV-tk (2 × 108 vector particles [vp]/μL) was injected intratumorally using the same coordinates. Anti–PD-1 antibody (200 μg) was administered intraperitoneally every 3 days from day 10 (4 injections). The murine anti-mouse PD-1 monoclonal antibody was generated in specific gene-deficient mice, in the laboratory of Dr Gordon Freeman (PD-1—29F.1A12, mouse IgG1). Twenty milligrams per kilogram GCV was administered twice daily intraperitoneally for 10 days starting one day after virus injection. For fluorescence microscopy, brains were perfused, fixed, and frozen before making 30-μm sections on a cryostat. Bioluminescence imaging (BLI) was performed using a Perkin-Elmer IVIS Lumina 3. Animals received 100 μL intraperitoneal injection of 30 mg/mL sterile D-luciferin in 0.9% NaCl. For imaging, animals were anesthetized with 1% isoflurane/oxygen and placed on a heated stage in the IVIS. The bioluminescent signal was captured over time, starting as soon as feasible after D-luciferin injection with 15 images collected every 2 min. Radiance was measured using the region of interest analysis tool to obtain maximum/plateau value of signal for each animal.
Rechallenge Experiments
GL261Luc2 cells (100000) were injected intracranially into the contralateral hemisphere in mice who survived over 100 days post initial tumor implantation as well as control tumor naïve, age-matched C57BL/6 mice. Tumor growth was monitored by BLI.
Characterization of Immune Response
Brains were harvested from treated mice at day 21 and homogenized using enzymatic (1.5 mg/mL collagenase IV, 200 U/mL DNase I, HBSS with calcium and magnesium) and mechanical tissue disaggregation. Cells were resuspended in 25% Percoll Plus (Sigma-Aldrich) for myelin removal and leukocyte isolation. Red blood cells were removed using a Ficoll gradient (GE Life Sciences). The following antibodies were used for flow cytometry: anti-CD45 (30-F11), anti-CD3 (17A2), anti-CD4 (RM4-5), anti-CD8 (53–6.7), anti–PD-L1 (10F.9G2), anti–Tim-3 (B8.2.C12), anti–CTLA-4 (UC10-4B9), anti-CD11c (N418), anti-CD11b (M1/70) (BioLegend), and anti–PD-1 (RMP1-30; eBioscience). Dead cells were excluded using the Zombie NIR Fixable Viability kit (BioLegend). For intracellular staining, cells were treated with the FoxP3 Fixation/Permeabilization kit (eBioscience). The following antibodies were used: anti-FoxP3 (MF-14), IFN-γ (XMG1.2), CTLA-4 (UC10-4B9) (BioLegend), and anti-Granzyme B (NGZB; eBioscience). Data acquisition and compensation were performed on an LSR Fortessa SORP HTS flow cytometer (BD Biosciences) and analyzed using FlowJo X 10.7.7r2 (Tree Star).
Statistics
Two sample t-tests (adjusted with Bonferroni’s method) and one-way ANOVA (with Greenhouse–Geisser correction) were used. Kaplan–Meier survival curves were compared using a log-rank (Mantel–Cox) test. P-values <0.05 were considered statistically significant. Quantitative analysis was performed with GraphPad Prism 6.
Study Approval
All mouse experiments and procedures described in this study were approved by BWH Institutional Animal Care and Use Committee.
Results
GMCI Induces DNA Damage, Cell Death, and PD-L1 Expression in Glioblastoma Cells
GMCI is a vector and prodrug-based therapy that causes double stranded DNA breaks and cell death. To confirm the effectiveness of GMCI in mouse glioblastoma cell lines, GL261Luc2 and CT-2ALuc cells were transduced with increasing concentrations of AdV-tk in the presence or absence of GCV. Both cell types showed dose-dependent cytotoxicity (Fig. 1A). GMCI, but not AdV-tk or GCV alone, induced high levels of DNA damage in GL261Luc2 and CT-2ALuc cells as measured by immunostaining for phosphorylated nuclear γH2AX (Fig. 1B).28
Fig. 1.

Cytotoxic effects and PD-L1 induction by GMCI in glioblastoma cell lines in vitro. (A) Mouse glioblastoma cell lines were transduced with AdV-tk at the indicated concentration and treated with 10 μg/mL of GCV/day for 4 days. Cytotoxicity was determined by the Presto blue (MTT) assay after 6 days treatment. (B) H2AX Ser139 quantification of 2 mouse glioblastoma cell lines (GL261Luc2 and CT-2ALuc) after AdV-tk, GCV, and GMCI treatments compared with mock 4 days after treatment. Confocal microscopic images of CT-2ALuc cells after AdV-tk, GCV, and GMCI treatments showing nuclear staining with phospho-histone H2AX (Ser139) in green and Hoechst in blue. Scale bars: 20 µm. (C–D) Human glioblastoma stemlike cells (hGSCs, G33, G35, G146, and G157) and mouse glioblastoma cell lines were infected with 10 vp/μL AdV-tk, and 10 μg/mL GCV was added daily for 4 days. (C) Representative flow cytometry contour plots from one human GSC (G35) (left) and aggregate plots from 4 human GSCs (right). (D) PD-L1 expression in mouse glioblastoma cell lines after GMCI compared with mock. Flow data are shown for CT-2ALuc cells (left). Graph shows a time-dependent increase in PD-L1 cell surface expression in CT-2ALuc and GL261Luc2 cells (right). One-way ANOVA was used to determine statistical significance (***P ≤ 0.001, **P ≤ 0.01, *P ≤ 0.05, ns, not statistically significant).
To determine whether GMCI could influence immune checkpoint expression, we investigated levels of PD-L1 in GMCI treated cells. PD-L1 is one of the major immune checkpoint ligands expressed on tumor cells and antigen-presenting cells.29 We examined PD-L1 levels on glioblastoma cells by flow cytometry after GMCI treatment in vitro. PD-L1 levels were upregulated in all cell lines examined (4 human and 2 mouse) (Fig. 1C, D). Flow cytometry after gating for live tumor cells showed an increase in PD-L1–expressing cells 4 days after GMCI treatment (from a median of 37.7% to 64.0% in human GSCs, and from 2.2% to 15.5% in mouse glioblastoma cells) which was time dependent (Fig. 1D).
Type I Interferon Signaling Is Upregulated by GMCI and Increases PD-L1 Levels
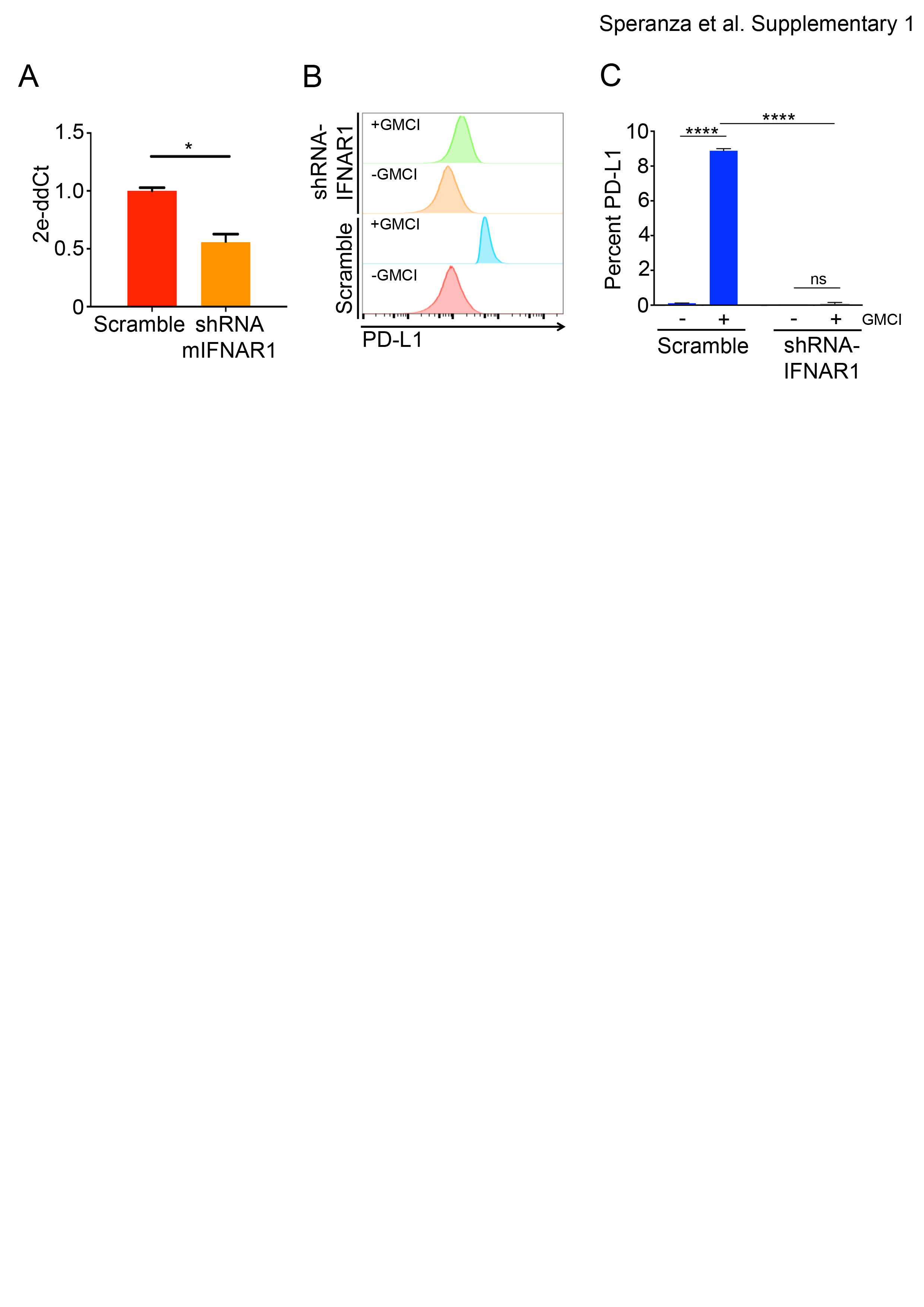
PD-L1 levels can be regulated by type I and type II interferon signaling in cancer cells.30,31 Additionally, type I interferons are known to be induced by viruses and by DNA damage.32,33 We observed upregulation of IFN-β in both GL261Luc2 and CT-2ALuc cell lines (Fig. 2A), and the effect was greatest with AdV-tk plus GCV, suggesting that DNA damage may be an important driver of this upregulation. In GL261Luc2 cells, no effect was observed with AdV-tk alone, with a moderately significant effect observed in CT-2ALuc cells. We then analyzed PD-L1 expression in response to exogenous IFN-α and IFN-β by flow cytometry. Upregulation of PD-L1 was observed in both GL261Luc2 and CT-2ALuc cells, with IFN-β showing the strongest effect (Fig. 2B). We also analyzed PD-L1 expression by immunofluorescence microscopy in the presence and absence of a monoclonal antibody that blocks type I IFN signaling via binding to the mouse type I interferon receptor IFNAR-1 (MAR1-5A3).34 Incubation with MAR1-5A3 completely blocked the IFN-β–mediated upregulation of PD-L1 on GL261Luc2 cells (Fig. 2C). However, upregulation of PD-L1 after GMCI treatment was not completely blocked by MAR1-5A3, indicating that type I interferon signaling may play a partial role in the autocrine upregulation of PD-L1 in response to GMCI. To provide further evidence of the involvement of type I interferon signaling in PD-L1 upregulation, we blocked interferon signaling via short hairpin RNA knockdown of the type I interferon receptor IFNAR1 in CT-2A cells. This led to a block of PD-L1 upregulation consistent with a role for type I interferon signaling (Supplementary Figure S1).
Fig. 2.

Type I interferon induction by GMCI and stimulation of PD-L1 expression. (A) IFN-β released from infected CT-2ALuc and GL261Luc2 tumor cells was detected 4 days after GMCI treatment by ELISA assay. (B) GL261Luc2 and CT-2ALuc cells were treated with 1000 U/mL IFN-α and IFN-β for 4 days followed by flow cytometry for cell surface PD-L1 detection. (C) Representative immunofluorescence images of PD-L1 expression (red) in GL261Luc2 cells after treatment with IFN decoy antibody MAR1-5A3. Ten micrograms per milliliter of MAR1-5A3 antibody and 1000 U/mL or 10 μg/mL/day GCV were added to the tumor cells 3 h after treatment with 10 vp/μL AdV-tk. Scale bar: 50 µm. One-way ANOVA was used to determine statistical significance (***P ≤ 0.001, **P ≤ 0.01).
Increased Intratumoral PD-L1 Expression Is Observed Post-GMCI
The in vivo impact of GMCI on PD-L1 expression was investigated in GL261Luc2 tumor-bearing animals. Animals were treated 7 days post intracranial tumor implantation and tumors were harvested 17 days post GMCI treatment. Immunostaining showed that PD-L1 was upregulated in GMCI treated tumors compared with controls (Fig. 3A). Flow cytometry of immune infiltrates showed a significant upregulation of PD-L1 on macrophages and microglia after GMCI, but not in controls (Fig. 3B). These data demonstrate that immune checkpoint pathways are induced in response to GMCI, supporting the concept of combining GMCI with anti–PD-1 in the treatment of glioblastoma.
Fig. 3.

GMCI induces an increase in PD-L1 levels in vivo. (A) Immunofluorescent staining of PD-L1 in untreated and GMCI treated GL261Luc2-bearing mice. Nuclei were stained with Hoechst (blue), tumor cells stained for vimentin (red) and PD-L1 (green). Scale bars: 20 µm. (B) Flow cytometry analysis of PD-L1 expression in infiltrating macrophages and microglia in the brains of mice bearing GL261Luc2 tumors at day 21. Macrophages were gated CD45highCd11c-Cd11b+ and microglia were gated CD45lowCd11c-C11b+. Long horizontal bars indicate the mean values. Shorter horizontal bars indicate the standard deviation. One-way ANOVA was used to determine statistical significance (***P ≤ 0.001, **P ≤ 0.01).
Combined GMCI and Anti–PD-1 Therapy Leads to Improved Survival in Syngeneic Intracranial Glioblastoma Models
To test the efficacy of GMCI combined with PD-1 immune checkpoint blockade, we performed a survival study using intracranially implanted GL261Luc2 cells in syngeneic C57BL/6 mice. This model has previously been shown to be moderately responsive to immune checkpoint blockade.35 Seven days post tumor implantation, and after confirmation of the presence of tumor by BLI, 6 × 108 AdV-tk particles were injected intratumorally in 3 μL PBS, followed by daily GCV administration. Ten days post tumor implantation, anti–PD-1 antibody was administered and tumor growth was monitored by BLI. This study showed that although no untreated animals survived beyond day 30, both GMCI alone and anti–PD-1 treatment alone resulted in long-term survival (LTS) (≥100 days) in 30%–50% of the animals, and the combination of GMCI and PD-1 blockade resulted in 88% (7/8) LTS mice (Fig. 4A), with no BLI signal in these animals (Fig. 4B). Similar outcomes were observed in repeat studies (data not shown). LTS and tumor-naïve age-matched control mice were rechallenged by injection of the same cells (GL261Luc2) into the contralateral hemisphere of the brain. Neither group in this study received any treatment. While none of the age-matched controls survived past the expected time point (30 days), all of the previously treated LTS mice (13/13) were protected and survived beyond 150 days after rechallenge (Fig. 4C). Lack of tumor growth in the LTS group was confirmed by BLI (Fig. 4D). This indicates that durable memory immunity was established in all long-term survivors (P ≤ 0.01). It has been suggested that GCV may alleviate T-cell exhaustion in vitro.36 However, incubation of isolated T cells with GCV did not change levels of PD-1 (Supplementary Figure S2A, B), and treatment of GL261-bearing mice with GCV alone had no effect on animal survival; combination with anti–PD-1 led to 1/6 long-term survivors, compared with anti–PD-1 alone, which led to 2/6 long-term survivors (Supplementary Figure S2C). Thus there was no independent effect of GCV in our models. A second survival study was performed using CT-2ALuc cells. Higher LTS numbers were also seen with the combination therapy in this model (Fig. 4E). Interestingly, the CT-2ALuc model was less responsive than GL261Luc2 to anti–PD-1 monotherapy. This may reflect differences in neoantigen load, although this is not known at present. Nonetheless, the combination of GMCI and anti–PD-1 was still very effective in this model.
Fig. 4.

GMCI/anti–PD-1 combination therapy improves outcome in the syngeneic GL261Luc2 mouse glioblastoma tumor model. (A) Treatment scheme and Kaplan–Meier survival curves following intratumoral delivery of AdV-tk at day 7 after GL261Luc2 injection (1 × 105 cells) in the right hemisphere of the brain. Twenty milligrams per kilogram GCV/twice daily for 10 days and 4 doses of 200 μg anti–PD-1 were systemically administered. (B) BLI data for 3 tumor-bearing mice from each treatment group at day 21. (C) Long-term tumor-free survivors (LTS, >100 days) initially treated with the GMCI, anti–PD-1, and combination, or treatment-naïve controls following intracranial inoculation of 1 × 105 GL261Luc2 cells. No treatment was given after tumor rechallenge in the left hemisphere. (D) BLI data for 3 rechallenged mice from each treatment group at day 15 post tumor implantation. All the LTS-mice show no residual tumor. (E) Kaplan–Meier survival curves following intratumoral delivery of AdV-tk at day 4 after CT-2ALuc injection (4 × 105 cells). The Mantel–Cox test was used to determine statistical significance (***P ≤ 0.0001, ***P ≤ 0.001, **P ≤ 0.01, *P ≤ 0.05).
GMCI Induces Increased IFN-α and Granzyme B+ T-Cell Infiltrates in Treated Animals
Differences in treatment-induced immune responses were evaluated by comparing immune cell infiltrates in each of the treatment groups (Fig. 5). Immune cells were extracted from each brain 21 days after tumor implantation and phenotypically characterized by flow cytometry (Supplementary Table S1). Gating examples areshown in Supplementary Figure S3A. This revealed an increase of infiltrating lymphocytes (CD3+ cells) and CD8+ cytotoxic T cells, but not CD4+ T cells after PD-1 blockade and combination therapy compared with controls (Fig. 5A, B, C). Importantly, an increase of IFN-γ was detected only in mice receiving GMCI, either alone or as combination therapy, suggesting an important role of GMCI in inducing an active immune response (Fig. 5D). The increase of IFN-γ was observed mainly on CD4+ T cells (Supplementary Figure S3B, C). Granzyme B was upregulated in all treatment groups, and most significantly in the combination group (Fig. 5E). Finally, due to the increase in cytotoxic T cells, the CD8+/regulatory T cell (Treg) ratio was also significantly increased in both GMCI and anti–PD-1 treatment groups compared with untreated controls (P ≤ 0.001 and P ≤ 0.0001, respectively) and was greater after GMCI therapy versus anti–PD-1 alone, both as a single agent and in combination (P ≤ 0.01) (Fig. 5F). There was no difference observed between GMCI and the GMCI/anti–PD-1 combination. These data suggest that GMCI plays an important role in inducing cytotoxic T-cell activity.
Fig. 5.

Immune cell infiltrates following GMCI, PD-1 blockade, and combination therapy. GL261Luc2 tumors were established and treated as described in Fig. 4. Tumor-infiltrating lymphocyte populations were prepared from brains on day 21. (A) CD3+ as a percentage of live CD45+ cells. (B) CD8+ effector T cells as a percentage of live CD45+CD3+ cells. (C) CD4+ T cells as a percentage of live CD45+CD3+ cells. (D) IFN-γ+ as a percentage of live CD45+CD3+ cells. (E) Quantification of CD8+/Granzyme B+ effector T cells as a percentage of live CD45+CD3+CD8+ cells. (F) Ratio of effector CD8+ T cells to Tregs. Long horizontal bars indicate the mean values. Shorter horizontal bars indicate the standard deviation. One-way ANOVA was used to determine statistical significance (***P ≤ 0.001, **P ≤ 0.01).
Influence of Combination Treatments on T-Cell Immune Checkpoint Molecules
Infiltrating lymphocyte populations were analyzed for the presence of the immune-inhibitory receptors PD-1, T-cell immunoglobulin and mucin-domain containing-3 (TIM-3), lymphocyte-activation gene 3 (LAG-3), CTLA-4, and T-cell immunoreceptor with immunoglobulin and immunoreceptor tyrosine-based inhibition motif domains (TIGIT) by flow cytometry. PD-1, LAG-3, and TIGIT expression were comparable among the different groups (Fig. 6A and data not shown). We observed changes in TIM-3 expression on T-cell infiltrates, which showed a trend toward higher levels after PD-1 blockade and were significantly reduced after GMCI treatment (Fig. 6B). Also, the T-cell population coexpressing PD-1 and TIM-3 significantly decreased after GMCI and combination therapy (Fig. 6C). This indicates a potential role of immunostimulation by GMCI in inhibiting TIM-3‒mediated T-cell dysfunction. In contrast, CTLA-4 upregulation was detected in all treatment groups, indicating an inhibitory effect of the different treatments and suggesting a rationale for additional combination therapies (Fig. 6D). No significant changes were observed in PD-1, TIM-3, and CTLA-4 in CD4+ cells (Supplementary Figure S3C–E). Overall, our immune characterization demonstrates that while each of the single therapies may induce activation of the adaptive immune response, the combination of GMCI with PD-1 blockade provides a reciprocal enhancement of the effectiveness of each agent by the other that leads to an increase in the proportion of responders compared with single-agent treatment.
Fig. 6.

Effect of GMCI and anti–PD-1 on immune checkpoint receptors in treated tumor bearing animals. (A) PD-1+ CD8+ T cells as a percentage of live CD45+CD3+ cells, and (B)TIM-3+ cells as a percentage of live CD45+CD3+ cells. (C) PD-1+/TIM-3+ T effector cells as a percentage of live CD45+CD3+CD8+ cells. (D) Representative images of CD8+/CTLA-4+ effector T cells as a percentage of live CD45+CD3+ cells for untreated, anti–PD-1, GMCI, and GMCI/anti–PD-1 combination. (D) Quantification of CD8+/CTLA-4+ effector T cells as a percentage of live CD45+CD3+CD8+ cells. Graphs include values for individually analyzed mice and the mean ± SD per treatment group. Long horizontal bars indicate the mean values. Shorter horizontal bars indicate the standard deviation. One-way ANOVA was used to determine statistical significance (***P ≤ 0.0001, ***P ≤ 0.001, **P ≤ 0.01, *P ≤ 0.05).
Discussion
In the current study, the combination of immunostimulatory GMCI with anti–PD-1 immune checkpoint blockade was investigated in syngeneic glioblastoma mouse models. The combination of GMCI plus anti–PD-1 increased the numbers of LTS animals compared with monotherapies. As single agents, GMCI and anti–PD-1 both increased the numbers of LTS mice (30%–50%) relative to untreated controls (0%). However, combination treatment significantly elevated the percentage of LTS animals (88%). Flow cytometry data suggest a reciprocal enhancement mechanism underlying these observations; GMCI and anti–PD-1 treatments both induced intratumoral accumulation of cytotoxic CD8+ T-cell infiltrates. However, these T cells appear to be more active in GMCI, as they express higher levels of IFN-γ, and CD8+/Treg ratios are higher in GMCI-treated animals. On the other hand, GMCI upregulates PD-L1 through INF signaling,37 which can be overcome with anti–PD-1 treatment. Anti–PD-1 T-cell infiltrates express lower levels of IFN-γ, suggesting they are less active. Anti–PD-1 driven tumor-infiltrating CD8+ T cells also have elevated TIM-3 levels, which appear to be strongly inhibited by GMCI. The significance of this observation is not clear at present, as TIM-3 is a marker of both T-cell activation and dysfunction. However, our observations would suggest that TIM-3 may be functioning as a redundant checkpoint system after treatment with anti–PD-1. Another immune checkpoint molecule, CTLA-4, was elevated in T-cell infiltrates in all treatment groups, suggesting the need to explore this in further combination studies.
The glioblastoma tumor microenvironment is highly immunosuppressive.38 Recently, focus has been placed on the expression of PD-L1 in glioblastoma.39 PD-L1 is a ligand expressed on tumor cells and antigen-presenting cells that activates PD-1 on T lymphocytes, suppressing immune responses during antigen-presenting cell/T-cell interactions.40 Similarly, activation of TIM-3 and CTLA-4 can suppress T-cell–mediated immune responses.41,42 In an immunohistochemical analysis, 61% of patients with glioblastoma had detectable tumor expression of PD-L1.43 In preclinical models of glioblastoma, blockade of PD-1 with an anti–PD-1 monoclonal antibody extended animal survival.44 Several clinical trials of this immune checkpoint therapy are under way for glioblastoma. The success of these therapies will be influenced by the balance of many factors systemically and in the tumor microenvironment. Combined immunostimulation with blocking of immunosuppressive mechanisms is attractive because of the potential for synergistic interactions.45
GMCI has shown promise in animal models and clinical trials, including in malignant gliomas.21 Viral components, immunogenic cell death, and tumor antigen release lead to activation of innate immunity (eg, IFN response) and adaptive immunity (eg, intratumoral lymphocytic infiltrate and systemic immune surveillance).21 Clinical evidence of increased intratumoral immune cell infiltrates after GMCI treatment has been demonstrated in various tumor types as well as increased PD-L1 expression.46 This supports the concept that combination of GMCI with immune PD-1 checkpoint blockade may provide enhanced activity.46 There are a number of additional similar types of prodrug metabolizing therapies which may benefit from this type of combination—for example, Toca511.47 Cell damage caused by these approaches may act as an immunostimulant and be readily combined with ICIs, although the viral component would likely have a greater influence on immune responses in these agents.47 Oncolytic agents have been shown to elicit antitumor immunity in the GL261 glioma model.48
GMCI elicits cancer cell death via DNA damage caused by local in situ chemotherapy. Chemotherapy has been increasingly linked to immunogenic cell death and immune stimulation.11,33 Mice that survived after GMCI, anti–PD-1, or the combination were protected from subsequent intracranial challenge with the same tumor. This may indicate that in the case of minimal residual disease, even if the therapy does not completely clear all of the tumor cells, the induced immune memory response can maintain vigilance against recurrence.
Our long-term question is whether the combination of GMCI with immune checkpoint inhibitors can overcome the potential resistance of newly diagnosed glioblastoma to immune checkpoint blockade. Indeed, primary glioblastoma has a lower neoantigen burden compared with responsive tumors, and exerts multiple mechanisms of immunosuppression.24,37 GMCI may address some of these hurdles. The current study did not investigate the effects of tumor burden or of combination with SOC, and was also performed in immunogenic chemically induced glioma cell lines. Future experiments will investigate combinations with SOC, and investigate less immunogenic genetically engineered glioblastoma mouse models. The present study presents encouraging safety and efficacy data in the animal models and supports the further investigation of this combination in animal models and the initiation of pertinent clinical trials.
Supplementary Material
Supplementary material is available at Neuro-Oncology online.
Funding
This work was supported by NIH PO1CA069246 (E.A.C., X.O.B., R.W.), the ABTA Medical Student Summer Fellowship in honor of Collegiate Charities Dropping the Puck on Cancer and Joggin for Jill (C.P.), and the DFCI Neurooncology Core (E.A.C.).
Conflict of interest statement. G.F. has patents/pending royalties on the PD-1 pathway from Bristol-Myers Squibb, Roche, Merck, EMD-Serono, AstraZeneca, and Novartis. G.F. has served on advisory boards in the last year for Roche, Seattle Genetics, Bethyl Laboratories, Xios, and Quiet. E.A.C. owns equity stock in DNAtrix and IP related to oncolytic viruses, owned by Partners, Inc. B.W.G., L.K.A., and E.A-C. are employees of Advantagene Inc.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
References
- 1. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell. 2015;27(4):450–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Anderson AC, Joller N, Kuchroo VK. Lag-3, Tim-3, and TIGIT: Co-inhibitory receptors with specialized functions in immune regulation. Immunity. 2016;44(5):989–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Robert C, Long GV, Brady B et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med. 2015;372(4):320–330. [DOI] [PubMed] [Google Scholar]
- 5. Reck M, Rodríguez-Abreu D, Robinson AG et al. ; KEYNOTE-024 Investigators Pembrolizumab versus chemotherapy for PD-L1-positive non-small-cell lung cancer. N Engl J Med. 2016;375(19):1823–1833. [DOI] [PubMed] [Google Scholar]
- 6. Rizvi NA, Hellmann MD, Snyder A et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348(6230):124–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gao J, Shi LZ, Zhao H et al. Loss of IFN-γ pathway genes in tumor cells as a mechanism of resistance to anti-CTLA-4 therapy. Cell. 2016;167(2):397–404.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sharma P, Allison JP. The future of immune checkpoint therapy. Science. 2015;348(6230):56–61. [DOI] [PubMed] [Google Scholar]
- 9. Formenti SC, Demaria S. Combining radiotherapy and cancer immunotherapy: a paradigm shift. J Natl Cancer Inst. 2013;105(4):256–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Postow MA, Chesney J, Pavlick AC et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med. 2015;372(21):2006–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pfirschke C, Engblom C, Rickelt S et al. Immunogenic chemotherapy sensitizes tumors to checkpoint blockade therapy. Immunity. 2016;44(2):343–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Trask TW, Trask RP, Aguilar-Cordova E et al. Phase I study of adenoviral delivery of the HSV-tk gene and ganciclovir administration in patients with current malignant brain tumors. Mol Ther. 2000;1(2):195–203. [DOI] [PubMed] [Google Scholar]
- 13. Chiocca EA, Aguilar LK, Bell SD et al. Phase IB study of gene-mediated cytotoxic immunotherapy adjuvant to up-front surgery and intensive timing radiation for malignant glioma. J Clin Oncol. 2011;29(27):3611–3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wheeler LA, Manzanera AG, Bell SD et al. Phase II multicenter study of gene-mediated cytotoxic immunotherapy as adjuvant to surgical resection for newly diagnosed malignant glioma. Neuro Oncol. 2016;18(8):1137–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Vile RG, Castleden S, Marshall J, Camplejohn R, Upton C, Chong H. Generation of an anti-tumour immune response in a non-immunogenic tumour: HSVtk killing in vivo stimulates a mononuclear cell infiltrate and a Th1-like profile of intratumoural cytokine expression. Int J Cancer. 1997;71(2):267–274. [DOI] [PubMed] [Google Scholar]
- 16. Curtin JF, Liu N, Candolfi M et al. HMGB1 mediates endogenous TLR2 activation and brain tumor regression. PLoS Med. 2009;6(1):e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Melcher A, Todryk S, Hardwick N, Ford M, Jacobson M, Vile RG. Tumor immunogenicity is determined by the mechanism of cell death via induction of heat shock protein expression. Nat Med. 1998;4(5):581–587. [DOI] [PubMed] [Google Scholar]
- 18. Moolten FL. Tumor chemosensitivity conferred by inserted herpes thymidine kinase genes: paradigm for a prospective cancer control strategy. Cancer Res. 1986;46(10):5276–5281. [PubMed] [Google Scholar]
- 19. Chen SH, Shine HD, Goodman JC, Grossman RG, Woo SL. Gene therapy for brain tumors: regression of experimental gliomas by adenovirus-mediated gene transfer in vivo. Proc Natl Acad Sci U S A. 1994;91(8):3054–3057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Perez-Cruet MJ, Trask TW, Chen SH et al. Adenovirus-mediated gene therapy of experimental gliomas. J Neurosci Res. 1994;39(4):506–511. [DOI] [PubMed] [Google Scholar]
- 21. Aguilar LK, Guzik BW, Aguilar-Cordova E. Cytotoxic immunotherapy strategies for cancer: mechanisms and clinical development. J Cell Biochem. 2011;112(8):1969–1977. [DOI] [PubMed] [Google Scholar]
- 22. Wen PY, Kesari S. Malignant gliomas in adults. N Engl J Med. 2008;359(5):492–507. [DOI] [PubMed] [Google Scholar]
- 23. Ostrom QT, Gittleman H, Fulop J et al. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2008–2012. Neuro Oncol. 2015;17(Suppl 4):iv1–iv62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015;348(6230):69–74. [DOI] [PubMed] [Google Scholar]
- 25. Reardon DA, Freeman G, Wu C et al. Immunotherapy advances for glioblastoma. Neuro Oncol. 2014;16(11):1441–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bouffet E, Larouche V, Campbell BB et al. Immune checkpoint inhibition for hypermutant glioblastoma multiforme resulting from germLine biallelic mismatch repair deficiency. J Clin Oncol. 2016;34(19):2206–2211. [DOI] [PubMed] [Google Scholar]
- 27. Garber ST, Hashimoto Y, Weathers SP et al. Immune checkpoint blockade as a potential therapeutic target: surveying CNS malignancies. Neuro Oncol. 2016;18(10):1357–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mah LJ, El-Osta A, Karagiannis TC. gammaH2AX: a sensitive molecular marker of DNA damage and repair. Leukemia. 2010;24(4):679–686. [DOI] [PubMed] [Google Scholar]
- 29. Preusser M, Lim M, Hafler DA, Reardon DA, Sampson JH. Prospects of immune checkpoint modulators in the treatment of glioblastoma. Nat Rev Neurol. 2015;11(9):504–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Liu J, Hamrouni A, Wolowiec D et al. Plasma cells from multiple myeloma patients express B7-H1 (PD-L1) and increase expression after stimulation with IFN-{gamma} and TLR ligands via a MyD88-, TRAF6-, and MEK-dependent pathway. Blood. 2007;110(1):296–304. [DOI] [PubMed] [Google Scholar]
- 31. Yang YQ, Dong WJ, Yin XF et al. Interferon-related secretome from direct interaction between immune cells and tumor cells is required for upregulation of PD-L1 in tumor cells. Protein Cell. 2016;7(7):538–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mühlbauer M, Fleck M, Schütz C et al. PD-L1 is induced in hepatocytes by viral infection and by interferon-alpha and -gamma and mediates T cell apoptosis. J Hepatol. 2006;45(4):520–528. [DOI] [PubMed] [Google Scholar]
- 33. Brzostek-Racine S, Gordon C, Van Scoy S, Reich NC. The DNA damage response induces IFN. J Immunol. 2011;187(10):5336–5345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sheehan KC, Lai KS, Dunn GP et al. Blocking monoclonal antibodies specific for mouse IFN-alpha/beta receptor subunit 1 (IFNAR-1) from mice immunized by in vivo hydrodynamic transfection. J Interferon Cytokine Res. 2006;26(11):804–819. [DOI] [PubMed] [Google Scholar]
- 35. Reardon DA, Gokhale PC, Klein SR et al. Glioblastoma eradication following immune checkpoint blockade in an orthotopic, immunocompetent model. Cancer Immunol Res. 2016;4(2):124–135. [DOI] [PubMed] [Google Scholar]
- 36. Chang C-M, Wu C-L, Chen I-H et al. In vitro treatment with ganciclovir restores the functionality of exhausted T cells from cancer patients. Int J Gerontol. 2013;(7):171–176. [Google Scholar]
- 37. Loke P, Allison JP. PD-L1 and PD-L2 are differentially regulated by Th1 and Th2 cells. Proc Natl Acad Sci U S A. 2003;100(9):5336–5341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Nduom EK, Weller M, Heimberger AB. Immunosuppressive mechanisms in glioblastoma. Neuro Oncol. 2015;17(Suppl 7):vii9–vii14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Preusser M, Berghoff AS, Wick W, Weller M. Clinical Neuropathology mini-review 6-2015: PD-L1: emerging biomarker in glioblastoma?Clin Neuropathol. 2015;34(6):313–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Freeman GJ, Long AJ, Iwai Y et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192(7):1027–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Anderson AC. Tim-3: an emerging target in the cancer immunotherapy landscape. Cancer Immunol Res. 2014;2(5):393–398. [DOI] [PubMed] [Google Scholar]
- 42. Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science. 1996;271(5256):1734–1736. [DOI] [PubMed] [Google Scholar]
- 43. Nduom EK, Wei J, Yaghi NK et al. PD-L1 expression and prognostic impact in glioblastoma. Neuro Oncol. 2016;18(2):195–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Reardon DA, Gokhale PC, Klein SR et al. Glioblastoma eradication following immune checkpoint blockade in an orthotopic, immunocompetent model. Cancer Immunol Res. 2016;4(2):124–135. [DOI] [PubMed] [Google Scholar]
- 45. Kong LY, Wei J, Fuller GN et al. Tipping a favorable CNS intratumoral immune response using immune stimulation combined with inhibition of tumor-mediated immune suppression. Oncoimmunology. 2016;5(5):e1117739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Aguilar LK, Shirley LA, Chung VM et al. Gene-mediated cytotoxic immunotherapy as adjuvant to surgery or chemoradiation for pancreatic adenocarcinoma. Cancer Immunol Immunother. 2015;64(6):727–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Cloughesy TF, Landolfi J, Hogan DJ et al. Phase 1 trial of vocimagene amiretrorepvec and 5-fluorocytosine for recurrent high-grade glioma. Sci Transl Med. 2016;8(341):341ra75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Jiang H, Clise-Dwyer K, Ruisaard KE et al. Delta-24-RGD oncolytic adenovirus elicits anti-glioma immunity in an immunocompetent mouse model. PLoS One. 2014;9(5):e97407. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.