

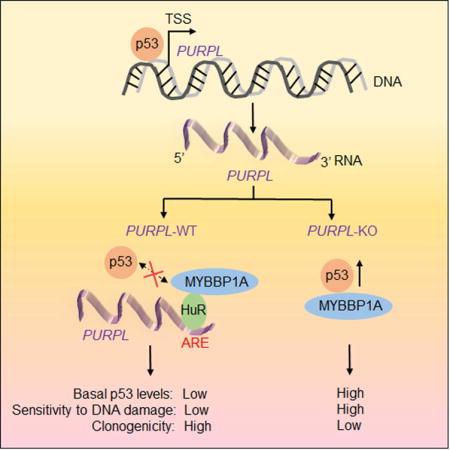
SUMMARY
Basal p53 levels are tightly suppressed under normal conditions. Disrupting this regulation results in elevated p53 levels to induce cell cycle arrest, apoptosis and tumor suppression. Here, we report the suppression of basal p53 levels by a nuclear, p53-regulated long noncoding RNA that we termed PURPL (p53 upregulated regulator of p53 levels). Targeted depletion of PURPL in colorectal cancer cells results in elevated basal p53 levels and induces growth defects in cell culture and in mouse xenografts. PURPL associates with MYBBP1A, a protein that binds to and stabilizes p53, and inhibits the formation of the p53-MYBBP1A complex. In the absence of PURPL, MYBBP1A interacts with and stabilizes p53. Silencing MYBBP1A significantly rescues basal p53 levels and proliferation in PURPL-deficient cells, suggesting that MYBBP1A mediates the effect of PURPL in regulating p53. These results reveal a p53-PURPL autoregulatory feedback loop and demonstrate a role for PURPL in maintaining basal p53 levels.
Keywords: lncRNA, lincRNA, PURPL, p53, HuR, MYBBP1A, LOC643401, LINC01021, RP11-46C20.1, CRC, CRISPR/Cas9
eTOC blurb
For a cell to divide, the tumor suppressor protein p53 must be kept at low levels. Li et al. find that a long noncoding RNA PURPL allows cancer cells to divide by keeping p53 levels low. PURPL binds to the p53-regulator MYBBP1A to suppress p53 levels and facilitate cell proliferation.
INTRODUCTION
Long noncoding RNAs (lncRNAs) are an emerging class of regulatory RNAs, >200 nucleotides (nt) long. The mammalian genome is transcribed into thousands of uncharacterized lncRNAs (Iyer et al., 2015, Ulitsky and Bartel, 2013) but only a few lncRNAs including HOTAIR, MALAT1, NORAD, XIST and CCAT2 have established biological functions (Lee et al., 2016, Lee, 2012, Tripathi et al., 2013, Tripathi et al., 2010, Fatica and Bozzoni, 2014, Engreitz et al., 2013, Arun et al., 2016, Ling et al., 2013, Redis et al., 2016, Mueller et al., 2015, Dey et al., 2014, Rinn et al., 2007, Li et al., 2013, Gupta et al., 2010).
Most lncRNAs are less conserved than mRNAs and are often expressed at low levels in a cell-type or tissue-specific manner (Tsoi et al., 2015). Therefore, it is difficult to predict whether a given lncRNA is functional or reflects transcriptional noise. LncRNAs are regulated by the same transcription factors that regulate protein-coding genes, and increasing evidence suggests that the master-regulatory transcription factor p53, controls the expression of subset of lncRNAs (Grossi et al., 2016, Chaudhary and Lal, 2016, Adriaens et al., 2016, Mello et al., 2017, Chaudhary et al., 2017). Coordinated regulation of even low-abundance lncRNAs indicates that many lncRNAs have undiscovered, yet critical functions in cell biology and disease.
The tumor suppressor p53 is mutated in more than 50% of human cancers (Vogelstein et al., 2000, Vousden and Lane, 2007). In response to stress such as DNA damage, p53 directly activates the transcription of a myriad of protein-coding genes that control a wide variety of cellular processes, including cell cycle arrest, apoptosis and senescence (Bieging and Attardi, 2012, Riley et al., 2008, Beckerman and Prives, 2010). We and others have shown that p53 also directly upregulates several microRNAs including miR-34a and miR-3189, which, in turn, suppress gene expression downstream of p53 (Chang et al., 2007, Lal et al., 2011, Hermeking, 2012, Jones et al., 2015, Raver-Shapira et al., 2007). More recently, we and others have demonstrated specific functions of a number of p53-regulated lncRNAs, including lincRNA-p21, PANDA, DINO, PINT, PR-lncRNA-1, LED, linc-475, NEAT1 and PINCR (Huarte et al., 2010, Dimitrova et al., 2014, Hung et al., 2011, Marin-Bejar et al., 2013, Sanchez et al., 2014, Leveille et al., 2015, Melo et al., 2016, Schmitt et al., 2016, Adriaens et al., 2016, Mello et al., 2017, Chaudhary et al., 2017). Although these studies illustrate the importance of lncRNAs in the p53 network as well as the functional heterogeneity of p53-regulated lncRNAs, the function of the vast majority of p53-regulated lncRNAs remains to be elucidated.
In this study, we investigated the function of a p53-regulated lncRNA that we named PURPL (p53 upregulated regulator of p53 levels). PURPL is an intergenic lncRNA that we identified by RNA-seq from multiple colorectal cancer (CRC) lines. We show that loss of PURPL results in elevated basal p53 levels and impaired cell growth in vitro and in vivo. PURPL regulates basal p53 levels by associating with MYBBP1A, a protein known to bind to and activate p53 (Ono et al., 2014, Kuroda et al., 2011, Kumazawa et al., 2015). Altogether, our study provides functional insights on PURPL, demonstrating a role of this lncRNA in suppressing basal p53 levels.
RESULTS
Identification and characterization of PURPL
To identify lncRNAs regulated by p53 in multiple CRC cell lines, we performed paired-end Ribo-zero RNA-seq from p53 wild-type (WT) and isogenic p53 knockout (KO) HCT116, RKO and SW48 cells under untreated condition or after DNA damage induced by Doxorubicin (DOXO) for 16 hr at a final concentration of 300 nM. Using a cut-off of 1.50-fold change, 511 transcripts were upregulated upon DOXO-treatment in a p53-dependent manner in all 3 CRC lines (Table S1 and Figure S1A). Although hundreds of annotated intergenic lncRNAs (lincRNAs) were induced in a p53-dependent manner in each cell line, only 33 were upregulated in all 3 lines. The overlap was more substantial between HCT116 and RKO; 230 lincRNAs were upregulated in both HCT116 and RKO. As positive controls, several well-established p53-regulated genes including p21 (CDKN1A), GDF15 and MDM2 were upregulated in all 3 lines (Figure 1A). The 33 lincRNAs included the p53-regulated PINCR, PR-lncRNA-1, LncRNA-4 and LncRNA-7 (Younger et al., 2015, Sanchez et al., 2014, Chaudhary et al., 2017); other p53-regulated lncRNAs such as NORAD, LED and linc-475 (Melo et al., 2016, Leveille et al., 2015, Lee et al., 2016) were upregulated in at least one of the 3 lines.
Figure 1. RNA-seq from multiple CRC lines identifies PURPL as a p53-regulated lncRNA.
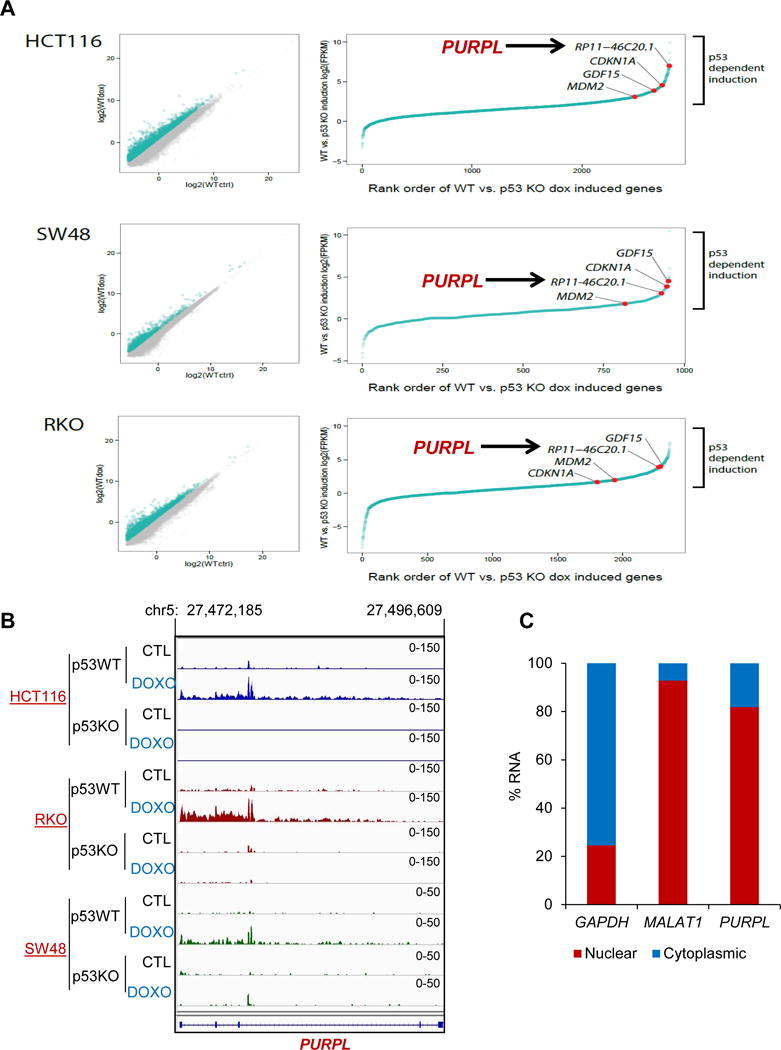
RNA-seq was performed from isogenic (p53WT and p53KO) HCT116, RKO and SW48 cells. (A) Scatter plot (left), Rank order of gene expression (right) and RNA-seq snapshot. (B) p53-dependent induction of PURPL (RP11-46C20.1) after DOXO treatment. (C) RT-qPCR for PURPL from nuclear and cytoplasmic fractions of untreated HCT116 cells. The cytoplasmic GAPDH mRNA and nuclear lncRNA MALAT1 were used as controls.
Our RNA-seq identified RP11-46C20.1/LOC643401/LINC01021, which we termed PURPL, an abundant lincRNA upregulated after DNA damage in a p53-dependent manner in all 3 lines (Figures 1A and 1B). Although RefSeq predicts a single multi-exonic transcript (LINC01021), according to GENCODE version 19 and ENSEMBL version 75, 6 transcripts are expressed from the PURPL locus (Figure S1B). In addition, we noticed a substantial signal from intron 2 (Figure 1B), also observed in a recent study from MCF7 cells (Leveille et al., 2015). RT-qPCR using exon-exon and exon-intron primers indicated that intron 2 may be retained and that exons 4 and 5 were expressed at low levels (Figure S1C), possibly due to co-transcriptional degradation of this lncRNA as recently reported (Schlackow et al., 2017).
We validated the p53-dependent upregulation of PURPL by RT-qPCR upon genetic loss of p53 (Figure S2A) or upon p53 knockdown (Figure S2B) in HCT116 in the presence or absence of DOXO. To determine if PURPL is a direct p53 target, we utilized published p53 ChIP-seq data from HCT116, MCF7 and U2OS cells (Nikulenkov et al., 2012, Sanchez et al., 2014, Menendez et al., 2013). We observed a strong p53 ChIP-seq signal (Figure S2C) located ~130 base pairs (bp) upstream of PURPL exon 1 and validated this result in HCT116 cells by ChIP-qPCR (Figure S2D). In contrast to these lines that express p53WT, knockdown of mutant p53 in HT29 and SW480 cells had no effect on PURPL levels (Figure S2E), demonstrating that PURPL expression is regulated by p53WT. During the course of our study, regulation of PURPL by p53 was also observed by others (Leveille et al., 2015, Hunten et al., 2015, Younger et al., 2015). Because the physiological function of PURPL was not known, we investigated a potential role of this lncRNA in the p53 network.
LncRNAs can be nuclear and/or cytoplasmic. RT-qPCR from nuclear and cytoplasmic fractions (Figure 1C) and RNA fluorescence in situ hybridization (RNA-FISH) (Figure S3A) suggested that a majority of PURPL is in the nucleus. Next, RT-qPCR from HCT116 total RNA and in vitro transcribed PURPL RNA revealed that PURPL is expressed at ~6 molecules per cell in untreated condition and ~60 molecules per cell after DOXO treatment (Figure S3B). We identified a canonical polyadenylation signal at the 3′end of PURPL (Figure S3C) and found that PURPL has extremely low coding potential (Figure S3D). To determine the stability of this lncRNA, we treated HCT116 cells with Actinomycin D (Act D) for 0, 2, 4 and 8 hr and measured PURPL levels by RT-qPCR. Unlike the very stable MALAT1 with a reported half-life of >9 hr (Tani et al., 2010, Tani et al., 2012), we found that the half-life of PURPL was ~2.5 hr (Figure S3E), suggesting that PURPL is an unstable lncRNA. As a positive control, more than 90% of MYC mRNA was lost by 2 hr of Act D treatment (Figure S3F).
Targeted depletion of PURPL using CRISPR/Cas9 uncovers its pro-survival function
In response to DNA damage, some p53 targets (e.g., PUMA, DINO and lincRNA-p21) are pro-apoptotic (Huarte et al., 2010, Dimitrova et al., 2014, Schmitt et al., 2016) whereas others such as p21, 14-3-3σ, PANDA and PINCR are pro-survival (Bunz et al., 1998, Chan et al., 1999, Hung et al., 2011, Villunger et al., 2003, Chaudhary et al., 2017). We therefore asked whether PURPL is a pro-apoptotic or a pro-survival lncRNA. Because even basal PURPL levels are controlled by p53 and there was a single p53 ChIP-seq peak in the PURPL promoter (Figure S2), we decided to generate mutations in this region in HCT116 using CRISPR/Cas9. Under this p53 ChIP-seq peak, there were two p53-response elements (p53REs), of which the left p53RE was much more similar to the canonical p53RE (Figures S4A–C). When we performed CRISPR/Cas9 followed by Sanger sequencing from 34 clones, we found 2 clones in which there were mutations in the p53 ChIP-seq peak region (Figure S4D). These clones were therefore designated as PURPL-KO#1 and PURPL-KO#2. For negative controls, we selected 2 wild-type clones in which the p53REs were intact and designated these as PURPL-WT#1 and PURPL-WT#2. In both KO clones, the left p53RE was intact but there were mutations in the right p53RE. Importantly, in the 2 KO clones, both basal and induced PURPL levels were dramatically (>90%) reduced (Figure 2A). In addition, binding of p53 to the PURPL promoter was completely lost after DOXO-treatment (Figure S4E) and deleting the left or the right p53RE resulted in reduced basal and induced luciferase expression in promoter reporter assays (Figure S4F).
Figure 2. Targeted disruption of the p53RE in PURPL uncovers a pro-survival function of PURPL.
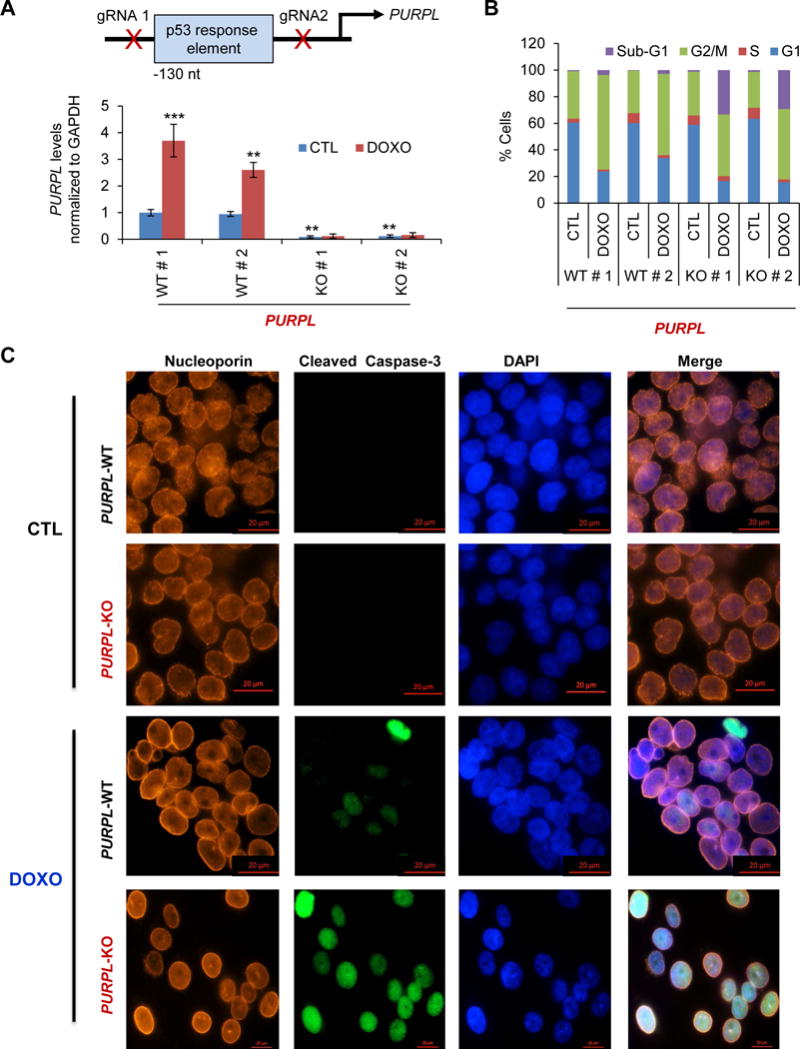
(A) (Top) Schematic showing the p53RE in the PURPL promoter and location of the guide RNAs. (Bottom) RT-qPCR analysis from HCT116 PURPL-WT cells (WT#1 and WT#2) and HCT116 PURPL-KO cells (KO#1 and KO#2) untreated or treated with DOXO for 16 hr. (B) PURPL-WT and PURPL-KO cells were untreated or treated with DOXO for 48 hr; cell death (sub-G1 cells) and effect on cell cycle was assessed by PI staining followed by FACS analysis. (C) Immunostaining for Nucleoporin and cleaved caspase-3 from PURPL-WT and PURPL-KO clones with or without DOXO treatment (72 hr). DNA was counterstained with DAPI. Error bars represent SD from 3 experiments. **p<0.01 and ***p<0.001.
We next treated PURPL-WT and PURPL-KO cells with DOXO and assessed the effect on cell cycle arrest and apoptosis by performing propidium iodide (PI) staining followed by flow cytometry (FACS) analysis. Both PURPL-KO clones displayed significantly increased cell death (>30% cells in sub-G1) and a modest decrease in the G2/M population after DNA damage (Figure 2B). Therefore, we speculated that loss of PURPL might result in a defective G2/M checkpoint that would disrupt the nuclear envelope and allow the cells to undergo apoptosis in M-phase by failing to arrest in G2. However, this was not the case. After DNA damage, the nuclear membrane was intact and the cells did not enter M-phase in both PURPL-WT and PURPL-KO cells as assessed by immunostaining for the nuclear envelope marker Nucleoporin (Figure 2C). Consistent with the increased sub-G1 population, a majority of the PURPL-KO cells stained positive for the apoptosis marker cleaved caspase-3 (Figure 2C). Thus, in the context of DNA damage, PURPL functions as a pro-survival p53-regulated gene.
PURPL suppresses basal p53 levels
To begin to investigate the mechanism involved, we performed microarrays from PURPL-WT and PURPL-KO cells (Table S2). A cut-off of 1.50-fold change and p<0.05 was used to identify differentially expressed genes. In our microarrays, the mRNA expression of the PURPL neighboring genes CDH6, CDH9 and CDH10 was not altered upon loss of PURPL. Surprisingly, loss of basal PURPL resulted in upregulation of several p53 target genes including CDKN1A, MDM2 and TP53INP1. Pathway analysis revealed over-representation of p53 signaling in the upregulated gene set (Figure S5A). and loss of PURPL resulted in significantly increased p53 transcriptional activity driven by a p53-responsive promoter luciferase vector containing 13 consensus p53 binding sites (el-Deiry et al., 1993) (Figure S5B). Moreover, basal p21, MDM2 and NOXA mRNAs and basal p53 and p21 protein levels were upregulated upon loss of PURPL (Figures 3A, 3B and S5C) suggesting that PURPL suppresses basal p53 levels.
Figure 3. Loss of PURPL results in upregulation of basal p53 levels.
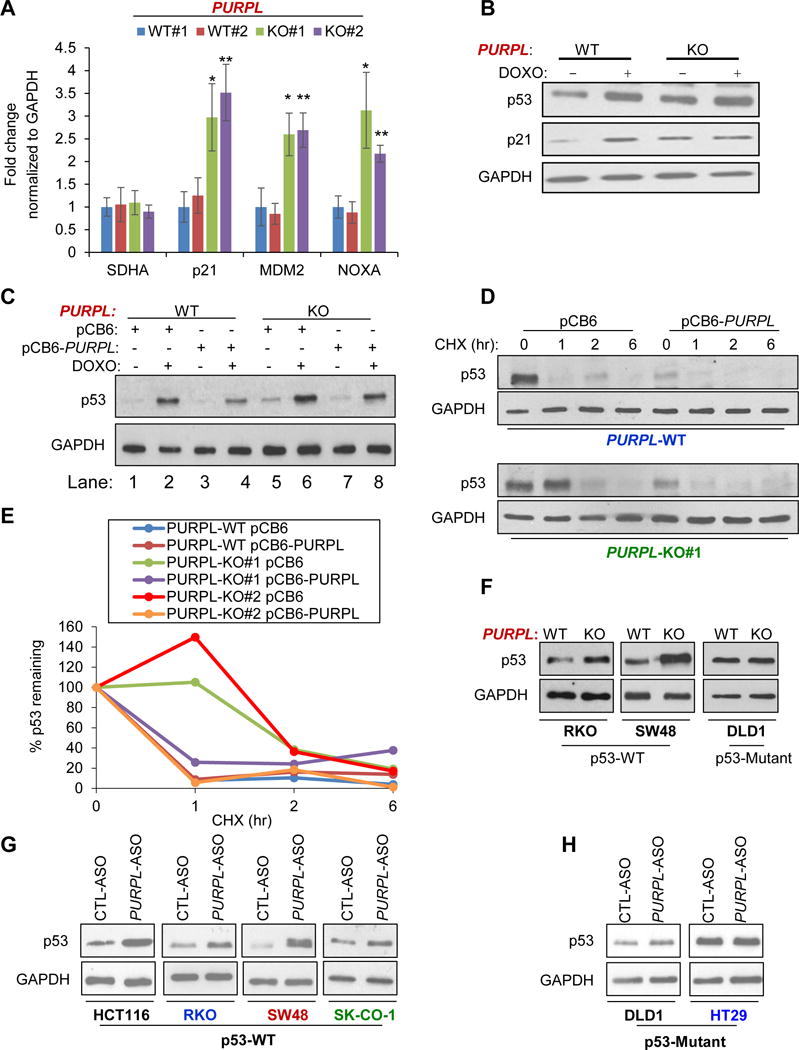
(A) RT-qPCR analysis for select p53-regulated mRNAs and the housekeeping mRNA SDHA from untreated PURPL-WT and PURPL-KO cells. (B) PURPL-WT and PURPL-KO cells were untreated or treated with DOXO for 24 hr and immunoblotting for p53, p21 and the loading control GAPDH was performed. (C) Immunoblotting was performed from PURPL-WT and PURPL-KO#1 cells transfected for 48 hr with pCB6 or pCB6-PURPL followed by DOXO for 16 hr. GAPDH was used as loading control. (D) PURPL-WT and PURPL-KO#1 cells were transfected for 48 hr with pCB6 or pCB6-PURPL and then treated with Cycloheximide (CHX) for the indicated times; immunoblotting for p53 and the loading control GAPDH was performed. (E) Decay curve for p53 protein quantitated by densitometry is shown for the immunoblot shown in Figures 3D and S9. (F) Immunoblotting for p53 and the loading control GAPDH was performed from PURPL-WT and PURPL-KO RKO, SW48 and DLD1 cells. (G, H) Parental HCT116, RKO, SW48, SK-CO-1, DLD1 and HT29 were transfected for 48 hr with CTL-ASO or PURPL-ASO and the levels of p53 and the loading control GAPDH were assessed by immunoblotting. Error bars represent SD from 3 experiments. *p<0.05 and **p<0.01.
We next performed a rescue experiment by re-introducing PURPL (LINC01021) in PURPL-KO cells. We transfected the cells for 48 hr with a mammalian expression vector (pCB6) or pCB6 in which we cloned PURPL using synthetic oligonucleotides (pCB6-PURPL). We observed up to 8-fold overexpression of PURPL (Figure S5D). Comparison of p53 levels normalized to GAPDH in lanes 5 vs 7 in the immunoblot in Figure 3C and lanes 1 vs 3 in Figure S5E (quantitated in Figure S5F) showed that reintroduction of PURPL in PURPL-KO cells decreased basal p53 levels in both KO clones. However, we did not observe a reproducible effect on the DOXO-induced p53 levels; compare p53 levels in lanes 6 vs 8 in Figure 3C and 2 vs 4 in Figure S5G. Interestingly, overexpression of PURPL in PURPL-WT cells decreased basal as well as DOXO-induced p53 levels (Figure 3C; quantitated in Figure S5G). Thus, basal p53 levels are consistently reduced upon overexpression of PURPL.
In our microarrays, we did not observe a significant difference in p53 mRNA levels between PURPL-WT and PURPL-KO cells (Table S2) indicating that the increased basal p53 levels is post-transcriptional. When we blocked protein synthesis using Cycloheximide (CHX), p53 was less rapidly degraded in PURPL-KO cells as compared to PURPL-WT cells (Figures 3D, 3E and S5H) indicating that the elevated basal p53 levels upon loss of PURPL is due to increased stability of the p53 protein.
To test if this regulation of basal p53 levels is not restricted to HCT116, we next mutated the p53RE in PURPL in 3 other CRC lines that included RKO, SW48 and DLD1. Unlike RKO and SW48 that are p53WT, DLD1 express mutant p53. Mutagenesis was confirmed by Sanger sequencing (Figure S6A). This mutagenesis decreased both basal and DOXO-induced PURPL expression in RKO and SW48 cells (Figure S6B). In DLD1, PURPL was not induced after DNA damage and mutation of the p53RE did not alter PURPL expression (Figure S6B). Moreover, basal p53 levels were elevated upon loss of PURPL in RKO and SW48 but not in DLD1 (Figures 3F and S6B). These data show that the observed loss of PURPL expression upon mutagenesis of the p53RE in its promoter, is dependent on p53WT.
We further validated these findings by knocking down this lncRNA with antisense oligos (ASOs) and performing RT-qPCR for p21 and PURPL in HCT116 and SKHep1 (liver cancer, p53WT) cells. In both lines, transient knockdown of PURPL resulted in significant upregulation of p21 mRNA (Figure S7A). Moreover, knockdown of PURPL using ASOs resulted in increased basal p53 levels (Figures 3G, 3H and S7B) and upregulated the p53-regulated p21 and TP53I3 mRNAs (Figure S7D) in multiple p53WT CRC lines that included HCT116, RKO, SW48 and SK-CO-1 but not in the mutant p53-expressing DLD1 and HT29 (Figures S7C and S7D). Of note, HCT116, RKO, SW48 and DLD1 cells are microsatellite unstable (MSI) whereas SK-CO-1 and HT29 cells are microsatellite stable (MSS). Thus, PURPL suppresses basal p53 levels in multiple CRC lines that express p53WT regardless of MSI status.
Loss of PURPL results in growth defects in vitro and impaired tumor growth in vivo
Consistent with the elevated basal p53 levels in PURPL-KO cells, these cells showed significant growth defects in vitro as compared to PURPL-WT cells (Figure 4A). This reduced growth was also observed upon transient knockdown of PURPL in HCT116 with ASOs (Figure 4B) and in colony formation assays (Figure 4C). Next, to investigate the effects of PURPL loss in an in vivo setting, we subcutaneously injected NOD-SCID mice with PURPL-WT and PURPL-KO cells. Loss of PURPL resulted in significantly reduced rate of xenograft tumor growth over a period of 4 weeks (Figures 4D–F). The difference in tumor volume between PURPL-WT and PURPL-KO tumors was ~2.5-fold (p<0.05) as early as 14 days after injection and reached ~6-fold (p<0.005) after 30 days (Figure 4E). These results indicate that PURPL plays a role in regulating tumor cell growth, both in vitro and in vivo.
Figure 4. PURPL-deficient cells show reduced proliferation in vitro and impaired tumor growth in vivo.
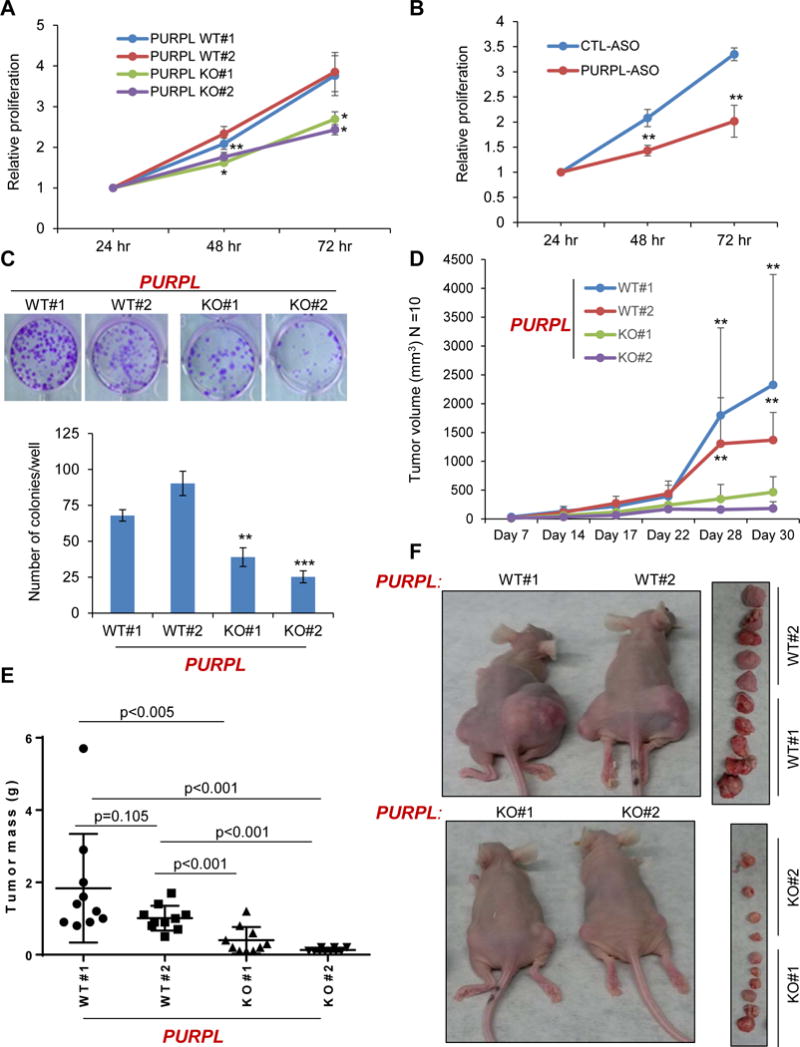
(A) Cell proliferation assays determined by Cell counting Kit-8 at the indicated time points were performed from untreated PURPL-WT and PURPL-KO cells 24 hr after seeding the cells in 96-well plates. (B) HCT116 cells were transfected with a CTL-ASO or PURPL-ASO (50 nM) and cell proliferation assays were performed as described in “(A)”. (C) Untreated PURPL-WT and PURPL-KO cells were seeded at low density in 12-well plates and colony formation assays were performed after 2 weeks. (D) Tumor volume (N=10 mice/each group) in mice was measured by caliper assessment after injecting PURPL-WT and PURPL-KO cells in mice. (E, F) Mice were euthanized after 30 days, tumors were excised and weighed. Average tumor mass at Day 30 is shown (E). Error bars represent SD from 3 experiments in A–C. *p<0.05, **p<0.01 and ***p<0.001.
To determine the contribution of increased p53 levels in the observed growth defects and hypersensitivity to DNA damage, we transfected PURPL-KO cells with CTL siRNA or p53 siRNAs. We confirmed efficient knockdown of p53 by RT-qPCR (Figure S8A). When we performed PI staining and FACS analysis in PURPL-KO cells, we observed marked reduction in cell death after p53 knockdown and DOXO treatment (Figure S8B). In contrast, upon p53 knockdown, PURPL-WT cells were more sensitive to DOXO (Figure S8C) consistent with literature (Bunz et al., 1999) where HCT116 were shown to be more sensitive to DOXO after p53 deletion. Thus, these results indicate a major role of p53 in the observed hypersensitivity of PURPL-KO cells to DOXO. Moreover, silencing p53 markedly rescued the proliferation defect of untreated PURPL-KO cells (Figure S8D) and transient co-depletion of PURPL and p53 in HCT116, partially restored cell proliferation in the absence of DNA damage (Figure S8E). These data indicate that elevated basal p53 levels play a role in the growth defects and hypersensitivity to DNA damage upon PURPL loss.
RNA pulldowns identify MYBBP1A as a PURPL-interacting protein
LncRNAs often mediate their effects by binding to RNA-binding proteins. To identify PURPL-interacting proteins, we incubated in vitro transcribed biotinylated PURPL (Bi-PURPL) or a control biotinylated luciferase (Bi-Luc) RNA with HCT116 whole cell lysates. We pulled down RNA-protein complexes using streptavidin beads and identified PURPL-associated proteins by mass spectrometry (Table S3). MYBBP1A, a protein known to stabilize p53, was strongly enriched (Figures 5A–C) in the PURPL pulldowns (Ono et al., 2014, Kuroda et al., 2011, Kumazawa et al., 2015, Akaogi et al., 2013). MYBBP1A is a predominantly nucleolar protein that associates with RNA and directly binds to p53 in the nucleoplasm resulting in p53 activation and stabilization (Ono et al., 2014, Kuroda et al., 2011, Hochstatter et al., 2012, George et al., 2015). Given the known role of MYBBP1A in regulating p53, we selected MYBBP1A for further analysis.
Figure 5. PURPL associates with MYBBP1A and prevents the formation of a p53-MYBBP1A complex.
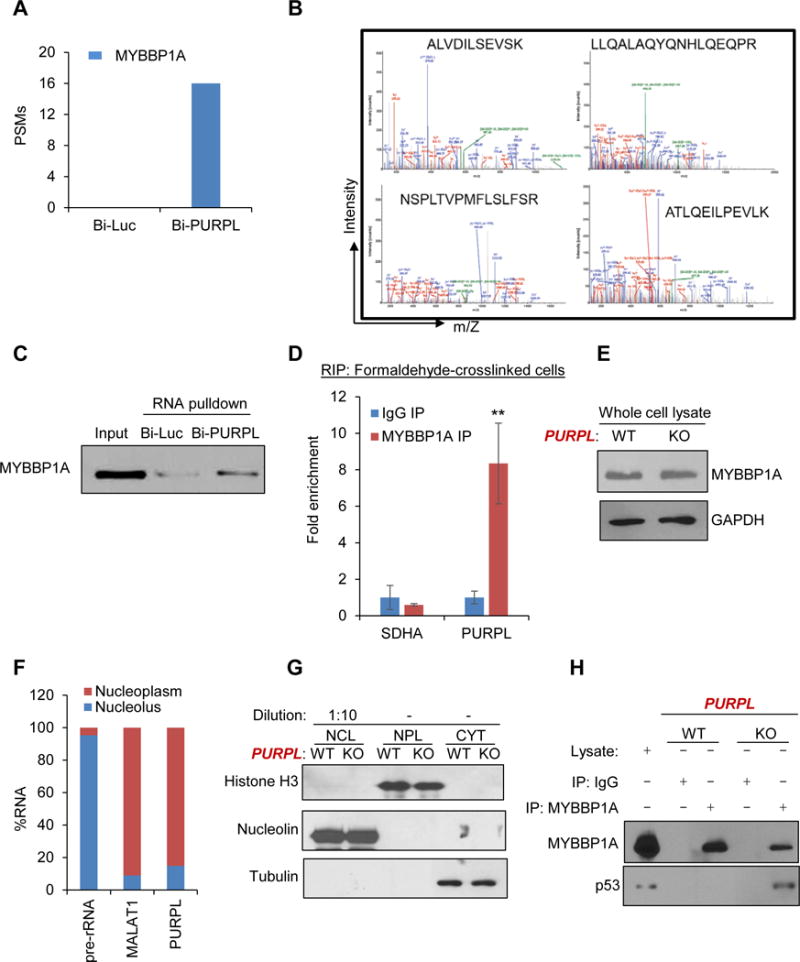
(A, B) RNA pulldowns were performed using in vitro-transcribed biotinylated PURPL (Bi-PURPL) RNA or biotinylated luciferase (Bi-Luc) RNA and HCT116 whole cell lysates. Associated proteins were pulled down with streptavidin beads and analyzed by SDS-PAGE and mass spectrometry. (A) Peptide spectrum matches (PSMs) corresponding to MYBBP1A in the Bi-Luc and Bi-PURPL pulldowns from mass spectrometry analysis, and (B) spectra, show the four MYBBP1A peptides that associate with PURPL in the RNA pulldowns and mass spectrometry. (C) RNA pulldowns were performed as described in ‘A” followed by immunoblotting for MYBBP1A. (D) The enrichment of PURPL was measured by RT-qPCR from MYBBP1A RNA IPs (RIP) performed from formaldehyde-crosslinked HCT116 cells. IgG IP and the housekeeping SDHA mRNA were used as negative controls. (E) Immunoblotting for MYBBP1A and the loading control GAPDH was performed from PURPL-WT and PURPL-KO HCT116 whole cell lysates. (F) Nucleoplasmic and nucleolar fractions were prepared from HCT116 cells and the levels of the nucleolar pre-rRNA, the nucleoplasmic MALAT1 and PURPL were measured by RT-qPCR. (G) Nucleoplasmic (NPL), nucleolar (NCL) and cytoplasmic (CYT) fractions were prepared from PURPL-WT and PURPL-KO cells and the levels of Histone H3, Nucleolin and Tubulin were assessed as controls for Nucleoplasmic, nucleolar and cytoplasmic fractions, respectively. (H) The interaction of MYBBP1A with p53 was determined by immunoblotting following co-IPs from PURPL-WT and PURPL-KO nucleoplasmic extracts. Lysate refers to whole cell extract prepared from PURPL-WT cells. Error bars represent SD from 3 experiments. **p<0.01.
To determine if MYBBP1A associates with PURPL in intact cells, we performed RNA-immunoprecipitation (RIP) from formaldehyde cross-linked HCT116. We observed significant enrichment (~8-fold) of PURPL but not the housekeeping mRNA SDHA, in the MYBBP1A IPs (Figure 5D). However, loss of PURPL did not alter MYBBP1A expression (Figure 5E), consistent with our microarray data. Given the known role of MYBBP1A in binding to and stabilizing p53 in the nucleoplasm, we hypothesized that PURPL associates with MYBBP1A in the nucleoplasm to prevent the MYBBP1A-p53 complex formation. If this is the case, MYBBP1A, p53 and PURPL should be localized in the nucleoplasm. MYBBP1A is a predominantly nucleolar protein but after its synthesis in the cytoplasm, it will pass through the nucleoplasm, to enter the nucleolus. p53 is also known to be present in the nucleoplasm.
To determine if PURPL is in the nucleoplasm, we performed subcellular fractionation in which we isolated nucleolar and nucleoplasmic fractions from HCT116 cells according to the well-established protocol from the Lamond lab (see experimental procedures). In the nucleus, majority of PURPL and the lncRNA MALAT1 were nucleoplasmic (Figure 5F); the control pre-rRNA, was mostly nucleolar. The fractionation was further confirmed by immunoblotting for Histone H3 (NPL (nucleoplasmic) marker), Nucleolin (NCL (nucleolar) marker) and tubulin (CYT (cytoplasmic) marker) (Figure 5G). Next, in co-IPs from nucleoplasmic extracts, p53 was enriched in the MYBBP1A IP in PURPL-KO cells but not in PURPL-WT cells (Figure 5H). Importantly, re-introduction of PURPL in PURPL-KO cells inhibited the p53-MYBBP1A interaction (Figure S19A) and partially rescued hypersensitivity to DNA damage (Figure S9B). These data indicate that PURPL inhibits the formation of a MYBBP1A-p53 complex in the nucleoplasm.
HuR directly binds to PURPL and acts as an adaptor to recruit MYBBP1A to PURPL
MYBBP1A does not possess canonical RNA-binding domains but has been shown to associate with RNA via yet, unidentified RNA-binding proteins (Kuroda et al., 2011). In a recent study, in vivo UV-crosslinking and RNA-IP (RIP) was utilized to show direct binding of p53 to DINO, a p53-regulated lncRNA (Schmitt et al., 2016). To test if PURPL directly binds to MYBBP1A, we used this approach. We did not observe enrichment of PURPL in the MYBBP1A RIPs (Figure 6A). This result together with the observed association of PURPL with MYBBP1A from formaldehyde crosslinked cells (Figure 5D), a reagent that can crosslink proteins that indirectly associate with the RNA, indicates that MYBBP1A associates with PURPL but does not directly bind to this lncRNA.
Figure 6. PURPL associates with MYBBP1A via the adaptor protein HuR.
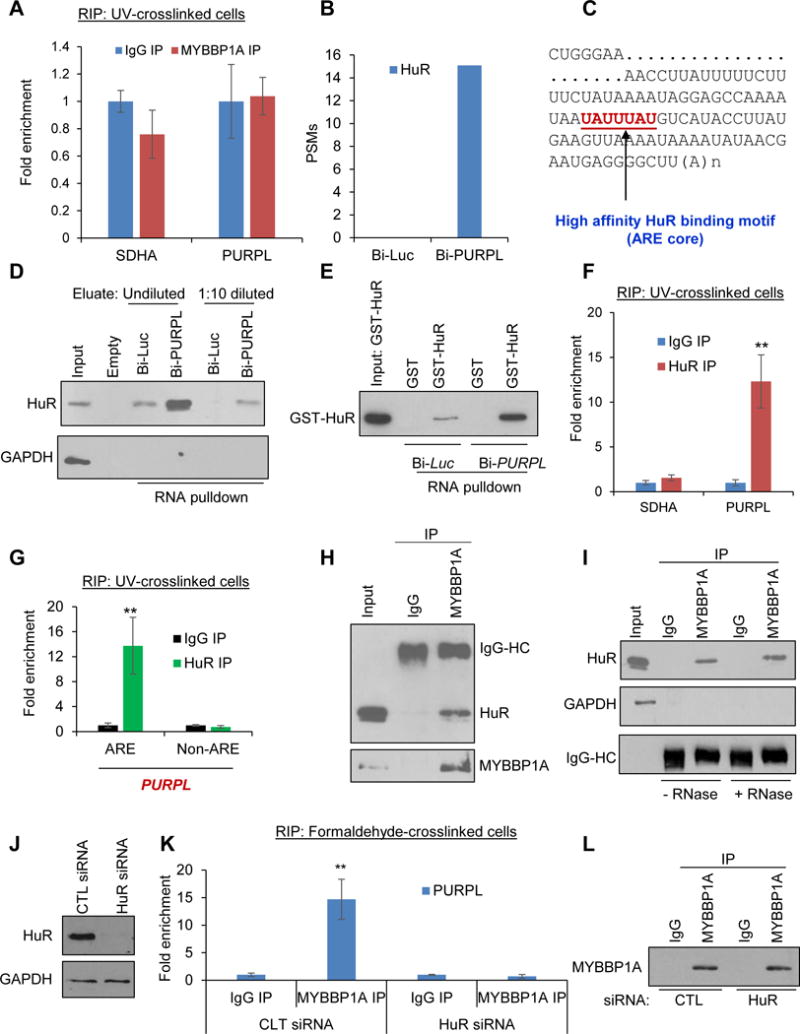
(A) In vivo UV-crosslinked HCT116 cells were subjected to RNA immunoprecipitation (RIP) with anti-MYBBP1A or IgG antibody. The levels of PURPL or the housekeeping SDHA mRNA were measured in the IP material by RT-qPCR. (B) Peptide spectrum matches (PSMs) corresponding to HuR in the Bi-Luc and Bi-PURPL pulldowns from mass spectrometry analysis is shown. (C) Partial sequence of PURPL RNA and the consensus high affinity HuR binding motif (UAUUUAU) at the 3′end is shown. (D) Streptavidin pulldowns were performed using HCT116 whole cell lysates and in vitro transcribed Bi-Luc or Bi-PURPL and the eluted material (undiluted or 1:10 diluted) was subjected to immunoblotting for HuR or the negative control GAPDH. (E) Bi-Luc or Bi-PURPL were incubated with GST or GST-HuR and the mixture was then added to streptavidin beads. Enrichment of GST-HuR in the RNA pulldowns was determined by immunoblotting for HuR. (F) RNA IPs (RIP) assays were performed from UV-crosslinked HCT116 cell lysates using HuR antibody or IgG. Enrichment of PURPL in the IP material was assessed by RT-qPCR. SDHA mRNA was used as negative control. (G) RIP assays using HuR antibody or IgG were performed after sonicating UV-crosslinked HCT116 cell lysates. Enrichment of PURPL-ARE and not the nearby Non-ARE sequence in PURPL in the IP material was assessed by RT-qPCR normalized to GAPDH using primers that span the ARE or Non-ARE region. (H) HuR-MYBBP1A interaction was examined in HCT116 cells by immunoprecipitating MYBBP1A from whole cell lysates followed by immunoblotting for HuR and MYBBP1A. IgG IP was used as control. “IgG-HC” refers to the IgG heavy chain. (I) MYBBP1A was immunoprecipitated from HCT116 whole cell lysates and the enrichment of HuR in the IP material was assessed by immunoblotting in the presence or absence of RNase A. GAPDH was used as negative control. IgG-HC refers to the IgG heavy chain. (J) HCT116 cells were transfected with CTL siRNA or HuR siRNAs for 48 hr and knockdown of HuR was determined by immunoblotting using GAPDH as loading control. (K) RIP assays were performed from formaldehyde-crosslinked HCT116 cell lysates transfected with CTL siRNA or HuR siRNAs. Enrichment of PURPL normalized to GAPDH mRNA in the IP material was assessed by RT-qPCR. (L) Immunoblotting for MYBBP1A was performed for the RIP assays in “K”. Error bars represent SD from 3 experiments. **p<0.01.
We therefore hypothesized that the MYBBP1A-PURPL complex is formed through an adaptor protein that directly binds to this lncRNA. From our RNA pulldowns and mass spectrometry, we selected RNA-binding protein HuR as a potential adaptor protein due to the following reasons. First, HuR was strongly enriched in the PURPL pulldowns (Figure 6B). Second, we and others have previously shown that HuR plays a role in the p53 pathway (Lopez de Silanes et al., 2004, Lal et al., 2004, Lal et al., 2005, Mazan-Mamczarz et al., 2003, Galban et al., 2003). Finally, HuR binds to AU-rich elements (AREs) in RNA (Lopez de Silanes et al., 2004, Lal et al., 2004) and we found a single consensus (Lebedeva et al., 2011) high affinity HuR binding motif (ARE core) at the 3′end of PURPL (Figure 6C).
When we performed in vitro RNA pulldowns. we found that Bi-PURPL but not Bi-Luc associates with HuR (Figure 6D); as a negative control, GAPDH was detected only in input (Figure 6D). Moreover, RNA pulldowns using purified recombinant GST-HuR, showed that GST-HuR directly binds to Bi-PURPL (Figure 6E). In RT-qPCR following HuR-RIP from UV-crosslinked HCT116 cells, ~12-fold enrichment of PURPL was observed in HuR RIPs indicating direct interaction of PURPL and HuR in intact cells (Figure 6F). To determine if HuR interacts with the ARE in PURPL, we performed HuR-RIP assays from UV-crosslinked HCT116 cells after sonicating the lysate before the RIP assay, to fragment the RNAs, a strategy used in the DINO paper (Schmitt et al., 2016). RT-qPCR with primers that span the PURPL-ARE or an adjacent Non-ARE region, showed that the PURPL-ARE was specifically enriched in the HuR IPs (Figure 6G). These data suggest that HuR directly binds to the ARE at the 3′end of PURPL RNA.
If HuR is the adaptor responsible for recruiting MYBBP1A to the PURPL RNA, HuR should interact with MYBBP1A, and silencing HuR should abrogate the PURPL-MYBBP1A interaction. Indeed, we found enrichment of HuR in the MYBBP1A IP (Figure 6H); the HuR-MYBBP1A complex was resistant to RNase, suggesting that HuR and MYBBP1A form a protein-protein complex (Figure 6I). Upon silencing HuR (Figure 6J) followed by MYBBP1A RIP and RT-qPCR from formaldehyde crosslinked cells, the PURPL-MYBBP1A interaction was lost (Figure 6K), although we pulled down comparable levels of MYBBP1A (Figure 6L). These data indicate that MYBBP1A associates with PURPL RNA via the adaptor protein HuR.
Silencing MYBBP1A partially rescues basal p53 levels and proliferation of PURPL-KO cells
We next silenced MYBBP1A in PURPL-WT and PURPL-KO cells and assessed the effect on p53 protein levels and cell proliferation. Silencing MYBBP1A partially reduced basal p53 protein levels (Figures 7A, S10A and S10B) and p21 mRNA levels (Figure 7B) and increased proliferation only in PURPL-KO cells (Figure 7C), suggesting that MYBBP1A mediates the effects of PURPL, at least in part, by regulating basal p53 levels and cell proliferation.
Figure 7. Silencing MYBBP1A in PURPL-KO cells partially rescues basal p53 levels and cell proliferation.
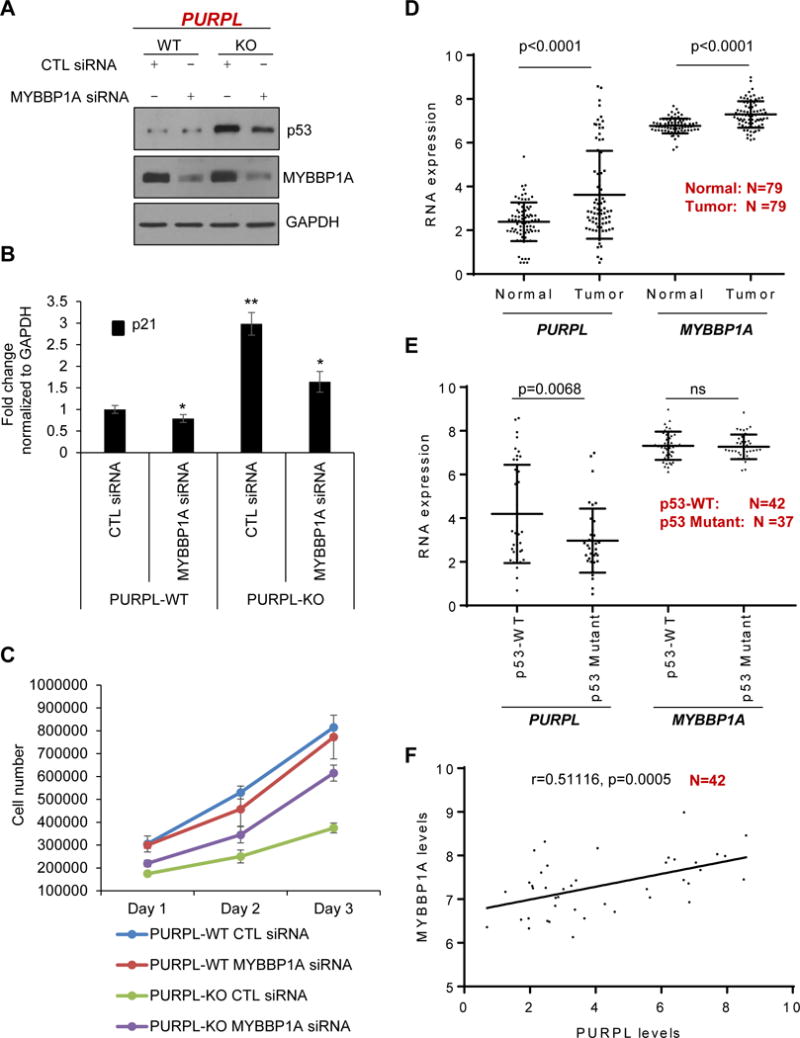
(A, B) PURPL-WT and PURPL-KO cells were transfected with CTL siRNA or MYBBP1A siRNAs for 48 hr. The levels of MYBBP1A, p53 and the loading control GAPDH were assessed by immunoblotting from whole cell extracts (A) and p21 mRNA levels were measured by RT-qPCR normalized to GAPDH mRNA (B). (C) PURPL-WT and PURPL-KO cells were transfected with CTL siRNA or MYBBP1A siRNAs and cell proliferation was assessed by trypan blue exclusion cell count assay from 2 independent experiments. (D–F) Analysis of PURPL and MYBBP1A mRNA levels in CRC patient samples in the microarrays from the UMMC cohort. PURPL and MYBBP1A mRNA levels (log2 transformed) were compared between normal (N=79) and tumor (N=79) samples (D) and between p53WT (N=42) and mutant p53 tumors (N=37) (E). (F) Significant positive correlation (correlation coefficient shown as “r”) between PURPL and MYBBP1A mRNA levels was observed in the p53WT CRC tumors. “N” represents the number of samples in D–F. Error bars in “B” represent SD from 3 experiments. *p<0.05 and **p<0.01.
To determine if our findings from cell lines are relevant to CRC, we first analyzed PURPL and MYBBP1A mRNA levels in a CRC cohort (UMMC cohort) where we have performed lncRNA arrays from 79 CRC patient samples and matched normal (Schetter et al., in preparation). Both PURPL RNA and MYBBP1A mRNA were significantly up-regulated (PURPL: 2.35-fold, p<0.0001; MYBBP1A mRNA: 1.44-fold p<0.0001) in the tumors when compared to normal tissue (Figure 7D). To determine if PURPL and MYBBP1A expression correlate with p53 mutation status, we compared PURPL levels between p53WT and mutant p53 tumors. Of the 79 tumor samples, 42 were p53WT and 37 had missense or nonsense TP53 mutations. As compared to p53WT tumors, PURPL was significantly down-regulated (2.39-fold, p=0.0068) in mutant p53 tumors (Figure 7E). In addition, PURPL was more significantly upregulated in p53WT CRC tumors vs matched normal (p<0.0001) as compared to Mutant p53 CRC tumors vs matched normal (p=0.036) (Figures S10A and S10B).
However, MYBBP1A mRNA levels were not significantly different between p53WT and p53 mutant tumors (Figure 7E). The higher PURPL levels and the unchanged MYBBP1A mRNA levels between p53WT tumors as compared to p53 mutant tumors is consistent with our RNA-seq data and other reports (Hunten et al., 2015, Leveille et al., 2015) where PURPL was identified as a p53-regulated lncRNA, whereas MYBBP1A is not a p53 target gene. Interestingly, when we looked at the correlation between PURPL and MYBBP1A mRNA levels in this CRC cohort, we observed significant positive correlation (correlation coefficient r=0.51, p=0.0005) between MYBBP1A mRNA and PURPL levels in p53WT tumors (Figure 7F) but not in the p53 mutant tumors (Figure S10C), which may be consistent with our data from cell lines indicating that both PURPL and MYBBP1A are involved in keeping basal p53 levels low. Collectively, our results identify an auto-regulatory feedback loop between p53 and PURPL that is mediated in part by recruitment of MYBBP1A to PURPL by the RNA-binding protein HuR.
DISCUSSION
In this study, we functionally characterized PURPL, a lncRNA directly regulated by p53 under basal conditions and after DNA damage. PURPL suppresses basal p53 levels, suggesting an auto-regulatory feedback loop.
It is becoming increasingly clear that even in actively dividing cells with minimal p53 activity, p53 directly activates the basal expression of many genes including CDKN1A, GDF15 and DDB2 (Allen et al., 2014, Espinosa et al., 2003, Tang et al., 1998). Using Global Run-on sequencing, it was proposed that many p53 targets are ‘primed’ before p53 activation to allow rapid upregulation of these genes upon p53 activation (Allen et al., 2014). Consistent with these findings, although PURPL is upregulated by p53 after DNA damage, its basal expression is also controlled by p53 in multiple CRC lines and in CRC tumors. Although p53 is inactivated in a majority of cancers, many tumors have intact p53 signaling, and therapeutic activation of p53 signaling through MDM2 inhibition is being investigated in clinical trials (Khoo et al., 2014). The significance of basal p53 signaling in cancer therapeutics was demonstrated in a recent study on a large panel of cell lines and patient-derived tumor xenografts (Espinosa and Sullivan, 2015, Jeay et al., 2015). The authors found that cell lines sensitive to p53-activating drugs had partially active p53 signaling even in the absence of the drug. PURPL may be a promising candidate for future translational research since PURPL loss can elevate basal p53 to impair proliferation, induce cell death in response to genotoxic agents and inhibit tumorigenicity in vivo. However, it will be important to determine the function of PURPL in normal cells.
Our study contributes to the emerging concept that lncRNAs acts as guides, decoys or scaffolds to control cellular processes. This has been shown previously in the context of p53: lincRNA-p21 functions as a transcriptional repressor in the p53 pathway and modulates the localization of hnRNPK (Huarte et al., 2010, Dimitrova et al., 2014). PANDA, another p53-regulated lncRNA transcribed from a locus upstream of p21, interacts with the transcription factor NF-YA to regulate the expression of pro-apoptotic genes during DNA damage (Hung et al., 2011). In addition, NORAD is indirectly regulated by p53 and plays a critical role in maintaining genomic stability by functioning as a decoy and regulates the association of the RNA-binding protein PUMILIO to its target mRNAs (Lee et al., 2016). A recent study showed that the p53-regulated lncRNA DINO directly binds to the C-terminal domain of p53 and regulates p53 levels in response to DNA damage (Schmitt et al., 2016).
PURPL suppresses basal p53 levels, in part, by preventing the p53-MYBBP1A complex formation in the nucleoplasm. To further support this regulation, it would be important to estimate, the number of molecules of PURPL, p53 and MYBBP1A in the nucleoplasm, to determine if they are present in stoichiometric amounts. Further experiments are needed to determine if this mechanism, the p53-dependence of PURPL expression and the p53-dependence of the chemosensitizing effects of PURPL inactivation are restricted to tumor cells that express p53WT or could also be observed in normal cells and mutant p53 cells. The role of PURPL in CRC and other cancers will require further analysis in multiple cohorts to determine if PURPL expression is associated with clinical outcome.
Future investigations on identification of other effectors of PURPL, determining if HuR and MYBBP1A interact directly or via other proteins, additional molecular mechanism(s) by which this lncRNA regulates p53, identifying and determining the role of other PURPL targets in mediating the effects of this lncRNA, will be necessary to fully understand how PURPL controls basal p53 levels and in determining its role in tumor initiation and progression. Finally, it will be interesting to see if depletion/deletion of p53 in PURPL-KO cells influences the tumorigenicity of these cells in mice.
EXPERIMENTAL PROCEDURES
Ribo-zero paired-end RNA-Seq and bioinformatic analysis
The isogenic p53WT and p53KO HCT116, RKO and SW48 cells were untreated or treated with DOXO (300 nM) for 16 hr and total RNA was isolated using RNeasy kit (Qiagen). Total RNA was fragmented and the cleaved RNA fragments were copied into first strand cDNA using reverse transcriptase and random primers, followed by second strand cDNA synthesis using DNA polymerase I and RNase H. The resulting double-strand cDNA was used as the input to a standard Illumina library prep with end-repair, adapter ligation and PCR amplification being performed to generate a library that would go on to the HiSeq2000 instrument for sequencing. The HiSeq Real Time Analysis software (RTA 1.18.64) was used for processing image files, the Illumina BCL2fastq1.8.4 was used for demultiplex and converting binary base calls and qualities to FASTQ format. The sequencing reads were trimmed adapters and low quality bases using Trimmomatic (version 0.3), the trimmed reads were aligned to human hg19 reference genome (GRCh37/UCSC hg19) and Ensembl annotation version 70 using TopHat_v2.0.8 software. Hundred bases long paired-end reads were assessed for quality using PICARD and FastQC. The generated FASTQ files were mapped using TopHat2 alignment algorithm. Differential gene expression analysis was performed using Cufflinks and Cuffdiff. Average read length was 110 nucleotides and we had ~ 150 million mapped reads per sample. For a non-zero-fold change, we added 0.01 to the FPKM of each gene.
UMMC (University of Maryland Medical Center) cohort analysis
Pairs of primary colon tumor and adjacent non-tumorous tissues came from 83 patients recruited from the University of Maryland Medical Center or Baltimore Veterans Affairs Medical Center between 1993 and 2002. Cases with familial adenomatous polyposis or human nonpolyposis colorectal cancer were excluded from this study. Tissues were flash frozen after surgery. Detailed backgrounds for each tissue donor, including age, sex, clinical staging, tumor location, and receipt of adjuvant chemotherapy have been collected. Tumor histopathology was classified according to the World Health Organization Classification of Tumor system. RNA from frozen tissue samples was extracted using standard TRIZOL (Invitrogen, Carlsbad, California) methods. TP53 mutation status was determined by sequencing all TP53 exons. The microarrays were SurePrint G3 Human Gene Expression 8×60K Microarray Kit and performed following the manufacturer’s instructions.
Targeted deletion using CRISPR/Cas9
An All-in-one construct expressing GFP, the human codon-optimized Streptococcus pyogenes Cas9 Nickase mutant and the 2 gRNAs targeting the p53RE in the PURPL promoter was designed and purchased from DNA2.0 (https://www.dna20.com/). To select PURPL-WT and PURPL-KO clones, HCT116 were transfected with 5 μg of the all-in-one construct and after 48 hr, GFP positive cells were FACS sorted. The GFP positive cells were seeded at 1 cell per well of 96-well plates in 100 μl of DMEM. After 3 weeks, clones were harvested and split into two 24-well plates. Total RNA was isolated from a 24-well plate and PURPL expression was measured by RT-qPCR normalized to GAPDH. Genomic DNA from individual clones with dramatically reduced PURPL expression was extracted, and the DNA flanking the p53RE of PURPL was PCR amplified and subjected to Sanger sequencing.
Xenograft assays
Animal protocols were approved by the National Cancer Institute Animal Care and Use Committee following AALAAC guidelines and policies. HCT116 PURPL-WT and PURPL-KO cells were trypsinized and washed with PBS. Live cells were counted with trypan blue exclusion and equal numbers of live cells were injected for each clone. Cells (1 × 106) were mixed with 30% matrigel in PBS on ice and the mixture was injected into the flanks of 6–8-week-old female athymic nude mice (Animal Production Program, Frederick, MD, USA) (each group N=10). Tumor volume was measured twice a week after 1 week of injection.
Supplementary Material
Table S1, Related to Figure S1: RNA-seq data from HCT116, RKO and SW48 cells.
Table S2, Related to Figure 3. Microarrays from PURPL-WT and PURPL-KO HCT116.
Table S3, Related to Figure 5. RNA-pulldown followed by mass spectrometry data.
Table S4, Related to Figures 1–7. Sequence of primers and ASOs used in this study.
Highlights.
PURPL is a p53-regulated lncRNA
p53 is upregulated upon loss of PURPL, inducing growth defects
PURPL associates with the p53-regulator, MYBBP1A
PURPL suppresses p53 levels by inhibiting the p53-MYBBP1A interaction
Acknowledgments
We thank Bert Vogelstein for the isogenic cell lines, Myriam Gorospe for the GST and GST-HuR proteins and Tom Misteli, Susan Gottesman, Shiv Grewal and Glenn Merlino for their comments on this manuscript. This research was supported by the Intramural Research Program (A.L., S. A., C. C. H) of the National Cancer Institute (NCI), Center for Cancer Research (CCR), NIH and the NIGMS/NIH grant 5SC1GM093999-06 (S.S). K.V.P lab is supported by grants from NIH [GM088252] and American Cancer Society [RSG-11-174-01-RMC].
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
ACCESSION NUMBERS
The accession number for the RNA-seq data deposited for this paper is GSE79249 and accession number for the microarray data is GSE79053.
AUTHOR CONTRIBUTIONS
Conceived and designed the experiments: Li.X., S.M., J.M.F., L.A. Performed experiments: S.D.K., Z.X., M.M., C.R., R.C., P.S., J.K., M.J., L.A. Contributed materials/tools and performed data analysis: G.B., S.S., S.A., W.X., K.D., J.L., T.W., E.F., H.M., Z.Y., C.M., A.A., H.C.C., D.A., F.S., R.F. Wrote the manuscript: Li.X., J.M.F., L. A. Edited the manuscript: Li, X, J.M.F., P.K.V., L.A.
References
- ADRIAENS C, STANDAERT L, BARRA J, LATIL M, VERFAILLIE A, KALEV P, BOECKX B, WIJNHOVEN PW, RADAELLI E, VERMI W, LEUCCI E, LAPOUGE G, BECK B, VAN DEN OORD J, NAKAGAWA S, HIROSE T, SABLINA AA, LAMBRECHTS D, AERTS S, BLANPAIN C, MARINE JC. p53 induces formation of NEAT1 lncRNA-containing paraspeckles that modulate replication stress response and chemosensitivity. Nat Med. 2016;22:861–8. doi: 10.1038/nm.4135. [DOI] [PubMed] [Google Scholar]
- AKAOGI K, ONO W, HAYASHI Y, KISHIMOTO H, YANAGISAWA J. MYBBP1A suppresses breast cancer tumorigenesis by enhancing the p53 dependent anoikis. BMC Cancer. 2013;13:65. doi: 10.1186/1471-2407-13-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ALLEN MA, ANDRYSIK Z, DENGLER VL, MELLERT HS, GUARNIERI A, FREEMAN JA, SULLIVAN KD, GALBRAITH MD, LUO X, KRAUS WL, DOWELL RD, ESPINOSA JM. Global analysis of p53-regulated transcription identifies its direct targets and unexpected regulatory mechanisms. Elife. 2014;3:e02200. doi: 10.7554/eLife.02200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ARUN G, DIERMEIER S, AKERMAN M, CHANG KC, WILKINSON JE, HEARN S, KIM Y, MACLEOD AR, KRAINER AR, NORTON L, BROGI E, EGEBLAD M, SPECTOR DL. Differentiation of mammary tumors and reduction in metastasis upon Malat1 lncRNA loss. Genes Dev. 2016;30:34–51. doi: 10.1101/gad.270959.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BECKERMAN R, PRIVES C. Transcriptional regulation by p53. Cold Spring Harb Perspect Biol. 2010;2:a000935. doi: 10.1101/cshperspect.a000935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BIEGING KT, ATTARDI LD. Deconstructing p53 transcriptional networks in tumor suppression. Trends Cell Biol. 2012;22:97–106. doi: 10.1016/j.tcb.2011.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BUNZ F, DUTRIAUX A, LENGAUER C, WALDMAN T, ZHOU S, BROWN JP, SEDIVY JM, KINZLER KW, VOGELSTEIN B. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science. 1998;282:1497–501. doi: 10.1126/science.282.5393.1497. [DOI] [PubMed] [Google Scholar]
- BUNZ F, HWANG PM, TORRANCE C, WALDMAN T, ZHANG Y, DILLEHAY L, WILLIAMS J, LENGAUER C, KINZLER KW, VOGELSTEIN B. Disruption of p53 in human cancer cells alters the responses to therapeutic agents. J Clin Invest. 1999;104:263–9. doi: 10.1172/JCI6863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHAN TA, HERMEKING H, LENGAUER C, KINZLER KW, VOGELSTEIN B. 14-3-3Sigma is required to prevent mitotic catastrophe after DNA damage. Nature. 1999;401:616–20. doi: 10.1038/44188. [DOI] [PubMed] [Google Scholar]
- CHANG TC, WENTZEL EA, KENT OA, RAMACHANDRAN K, MULLENDORE M, LEE KH, FELDMANN G, YAMAKUCHI M, FERLITO M, LOWENSTEIN CJ, ARKING DE, BEER MA, MAITRA A, MENDELL JT. Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol Cell. 2007;26:745–52. doi: 10.1016/j.molcel.2007.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHAUDHARY R, GRYDER B, WOODS WS, SUBRAMANIAN M, JONES MF, LI XL, JENKINS LM, SHABALINA SA, MO M, DASSO M, YANG Y, WAKEFIELD LM, ZHU Y, FRIER SM, MORIARITY BS, PRASANTH KV, PEREZ-PINERA P, LAL A. Prosurvival long noncoding RNA PINCR regulates a subset of p53 targets in human colorectal cancer cells by binding to Matrin 3. Elife. 2017:6. doi: 10.7554/eLife.23244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHAUDHARY R, LAL A. Long noncoding RNAs in the p53 network. Wiley Interdiscip Rev RNA. 2016 doi: 10.1002/wrna.1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DEY BK, PFEIFER K, DUTTA A. The H19 long noncoding RNA gives rise to microRNAs miR-675-3p and miR-675-5p to promote skeletal muscle differentiation and regeneration. Genes Dev. 2014;28:491–501. doi: 10.1101/gad.234419.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DIMITROVA N, ZAMUDIO JR, JONG RM, SOUKUP D, RESNICK R, SARMA K, WARD AJ, RAJ A, LEE JT, SHARP PA, JACKS T. LincRNA-p21 activates p21 in cis to promote Polycomb target gene expression and to enforce the G1/S checkpoint. Mol Cell. 2014;54:777–90. doi: 10.1016/j.molcel.2014.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EL-DEIRY WS, TOKINO T, VELCULESCU VE, LEVY DB, PARSONS R, TRENT JM, LIN D, MERCER WE, KINZLER KW, VOGELSTEIN B. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993;75:817–25. doi: 10.1016/0092-8674(93)90500-p. [DOI] [PubMed] [Google Scholar]
- ENGREITZ JM, PANDYA-JONES A, MCDONEL P, SHISHKIN A, SIROKMAN K, SURKA C, KADRI S, XING J, GOREN A, LANDER ES, PLATH K, GUTTMAN M. The Xist lncRNA exploits three-dimensional genome architecture to spread across the X chromosome. Science. 2013;341:1237973. doi: 10.1126/science.1237973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ESPINOSA JM, SULLIVAN KD. A signature for success. Elife. 2015:4. doi: 10.7554/eLife.08773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ESPINOSA JM, VERDUN RE, EMERSON BM. p53 functions through stress- and promoter-specific recruitment of transcription initiation components before and after DNA damage. Mol Cell. 2003;12:1015–27. doi: 10.1016/s1097-2765(03)00359-9. [DOI] [PubMed] [Google Scholar]
- FATICA A, BOZZONI I. Long non-coding RNAs: new players in cell differentiation and development. Nat Rev Genet. 2014;15:7–21. doi: 10.1038/nrg3606. [DOI] [PubMed] [Google Scholar]
- GALBAN S, MARTINDALE JL, MAZAN-MAMCZARZ K, LOPEZ DE SILANES I, FAN J, WANG W, DECKER J, GOROSPE M. Influence of the RNA-binding protein HuR in pVHL-regulated p53 expression in renal carcinoma cells. Mol Cell Biol. 2003;23:7083–95. doi: 10.1128/MCB.23.20.7083-7095.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GEORGE B, HORN D, BAYO P, ZAOUI K, FLECHTENMACHER C, GRABE N, PLINKERT P, KRIZHANOVSKY V, HESS J. Regulation and function of Myb-binding protein 1A (MYBBP1A) in cellular senescence and pathogenesis of head and neck cancer. Cancer Lett. 2015;358:191–9. doi: 10.1016/j.canlet.2014.12.042. [DOI] [PubMed] [Google Scholar]
- GROSSI E, SANCHEZ Y, HUARTE M. Expanding the p53 regulatory network: LncRNAs take up the challenge. Biochim Biophys Acta. 2016;1859:200–8. doi: 10.1016/j.bbagrm.2015.07.011. [DOI] [PubMed] [Google Scholar]
- GUPTA RA, SHAH N, WANG KC, KIM J, HORLINGS HM, WONG DJ, TSAI MC, HUNG T, ARGANI P, RINN JL, WANG Y, BRZOSKA P, KONG B, LI R, WEST RB, VAN DE VIJVER MJ, SUKUMAR S, CHANG HY. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature. 2010;464:1071–6. doi: 10.1038/nature08975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HERMEKING H. MicroRNAs in the p53 network: micromanagement of tumour suppression. Nat Rev Cancer. 2012 doi: 10.1038/nrc3318. [DOI] [PubMed] [Google Scholar]
- HOCHSTATTER J, HOLZEL M, ROHRMOSER M, SCHERMELLEH L, LEONHARDT H, KEOUGH R, GONDA TJ, IMHOF A, EICK D, LANGST G, NEMETH A. Myb-binding protein 1a (Mybbp1a) regulates levels and processing of pre-ribosomal RNA. J Biol Chem. 2012;287:24365–77. doi: 10.1074/jbc.M111.303719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HUARTE M, GUTTMAN M, FELDSER D, GARBER M, KOZIOL MJ, KENZELMANN-BROZ D, KHALIL AM, ZUK O, AMIT I, RABANI M, ATTARDI LD, REGEV A, LANDER ES, JACKS T, RINN JL. A large intergenic noncoding RNA induced by p53 mediates global gene repression in the p53 response. Cell. 2010;142:409–19. doi: 10.1016/j.cell.2010.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HUNG T, WANG Y, LIN MF, KOEGEL AK, KOTAKE Y, GRANT GD, HORLINGS HM, SHAH N, UMBRICHT C, WANG P, WANG Y, KONG B, LANGEROD A, BORRESEN-DALE AL, KIM SK, VAN DE VIJVER M, SUKUMAR S, WHITFIELD ML, KELLIS M, XIONG Y, WONG DJ, CHANG HY. Extensive and coordinated transcription of noncoding RNAs within cell-cycle promoters. Nat Genet. 2011;43:621–9. doi: 10.1038/ng.848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HUNTEN S, KALLER M, DREPPER F, OELJEKLAUS S, BONFERT T, ERHARD F, DUECK A, EICHNER N, FRIEDEL CC, MEISTER G, ZIMMER R, WARSCHEID B, HERMEKING H. p53-Regulated Networks of Protein, mRNA, miRNA, and lncRNA Expression Revealed by Integrated Pulsed Stable Isotope Labeling With Amino Acids in Cell Culture (pSILAC) and Next Generation Sequencing (NGS) Analyses. Mol Cell Proteomics. 2015;14:2609–29. doi: 10.1074/mcp.M115.050237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IYER MK, NIKNAFS YS, MALIK R, SINGHAL U, SAHU A, HOSONO Y, BARRETTE TR, PRENSNER JR, EVANS JR, ZHAO S, POLIAKOV A, CAO X, DHANASEKARAN SM, WU YM, ROBINSON DR, BEER DG, FENG FY, IYER HK, CHINNAIYAN AM. The landscape of long noncoding RNAs in the human transcriptome. Nat Genet. 2015;47:199–208. doi: 10.1038/ng.3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JEAY S, GAULIS S, FERRETTI S, BITTER H, ITO M, VALAT T, MURAKAMI M, RUETZ S, GUTHY DA, RYNN C, JENSEN MR, WIESMANN M, KALLEN J, FURET P, GESSIER F, HOLZER P, MASUYA K, WURTHNER J, HALILOVIC E, HOFMANN F, SELLERS WR, GRAUS PORTA D. A distinct p53 target gene set predicts for response to the selective p53-HDM2 inhibitor NVP-CGM097. Elife. 2015:4. doi: 10.7554/eLife.06498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JONES MF, LI XL, SUBRAMANIAN M, SHABALINA SA, HARA T, ZHU Y, HUANG J, YANG Y, WAKEFIELD LM, PRASANTH KV, LAL A. Growth differentiation factor-15 encodes a novel microRNA 3189 that functions as a potent regulator of cell death. Cell Death Differ. 2015;22:1641–53. doi: 10.1038/cdd.2015.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KHOO KH, VERMA CS, LANE DP. Drugging the p53 pathway: understanding the route to clinical efficacy. Nat Rev Drug Discov. 2014;13:217–36. doi: 10.1038/nrd4236. [DOI] [PubMed] [Google Scholar]
- KUMAZAWA T, NISHIMURA K, KATAGIRI N, HASHIMOTO S, HAYASHI Y, KIMURA K. Gradual reduction in rRNA transcription triggers p53 acetylation and apoptosis via MYBBP1A. Sci Rep. 2015;5:10854. doi: 10.1038/srep10854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KURODA T, MURAYAMA A, KATAGIRI N, OHTA YM, FUJITA E, MASUMOTO H, EMA M, TAKAHASHI S, KIMURA K, YANAGISAWA J. RNA content in the nucleolus alters p53 acetylation via MYBBP1A. EMBO J. 2011;30:1054–66. doi: 10.1038/emboj.2011.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LAL A, KAWAI T, YANG X, MAZAN-MAMCZARZ K, GOROSPE M. Antiapoptotic function of RNA-binding protein HuR effected through prothymosin alpha. EMBO J. 2005;24:1852–62. doi: 10.1038/sj.emboj.7600661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LAL A, MAZAN-MAMCZARZ K, KAWAI T, YANG X, MARTINDALE JL, GOROSPE M. Concurrent versus individual binding of HuR and AUF1 to common labile target mRNAs. EMBO J. 2004;23:3092–102. doi: 10.1038/sj.emboj.7600305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LAL A, THOMAS MP, ALTSCHULER G, NAVARRO F, O’DAY E, LI XL, CONCEPCION C, HAN YC, THIERY J, RAJANI DK, DEUTSCH A, HOFMANN O, VENTURA A, HIDE W, LIEBERMAN J. Capture of microRNA-bound mRNAs identifies the tumor suppressor miR-34a as a regulator of growth factor signaling. PLoS Genet. 2011;7:e1002363. doi: 10.1371/journal.pgen.1002363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEBEDEVA S, JENS M, THEIL K, SCHWANHAUSSER B, SELBACH M, LANDTHALER M, RAJEWSKY N. Transcriptome-wide analysis of regulatory interactions of the RNA-binding protein HuR. Mol Cell. 2011;43:340–52. doi: 10.1016/j.molcel.2011.06.008. [DOI] [PubMed] [Google Scholar]
- LEE JT. Epigenetic regulation by long noncoding RNAs. Science. 2012;338:1435–9. doi: 10.1126/science.1231776. [DOI] [PubMed] [Google Scholar]
- LEE S, KOPP F, CHANG TC, SATALURI A, CHEN B, SIVAKUMAR S, YU H, XIE Y, MENDELL JT. Noncoding RNA NORAD Regulates Genomic Stability by Sequestering PUMILIO Proteins. Cell. 2016;164:69–80. doi: 10.1016/j.cell.2015.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEVEILLE N, MELO CA, ROOIJERS K, DIAZ-LAGARES A, MELO SA, KORKMAZ G, LOPES R, AKBARI MOQADAM F, MAIA AR, WIJCHERS PJ, GEEVEN G, DEN BOER ML, KALLURI R, DE LAAT W, ESTELLER M, AGAMI R. Genome-wide profiling of p53-regulated enhancer RNAs uncovers a subset of enhancers controlled by a lncRNA. Nat Commun. 2015;6:6520. doi: 10.1038/ncomms7520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LI L, LIU B, WAPINSKI OL, TSAI MC, QU K, ZHANG J, CARLSON JC, LIN M, FANG F, GUPTA RA, HELMS JA, CHANG HY. Targeted disruption of Hotair leads to homeotic transformation and gene derepression. Cell Rep. 2013;5:3–12. doi: 10.1016/j.celrep.2013.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LING H, SPIZZO R, ATLASI Y, NICOLOSO M, SHIMIZU M, REDIS RS, NISHIDA N, GAFA R, SONG J, GUO Z, IVAN C, BARBAROTTO E, DE VRIES I, ZHANG X, FERRACIN M, CHURCHMAN M, VAN GALEN JF, BEVERLOO BH, SHARIATI M, HADERK F, ESTECIO MR, GARCIA-MANERO G, PATIJN GA, GOTLEY DC, BHARDWAJ V, SHUREIQI I, SEN S, MULTANI AS, WELSH J, YAMAMOTO K, TANIGUCHI I, SONG MA, GALLINGER S, CASEY G, THIBODEAU SN, LE MARCHAND L, TIIRIKAINEN M, MANI SA, ZHANG W, DAVULURI RV, MIMORI K, MORI M, SIEUWERTS AM, MARTENS JW, TOMLINSON I, NEGRINI M, BERINDAN-NEAGOE I, FOEKENS JA, HAMILTON SR, LANZA G, KOPETZ S, FODDE R, CALIN GA. CCAT2, a novel noncoding RNA mapping to 8q24, underlies metastatic progression and chromosomal instability in colon cancer. Genome Res. 2013;23:1446–61. doi: 10.1101/gr.152942.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LOPEZ DE SILANES I, ZHAN M, LAL A, YANG X, GOROSPE M. Identification of a target RNA motif for RNA-binding protein HuR. Proc Natl Acad Sci U S A. 2004;101:2987–92. doi: 10.1073/pnas.0306453101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MARIN-BEJAR O, MARCHESE FP, ATHIE A, SANCHEZ Y, GONZALEZ J, SEGURA V, HUANG L, MORENO I, NAVARRO A, MONZO M, GARCIA-FONCILLAS J, RINN JL, GUO S, HUARTE M. Pint lincRNA connects the p53 pathway with epigenetic silencing by the Polycomb repressive complex 2. Genome Biol. 2013;14:R104. doi: 10.1186/gb-2013-14-9-r104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MAZAN-MAMCZARZ K, GALBAN S, LOPEZ DE SILANES I, MARTINDALE JL, ATASOY U, KEENE JD, GOROSPE M. RNA-binding protein HuR enhances p53 translation in response to ultraviolet light irradiation. Proc Natl Acad Sci U S A. 2003;100:8354–9. doi: 10.1073/pnas.1432104100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MELLO SS, SINOW C, RAJ N, MAZUR PK, BIEGING-ROLETT K, BROZ DK, IMAM JFC, VOGEL H, WOOD LD, SAGE J, HIROSE T, NAKAGAWA S, RINN J, ATTARDI LD. Neat1 is a p53-inducible lincRNA essential for transformation suppression. Genes Dev. 2017;31:1095–1108. doi: 10.1101/gad.284661.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MELO CA, LEVEILLE N, ROOIJERS K, WIJCHERS PJ, GEEVEN G, TAL A, MELO SA, DE LAAT W, AGAMI R. A p53-bound enhancer region controls a long intergenic noncoding RNA required for p53 stress response. Oncogene. 2016 doi: 10.1038/onc.2015.502. [DOI] [PubMed] [Google Scholar]
- MENENDEZ D, NGUYEN TA, FREUDENBERG JM, MATHEW VJ, ANDERSON CW, JOTHI R, RESNICK MA. Diverse stresses dramatically alter genome-wide p53 binding and transactivation landscape in human cancer cells. Nucleic Acids Res. 2013;41:7286–301. doi: 10.1093/nar/gkt504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MUELLER AC, CICHEWICZ MA, DEY BK, LAYER R, REON BJ, GAGAN JR, DUTTA A. MUNC, a long noncoding RNA that facilitates the function of MyoD in skeletal myogenesis. Mol Cell Biol. 2015;35:498–513. doi: 10.1128/MCB.01079-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NIKULENKOV F, SPINNLER C, LI H, TONELLI C, SHI Y, TURUNEN M, KIVIOJA T, IGNATIEV I, KEL A, TAIPALE J, SELIVANOVA G. Insights into p53 transcriptional function via genome-wide chromatin occupancy and gene expression analysis. Cell Death Differ. 2012;19:1992–2002. doi: 10.1038/cdd.2012.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ONO W, HAYASHI Y, YOKOYAMA W, KURODA T, KISHIMOTO H, ITO I, KIMURA K, AKAOGI K, WAKU T, YANAGISAWA J. The nucleolar protein Myb-binding protein 1A (MYBBP1A) enhances p53 tetramerization and acetylation in response to nucleolar disruption. J Biol Chem. 2014;289:4928–40. doi: 10.1074/jbc.M113.474049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RAVER-SHAPIRA N, MARCIANO E, MEIRI E, SPECTOR Y, ROSENFELD N, MOSKOVITS N, BENTWICH Z, OREN M. Transcriptional activation of miR-34a contributes to p53-mediated apoptosis. Mol Cell. 2007;26:731–43. doi: 10.1016/j.molcel.2007.05.017. [DOI] [PubMed] [Google Scholar]
- REDIS RS, VELA LE, LU W, FERREIRA DE OLIVEIRA J, IVAN C, RODRIGUEZ-AGUAYO C, ADAMOSKI D, PASCULLI B, TAGUCHI A, CHEN Y, FERNANDEZ AF, VALLEDOR L, VAN ROOSBROECK K, CHANG S, SHAH M, KINNEBREW G, HAN L, ATLASI Y, CHEUNG LH, HUANG GY, MONROIG P, RAMIREZ MS, CATELA IVKOVIC T, VAN L, LING H, GAFA R, KAPITANOVIC S, LANZA G, BANKSON JA, HUANG P, LAI SY, BAST RC, ROSENBLUM MG, RADOVICH M, IVAN M, BARTHOLOMEUSZ G, LIANG H, FRAGA MF, WIDGER WR, HANASH S, BERINDAN-NEAGOE I, LOPEZ-BERESTEIN G, AMBROSIO AL, GOMES DIAS SM, CALIN GA. Allele-Specific Reprogramming of Cancer Metabolism by the Long Non-coding RNA CCAT2. Mol Cell. 2016;61:520–34. doi: 10.1016/j.molcel.2016.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RILEY T, SONTAG E, CHEN P, LEVINE A. Transcriptional control of human p53-regulated genes. Nat Rev Mol Cell Biol. 2008;9:402–12. doi: 10.1038/nrm2395. [DOI] [PubMed] [Google Scholar]
- RINN JL, KERTESZ M, WANG JK, SQUAZZO SL, XU X, BRUGMANN SA, GOODNOUGH LH, HELMS JA, FARNHAM PJ, SEGAL E, CHANG HY. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell. 2007;129:1311–23. doi: 10.1016/j.cell.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SANCHEZ Y, SEGURA V, MARIN-BEJAR O, ATHIE A, MARCHESE FP, GONZALEZ J, BUJANDA L, GUO S, MATHEU A, HUARTE M. Genome-wide analysis of the human p53 transcriptional network unveils a lncRNA tumour suppressor signature. Nat Commun. 2014;5:5812. doi: 10.1038/ncomms6812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHLACKOW M, NOJIMA T, GOMES T, DHIR A, CARMO-FONSECA M, PROUDFOOT NJ. Distinctive Patterns of Transcription and RNA Processing for Human lincRNAs. Mol Cell. 2017;65:25–38. doi: 10.1016/j.molcel.2016.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHMITT AM, GARCIA JT, HUNG T, FLYNN RA, SHEN Y, QU K, PAYUMO AY, PERES-DA-SILVA A, BROZ DK, BAUM R, GUO S, CHEN JK, ATTARDI LD, CHANG HY. An inducible long noncoding RNA amplifies DNA damage signaling. Nat Genet. 2016;48:1370–1376. doi: 10.1038/ng.3673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TANG HY, ZHAO K, PIZZOLATO JF, FONAREV M, LANGER JC, MANFREDI JJ. Constitutive expression of the cyclin-dependent kinase inhibitor p21 is transcriptionally regulated by the tumor suppressor protein p53. J Biol Chem. 1998;273:29156–63. doi: 10.1074/jbc.273.44.29156. [DOI] [PubMed] [Google Scholar]
- TANI H, MIZUTANI R, SALAM KA, TANO K, IJIRI K, WAKAMATSU A, ISOGAI T, SUZUKI Y, AKIMITSU N. Genome-wide determination of RNA stability reveals hundreds of short-lived noncoding transcripts in mammals. Genome Res. 2012;22:947–56. doi: 10.1101/gr.130559.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TANI H, NAKAMURA Y, IJIRI K, AKIMITSU N. Stability of MALAT-1, a nuclear long non-coding RNA in mammalian cells, varies in various cancer cells. Drug Discov Ther. 2010;4:235–9. [PubMed] [Google Scholar]
- TRIPATHI V, ELLIS JD, SHEN Z, SONG DY, PAN Q, WATT AT, FREIER SM, BENNETT CF, SHARMA A, BUBULYA PA, BLENCOWE BJ, PRASANTH SG, PRASANTH KV. The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Mol Cell. 2010;39:925–38. doi: 10.1016/j.molcel.2010.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TRIPATHI V, SHEN Z, CHAKRABORTY A, GIRI S, FREIER SM, WU X, ZHANG Y, GOROSPE M, PRASANTH SG, LAL A, PRASANTH KV. Long noncoding RNA MALAT1 controls cell cycle progression by regulating the expression of oncogenic transcription factor B-MYB. PLoS Genet. 2013;9:e1003368. doi: 10.1371/journal.pgen.1003368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TSOI LC, IYER MK, STUART PE, SWINDELL WR, GUDJONSSON JE, TEJASVI T, SARKAR MK, LI B, DING J, VOORHEES JJ, KANG HM, NAIR RP, CHINNAIYAN AM, ABECASIS GR, ELDER JT. Analysis of long non-coding RNAs highlights tissue-specific expression patterns and epigenetic profiles in normal and psoriatic skin. Genome Biol. 2015;16:24. doi: 10.1186/s13059-014-0570-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ULITSKY I, BARTEL DP. lincRNAs: Genomics, Evolution, and Mechanisms. Cell. 2013;154:26–46. doi: 10.1016/j.cell.2013.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VILLUNGER A, MICHALAK EM, COULTAS L, MULLAUER F, BOCK G, AUSSERLECHNER MJ, ADAMS JM, STRASSER A. p53- and drug-induced apoptotic responses mediated by BH3-only proteins puma and noxa. Science. 2003;302:1036–8. doi: 10.1126/science.1090072. [DOI] [PubMed] [Google Scholar]
- VOGELSTEIN B, LANE D, LEVINE AJ. Surfing the p53 network. Nature. 2000;408:307–10. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- VOUSDEN KH, LANE DP. p53 in health and disease. Nat Rev Mol Cell Biol. 2007;8:275–83. doi: 10.1038/nrm2147. [DOI] [PubMed] [Google Scholar]
- YOUNGER ST, KENZELMANN-BROZ D, JUNG H, ATTARDI LD, RINN JL. Integrative genomic analysis reveals widespread enhancer regulation by p53 in response to DNA damage. Nucleic Acids Res. 2015;43:4447–62. doi: 10.1093/nar/gkv284. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1, Related to Figure S1: RNA-seq data from HCT116, RKO and SW48 cells.
Table S2, Related to Figure 3. Microarrays from PURPL-WT and PURPL-KO HCT116.
Table S3, Related to Figure 5. RNA-pulldown followed by mass spectrometry data.
Table S4, Related to Figures 1–7. Sequence of primers and ASOs used in this study.