

Abstract
The emerging standard of care for patients with inoperable pancreatic cancer is a combination of cytotoxic drugs gemcitabine and Abraxane, but patient response remains moderate. Pancreatic cancer development and metastasis occur in complex settings, with reciprocal feedback from microenvironmental cues influencing both disease progression and drug response. Little is known about how sequential dual targeting of tumor tissue tension and vasculature before chemotherapy can affect tumor response. We used intravital imaging to assess how transient manipulation of the tumor tissue, or “priming,” using the pharmaceutical Rho kinase inhibitor Fasudil affects response to chemotherapy. Intravital Förster resonance energy transfer imaging of a cyclin-dependent kinase 1 biosensor to monitor the efficacy of cytotoxic drugs revealed that priming improves pancreatic cancer response to gemcitabine/Abraxane at both primary and secondary sites. Transient priming also sensitized cells to shear stress and impaired colonization efficiency and fibrotic niche remodeling within the liver, three important features of cancer spread. Last, we demonstrate a graded response to priming in stratified patient-derived tumors, indicating that fine-tuned tissue manipulation before chemotherapy may offer opportunities in both primary and metastatic targeting of pancreatic cancer.
INTRODUCTION
A number of new therapeutics have been exploited to improve upon gemcitabine (Gem) in pancreatic cancer (PC). Recently, the addition of nab-paclitaxel (Abraxane) to Gem improved patient survival from 6.6 to 8.7 months (1). Although the improvements found with the antimitotic Abraxane are encouraging and the combination is rapidly becoming a first-line treatment in this aggressive disease (2), there is an urgent need to improve upon this moderate shift in patient survival.
The actin cytoskeleton and its prototypical regulatory proteins Rhoguanosine triphosphatases (GTPases) are commonly hijacked by many cancers to drive tumor progression (3-6). In particular, altering cytoskeleton-based cell contractility affects not only coordinated cancer cell protrusion during invasion but also the bidirectional interaction between stromal and cancer cells to induce tissue stiffening and drive tumor survival, proliferation, and progression (7-10). Hence, targeting tissue architecture via Rho GTPase inhibition to alter tissue stiffness, cellular rheology, vasodilation, or mechanoplasticity is an emerging area of potential therapeutic intervention in cancer (3, 5,11-14).
Optimizing preclinical disease models in drug discovery requires innovative approaches to assess drug response in live tissue at the single-cell and molecular levels. Intravital imaging offers insights into how cells behave in their native environment and provides a dynamic four-dimensional (4D) molecular readout of therapeutic response, undetectable in vitro (15,16). Here, we derived primary and invasive cells from both the Pdxl-Cre, LSL-KrasG12D/+, LSL-Trp53R172H/+ (KPC) mouse model (17-21) and pancreatic patient-derived xenografts (PDXs) and engineered them to express the highly validated cyclin-dependent kinase 1 (CDK1) Forster resonance energy transfer (FRET) biosensor (22,23). We monitored CDK1 activity as a surrogate marker of M-phase cell cycle arrest induced by Gem/Abraxane (22-27). Upon investigating the ultrastructure, integrity, and stiffness of the extracellular matrix (ECM) in complex organotypic matrices in response to transient priming with the ROCK inhibitor Fasudil (HA-1077) (28), we mapped PC cell response to Gem/Abraxane in live tumor tissues during disease progression. First, this was achieved through spatiotemporal monitoring of the response to Gem/Abraxane at distinct sites within live primary tumors and PDXs, whereas assessment in liver metastases revealed a multisite improvement in response to chemotherapy after transient dual manipulation of tissue tension and tumor vasculature with Fasudil. Second, priming impaired tumor cell extravasation efficiency, resistance to shear stress, and metastatic niche remodeling within the liver, reducing spread in secondary sites. Last, we reveal a graded response to transient priming regimens relative to ECM found in PDXs from the Australian Pancreatic Cancer Genome Initiative (APGI) cohort (29). Collectively, we present preclinical evidence that tailored, fine-tuned tissue priming before chemotherapy may offer opportunities in both primary and metastatic targeting in PC.
RESULTS
Priming the stroma via ROCK inhibition disrupts ECM integrity
The effect of ROCK inhibition on ECM integrity was assessed using fibroblast-driven contraction assays, which revealed a decrease in contraction upon treatment with Fasudil (Fig. 1A). Immunohistochemical (IHC) analysis of ROCK signaling in matrices confirmed that Fasudil inhibits the downstream targets of ROCK, pMLC2, and pMYPT1 (fig. S1, A and B) (30), whereas 2D and 3D cell proliferation analyses indicated that impairment of matrix contraction was independent of changes in fibroblast proliferation and survival (fig. S1, C to E).
Fig. 1. ROCK inhibition disrupts collagen matrix integrity.
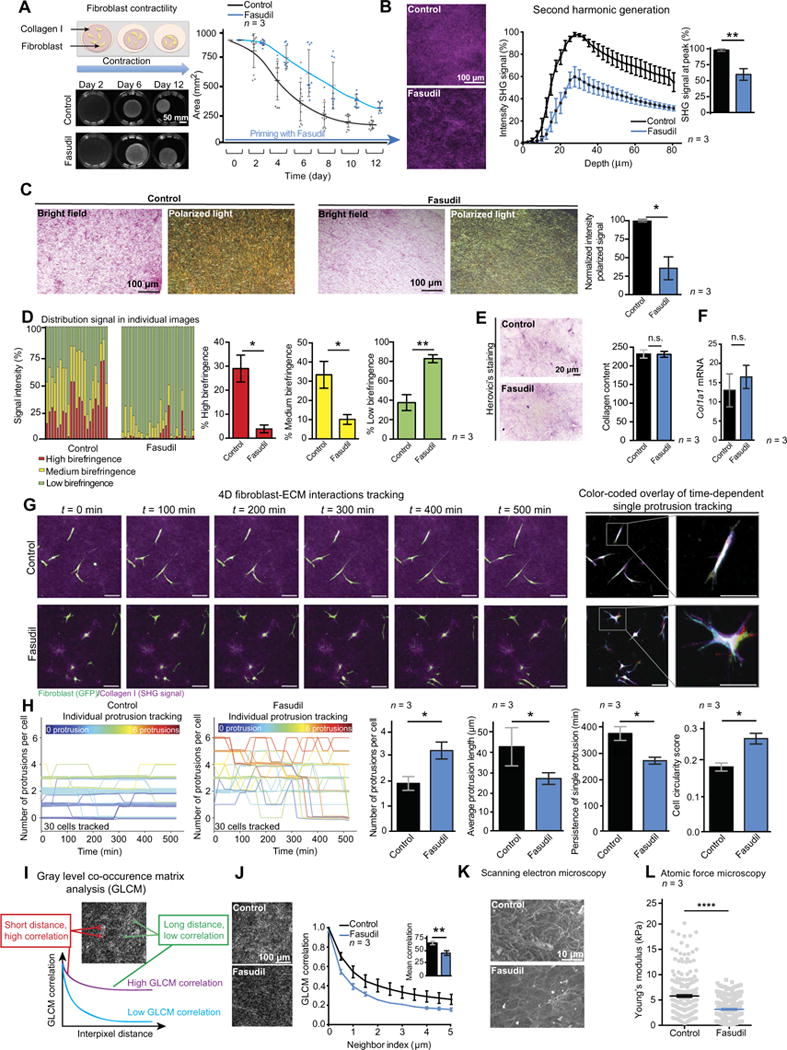
(A) Schematic of contraction assay (left), representative images of telomerase-immortalized fibroblast (TIF)-collagen matrices, and quantification of matrix area ± Fasudil over time. n = 3 biological repeats with three matrices per condition and per repeat. All individual data points are represented with SD, and statistical analysis was performed using the average values of the three biological repeats. (B) Representative maximum intensity projections of SHG signal and quantification of SHG signal intensity by depth and at peak in TIF-collagen matrices after 12 days of contraction ± Fasudil. n = 3 biological repeats with three matrices per condition and per repeat. (C) Bright-field and polarized light imaging of picrosirius red-stained organotypic matrices primed ± Fasudil and quantification of the intensity of the signal acquired via polarized light. n = 3 biological repeats with three matrices per condition and per repeat. (D) Contribution and quantification of signal emitted from fibers with high, medium, and low birefringence normalized to total signal acquired via polarized imaging of picrosirius red-stained collagen matrices ± Fasudil. Thick remodeled collagen fibers are highly birefringent (red-orange), whereas less remodeled fibers have a lower birefringence (green). n = 3 biological repeats with three or two matrices per condition and per repeat. (E) Herovici’s staining and quantification of collagen content in collagen matrices ± Fasudil. n = 3 biological repeats with three matrices per condition and per repeat. ns., not significant. (F) Rea-time quantitative polymerase chain reaction analysis of Col1a1 mRNA in TIFs ± Fasudil exposure for 72 hours. n = 3 biological repeats. (G) Time-lapse tracking of fibroblast-ECM interactions on day 6 of contraction ± Fasudil and color-coded overlay of single protrusion tracking (red, t =0 min;light green, t =100 min;yellow, t = 200 min;blue, t = 300 min;dark green, t = 400 min; purple, t = 500 min). Scale bars, 100 μm. (H) Number of cell protrusions over time in control (left) and Fasudil-primed (right) matrices. Traces are color-coded according to the initial protrusion number (color key informs on initial number of protrusions at t =0 min). The graphs on the right indicate protrusion number, length, persistence, and cell circularity in collagen matrices ± priming with Fasudil (30 cells per group and n = 3 independent matrices). (I) Schematic of GLCM analysis of collagen texture. (J) Representative single-plane images of SHG signal acquired for GLCM analysis (left) and quantification of GLCM correlation in matrices (right). n = 3 biological repeats with three matrices per condition and per repeat. (K) Representative images of scanning electron microscopy. (L) Atomic force microscopy analysis of matrix Young’s modulus. n = 3 biological repeats with three matrices per condition and per repeat. Results are means ± SEM, unless stated otherwise. P values were determined by unpaired, nonparametric t test with Mann-Whitney U correction.
Second harmonic generation (SHG) imaging (31) and polarized light microscopy of picrosirius red staining (32, 33) of contracted collagen matrices revealed a reduction in fibrillar collagen coverage upon ROCK inhibition compared to control (Fig. 1, B to D, and fig. S1F), whereas collagen production was unaltered (Fig. 1, E and F). Live monitoring of fibroblast-ECM interactions (Fig. 1G and movie S1) (34) demonstrated uncoordinated, shorter, and unstable fibroblast protrusions in Fasudil-treated conditions, as well as increased cell circularity (Fig. 1H). Analysis of matrix ultrastructure using gray-level cooccurrence matrix (GLCM) (35) and scanning electron microscopy confirmed a disorganized ECM network in Fasudil-treated condition (Fig. 1, I to K), and atomic force microscopy measurements revealed a reduction in matrix stiffness for primed matrices (Fig. 1L). Similar results were observed for matrices treated with the structurally unrelated ROCK inhibitor Y-27632 (fig. S1, G to L), confirming that ROCK targeting disrupts matrix integrity.
Recent studies assessing the effect of stromal manipulation via longterm interference with ECM remodeling have yielded conflicting data on the utility of stromal targeting in limiting the progression of PC (10, 12, 36-43). We therefore sought to assess whether fine-tuned transient interference with stromal tension affects PC progression and drug response (10).
The scheduling of priming regimens influences PC cell invasion and Gem/Abraxane efficacy
We examined cancer cell invasion in a 3D organotypic model using primary PC cells established from KPC mice (17, 18, 44, 45). 3D organotypic matrices were treated with Fasudil either during contraction (priming) or during invasion (later treatment) (Fig. 2A). KPC cell invasion was decreased in matrices primed with Fasudil compared to control (Fig. 2, B and C). Later Fasudil treatment alone did not significantly affect KPC invasion, and continuous treatment during both contraction and invasion had no advantage over priming of the matrix alone (Fig. 2, B and C), demonstrating that transient ROCK targeting can impair PC invasion and could minimize the need for long-term treatments.
Fig. 2. Scheduling of priming regimens influences PC cell invasion and Gem/Abraxane efficacy.
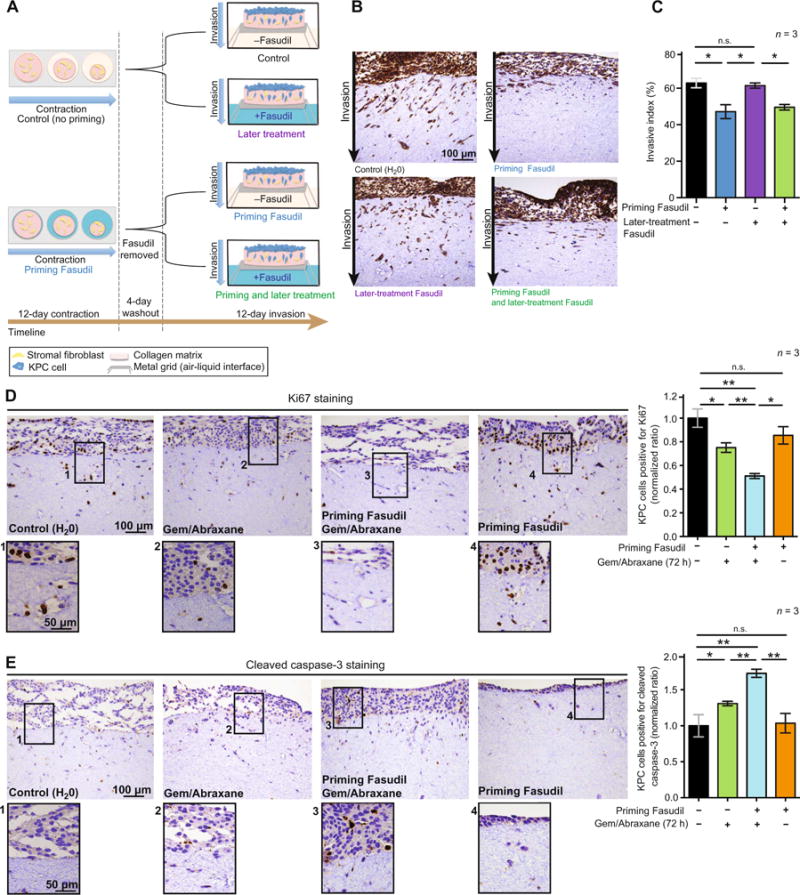
(A) Schematic representation of organotypic invasion assay with KPC cells. Matrices were primed with Fasudil during contraction and/or treated with Fasudil during KPC cell invasion. (B) Representative images of pancytokeratin staining in organotypic matrices. (C) Quantification of KPC cell invasive index upon priming, later, and priming + later treatment with Fasudil. (D) Representative images of Ki67 staining as a marker of cell proliferation and quantification of KPC cells positive for Ki67 (ratio normalized to control) in organotypic matrices after 72 hours of treatment with Gem/Abraxane. (E) Representative images of cleaved caspase-3 staining as a marker of cell apoptosis and quantification of KPC cells positive for cleaved caspase-3 (ratio normalized to control) in organotypic matrices after 72 hours of treatment with Gem/Abraxane. Results are means ± SEM. n = 3 biological repeats with three matrices per condition and per repeat. P values were determined by nonparametric analysis of variance (ANOVA) test with Holm-Sidak correction for multiple comparisons.
Because mechanical cues provided by the ECM have been suggested to limit response to chemotherapy (46, 47), we next investigated whether transient Fasudil priming would increase KPC cell response to subsequent Gem/Abraxane treatment in organotypic matrices. Ki67 and cleaved caspase-3 staining demonstrated that Gem/Abraxane treatment alone reduced cell proliferation and induced cell apoptosis (Fig. 2, D and E, black bars versus green bars), as expected given their clinical use (2). This was improved in Fasudil-primed settings (Fig. 2, D and E, green bars versus blue bars), although Fasudil priming alone had no effect on KPC cell proliferation and apoptosis in this context (Fig. 2, D and E, black bars versus orange bars).
Although these static markers of tumor behavior provide an initial insight into tumor response, they lack the capacity to monitor more detailed temporal and spatial information. We therefore opted to longitudinally map Gem/Abraxane therapy using a CDK1-FRET biosensor (22, 23) as a molecular readout of cell cycle arrest induced by chemotherapy (27).
A CDK1 biosensor monitors PC cell response to Gem/Abraxane
Primary KPC cells were engineered to express a CDK1-FRET biosensor, and the activity of CDK1 was measured by fluorescence lifetime imaging microscopy of the FRET signal (FLIM-FRET; Fig. 3A). In the lifetime color maps, low CDK1 activity is represented by longer lifetimes and yellow/red colors, whereas high CDK1 activity is represented by shorter lifetimes and blue/green colors, and areas with no signal are black (Fig. 3B and fig. S2A).
Fig. 3. Spatiotemporal analyses of combination therapy are performed using the CDK1 biosensor.
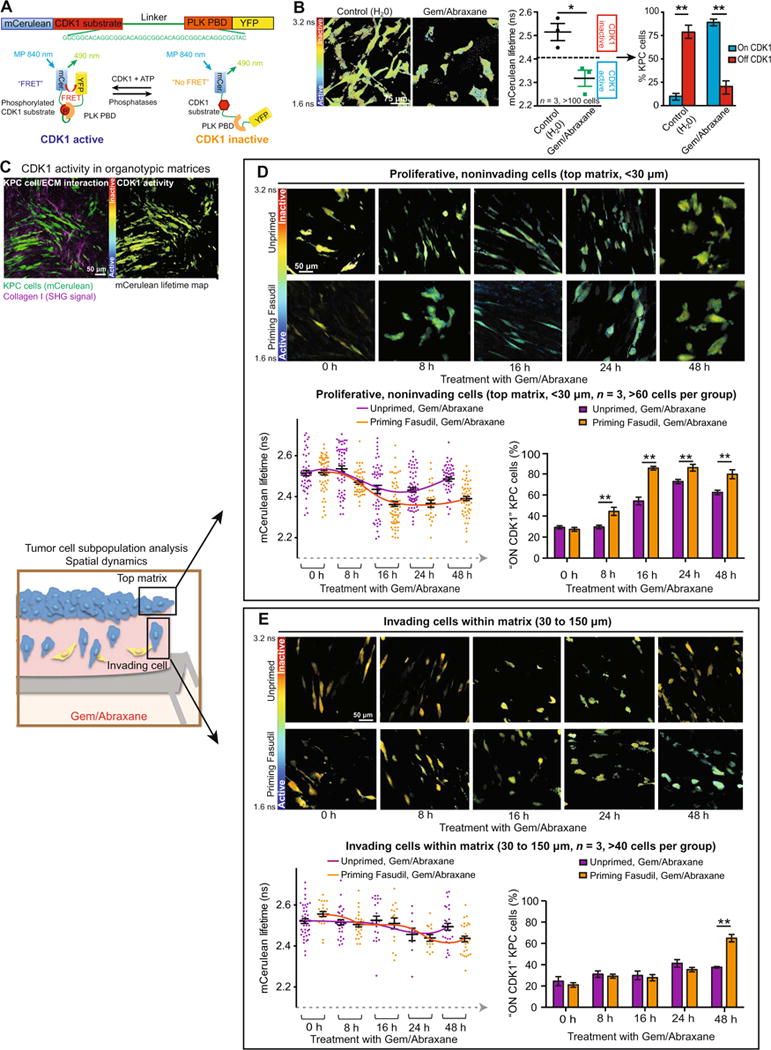
(A) Schematic representation of the CDK1-FRET biosensor conformation upon activation and inactivation of CDK1 (23). PLK, Polo-like kinase;PBD, Polo-box domain; ATP, adenosine 5′-triphosphate;YFP, yellow fluorescent protein; MP, multiphoton. (B) Representative mCerulean lifetime maps, quantification of mCerulean lifetimes, and stratification of CDK1 activity in KPC cells upon treatment with Gem/Abraxane for 24 hours in 2D settings. n = 3 independent biological repeats with one technical replicate per repeat. (C) Representative intensity images and mCerulean lifetime maps of KPC-CDK1 cells imaged invading in organotypic matrices. (D) Monitoring of CDK1 activity in cells on top of the collagen matrix and in response to Gem/Abraxane over time. Representative mCerulean lifetime maps (top), quantification of mCerulean lifetimes (bottom, left) (60 cells per group represented), and CDK1 accumulation overtime (bottom, right) (average of three matrices represented) in KPC cells seeded on top of primed (orange) and unprimed (purple) matrices. n = 3 independent matrices, >60 cells per group. (E) Monitoring of CDK1 activity in cells invading at depth intocollagen matrix and in response to Gem/Abraxane over time. Representative mCerulean lifetime maps (top), quantification of mCerulean lifetimes (bottom, left) (60 cells per group represented), and CDK1 accumulation over time (bottom, right) (average of three matrices represented) in KPC cells invading into primed (orange) and unprimed (purple) matrices. n = 3 independent matrices, >40 cells per group. Results are means ± SEM. P values were determined by unpaired, nonparametric t test with Mann-Whitney U correction.
To confirm that the CDK1-FRET biosensor reports changes in CDK1 activity, KPC cells were treated with Abraxane to arrest them in mitosis, where CDK1 activity is high (22-27), followed by treatment with the specific CDK1 inhibitor RO3306 (fig. S2, B and C) (48). Abraxane induced a decrease in the fluorescence lifetimes, indicating active CDK1, and treatment with RO3306 reverted the lifetimes to values similar to control, confirming CDK1 inhibition (fig. S2, C and D, and movie S2). Using the lifetime distribution, we stratified cells as “CDK1 ON” or “CDK1 OFF” and confirmed changes in CDK1 activity by Western blot analysis of pY15-CDK1 (fig. S2, E and F). Similarly, FLIM-FRET analysis of cells in different stages of the cell cycle confirmed accumulation of active CDK1 in mitotic cells compared to cells in interphase (fig. S2, G to I, and movie S3). We also confirmed that expression of the CDK1 biosensor did not affect KPC cell response to Gem/Abraxane (fig. S2J). Next, monitoring changes in CDK1 activity upon treatment with Gem/Abraxane demonstrated a decrease of lifetime and increased activation of CDK1 compared to control (Fig. 3B). This aligned with CDK1 activation and mitotic arrest as confirmed by FACS (fluorescence- activated cell sorting) and Western blot analyses (fig. S2, K and L) (49). After validating the CDK1 biosensor, we monitored individual KPC cell response to chemotherapy within a 3D microenvironment (Fig. 3C).
Priming with Fasudil improves the spatiotemporal response of Gem/Abraxane in organotypic matrices
Cancer progression and response to drug treatment can be regulated by environmental cues within tumor subpopulations, such as areas of invasion (50-53). Hence, we assessed CDK1 activity in cells invading into the matrix versus cells on top of the matrix over time (Fig. 3, C to E, and movie S4). For cells on top of the matrix, we observed a decrease in FRET lifetime and corresponding activation of CDK1 after 16 hours of treatment with Gem/Abraxane. This was sustained for 24 hours and then reverted to higher lifetimes at 48 hours (Fig. 3D, purple line). An earlier activation of CDK1 in matrices that had been primed with Fasudil was evident after 8 hours of Gem/Abraxane treatment, and the activation of CDK1 was prolonged (Fig. 3D, orange line). Monitoring of CDK1 at depth demonstrated a distinctly delayed pattern of response to Gem/Abraxane in invading cells (compare the purple line in Fig. 3D to that in Fig. 3E); however, Fasudil priming increased the response of invasive cells at later time points compared to Gem/Abraxane treatment alone (Fig. 3E). The ability to detect differences in drug efficacy in a spatial manner allowed us to uncover that proliferating cells on top of the matrix respond well to Gem/Abraxane therapy alone, whereas invading and less proliferative cells (confirmed in fig. S2M) (50) are less susceptible to cytotoxic drugs. Thus, although Fasudil interference with stromatumor feedback improves targeting of cells on top of the matrix, it may also have additional, yet somewhat delayed, benefits by rendering invasive cells more vulnerable to chemotherapy.
Priming live primary tumors enhances Gem/Abraxane efficacy
We next generated subcutaneous KPC-CDK1 xenografts, and once primary tumors were palpable, mice were primed with Fasudil for 3 days before treatment with Gem/Abraxane (Fig. 4A). Twenty-four hours after administration of Gem/Abraxane, live tumor tissues were exposed using a skin flap technique for intravital imaging (Fig. 4B). SHG imaging and polarized light microscopy of picrosirius red staining of KPC xenografts confirmed that Fasudil priming reduces fibrillar collagen organization in this setting, whereas no significant change in collagen content was observed (Fig. 4, C and D; fig. S3, A and B; and movie S5). In vivo FLIM-FRET analysis of the CDK1-FRET biosensor (movie S6) demonstrated activation of CDK1 upon Gem/Abraxane treatment alone, which was enhanced in Fasudil-primed conditions (Fig. 4E, green bar versus blue bar), in line with findings in organotypic matrices (Figs. 2 and 3). Fasudil priming alone had no significant effect on CDK1 activity in live tumors (Fig. 4E, black bar versus orange bar). IHC analyses of pMYPT1 in KPC xenografts confirmed inhibition of ROCK signaling upon Fasudil priming (fig. S3C), whereas Ki67 and cleaved caspase-3 analyses confirmed that although Fasudil priming alone had no significant effect on cell proliferation or survival, it improved the effectiveness of Gem/Abraxane in this setting (fig. S3, D and E). To assess whether ECM-independent mechanisms may also play a role in the enhanced response to chemotherapy, we examined the known role of Fasudil as a vasodilator in vivo (28) by evaluating vasculature abundance using anti-CD31 staining. This demonstrated an increase in CD31-positive cells upon treatment with Fasudil (Fig. 4F). We next injected quantum dots into mice bearing subcutaneous KPC tumors to assess whether vascular patency was altered in Fasudil-treated conditions (39, 54). Here, an increase in quantum dot signal was observed outside blood vessels, with quantum dots leaking into the tumor tissue in Fasudil-primed conditions compared to control (Fig. 4, G and H, and movie S7). Because PC patients often present with locally invasive and metastatic disease at diagnosis (55), we next investigated whether Fasudil priming alone or in combination with chemotherapy alters metastatic colonization of secondary sites.
Fig. 4. Priming live primary tumors enhances Gem/ Abraxane efficacy.
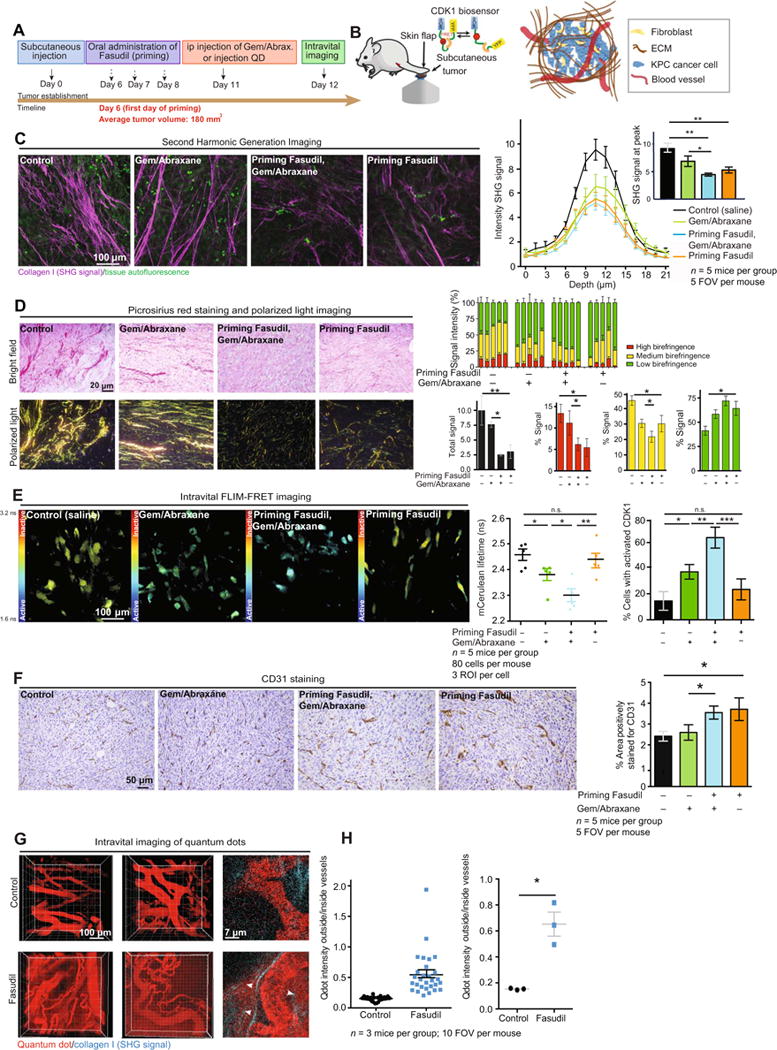
(A) Schematic representation of subcutaneous injection of KPC cells, tumor development, and treatment timeline. IP, intraperitoneal; QD, quantum dots. (B) Schematic of intravital imaging of subcutaneous KPC-CDK1 xenografts using a skin flap technique. (C) Representative maximum intensity projections of ex vivo SHG imaging of tumor tissue and quantification of SHG signal acquired in subcutaneous KPC xenografts upon treatment with control (saline); Gem/Abraxanealone;Fasudil, then Gem/Abraxane; and Fasudil alone. n = 5 mice per group and five fields of view (FOVs) per mouse. (D) Left: Picrosirius red staining and polarized light imaging of tumor tissue. Right: Quantification of total signal (black) and contribution to signal emitted from collagen fibers with high (red), medium (yellow), and low (green) birefringence acquired via polarized light imaging. n = 5 mice per group and five FOVs per mouse. (E) Representative mCerulean lifetime maps, quantification of mCerulean lifetimes, and CDK1 activity in subcutaneous KPC-CDK1 tumors upon treatment with control (saline); Gem/Abraxane alone; Fasudil, then Gem/ Abraxane; and Fasudil alone. n = 5 mice per group, 80 cells per mouse, and three regions of interest (ROIs) per cell. (F) CD31 staining and quantification of area positively stained in subcutaneous KPC-CDK1 tumors upon treatment with control (saline); Gem/ Abraxane alone; Fasudil, then Gem/Abraxane; and Fasudil alone. n = 5 mice per group and five FOVs per mouse. (G) Intravital imaging ofquan tum dots (655 nm) circulating through tumor-associated blood vessels and diffusing into tumor tissue. (H) Quantification of quantum dot (Qdot) diffusion in subcutaneous KPC tumors upon treatment with control (saline) or Fasudil. n = 3 mice per group and 10 FOVs per mouse. Results are means ± SEM. P values were determined by nonparametric ANOVA test with Holm-Sidak correction for multiple comparisons and by unpaired, nonparametric t test with Mann-Whitney U correction for comparisons between two groups.
Priming with Fasudil enhances chemotherapy response at secondary sites and reduces metastatic spread
To mimic systemic ROCK inhibition or adjuvant therapy in the presence of circulating tumor cells, we performed intrasplenic injections of KPC cells expressing the CDK1-FRET biosensor in parallel with Fasudil administration (Fig. 5, A and B, and movie S8). FLIM-FRET measurements of the CDK1 biosensor in liver metastases revealed that priming with Fasudil enhanced Gem/Abraxane efficacy at this secondary site (Fig. 5, C to E). In line with our results in primary tumors (Fig. 4), we observed enhanced CD31 staining and increased number of microvessels with detectable lumens in the livers of Fasudil-treated mice (Fig. 5F and fig. S4A), which could partly explain the observed enhanced drug response. Pathological and IHC analyses on serial sections of metastases (Fig. 5, G and H) confirmed that mice treated with Fasudil and Gem/Abraxane showed a marked reduction in metastatic spread compared to Gem/Abraxane treatment alone. This is in line with recent work regarding the establishment of the metastatic niche (56, 57) and prompted us to assess micrometastatic events, such as cancer cell extravasation, adhesion, response to shear stress, and colonization after Fasudil priming.
Fig. 5. Priming with Fasudil improves chemotherapy at secondary sites and decreases metastatic burden.
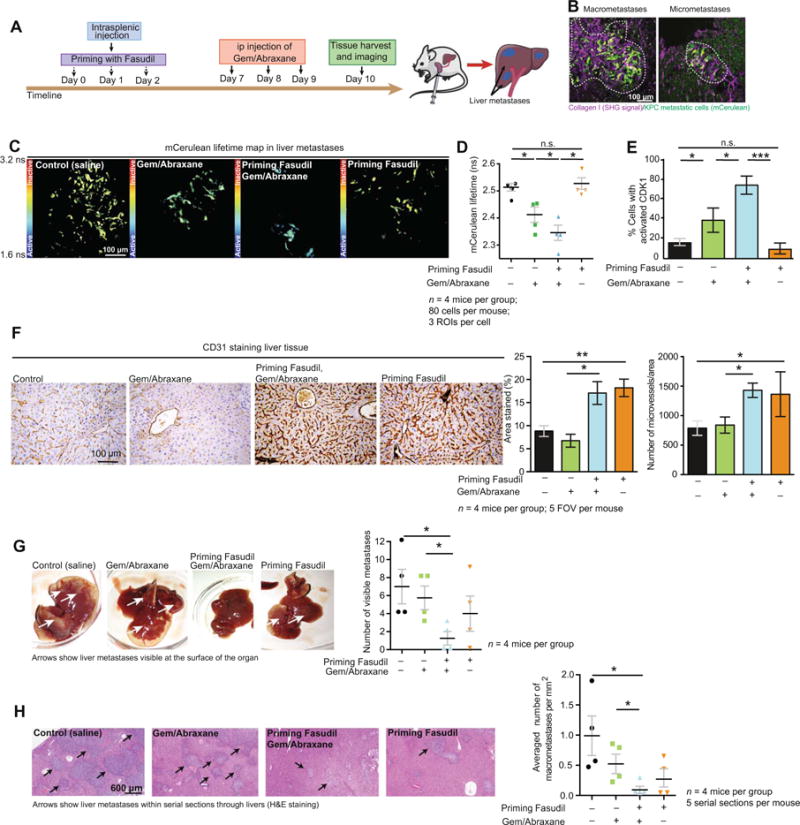
(A) Schematic representation of intrasplenic injection of KPC cells and treatment timeline. (B) Representative pictures of macrometastases (diameter, >200 μm) and micrometastases (diameter, <200 μm) identified via live imaging in liver metastases at the experimental end point. White dashed lines delimitate macrometastases and micrometastases. (C) Representative mCerulean lifetime maps in liver metastases. (D) Quantification of mCerulean lifetime. (E) CDK1 activity in metastatic KPC cells upon treatment with control (saline);Gem/Abraxane alone; Fasudil, then Gem/Abraxane;and Fasudil alone. n = 4 mice per group, 80 cells per mouse, three ROIs per cell. (F) CD31 staining and quantification of area positively stained and microvessel density in liver tissue upon treatment with control (saline);Gem/Abraxane alone;Fasudil, then Gem/Abraxane;and Fasudil alone. n = 4 mice per group and five FOVs per mouse. (G) Pathological examination of liver metastases and quantification of visible metastases on the surface of the tissue. White arrows point at KPC metastases visible at the surface of the organ. (H) Quantification of liver macrometastases normalized to liver surface area in hematoxylin and eosin (H&E) serial sections. Black arrows point at macrometastases in the liver tissue. Results are means ± SEM. n = 4 mice per group and five serial sections per organ (100-mm step). P values were determined by nonparametric ANOVA test with Holm-Sidak correction for multiple comparisons.
Priming hinders extravasation and spread in the liver while sensitizing cells to shear stress and reducing anchorage-independent growth
To identify pancreatic tumor cells that had recently extravasated in the liver and breached the matrix surrounding blood vessels, we stained liver tissues with Elastica van Gieson and PDX-1 (Fig. 6, A and B). Quantification of extravasation events revealed that priming with Fasudil impaired extravasation compared to control and Gem/Abraxane treatment (Fig. 6, A to C), suggesting that Fasudil may act as a potential antiadhesion drug. This mirrors recent work showing that targeting early “homing” events in the liver using anti-adhesion drugs can have additional advantages over cytotoxic targeting alone (58,59). To address the anti-adhesion effect of Fasudil in a more controlled platform, we used cell-derived matrices (CDMs) (60, 61). Here, priming with Fasudil during ECM deposition and remodeling reduced ECM integrity (fig. S4, B to D). KPC cells were then seeded onto CDMs in the presence or absence of Gem/Abraxane (Fig. 6, D and E), and time-lapse tracking (fig. S4E) revealed a marked reduction in KPC cell adhesion in Fasudil-primed conditions over time compared to control and Gem/Abraxane counterparts (Fig. 6E, compare black and orange lines). In addition, the cytotoxic effects of Gem/Abraxane combined with Fasudil’s anti-adhesion effect increased the reduction in KPC attachment and viability (Fig. 6E, compare green to blue lines at 24 hours). This was in line with FLIM-FRET analysis of CDK1 on CDMs and in the liver in vivo (Fig. 5C and fig. S4F), where Fasudil priming enhanced response to Gem/Abraxane.
Fig. 6. Priming with Fasudil decreases KPC cell extravasation and adhesion in secondary sites and makes KPC cells more sensitive to shear stress.
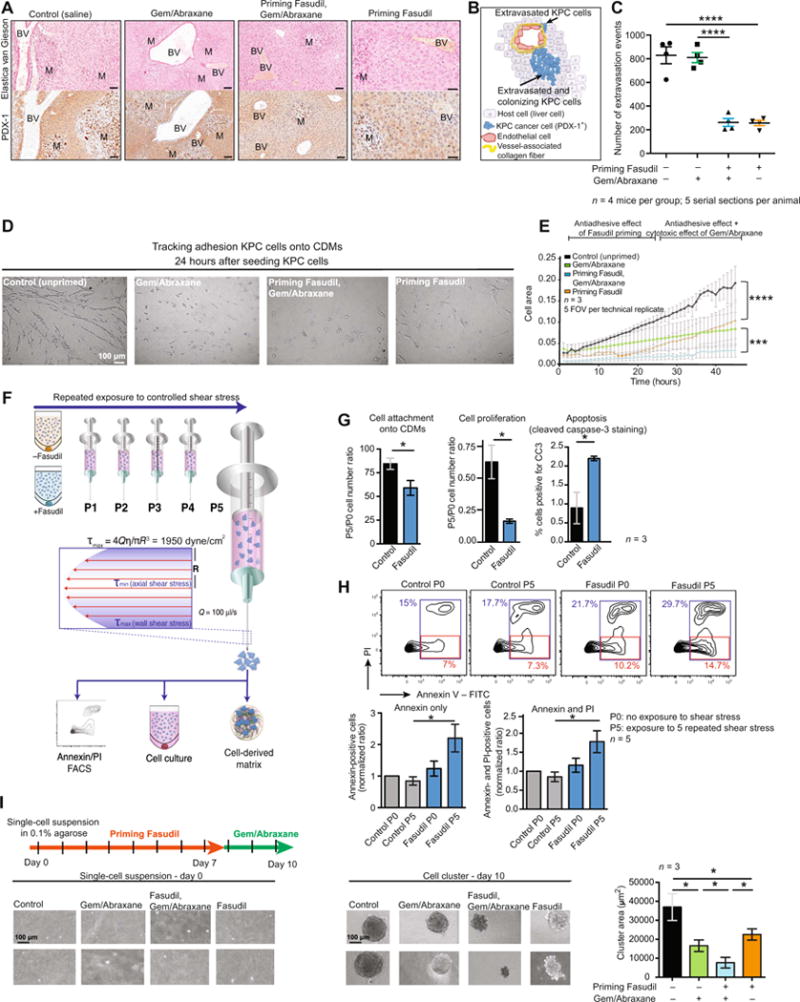
(A) Representative images of Elastica van Gieson (marker of vessel-associated collagen fibers) staining and PDX-1 (marker of PC cells) IHC staining in liver tissues after treatment with control (saline); Gem/Abraxane alone; Fasudil, then Gem/Abraxane; and Fasudil alone. BV, blood vessel; M, metastasis. Scale bars, 100 mm. (B) Schematic representation of KPC cells that breached through the matrix surrounding blood vessels observed in liver tissue.(C) Quantification of KPC extravasation in the liver upon treatment with control (saline); Gem/Abraxane alone; Fasudil, then Gem/Abraxane; and Fasudil alone. n = 4 mice per group and five serial sections per mouse (100-mm step). (D) Representative phase images of KPC cells 24 hours after seeding onto CDMs primed or unprimed with Fasudil and treated with or without Gem/Abraxane. (E) Automated quantification of KPC cell attachment to CDMs over time. n = 3 biological repeats with three CDMs per condition and per repeat and five FOVs per CDM. (F) Schematic representation of fluid shear stress assay, adapted from (63). (G) Quantification of cell attachment onto CDMs, growth, and apoptosis after shear stress for KPC cells ± Fasudil pretreatment. P0, cells not subjected to shear stress; P5, cells subjected to five consecutive exposures to shear stress. Y-axis values (P5/P0 ratios) are values for cells subjected to five exposures to shear stress divided by values for cells not exposed to shear stress. n = 3 biological repeats with one CDM per condition and per repeat. (H) FACS analysis of annexin and propidium iodide (PI) amounts in KPC cells 24 hours after shear stress. Representative FACS plots for one of the replicates (top) and quantification of five replicates with means ± SEM (bottom). Annexin V-positive population was defined as FITC+ (fluorescein isothiocyanate-positive)/PI- (red gate), and all dead cells were defined as FITC+ and/or PI+ (blue gate). (I) Schematic representation of treatment timeline for AIG assay with representative images and quantification of AIG of KPC cells cultured ± Fasudil and ± Gem/Abraxane. n = 3 biological repeats with three wells per condition and per repeat. Results are means ± SEM. P values were determined by unpaired, nonparametric t test with Mann-Whitney U correction (comparison between two groups) and nonparametric ANOVA test with Holm-Sidak correction for multiple comparisons.
To further assess the observed reduction of liver colonization upon Fasudil priming, we examined the viability of cells in response to shear stress and anchorage-independent growth (AIG), which circulating tumor cells are subjected to in vivo. KPC cells were primed with Fasudil and exposed to controlled shear stress as previously described (Fig. 6F) (62) and then plated on CDMs to measure their ability to attach, survive, or proliferate after shear stress. Cell attachment on CDMs and cell growth were reduced upon treatment with Fasudil, with a concomitant increase in apoptosis (Fig. 6G), as confirmed by FACS analyses (Fig. 6H). These data, along with reduced long-term AIG in Fasudil-primed conditions (Fig. 6I), point to a role of ROCK inhibition in sensitizing cells to shear stress in the systemic circulation and suggest an additional way in which Fasudil can impinge on metastasis in PC (10,13, 63, 64).
Fasudil priming reduces cell streaming and metastatic niche remodeling
To assess whether priming with Fasudil affects KPC cell movement, we performed time-lapse monitoring of KPC cell streaming on CDMs primed with or without Fasudil and quantified cell movement anisotropy as a readout of coordinated cell movement (Fig. 7A and movie S9) (65). In control conditions, concerted streams of cells moving in a coordinated, organized manner were observed (Fig. 7A, green arrows) (65, 66). This was reduced in cells seeded on Fasudil-primed matrices, where noncoordinated cell movement was evident over time (Fig. 7, A and B), and was further supported by a clear decrease in cell stream width compared to control (Fig. 7C). We next studied whether Fasudil priming altered metastatic spread patterns in the liver using IHC analysis of PDX-1-stained serial sections. Here, we observed different morphology of metastases upon priming with Fasudil. In control and Gem/Abraxane conditions, we identified elongated and malleable emboli of KPC cells spreading throughout the liver tissue [Fig. 7, D (top) and E], whereas in Fasudil and Gem/Abraxane or Fasudil alone conditions, metastases were round, and elongated emboli were ablated [Fig. 7, D (top) and E]. This observation suggests that Fasudil priming and manipulation of the ECM during the early stages of metastatic colonization (59) alter spread in the host tissue, as evidenced by an overall reduction in metastatic burden in these conditions (Fig. 5, G and H).
Fig. 7. Priming with Fasudil impairs metastatic spread and remodeling of secondary sites.
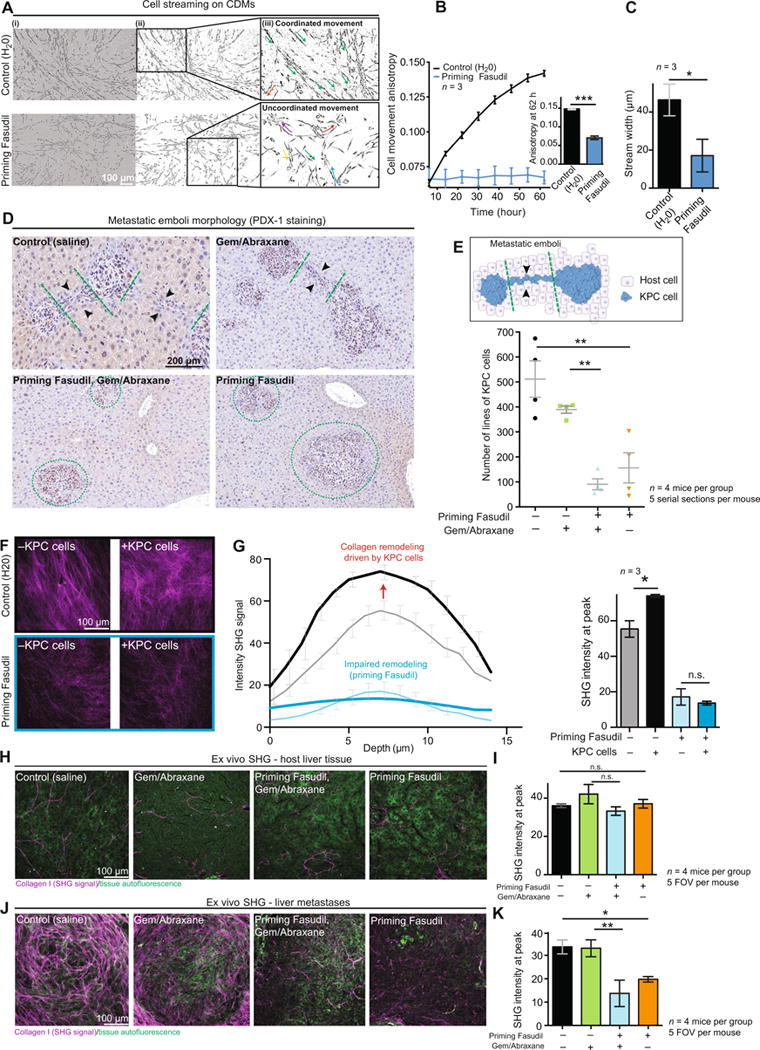
(A) Representative images of KPC cell streams on CDMs primed with control or with Fasudil 62 hours after seeding. (i) Phase image, (ii) cell shape mask, and (iii) inset with arrows showing cell stream directionality. (B) Quantification of KPC cell movement anisotropy over time using FibrilTool (65). (C) Cell stream width 62 hours after seeding on CDMs. n = 3 biological repeats with three CDMs per condition and per repeat. (D) Representative images of PDX-1 staining, identifying different morphologies of metastatic emboli upon treatment with control (saline); Gem/Abraxane; Fasudil, then Gem/Abraxane; or Fasudil alone. Black arrowheads point at lines of KPC cells. Green dashed lines delimit individual metastases. (E) Schematic representation of lines of KPC cells observed by IHC analysis in the liver (top) and quantification of the number of lines on serial sections upon treatment with control (saline); Gem/Abraxane; Fasudil, then Gem/Abraxane; or Fasudil alone. n = 4 mice and five serial sections per mouse (100-μm step). (F) Representative maximum projection of SHG imaging. (G) Quantification of SHG signal derived from primed (blue lines) or unprimed (black lines) CDMs before seeding KPC cells (thin lines) and 48 hours after seeding KPC cells (bold lines). n = 3 independent biological repeats with one CDM per condition and per repeat. (H) Representative maximum intensity projections of SHG imaging. (I) Quantification of SHG signal intensity in host liver tissue upon treatment with control (saline); Gem/Abraxane; Fasudil, then Gem/Abraxane; or Fasudil alone. n = 4 mice per group and five FOVs per mouse. (J) Representative maximum intensity projections of SHG imaging. (K) Quantification of SHG signal intensity in liver metastases upon treatment with control (saline); Gem/Abraxane; Fasudil, then Gem/Abraxane; or Fasudil alone. n = 4 mice per group and five FOVs per mouse. Results are means ± SEM. P values were determined by unpaired, nonparametric t test with Mann-Whitney U correction (comparison between two groups) and nonparametric ANOVA test with Holm-Sidak correction for multiple comparisons.
ECM remodeling can also provide cancer cells with a “fibrotic refuge” (67), supporting tumor growth and limiting chemotherapeutic targeting (68). Because Fasudil priming improved Gem/Abraxane effectiveness in the liver (Fig. 5, C to E), we assessed the ability of KPC cells to remodel the ECM upon priming with Fasudil. Using CDMs as a model system, we found that KPC cells can remodel the matrix of unprimed CDMs, as measured by increased SHG signal over time (Fig. 7, F and G, black lines). This was impaired for KPC cells seeded on Fasudil-primed CDMs, where SHG intensity did not significantly increase after seeding of KPC cells (Fig. 7, F and G, blue lines). Moreover, KPC cell protrusions on primed CDMs were also disrupted compared to control (fig. S4, G to I), in line with earlier observations, and this prompted us to assess the amount of fibrillar collagen within liver metastases. Here, we found that remodeling of the metastatic niche was impaired upon priming with Fasudil (Fig. 7, H to K, and fig. S4J). Hence, priming may make secondary tissues less permissive to the establishment of metastases and expose cancer cells to a less favorable environment (67). This may partly explain the enhanced response to chemotherapy at secondary sites observed in Fasudil-primed conditions.
Disruption of SRC signaling and actomyosin cytoskeleton dynamics with Fasudil may sensitize KPC cells to chemotherapy
In line with the reported role of ECM-integrin outside-in signaling in cancer (47, 69), we assessed whether the integrin-SRC signaling axis is altered upon priming with Fasudil. Using a SRC-FRET biosensor (fig. S5A) (70) in 3D organotypic matrices, we found that SRC activity is reduced after manipulation of the ECM with Fasudil (fig. S5, B and C) (70). Intravital imaging in subcutaneous KPC tumors also confirmed inhibition of SRC signaling in response to Fasudil priming in vivo (fig. S5, D and E). This is in line with the recent role that SRC has been proposed to play in both sensitizing cancer cells to chemotherapy (71-74) and driving ECM-dependent events in PC (75-77). Additionally, ROCK inhibitors can induce mitotic defects (78-81), and we therefore assessed early-to-mid and late mitosis in KPC subcutaneous tumors. This revealed that although we do not see any significant change of cell proliferation or death upon Fasudil priming (Fig. 4E and fig. S3, D and E), ROCK inhibition induces an accumulation of cells in late mitosis and multinuclear cells (fig. S5, F and G), thereby potentially making cells more responsive to subsequent treatment with antimicrotubule chemotherapy. Together, our data in cells from the KPC model suggest that priming delays PC progression in both primary and secondary sites. We therefore sought to translate our findings to patient-derived tumors.
Patient-derived models reveal potential for tailored, short-term priming strategy in PC
Because PC is highly heterogeneous (29, 82), we next assessed whether fibrillar collagen abundance could influence tumor response to priming with Fasudil before chemotherapy. SHG analysis of PDX samples from the APGI cohort (29) allowed us to identify TKCC2 as an example of a –low ECM PDX” and TKCC5 as an example of a “high ECM PDX” (Fig. 8A). Patient-derived cancer cell lines (PDCLs) isolated from the respective PDXs (29) were then subcutaneously reinjected into mice (Fig. 8B). Subsequent SHG imaging revealed that reinjected PDCLs recapitulated many of the original features of the collagen matrix (Fig. 8B), demonstrating the capacity of PDCLs to remodel the tissue and to “activate” host fibroblasts to maintain inherent features of the tumor.
Fig. 8. ECM-stratified patient- derived tumors respond in a graded manner to priming strategy.
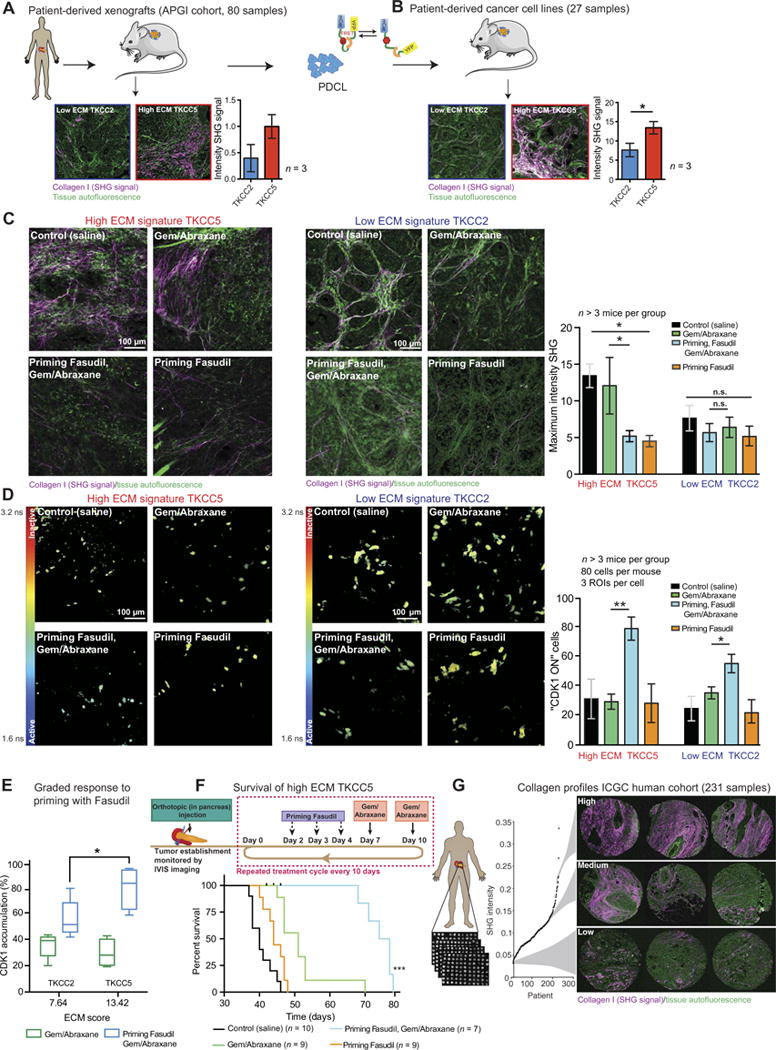
(A) Schematic representation of PDX establishment and representative maximum projections of SHG imaging in high ECM and low ECM PDXs from the APGI PDX cohort. n = 3 cores per group. (B) Patient-derived cell lines were isolated from PDXs, purified, and reinjected subcutaneously into nonobese diabetic-severe combined immunodeficient mice. Representative maximum intensity projections of SHG imaging and quantification of high ECM and low ECM PDCL xenografts. n = 3 tumors per group. (C) Representative maximum intensity projections of ex vivo SHG imaging in subcutaneous TKCC5 (left) and TKCC2 (right) xenografts. Quantification of SHG signal intensity in subcutaneous TKCC5 and TKCC2 xenografts upon treatment with control (saline), Gem/ Abraxane, Fasudil and Gem/ Abraxane, or Fasudil alone. (D) Rep resentative images and quantification of in vivo measurements of mCerulean lifetime and CDK1 activity in subcutaneous TKCC5 (left) and TKCC2 (right) xenografts upon treatment with control (saline), Gem/Abraxane, Fasudil and Gem/ Abraxane, or Fasudil alone. (E) Graded response to priming before Gem/ Abraxane compared to Gem/ Abraxane alone in TKCC2 and TKCC5 xenografts. Results are means ± SEM. TKCC2: n = 5 mice per group; TKCC5: n = 4 for control, n = 4 for Gem/Abraxane, n = 4 for Fasudil, then Gem/ Abraxane, n = 3 for priming with Fasudil. For SHG analysis, five FOVs per mouse were imaged; for FLIM-FRET analysis, 80 cells per mouse with three ROIs per cell were quantified. (F) Representative schematic of orthotopic injection of TKCC5-luciferase cells and treatment cycle. Kaplan-Meier analysis of survival for mice orthotopically injected with TKCC5 cancer cells and undergoing treatment cycles with control (saline); Gem/Abraxane; Fasudil, then Gem/Abraxane; and Fasudil alone. Control, n = 10 mice; Fasudil priming alone, n = 9 mice; Gem/Abraxane, n = 9 mice; Fasudil, then Gem/Abraxane, n = 7 mice. (G) Schematic representation of in-house automated SHG analysis of the ECM in the ICGC human tissue microarray (TMA) cohort, with examples of SHG signal in cores (triplicates) from patients with high, medium, and low SHG signal. Results are means ± SEM. P values were determined by unpaired, nonparametric t test with Mann-Whitney U correction (comparison etween two groups) and nonparametric ANOVA test with Holm-Sidak correction for multiple comparisons.
To assess PDCL response to the priming regimen, we first established patient-personalized organotypic matrices, where cancer-associated fibroblasts (CAFs), “activated” by the respective patient-derived tumor cells in vivo, were isolated from PDXs, embedded in collagen, and characterized by IHC staining with pancytokeratin, a-smooth muscle actin, and fibroblast activation protein (figs. S6 and S7). After remodeling of the matrix by CAFs, matched patient-derived cancer cells were seeded on top of the contracted matrices (figs. S6, A and C, and S7, A and C), as previously performed with KPC cells (Figs. 2 and 3). Priming with Fasudil impaired the ability of the “high ECM” TKCC5 CAFs to remodel collagen, as demonstrated by SHG analysis (fig. S6, B and D to F), and resulted in decreased cancer cell invasion (fig. S6, G and H) and increased response to Gem/Abraxane treatment (fig. S6, I and J). Conversely, priming had no effect on “low ECM” TKCC2 CAF-driven collagen contraction (fig. S7, B and D to F) and did not modify response to chemotherapy (fig. S7, G and H). This prompted us to assess TKCC2 and TKCC5 response to priming with Fasudil in live tumors.
PDCLs were engineered to express the CDK1-FRET biosensor, and in vitro FLIM-FRET measurements confirmed that the biosensor can distinguish changes in CDK1 activity in both lines (figs. S6, K and L, and S7, I and J), as previously achieved with KPC cells (fig. S2). CDK1-PDCLs were subcutaneously injected into mice, and when tumors were fully established (average tumor volume, 180 mm3, as previously described for KPC tumors; figs. S6M and S7K), mice were subjected to 3 days of priming with Fasudil before administration of Gem/Abraxane and intravital imaging (figs. S6M and S7K, timelines). Although SHG analysis demonstrated a reduction of fibrillar collagen abundance in the high ECM TKCC5 upon manipulation with Fasudil (Fig. 8C), priming had no effect on the matrix of low ECM TKCC2 (Fig. 8C). Intravital analysis of CDK1 activity in response to chemotherapy revealed that priming with Fasudil induces a robust increase of TKCC5 response to Gem/Abraxane in vivo (Fig. 8D, left). A moderate improvement of TKCC2 response to chemotherapy was observed (Fig. 8D, right), demonstrating a graded response to the priming strategy in relation to initial PDX collagen status (Fig. 8E).
Last, given the enhanced response to priming found in the high ECM TKCC5 model, we assessed the long-term effects of priming on therapeutic response in an orthotopic patient-derived model. Orthotopic injections of TKCC5 cells were conducted, and primary tumors were allowed to grow until average in vivo imaging system (IVIS) luciferase signal reached 5 × 107 photons/s (fig. S8A). Mice were subjected to cycles of priming with Fasudil before administration of Gem/Abraxane until the experimental end point (Fig. 8F and fig. S8A). Survival was increased by the priming regimen compared to treatment with Gem/ Abraxane alone (mean survival: Gem/Abraxane, 51 days; Fasudil priming and Gem/Abraxane, 75 days) (Fig. 8F). Mice receiving Fasudil priming before Gem/Abraxane also had a reduced primary tumor burden at the ethical end point (fig. S8B). Time to detection of metastasis observed via whole-body IVIS imaging was also delayed by the priming regimen compared to control (fig. S8, A and C).
Finally, we created an in-house automated SHG tool to analyze the International Cancer Genome Consortium (ICGC) cohort of patient biopsy samples (231ICGC samples) (Fig. 8G and fig. S8, D and E). Although we found no significant differences in survival or clinicopathological variables with regard to bulk patient ECM content (fig. S8F), in line with recent work (10), this rapid and label-free biopsy imaging approach could be used as a companion personalized biomarker for current stromal-based combination therapies in PC, where the initial amount of tumor ECM could guide whether a patient would be suitable for “transient” stromal intervention (Fig. 8, D to F) (12, 83).
DISCUSSION
Combined therapies and multimodal targeting have improved outcomes in multiple cancers (84), but little is known on how to streamline these methods to provide maximum benefit. Here, we demonstrate that transient priming of primary and secondary sites via ROCK inhibition improves chemotherapy efficacy and retards the onset of metastasis in PC.
Mechanoreciprocity and host tissue properties play a vital role in nascent tumors, from providing initial survival signals to driving cancer cell proliferation, invasion, and metastasis (85, 86). Here, we use a combination of 3D in vitro and in vivo platforms with mouse- and patient-derived PC cells to guide and optimize tissue manipulation scheduling in the context of Gem/Abraxane therapy. Our results suggest that shortterm inhibition of cellular tension in the whole tumor tissue before chemotherapy reduces cancer progression and improves response to treatment. Despite the distinct composition of the ECM in our different in vitro and in vivo models, our data reveal a similar effect in all settings, suggesting that this is a general phenomenon for multiple microenvironments. This suggests that depriving cancer cells of cues provided by the surrounding matrix at both primary and secondary sites renders them more sensitive to cytotoxic drugs, partly via impaired integrin signaling, as previously demonstrated in melanoma (67). This is in line with our recent findings that inhibition of collagen cross-linking via antibody-targeted therapy against lysyl oxidase in combination with Gem effectively impaired PC progression (12). ROCK inhibition also seems to induce accumulation of mitotic defects in vivo, potentially making cells more susceptible to subsequent chemotherapy (3).
The molecular fingerprint of human PC is highly heterogeneous (29, 82). High-throughput SHG analysis of the human ICGC cohort revealed heterogeneity in collagen profiles across patients. Using intravital monitoring of chemotherapy efficacy in patient-derived samples, we showed a graded response to Fasudil priming strategies relative to the initial fibrillar collagen profile in tissue biopsies. Our findings thereby suggest that high amounts of fibrillar collagen could potentially serve as a companion biomarker to identify patients who are likely to benefit from transient ECM manipulation before chemotherapy. This is in line with current clinical stroma-targeting therapies, where such companion biomarkers are being used for hyaluronic acid targeting, supporting matrix manipulation therapy in a subpopulation of patients (83). Fine-tuned ECM-based stratification could therefore facilitate a more tailored therapeutic approach to matrix manipulation therapy in PC and could be used as an alternative to the “all patients” chronic stromal targeting approach in this disease.
Although we demonstrate disruption of the physical properties of the ECM upon manipulation with Fasudil, we also reveal that repurposing Fasudil’s clinical vasodilatory effects (28) through transient priming before chemotherapy improves short-term tumor blood perfusion and, thereby, drug delivery. This is in line with its current use for the treatment of cerebral vasospasm after subarachnoid hemorrhage in Japan (87-89) while avoiding any long-term toxic effects that could arise from chronic treatment (36, 39). Therefore, targeting ECM-dependent and ECM-independent changes in cancer appears to be an important advantage of transient ROCK inhibition compared to chronic ECM targeting alone (39, 90, 91) and warrants further consideration with regard to repurposing the off-patent drugs in PC (92).
Monitoring the onset of hepatic micrometastasis revealed that manipulation of tissue at secondary sites impairs liver colonization. Analyses on CDMs and in the liver demonstrated that priming with Fasudil alters KPC cell attachment, coordinated movement, and malleable spread of the tumor emboli, potentially via changes of the mechanical and biochemical properties of the host ECM, thereby altering ECM-cell interactions and durotaxis (93, 94). Furthermore, the Rho-ROCK-LIMK pathway drives path generation by leading tumor cells (66, 95), and hence, ROCK inhibition with Fasudil may inhibit path generation and coordinated cell movement of tumor cells in this context (95, 96). Additionally, priming with Fasudil may decrease cell survival in the bloodstream via impaired tolerance to shear stress (97). Whereas previous studies have reported conflicting findings on the effects of ROCK inhibition on cell invasion (98-100), short-term treatment with Fasudil did not show adverse effects, unlike chronic ROCK targeting. We also reveal that Fasudil priming impairs the ability of metastasized cells to create a fibrotic refuge, potentially enhancing their vulnerability to subsequent chemotherapy (67). We propose that instead of completely ablating stroma–cancer cell feedback, transient priming of tissue by Fasudil before chemotherapy can impair cancer cell establishment, survival, growth, and spread at multiple sites in vivo while minimizing negative effects that may arise from chronic treatment.
Although Fasudil monotherapy or combinations with selected cardiovascular agents have previously been shown to be safe in humans by numerous large-scale studies (87-89), this agent is yet to be combined with chemotherapies in the clinic. A phase 1 clinical trial would determine the safety of transient “priming” regimen with Fasudil before treatment with Gem/Abraxane in patients. Our study used patient TMA material from the ICGC cohort (82), which is composed of largely early-stage tumors (I, IIa, and IIb) and hence does not equally cover the full spectrum of the disease. Validation studies would ideally include a cohort of more metastatic disease to fully appreciate the utility of our proposed SHG companion biomarker. Additionally, given the dual effects of Fasudil priming on both the ECM and the tumor vasculature, combination of both SHG and CD31-based biomarkers might further facilitate the identification of patients who could benefit from priming before chemotherapy.
Newer ROCK inhibitors such as K-115 (Ripasudil), which have recently entered the clinic for the treatment of glaucoma (101,102) or are currently in the clinical testing pipeline, including phase 2 clinical trial for glaucoma (AMA0076) (103), or CCT129254 or AT13148, which reduce metastasis in other cancers (104), could also have similar applications (105). Patient presentation in PC ranges from early local invasion at the time of diagnosis to late metastatic disease (106). We have demonstrated multisite benefits using priming in combination with standard-of-care therapy (summarized in fig. S9) and envision its potential use to treat PC at several stages.
MATERIALS AND METHODS
Study design
This study interrogates the effect of transient manipulation of tissue tension via ROCK inhibition on PC progression and response to Gem/Abraxane. In vitro organotypic and CDM experiments were performed in independent biological triplicates, with three technical replicates per repeat and per treatment group. For in vivo experiments, numbers of mice used for each model are outlined in corresponding figures and figure legends. In vivo priming started when tumor volume reached 180 mm3 (average size) or 7 × 105 photons/s (average IVIS signal). Mice for which tumor volume or IVIS signal was 10% lower or higher than the average value before enrollment were excluded from analysis.
FLIM-FRET analysis of CDK1 and SRC activity in vitro was conducted in >30 cells per group in three independent biological repeats. In vivo analysis of CDK1 and SRC was performed in 80 cells per mouse, with measurements in three subcellular areas per cell to generate an average value for CDK1 specifically, whereas measurements of lifetime in the whole cell (one value per cell) were performed for analysis of SRC activity.
IHC, SHG, picrosirius red, and GLCM analyses were conducted on three representative FOVs in organotypic matrices and CDMs and in five representative FOVs in subcutaneous xenograft and intrasplenic experiments. Metastatic burden, extravasation, and metastasis morphology in the liver were analyzed in serial sections (five sections per organ with a 100-μm step). Experimental end points for survival experiments were in compliance with Garvan Ethics Committee guidelines (13/17, 14/06, 14/11, and 16/13 protocols).
Statistical analysis
Unless stated otherwise, P values were determined by unpaired, nonparametric t test with Mann-Whitney U correction (comparison between two groups) or nonparametric ANOVA test with Holm-Sidak correction for multiple comparison (more than two groups). Kaplan-Meier curves were compared using a log-rank Mantel-Cox test. Exact P values for all figures are provided in table S1.
Supplementary Material
Fig. S1. ROCK inhibition with Fasudil and Y-27632 impairs ECM integrity.
Fig. S2. The CDK1-FRET biosensor distinguishes changes in CDK1 activity and is a surrogate for cell cycle arrest.
Fig. S3. Priming with Fasudil disrupts ECM remodeling, inhibits ROCK signaling, and improves Gem/Abraxane efficacy in vivo.
Fig. S4. Priming with Fasudil influences liver vasculature, cell attachment to CDMs, response to chemotherapy, and remodeling of the ECM.
Fig. S5. Priming with Fasudil results in decreased SRC activity and defects in mitosis.
Fig. S6. High ECM TKCC5 patient model responds to priming strategies in a 3D patient-personalized organotypic matrix.
Fig. S7. Low ECM TKCC2 patient model does not respond to priming strategies in a 3D patient-personalized organotypic matrix.
Fig. S8. TKCC5 orthotopic tumors respond to priming, and SHG does not predict survival.
Fig. S9. Priming with Fasudil uncouples PC progression.
Table S1. List of P values.
Table S2. Details of antibodies used for the study.
Movie S1. 4D monitoring of fibroblast-ECM interactions upon treatment with vehicle and Fasudil.
Movie S2. Live FLIM-FRET imaging of the CDK1 biosensor in KPC cells in response to Abraxane and Abraxane + RO3306.
Movie S3. Live FLIM-FRET imaging of the CDK1 biosensor in KPC cells in interphase and mitosis.
Movie S4. Live FLIM-FRET monitoring of CDK1 activity in KPC cells actively interacting with an organotypic matrix.
Movie S5. Intravital FLIM-FRET imaging of subcutaneous xenografts with KPC-CDK1 cells and imaging of fibrillar collagen.
Movie S6. Intravital monitoring of CDK1 accumulation in subcutaneous KPC tumors.
Movie S7. Intravital imaging of quantum dots circulating through tumor-associated vasculature and diffusing into tumor tissue upon priming with Fasudil.
Movie S8. Imaging of liver tissue with metastatic KPC cells expressing the CDK1 biosensor forming macrometastases and micrometastases.
Movie S9. Time-lapse tracking of collective cell streaming on CDMs unprimed or primed with Fasudil.
Acknowledgments
We thank H. Bennett and K. Murphy for critical reading of the manuscript.
Funding:
This study was supported by the National Health and Medical Research Council (NHMRC), Cancer Council NSW, Cancer Australia, Tour de Cure grants, Cancer Institute NSW, the Australian Research Council Future Fellowships, The Avner Pancreatic Cancer Foundation, Lens Ainsworth and Philip Hemstritch Pancreatic Cancer Fellowships, Sydney Catalyst scholarships, Royal Australasian College of Physicians Research Foundation scholarships, Pancare Australia stipend, NHMRC scholarships, a Cancer Research UK core grant, and a Patricia Helen Guest Fellowship. T.R.J.E. received funding for clinical trials (payable to the Institution) from Celgene, Bristol-Myers Squibb, AstraZeneca, Bayer, Roche, and Basilea.
Footnotes
Author contributions:
Investigation, validation, and formal analysis: C.V., V.T.C., S.C.W., M.C.L., D.H., P.M., S.N.W., G.d.M.-N., A.A.S.A., S.T.G., A.M., A.H.A., R A.C., J.R.W.C., M.N., M. Pinese, A. Boulghourjian, A.Z., A.M.N., N.C., R.J., W.W., A.C., A.S., A.D., D.F., C.H., M.G.-L., D.G.-O., N.L.E.H., R.W., A.L.J., J.S.S., L.C., T.R.C., A.J.G., A. Burgess, and APGI. Conceptualization and funding acquisition: C.V., S.C.W., D.H., T.P., A. Burgess, O.J.S., A.V.B., M.K.-C., Y.W., M.S.S., T.R.C., J.P.M., C.J.O., M. Pajic, and P.T. Methodology: C.V., S.C.W., M.C.L., D.H., A.M., S.N.W., G.d.M.-N., A. Boulghourjian, A.Z., E.J.G., S.T.G., R.P.H., Y.W., M.S.S., T.R.J.E., K.I.A., A. Burgess,T.R.C.,J.P.M., M. Pajic, and P.T. Writing and visualization: C.V., P.T., and M. Pajic.
Competing interests:
The authors declare that they have no competing interests.
Members of APGI Consortium:
Amber L. Johns1, Anthony J. Gill1,2, David K. Chang1,3, Christopher J. Scarlett1, Skye H. McKay1, Lorraine A. Chantrill1,4, Venessa T. Chin1, Angela Chou1, Mark J. Cowley1, Jeremy L. Humphris1, Marina Pajic1, Angela Steinmann1, Mehreen Arshi1, Ali Drury1, Danielle Froio1, Ashleigh Morgan1, Paul Timpson1, David Herrmann1, Claire Vennin1, Sean C. Warren1, Mark Pinese1,Jianmin Wu1, Andreia V. Pinho1, Nicola Waddell5, Ann-Marie Patch5, John V. Pearson5, Sarah Song5, Suzanne Manning5, Shivangi Wani5, Milena Gongora5, Matthew Anderson5, Oliver Holmes5, Conrad Leonard5, Darrin Taylor5, Scott Wood5, Christina Xu5, Katia Nones5, J. Lynn Fink5, Felicity Newell5, Michael Quinn5, Stephen Kazakoff5, Nick Waddell5, Keerthana Krisnan5, David Wood5, Sean M. Grimmond2, Oliver Hofmann2, Peter J. Wilson6, Angelika Christ6,Tim Bruxner6, Jaswinder S. Samra7, AnthonyJ. Gill7, Nick Pavlakis7, Hilda A. High7, Ray Asghari8, Neil D. Merrett8, Darren Pavey8, Amitabha Das8, Peter H. Cosman9, Kasim Ismail9, Chelsie O’Connnor9, Alina Stoita4, David Williams4, Allan Spigellman4, Vincent W. Lam10, Duncan McLeod10, Henry C. Pleass10, Adnan M. Nagrial1,10, Judy Kirk10, Virginia James10, James G. Kench11, Peter Grimison11, Caroline L. Cooper11, Charbel Sandroussi11, Michael Crawford11, Annabel Goodwin9,11, R. Scott Mead1,12, Katherine Tucker12, Lesley Andrews12, Michael Texler13, Cindy Forest13, Andrew Laycock13, Krishna P. Epari13, Mo Ballal13, David R. Fletcher13, Sanjay Mukhedkar13, Nigel A. Spry14, Bastiaan DeBoer14, Ming Chai14, Nikolajs Zeps15, Maria Beilin15, Kynan Feeney15, Nan Q. Nguyen16, Andrew R. Ruszkiewicz16, Chris Worthley16, John Chen17, Mark E. Brooke-Smith17, Virginia Papangelis17, Andrew D. Clouston18, Patrick Martin18, Andrew P. Barbour19, Thomas J. O’Rourke19, Jonathan W. Fawcett19, Kellee Slater19, Michael Hatzifotis19, Peter Hodgkinson19, Christopher Christophi20, Mehrdad Nikfarjam20, James R. Eshleman21, Ralph H. Hruban21, Richard D. Schulick21, Christopher L. Wolfgang21, Richard A. Morgan21, Mary Hodgin21, Aldo Scarpa3, Rita T. Lawlor3, Stefania Beghelli3, Vincenzo Corbo3, Maria Scardoni3, Claudio Bassi3, Andrew V. Biankin1,22, David K. Chang1,22, Peter Bailey22, Sancha Martin22, Elizabeth A. Musgrove22, Marc D. Jones22, Craig Nourse22, Nigel B. Jamieson22
SUPPLEMENTARY MATERIALS
www.sciencetranslationalmedicine.org/cgi/content/full/9/384/eaai8504/DC1(/p)(p)Materials and Methods
REFERENCES AND NOTES
- 1.Goldstein D, El-Maraghi RH, Hammel P, Heinemann V, Kunzmann V, Sastre J, Scheithauer W, Siena S, Tabernero J, Teixeira L, Tortora G, Van Laethem JL, Young R, Penenberg DN, Lu B, Romano A, Von Hoff DD. nab-Paclitaxel plus gemcitabine for metastatic pancreatic cancer: Long-term survival from a phase III trial. J Natl Cancer Inst. 2015;107:dju413. doi: 10.1093/jnci/dju413. [DOI] [PubMed] [Google Scholar]
- 2.Von Hoff DD, Ervin T, Arena FP, Chiorean EG, Infante J, Moore M, Seay T, Tjulandin SA, Ma WW, Saleh MN, Harris M, Reni M, Dowden S, Laheru D, Bahary N, Ramanathan RK, Tabernero J, Hidalgo M, Goldstein D, Van Cutsem E, Wei X, Iglesias J, Renschler MF. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med. 2013;369:1691–1703. doi: 10.1056/NEJMoa1304369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rath N, Olson MF. Rho-associated kinases in tumorigenesis: Re-considering ROCK inhibition for cancer therapy. EMBO Rep. 2012;13:900–908. doi: 10.1038/embor.2012.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sanz-Moreno V, Gaggioli C, Yeo M, Albrengues J, Wallberg F, Viros A, Hooper S, Mitter R, Feral CC, Cook M, Larkin J, Marais R, Meneguzzi G, Sahai E, Marshall CJ. ROCK and JAK1 signaling cooperate to control actomyosin contractility in tumor cells and stroma. Cancer Cell. 2011;20:229–245. doi: 10.1016/j.ccr.2011.06.018. [DOI] [PubMed] [Google Scholar]
- 5.Rodriguez-Hernandez I, Cantelli G, Bruce F, Sanz-Moreno V. Rho, ROCK and actomyosin contractility in metastasis as drug targets. F1000Res. 2016;5:783. doi: 10.12688/f1000research.7909.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kagawa Y, Matsumoto S, Kamioka Y, Mimori K, Naito Y, Ishii T, Okuzaki D, Nishida N, Maeda S, Naito A, Kikuta J, Nishikawa K, Nishimura J, Haraguchi N, Takemasa I, Mizushima T, Ikeda M, Yamamoto H, Sekimoto M, Ishii H, Doki Y, Matsuda M, Kikuchi A, Mori M, Ishii M. Cell cycle-dependent Rho GTPase activity dynamically regulates cancer cell motility and invasion in vivo. PLOS ONE. 2013;8:e83629. doi: 10.1371/journal.pone.0083629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Paszek MJ, Zahir N, Johnson KR, Lakins JN, Rozenberg GI, Gefen A, Reinhart-King CA, Margulies SS, Dembo M, Boettiger D, Hammer DA, Weaver VM. Tensional homeostasis and the malignant phenotype. Cancer Cell. 2005;8:241–254. doi: 10.1016/j.ccr.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 8.Sadok A, Marshall CJ. Rho GTPases: Masters of cell migration. Small GTPases. 2014;5:e29710. doi: 10.4161/sgtp.29710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Samuel MS, Lopez JI, McGhee EJ, Croft DR, Strachan D, Timpson P, Munro J, Schröder E, Zhou J, Brunton VG, Barker N, Clevers H, Sansom OJ, Anderson KI, Weaver VM, Olson MF. Actomyosin-mediated cellular tension drives increased tissue stiffness and β-catenin activation to induce epidermal hyperplasia and tumor growth. Cancer Cell. 2011;19:776–791. doi: 10.1016/j.ccr.2011.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Laklai H, Miroshnikova YA, Pickup MW, Collisson EA, Kim GE, Barrett AS, Hill RC, Lakins JN, Schlaepfer DD, Mouw JK, LeBleu VS, Roy N, Novitskiy SV, Johansen JS, Poli V, Kalluri R, Iacobuzio-Donahue CA, Wood LD, Hebrok M, Hansen K, Moses HL, Weaver VM. Genotype tunes pancreatic ductal adenocarcinoma tissue tension to induce matricellular fibrosis and tumor progression. Nat Med. 2016;22:497–505. doi: 10.1038/nm.4082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pickup MW, Mouw JK, Weaver VM. The extracellular matrix modulatesthe hallmarks of cancer. EMBO Rep. 2014;15:1243–1253. doi: 10.15252/embr.201439246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miller BW, Morton JP, Pinese M, Saturno G, Jamieson NB, McGhee E, Timpson P, Leach J, McGarry L, Shanks E, Bailey P, Chang D, Oien K, Karim S, Au A, Steele C, Carter CR, McKay C, Anderson K, Evans TRJ, Marais R, Springer C, Biankin A, Erler JT, Sansom OJ. Targeting the LOX/hypoxia axis reverses many of the features that make pancreatic cancer deadly: Inhibition of LOX abrogates metastasis and enhances drug efficacy. EMBO Mol Med. 2015;7:1063–1076. doi: 10.15252/emmm.201404827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Johansson-Percival A, Li ZJ, Lakhiani DD, He B, Wang X, Hamzah J, Ganss R. Intratumoral LIGHT restores pericyte contractile properties and vessel integrity. Cell Rep. 2015;13:2687–2698. doi: 10.1016/j.celrep.2015.12.004. [DOI] [PubMed] [Google Scholar]
- 14.Winkler F, Kozin SV, Tong RT, Chae SS, Booth MF, Garkavtsev I, Xu L, Hicklin DJ, Fukumura D, di Tomaso E, Munn LL, Jain RK. Kinetics of vascular normalization by VEGFR2 blockade governs brain tumor response to radiation: Role of oxygenation, angiopoietin-1, and matrix metalloproteinases. Cancer Cell. 2004;6:553–563. doi: 10.1016/j.ccr.2004.10.011. [DOI] [PubMed] [Google Scholar]
- 15.Ellenbroek SIJ, van Rheenen J. Imaging hallmarks of cancer in living mice. Nat Rev Cancer. 2014;14:406–418. doi: 10.1038/nrc3742. [DOI] [PubMed] [Google Scholar]
- 16.Conway JRW, Carragher NO, Timpson P. Developments in preclinical cancer imaging: Innovating the discovery of therapeutics. Nat Rev Cancer. 2014;14:314–328. doi: 10.1038/nrc3724. [DOI] [PubMed] [Google Scholar]
- 17.Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, Rustgi AK, Chang S, Tuveson DA. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005;7:469–483. doi: 10.1016/j.ccr.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 18.Hingorani SR, Petricoin EF, III, Maitra A, Rajapakse V, King C, Jacobetz MA, Ross S, Conrads TP, Veenstra TD, Hitt BA, Kawaguchi Y, Johann D, Liotta LA, Crawford HC, Putt ME, Jacks T, Wright CVE, Hruban RH, Lowy AM, Tuveson DA. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. 2003;4:437–450. doi: 10.1016/s1535-6108(03)00309-x. [DOI] [PubMed] [Google Scholar]
- 19.Kennedy AL, Morton JP, Manoharan I, Nelson DM, Jamieson NB, Pawlikowski JS, McBryan T, Doyle B, McKay C, Oien KA, Enders GH, Zhang R, Sansom OJ, Adams PD. Activation of the PIK3CA/AKT pathway suppresses senescence induced by an activated RAS oncogene to promote tumorigenesis. Mol Cell. 2011;42:36–49. doi: 10.1016/j.molcel.2011.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Walz S, Lorenzin F, Morton J, Wiese KE, von Eyss B, Herold S, Rycak L, Dumay-Odelot H, Karim S, Bartkuhn M, Roels F, Wustefeld T, Fischer M, Teichmann M, Zender L, Wei CL, Sansom O, Wolf E, Eilers M. Activation and repression by oncogenic MYC shape tumour-specific gene expression profiles. Nature. 2014;511:483–487. doi: 10.1038/nature13473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morran DC, Wu J, Jamieson NB, Mrowinska A, Kalna G, Karim SA, Au AYM, Scarlett CJ, Chang DK, Pajak MZ, Australian Pancreatic Cancer Genome Initiative. Oien KA, McKay CJ, Carter CR, Gillen G, Champion S, Pimlott SL, Anderson KI, Evans TRJ, Grimmond SM, Biankin AV, Sansom OJ, Morton JP. Targeting mTOR dependency in pancreatic cancer. Gut. 2014;63:1481–1489. doi: 10.1136/gutjnl-2013-306202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gavet O, Pines J. Activation of cyclin B1-Cdk1 synchronizes events in the nucleus and the cytoplasm at mitosis. J Cell Biol. 2010;189:247–259. doi: 10.1083/jcb.200909144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gavet O, Pines J. Progressive activation of CyclinB1-Cdk1 coordinates entry to mitosis. Dev Cell. 2010;18:533–543. doi: 10.1016/j.devcel.2010.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bekier ME, Fischbach R, Lee J, Taylor WR. Length of mitotic arrest induced by microtubule-stabilizing drugs determines cell death after mitotic exit. Mol Cancer Ther. 2009;8:1646–1654. doi: 10.1158/1535-7163.MCT-08-1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang Z, Kenny AE, Brito DA, Rieder CL. Cells satisfy the mitotic checkpoint in Taxol, and do so faster in concentrations that stabilize syntelic attachments. J Cell Biol. 2009;186:675–684. doi: 10.1083/jcb.200906150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Magidson V, He J, Ault JG, O’Connell CB, Yang N, Tikhonenko I, McEwen BF, Sui H, Khodjakov A. Unattached kinetochores rather than intrakinetochore tension arrest mitosis in taxol-treated cells. J Cell Biol. 2016;212:307–319. doi: 10.1083/jcb.201412139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Castedo M, Perfettini JL, Roumier T, Kroemer G. Cyclin-dependent kinase-1: Linking apoptosis to cell cycle and mitotic catastrophe. Cell Death Differ. 2002;9:1287–1293. doi: 10.1038/sj.cdd.4401130. [DOI] [PubMed] [Google Scholar]
- 28.Gupta V, Gupta N, Shaik IH, Mehvar R, McMurtry IF, Oka M, Nozik-Grayck E, Komatsu M, Ahsan F. Liposomal fasudil, a rho-kinase inhibitor, for prolonged pulmonary preferential vasodilation in pulmonary arterial hypertension. J Control Release. 2013;167:189–199. doi: 10.1016/j.jconrel.2013.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Waddell N, Pajic M, Patch A-M, Chang DK, Kassahn KS, Bailey P, Johns AL, Miller D, Nones K, Quek K, Quinn MCJ, Robertson AJ, Fadlullah MZH, Bruxner TJC, Christ AN, Harliwong I, Idrisoglu S, Manning S, Nourse C, Nourbakhsh E, Wani S, Wilson PJ, Markham E, Cloonan N, Anderson MJ, Fink JL, Holmes O, Kazakoff SH, Leonard C, Newell F, Poudel B, Song S, Taylor D, Waddell N, Wood S, Xu Q, Wu J, Pinese M, Cowley MJ, Lee HC, Jones MD, Nagrial AM, Humphris J, Chantrill LA, Chin V, Steinmann AM, Mawson A, Humphrey ES, Colvin EK, Chou A, Scarlett CJ, Pinho AV, Giry-Laterriere M, Rooman I, Samra JS, Kench JG, Pettitt JA, Merrett ND, Toon C, Epari K, Nguyen NQ, Barbour A, Zeps N, Jamieson NB, Graham JS, Niclou SP, Bjerkvig R, Grutzmann R, Aust D, Hruban RH, Maitra A, Iacobuzio-Donahue CA, Wolfgang CL, Morgan RA, Lawlor RT, Corbo V, Bassi C, Falconi M, Zamboni G, Tortora G, Tempero MA, Australian Pancreatic Cancer Genome Initiative. Gill AJ, Eshleman JR, Pilarsky C, Scarpa A, Musgrove EA, Pearson JV, Biankin AV, Grimmond SM. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature. 2015;518:495–501. doi: 10.1038/nature14169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Olson MF, Sahai E. The actin cytoskeleton in cancer cell motility. Clin Exp Metastasis. 2009;26:273–287. doi: 10.1007/s10585-008-9174-2. [DOI] [PubMed] [Google Scholar]
- 31.Marei H, Carpy A, Woroniuk A, Vennin C, White G, Timpson P, Macek B, Malliri A. Differential Rac1 signalling by guanine nucleotide exchange factors implicates FLII in regulating Rac1-driven cell migration. Nat Commun. 2016;7:10664. doi: 10.1038/ncomms10664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cox TR, Bird D, Baker AM, Barker HE, Ho MWY, Lang G, Erler JT. LOX-mediated collagen crosslinking is responsible for fibrosis-enhanced metastasis. Cancer Res. 2013;73:1721–1732. doi: 10.1158/0008-5472.CAN-12-2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rawlins JM, Lam WL, Karoo RO, Naylor IL, Sharpe DT. Quantifying collagen type in mature burn scars: A novel approach using histology and digital image analysis. J Burn Care Res. 2006;27:60–65. doi: 10.1097/01.bcr.0000192266.14329.7b. [DOI] [PubMed] [Google Scholar]
- 34.Kular J, Scheer KG, Pyne NT, Allam AH, Pollard AN, Magenau A, Wright RL, Kolesnikoff N, Moretti PA, Wullkopf L, Stomski FC, Cowin AJ, Woodcock JM, Grimbaldeston MA, Pitson SM, Timpson P, Ramshaw HS, Lopez AF, Samuel MS. A negative regulatory mechanism involving 14-3-3ζ limits signaling downstream of ROCK to regulate tissue stiffness in epidermal homeostasis. Dev Cell. 2015;35:759–774. doi: 10.1016/j.devcel.2015.11.026. [DOI] [PubMed] [Google Scholar]
- 35.Cicchi R, Kapsokalyvas D, De Giorgi V, Maio V, Van Wiechen A, Massi D, Lotti T, Pavone FS. Scoring of collagen organization in healthyand diseased human dermis by multiphoton microscopy. J Biophotonics. 2010;3:34–43. doi: 10.1002/jbio.200910062. [DOI] [PubMed] [Google Scholar]
- 36.Provenzano PP, Cuevas C, Chang AE, Goel VK, Von Hoff DD, Hingorani SR. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell. 2012;21:418–429. doi: 10.1016/j.ccr.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Neesse A, Michl P, Frese KK, Feig C, Cook N, Jacobetz MA, Lolkema MP, Buchholz M, Olive KP, Gress TM, Tuveson DA. Stromal biology and therapy in pancreatic cancer. Gut. 2011;60:861–868. doi: 10.1136/gut.2010.226092. [DOI] [PubMed] [Google Scholar]
- 38.Cox TR, Erler JR. Fibrosis and cancer: Partner in crime or opposing forces? Trends Cancer. 2016;2:279–282. doi: 10.1016/j.trecan.2016.05.004. [DOI] [PubMed] [Google Scholar]
- 39.Jacobetz MA, Chan DS, Neesse A, Bapiro TE, Cook N, Frese KK, Feig C, Nakagawa T, Caldwell ME, Zecchini HI, Lolkema MP, Jiang P, Kultti A, Thompson CB, Maneval DC, Jodrell DI, Frost GI, Shepard HM, Skepper JN, Tuveson DA. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut. 2013;62:112–120. doi: 10.1136/gutjnl-2012-302529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, Madhu B, Goldgraben MA, Caldwell ME, Allard D, Frese KK, DeNicola G, Feig C, Combs C, Winter SP, Ireland-Zecchini H, Reichelt S, Howat WJ, Chang A, Dhara M, Wang L, Rückert F, Grutzmann R, Pilarsky C, Izeradjene K, Hingorani SR, Huang P, Davies SE, Plunkett W, Egorin M, Hruban RH, Whitebread N, McGovern K, Adams J, Iacobuzio-Donahue C, Griffiths J, Tuveson DA. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324:1457–1461. doi: 10.1126/science.1171362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rhim AD, Oberstein PE, Thomas DH, Mirek ET, Palermo CF, Sastra SA, Dekleva EN, Saunders T, Becerra CP, Tattersall IW, Westphalen CB, Kitajewski J, Fernandez-Barrena MG, Fernandez-Zapico ME, Iacobuzio-Donahue C, Olive KP, Stanger BZ. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell. 2014;25:735–747. doi: 10.1016/j.ccr.2014.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Özdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu CC, Simpson TR, Laklai H, Sugimoto H, Kahlert C, Novitskiy SV, De Jesus-Acosta A, Sharma P, Heidari P, Mahmood U, Chin L, Moses HL, Weaver VM, Maitra A, Allison JP, LeBleu VS, Kalluri R. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell. 2014;25:719–734. doi: 10.1016/j.ccr.2014.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee JJ, Perera RM, Wang H, Wu DC, Liu XS, Han S, Fitamant J, Jones PD, Ghanta KS, Kawano S, Nagle JM, Deshpande V, Boucher Y, Kato T, Chen JK, Willmann JK, Bardeesy N, Beachy PA. Stromal response to Hedgehog signaling restrains pancreatic cancer progression. Proc Natl Acad Sci U.S.A. 2014;111:E3091–E3100. doi: 10.1073/pnas.1411679111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Morton JP, Timpson P, Karim SA, Ridgway RA, Athineos D, Doyle B, Jamieson NB, Oien KA, Lowy AM, Brunton VG, Frame MC, Evans TRJ, Sansom OJ. Mutant p53 drives metastasis and overcomes growth arrest/senescence in pancreatic cancer. Proc Natl Acad Sci USA. 2010;107:246–251. doi: 10.1073/pnas.0908428107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Erami Z, Herrmann D, Warren SC, Nobis M, McGhee EJ, Lucas MC, Leung W, Reischmann N, Mrowinska A, Schwarz JP, Kadir S, Conway JRW, Vennin C, Karim SA, Campbell AD, Gallego-Ortega D, Magenau A, Murphy KJ, Ridgway RA, Law AM, Walters SN, Grey ST, Croucher DR, Zhang L, Herzog H, Hardeman EC, Gunning PW, Ormandy CJ, Evans TRJ, Strathdee D, Sansom OJ, Morton JP, Anderson KI, Timpson P. Intravital FRAP imaging using an E-cadherin-GFP mouse reveals disease- and drug-dependent dynamic regulation of cell-cell junctions in live tissue. Cell Rep. 2016;14:152–167. doi: 10.1016/j.celrep.2015.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ibbetson SJ, Pyne NT, Pollard AN, Olson MF, Samuel MS. Mechanotransduction pathways promoting tumor progression are activated in invasive human squamous cell carcinoma. Am J Pathol. 2013;183:930–937. doi: 10.1016/j.ajpath.2013.05.014. [DOI] [PubMed] [Google Scholar]
- 47.Levental KR, Yu H, Kass L, Lakins JN, Egeblad M, Erler JT, Fong SFT, Csiszar K, Giaccia A, Weninger W, Yamauchi M, Gasser DL, Weaver VM. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell. 2009;139:891–906. doi: 10.1016/j.cell.2009.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vassilev LT, Tovar C, Chen S, Knezevic D, Zhao X, Sun H, Heimbrook DC, Chen L. Selective small-molecule inhibitor reveals critical mitotic functions of human CDK1. Proc Natl Acad Sci U.S.A. 2006;103:10660–10665. doi: 10.1073/pnas.0600447103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hans F, Dimitrov S. Histone H3 phosphorylation and cell division. Oncogene. 2001;20:3021–3027. doi: 10.1038/sj.onc.1204326. [DOI] [PubMed] [Google Scholar]
- 50.Alexander S, Friedl P. Cancer invasion and resistance: Interconnected processes of disease progression and therapy failure. Trends Mol Med. 2012;18:13–26. doi: 10.1016/j.molmed.2011.11.003. [DOI] [PubMed] [Google Scholar]
- 51.Sahai E, Marshall CJ. Differing modes of tumour cell invasion have distinct requirements for Rho/ROCK signalling and extracellular proteolysis. Nat Cell Biol. 2003;5:711–719. doi: 10.1038/ncb1019. [DOI] [PubMed] [Google Scholar]
- 52.Charras G, Sahai E. Physical influences of the extracellular environment on cell migration. Nat Rev Mol Cell Biol. 2014;15:813–824. doi: 10.1038/nrm3897. [DOI] [PubMed] [Google Scholar]
- 53.Timpson P, McGhee EJ, Morton JP, von Kriegsheim A, Schwarz JP, Karim SA, Doyle B, Quinn JA, Carragher NO, Edward M, Olson MF, Frame MC, Brunton VG, Sansom OJ, Anderson KI. Spatial regulation of RhoA activity during pancreatic cancer cell invasion driven by mutant p53. Cancer Res. 2011;71:747–757. doi: 10.1158/0008-5472.CAN-10-2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gallego-Ortega D, Ledger A, Roden DL, Law AMK, Magenau A, Kikhtyak Z, Cho C, Allerdice SL, Lee HJ, Valdes-Mora F, Herrmann D, Salomon R, Young AIJ, Lee BY, Sergio CM, Kaplan W, Piggin C, Conway JRW, Rabinovich B, Millar EKA, Oakes SR, Chtanova T, Swarbrick A, Naylor MJ, O’Toole S, Green AR, Timpson P, Gee JMW, Ellis IO, Clark SJ, Ormandy CJ. ELF5 drives lung metastasis in luminal breast cancer through recruitment of Gr1+ CD11b+ myeloid-derived suppressor cells. PLOS Biol. 2015;13:e1002330. doi: 10.1371/journal.pbio.1002330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hidalgo M. Pancreatic cancer. N Engl J Med. 2010;362:1605–1617. doi: 10.1056/NEJMra0901557. [DOI] [PubMed] [Google Scholar]
- 56.Costa-Silva B, Aiello NM, Ocean AJ, Singh S, Zhang H, Thakur BK, Becker A, Hoshino A, Mark MT, Molina H, Xiang J, Zhang T, Theilen TM, Garcia-Santos G, Williams C, Ararso Y, Huang Y, Rodrigues G, Shen TL, Labori KJ, Lothe IMB, Kure EH, Hernandez J, Doussot A, Ebbesen SH, Grandgenett PM, Hollingsworth MA, Jain M, Mallya K, Batra SK, Jarnagin WR, Schwartz RE, Matei I, Peinado H, Stanger BZ, Bromberg J, Lyden D. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat Cell Biol. 2015;17:816–826. doi: 10.1038/ncb3169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hoshino A, Costa-Silva B, Shen T-L, Rodrigues G, Hashimoto A, Mark MTesic, Molina H, Kohsaka S, Giannatale ADi, Ceder S, Singh S, Williams C, Soplop N, Uryu K, Pharmer L, King T, Bojmar L, Davies AE, Ararso Y, Zhang T, Zhang H, Hernandez J, Weiss JM, Dumont-Cole VD, Kramer K, Wexler LH, Narendran A, Schwartz GK, Healey JH, Sandstrom P, Labori KJ, Kure EH, Grandgenett PM, Hollingsworth MA, Sousa Mde, Kaur S, Jain M, Mallya K, Batra SK, Jarnagin WR, Brady MS, Fodstad O, Muller V, Pantel K, Minn AJ, Bissell MJ, Garcia BA, Kang Y, Rajasekhar VK, Ghajar CM, Matei I, Peinado H, Bromberg J, Lyden D. Tumour exosome integrins determine organotropic metastasis. Nature. 2015;527:329–335. doi: 10.1038/nature15756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Manegold C, Vansteenkiste J, Cardenal F, Schuette W, Woll PJ, Ulsperger E, Kerber A, Eckmayr J, von Pawel J. Randomized phase II study of three doses of the integrin inhibitor cilengitide versus docetaxel as second-line treatment for patients with advanced non-small-cell lung cancer. Invest New Drugs. 2013;31:175–182. doi: 10.1007/s10637-012-9842-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ritsma L, Steller EJA, Beerling E, Loomans CJM, Zomer A, Gerlach C, Vrisekoop N, Seinstra D, van Gurp L, Schäfer R, Raats DA, de Graaff A, Schumacher TN, de Koning EJP, Rinkes IHB, Kranenburg O, van Rheenen J. Intravital microscopy through an abdominal imaging window reveals a pre-micrometastasis stage during liver metastasis. Sci Transl Med. 2012;4:158ra145. doi: 10.1126/scitranslmed.3004394. [DOI] [PubMed] [Google Scholar]
- 60.Cukierman E, Pankov R, Stevens DR, Yamada KM. Taking cell-matrix adhesions to the third dimension. Science. 2001;294:1708–1712. doi: 10.1126/science.1064829. [DOI] [PubMed] [Google Scholar]
- 61.Herrmann D, Conway JRW, Vennin C, Magenau A, Hughes WE, Morton JP, Timpson P. Three-dimensional cancer models mimic cell–matrix interactions in the tumour microenvironment. Carcinogenesis. 2014;35:1671–1679. doi: 10.1093/carcin/bgu108. [DOI] [PubMed] [Google Scholar]
- 62.Barnes JM, Nauseef JT, Henry MD. Resistance to fluid shear stress is a conserved biophysical property of malignant cells. PLOS ONE. 2012;7:e50973. doi: 10.1371/journal.pone.0050973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Frentzas S, Simoneau E, Bridgeman VL, Vermeulen PB, Foo S, Kostaras E, Nathan MR, Wotherspoon A, Gao Z-h, Shi Y, Van den Eynden G, Daley F, Peckitt C, Tan X, Salman A, Lazaris A, Gazinska P, Berg TJ, Eltahir Z, Ritsma L, Rheenen Jvan, Khashper A, Brown G, Nyström H, Sund M, Laere SVan, Loyer E, Dirix L, Cunningham D, Metrakos P, Reynolds AR. Vessel co-option mediates resistance to anti-angiogenic therapy in liver metastases. Nat Med. 2016;22:1294–1302. doi: 10.1038/nm.4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wirtz D, Konstantopoulos K, Searson PC. The physics of cancer: The role of physical interactions and mechanical forces in metastasis. Nat Rev Cancer. 2011;11:512–522. doi: 10.1038/nrc3080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Boudaoud A, Burian A, Borowska-Wykręt D, Uyttewaal M, Wrzalik R, Kwiatkowska D, Hamant O. FibrilTool, an ImageJ plug-in to quantify fibrillar structures in raw microscopy images. Nat Protoc. 2014;9:457–463. doi: 10.1038/nprot.2014.024. [DOI] [PubMed] [Google Scholar]
- 66.Scott RW, Hooper S, Crighton D, Li A, Konig I, Munro J, Trivier E, Wickman G, Morin P, Croft DR, Dawson J, Machesky L, Anderson KI, Sahai EA, Olson MF. LIM kinases are required for invasive path generation by tumor and tumor-associated stromal cells. J CellBiol. 2010;191:169–185. doi: 10.1083/jcb.201002041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hirata E, Girotti MR, Viros A, Hooper S, Spencer-Dene B, Matsuda M, Larkin J, Marais R, Sahai E. Intravital imaging reveals how BRAF inhibition generates drug-tolerant microenvironments with high integrin β1/FAK signaling. Cancer Cell. 2015;27:574–588. doi: 10.1016/j.ccell.2015.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Duda DG, Ancukiewicz M, Isakoff SJ, Krop IE, Jain RK. Seeds and soil: Unraveling the roleoflocal tumorstroma in distant metastasis. J Natl CancerInst. 2014;106:dju187. doi: 10.1093/jnci/dju187. [DOI] [PubMed] [Google Scholar]
- 69.Kumar A, Ouyang M, Van den Dries K, McGhee EJ, Tanaka K, Anderson MD, Groisman A, Goult BT, Anderson KI, Schwartz MA. Talin tension sensor reveals novel features of focal adhesion force transmission and mechanosensitivity. J Cell Biol. 2016;213:371–383. doi: 10.1083/jcb.201510012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wang Y, Botvinick EL, Zhao Y, Berns MW, Usami S, Tsien RY, Chien S. Visualizing the mechanical activation of Src. Nature. 2005;434:1040–1045. doi: 10.1038/nature03469. [DOI] [PubMed] [Google Scholar]
- 71.Chang C-W, Chen Y-S, Chen C-C, Chan I-O, Chen C-C, Lin S-J, Sheu T-W, Chou S-H, Liu C-J, Lee T-C, Lo J-F. Targeting cancer initiating cells by promoting cell differentiation and restoring chemosensitivity via dual inactivation of STAT3 and Src activity using an active component of Antrodia cinnamomea mycelia. Oncotarget. 2016;7:73016–73031. doi: 10.18632/oncotarget.12194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tan Q, Wang H, Hu Y, Hu M, Li,Aodengqimuge X, Ma Y, Wei C, Song L. Src/STAT3-dependent heme oxygenase-1 induction mediates chemoresistance of breast cancer cells to doxorubicin by promoting autophagy. Cancer Sci. 2015;106:1023–1032. doi: 10.1111/cas.12712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Perez M, Lucena-Cacace A, Marin-Gomez LM, Padillo-Ruiz J, Robles-Frias MJ, Saez C, Garcia-Carbonero R, Carnero A. Dasatinib, a Src inhibitor, sensitizes liver metastatic colorectal carcinoma to oxaliplatin in tumors with high levels of phospho-Src. Oncotarget. 2016;7:33111–33124. doi: 10.18632/oncotarget.8880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Qin S, Zhang B, Xiao G, Sun X, Li G, Huang G, Gao X, Li X, Wang H, Yang C, Ren H. Fibronectin protects lung cancer cells against docetaxel-induced apoptosis by promoting Src and caspase-8 phosphorylation. Tumour Biol. 2016;37:13509–13520. doi: 10.1007/s13277-016-5206-8. [DOI] [PubMed] [Google Scholar]
- 75.Yeatman TJ. A renaissance for SRC. Nat Rev Cancer. 2004;4:470–480. doi: 10.1038/nrc1366. [DOI] [PubMed] [Google Scholar]
- 76.Zhang S, Yu D. Targeting Src family kinases in anti-cancer therapies: Turning promise into triumph. Trends Pharmacol Sci. 2012;33:122–128. doi: 10.1016/j.tips.2011.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Morton JP, Karim SA, Graham K, Timpson P, Jamieson N, Athineos D, Doyle B, McKay C, Heung MY, Oien KA, Frame MC, Evans TRJ, Sansom OJ, Brunton VG. Dasatinib inhibits the development of metastases in a mouse model of pancreatic ductal adenocarcinoma. Gastroenterology. 2010;139:292–303. doi: 10.1053/j.gastro.2010.03.034. [DOI] [PubMed] [Google Scholar]
- 78.Yokoyama T, Goto H, Izawa I, Mizutani H, Inagaki M. Aurora-B and Rho-kinase/ROCK, the two cleavage furrow kinases, independently regulate the progression of cytokinesis: Possible existence of a novel cleavage furrow kinase phosphorylates ezrin/radixin/ moesin (ERM) Genes Cells. 2005;10:127–137. doi: 10.1111/j.1365-2443.2005.00824.x. [DOI] [PubMed] [Google Scholar]
- 79.Kosako H, Yoshida T, Matsumura F, Ishizaki T, Narumiya S, Inagaki M. Rho-kinase/ROCK is involved in cytokinesis through the phosphorylation of myosin light chain and not ezrin/radixin/moesin proteins at the cleavage furrow. Oncogene. 2000;19:6059–6064. doi: 10.1038/sj.onc.1203987. [DOI] [PubMed] [Google Scholar]
- 80.Madaule P, Eda M, Watanabe N, Fujisawa K, Matsuoka T, Bito H, Ishizaki T, Narumiya S. Role of citron kinase as a target of the small GTPase Rho in cytokinesis. Nature. 1998;394:491–494. doi: 10.1038/28873. [DOI] [PubMed] [Google Scholar]
- 81.Roy A, Lordier L, Mazzi S, Chang Y, Lapierre V, Larghero J, Debili N, Raslova H, Vainchenker W. Activity of nonmuscle myosin II isoforms determines their localization at the cleavage furrow of megakaryocytes. Blood. 2016;128:3137–3145. doi: 10.1182/blood-2016-04-711630. [DOI] [PubMed] [Google Scholar]
- 82.Biankin AV, Waddell N, Kassahn KS, Gingras M-C, Muthuswamy LB, Johns AL, Miller DK, Wilson PJ, Patch A-M, Wu J, Chang DK, Cowley MJ, Gardiner BB, Song S, Harliwong I, Idrisoglu S, Nourse C, Nourbakhsh E, Manning S, Wani S, Gongora M, Pajic M, Scarlett CJ, Gill AJ, Pinho AV, Rooman I, Anderson M, Holmes O, Leonard C, Taylor D, Wood S, Xu Q, Nones K, Fink JL, Christ A, Bruxner T, Cloonan N, Kolle G, Newell F, Pinese M, Mead RS, Humphris JL, Kaplan W, Jones MD, Colvin EK, Nagrial AM, Humphrey ES, Chou A, Chin VT, Chantrill LA, Mawson A, Samra JS, Kench JG, Lovell JA, Daly RJ, Merrett ND, Toon C, Epari K, Nguyen NQ, Barbour A, Zeps N, Australian Pancreatic Cancer Genome Initiative. Kakkar N, Zhao F, Wu YQ, Wang M, Muzny DM, Fisher WE, Brunicardi FC, Hodges SE, Reid JG, Drummond J, Chang K, Han Y, Lewis LR, Dinh H, Buhay CJ, Beck T, Timms L, Sam M, Begley K, Brown A, Pai D, Panchal A, Buchner N, Borja RDe, Denroche RE, Yung CK, Serra S, Onetto N, Mukhopadhyay D, Tsao M-S, Shaw PA, Petersen GM, Gallinger S, Hruban RH, Maitra A, Iacobuzio-Donahue CA, Schulick RD, Wolfgang CL, Morgan RA, Lawlor RT, Capelli P, Corbo V, Scardoni M, Tortora G, Tempero MA, Mann KM, Jenkins NA, Perez-Mancera PA, Adams DJ, Largaespada DA, Wessels LFA, Rust AG, Stein LD, Tuveson DA, Copeland NG, Musgrove EA, Scarpa A, Eshleman JR, Hudson TJ, Sutherland RL, Wheeler DA, Pearson JV, McPherson JD, Gibbs RA, Grimmond SM. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature. 2012;491:399–405. doi: 10.1038/nature11547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hingorani SR, Harris WP, Hendifar AE, Bullock AJ, Wu XW, Huang Y, Jiang P. High response rate and PFS with PEGPH20 added to nab-paclitaxel/gemcitabine in stage IV previously untreated pancreatic cancer patients with high-HA tumors: Interim results of a randomized phase II study. J Clin Oncol. 2015;33:4006. abstract. [Google Scholar]
- 84.Carragher NO, Unciti-Broceta A, Cameron DA. Advancing cancer drug discovery towards more agile development of targeted combination therapies. Future Med Chem. 2012;4:87–105. doi: 10.4155/fmc.11.169. [DOI] [PubMed] [Google Scholar]
- 85.Lu P, Weaver VM, Werb Z. The extracellular matrix: A dynamic niche in cancer progression. J Cell Biol. 2012;196:395–406. doi: 10.1083/jcb.201102147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kai F, Laklai H, Weaver VM. Force matters: Biomechanical regulation of cell invasion and migration in disease. Trends Cell Biol. 2016;26:486–497. doi: 10.1016/j.tcb.2016.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Vicari RM, Chaitman B, Keefe D, Smith WB, Chrysant SG, Tonkon MJ, Bittar N, Weiss RJ, Morales-Ballejo H, Thadani U, Fasudil Study Group Efficacy and safety of fasudil in patients with stable angina: A double-blind, placebo-controlled, phase 2 trial. J Am Coll Cardiol. 2005;46:1803–1811. doi: 10.1016/j.jacc.2005.07.047. [DOI] [PubMed] [Google Scholar]
- 88.Liu GJ, Wang ZJ, Wang YF, Xu LL, Wang XL, Liu Y, Luo GJ, He GH, Zeng YJ. Systematic assessment and meta-analysis of the efficacy and safety of fasudil in the treatment of cerebral vasospasm in patients with subarachnoid haemorrhage. Eur J Clin Pharmacol. 2012;68:131–139. doi: 10.1007/s00228-011-1100-x. [DOI] [PubMed] [Google Scholar]
- 89.Braybrook C. Fasudil as an adjunctive therapy for patients with stable angina. Nat Clin Pract Cardiovasc Med. 2006;3:63–64. [Google Scholar]
- 90.Rahbari NN, Kedrin D, Incio J, Liu H, Ho WW, Nia HT, Edrich CM, Jung K, Daubriac J, Chen I, Heishi T, Martin JD, Huang Y, Maimon N, Reissfelder C, Weitz J, Boucher Y, Clark JW, Grodzinsky AJ, Duda DG, Jain RK, Fukumura D. Anti-VEGF therapy induces ECM remodeling and mechanical barriers to therapy in colorectal cancer liver metastases. Sci Transl Med. 2016;8:360ra135. doi: 10.1126/scitranslmed.aaf5219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Boucher SM, Tolaney Y, Duda DG, Martin JD, Seano G, Barry M, Ancukiewicz WT, Goel S, Lahdenrata J, Isakoff SJ, Yeh ED, Jain SR, Golshan M, Brock J, Snuderl M, Winer EP, Krop IE, Jain RK. Role of vascular density and normalization in response to neoadjuvant bevacizumab and chemotherapy in breast cancer patients. Proc Natl Acad Sci U.S.A. 2015;112:14325–14330. doi: 10.1073/pnas.1518808112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Pantziarka P, Cairns L. Recycling existing drugs for cancer therapy: Delivering low cost cancer care. Ecancermedicalscience. 2014;8:ed40. doi: 10.3332/ecancer.2014.ed40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Erler JT, Bennewith KL, Cox TR, Lang G, Bird D, Koong A, Le QT, Giaccia AJ. Hypoxia-induced lysyl oxidase is a critical mediator of bone marrow cell recruitment to form the premetastatic niche. Cancer Cell. 2009;15:35–44. doi: 10.1016/j.ccr.2008.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Psaila B, Lyden D. The metastatic niche: Adapting the foreign soil. Nat Rev Cancer. 2009;9:285–293. doi: 10.1038/nrc2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Rath N, Morton JP, Julian L, Helbig L, Kadir S, McGhee EJ, Anderson KI, Kalna G, Mullin M, Pinho AV, Rooman I, Samuel MS, Olson MF. ROCK signaling promotes collagen remodeling to facilitate invasive pancreatic ductal adenocarcinoma tumor cell growth. EMBO Mol Med. 2017;9:198–218. doi: 10.15252/emmm.201606743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Patsialou A, Bravo-Cordero JJ, Wang Y, Entenberg D, Liu H, Clarke M, Condeelis JS. Intravital multiphoton imaging reveals multicellular streaming as a crucial component of in vivo cell migration in human breast tumors. Intravital. 2013;2:e25294. doi: 10.4161/intv.25294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wojciak-Stothard B, Ridley AJ. Shear stress-induced endothelial cell polarization is mediated by Rho and Rac but not Cdc42 or PI 3-kinases. J Cell Biol. 2003;161:429–439. doi: 10.1083/jcb.200210135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Vishnubhotla R, Bharadwaj S, Sun S, Metlushko V, Glover SC. Treatment with Y-27632, a ROCK inhibitor, increases the proinvasive nature of SW620 cells on 3D collagen type 1 matrix. Int J Cell Biol. 2012;2012:259142. doi: 10.1155/2012/259142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Torka R, Thuma F, Herzog V, Kirfel G. ROCK signaling mediates the adoption of different modes of migration and invasion in human mammary epithelial tumor cells. Exp Cell Res. 2006;312:3857–3871. doi: 10.1016/j.yexcr.2006.08.025. [DOI] [PubMed] [Google Scholar]
- 100.Bhandary L, Whipple RA, Vitolo MI, Charpentier MS, Boggs AE, Chakrabarti KR, Thompson KN, Martin SS. ROCK inhibition promotes microtentacles that enhance reattachment of breast cancer cells. Oncotarget. 2015;6:6251–6266. doi: 10.18632/oncotarget.3360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Garnock-Jones KP. Ripasudil: First global approval. Drugs. 2014;74:2211–2215. doi: 10.1007/s40265-014-0333-2. [DOI] [PubMed] [Google Scholar]
- 102.Kaneko N, Mashiko T, Ohnishi T, Ohta M, Namba K, Watanabe E, Kawai K. Manufacture of patient-specific vascular replicas for endovascular simulation using fast, low-cost method. Sci Rep. 2016;6:39168. doi: 10.1038/srep39168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Boland S, Bourin A, Alen J, Geraets J, Schroeders P, Castermans K, Kindt N, Boumans N, Panitti L, Fransen S, Vanormelingen J, Stassen JM, Leysen D, Defert O. Design, synthesis, and biological evaluation of novel, highly active soft ROCK inhibitors. J Med Chem. 2015;58:4309–4324. doi: 10.1021/acs.jmedchem.5b00308. [DOI] [PubMed] [Google Scholar]
- 104.Sadok A, McCarthy A, Caldwell J, Collins I, Garrett MD, Yeo M, Hooper S, Sahai E, Kuemper S, Mardakheh FK, Marshall CJ. Rho kinase inhibitors block melanoma cell migration and inhibit metastasis. Cancer Res. 2015;75:2272–2284. doi: 10.1158/0008-5472.CAN-14-2156. [DOI] [PubMed] [Google Scholar]
- 105.Bertolini F, Sukhatme VP, Bouche G. Drug repurposing in oncology—Patient and health systems opportunities. Nat Rev Clin Oncol. 2015;12:732–742. doi: 10.1038/nrclinonc.2015.169. [DOI] [PubMed] [Google Scholar]
- 106.Steeg PS. Targeting metastasis. Nat Rev Cancer. 2016;16:201–218. doi: 10.1038/nrc.2016.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Pajic M, Herrmann D, Vennin C, Conway JRW, Chin VT, Johnsson AKE, Welch HCE, Timpson P. The dynamics of Rho GTPase signaling and implications for targeting cancer and the tumor microenvironment. Small GTPases. 2015;6:123–133. doi: 10.4161/21541248.2014.973749. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. ROCK inhibition with Fasudil and Y-27632 impairs ECM integrity.
Fig. S2. The CDK1-FRET biosensor distinguishes changes in CDK1 activity and is a surrogate for cell cycle arrest.
Fig. S3. Priming with Fasudil disrupts ECM remodeling, inhibits ROCK signaling, and improves Gem/Abraxane efficacy in vivo.
Fig. S4. Priming with Fasudil influences liver vasculature, cell attachment to CDMs, response to chemotherapy, and remodeling of the ECM.
Fig. S5. Priming with Fasudil results in decreased SRC activity and defects in mitosis.
Fig. S6. High ECM TKCC5 patient model responds to priming strategies in a 3D patient-personalized organotypic matrix.
Fig. S7. Low ECM TKCC2 patient model does not respond to priming strategies in a 3D patient-personalized organotypic matrix.
Fig. S8. TKCC5 orthotopic tumors respond to priming, and SHG does not predict survival.
Fig. S9. Priming with Fasudil uncouples PC progression.
Table S1. List of P values.
Table S2. Details of antibodies used for the study.
Movie S1. 4D monitoring of fibroblast-ECM interactions upon treatment with vehicle and Fasudil.
Movie S2. Live FLIM-FRET imaging of the CDK1 biosensor in KPC cells in response to Abraxane and Abraxane + RO3306.
Movie S3. Live FLIM-FRET imaging of the CDK1 biosensor in KPC cells in interphase and mitosis.
Movie S4. Live FLIM-FRET monitoring of CDK1 activity in KPC cells actively interacting with an organotypic matrix.
Movie S5. Intravital FLIM-FRET imaging of subcutaneous xenografts with KPC-CDK1 cells and imaging of fibrillar collagen.
Movie S6. Intravital monitoring of CDK1 accumulation in subcutaneous KPC tumors.
Movie S7. Intravital imaging of quantum dots circulating through tumor-associated vasculature and diffusing into tumor tissue upon priming with Fasudil.
Movie S8. Imaging of liver tissue with metastatic KPC cells expressing the CDK1 biosensor forming macrometastases and micrometastases.
Movie S9. Time-lapse tracking of collective cell streaming on CDMs unprimed or primed with Fasudil.