

Abstract
The study and development of azole-based CYP51 inhibitors is an active area of research across disciplines of biochemistry, pharmacology and infectious disease. Support of in vitro and in vivo studies require the development of robust asymmetric routes to single enantiomer products of this class of compounds. Herein, we describe a scalable and enantioselective synthesis to VNI and VFV, the two potent inhibitors of protozoan sterol 14α-demethylase (CYP51) that are currently under consideration for clinical trials for Chagas disease. A key transformation is the Jacobsen Hydrolytic Kinetic Resolution (HKR) reaction. The utility of the synthetic route is illustrated by the preparation of >25 g quantities of single enantiomers of VNI and VFV.
Keywords: Kinetic resolution, Stereospecific, Enzyme inhibitor, Azole
The cytochrome P450 sterol 14α-demethylase (CYP51) is a conserved metabolic enzyme required for sterol biosynthesis in eukaryotes, validated target for clinical and agricultural antifungals, and a target in trials for human infections with protozoan parasites. 1 Inhibition of CYP51 is effective for the treatment of these infections as the product of the pathway ergosterol is an essential component of membrane assembly in these pathogens.2 Recently, CYP51 inhibition has attracted interest as a potential anticancer target as rapidly proliferating tumor cells have a high demand for cholesterol to support their membrane assembly.3 The design, synthesis and evaluation of small molecule CYP51 inhibitors is an active area of research.4
Lepesheva and co-workers have pursued the development of several azole-based CYP51 inhibitors including VNI and VFV (Fig. 1) using both biochemical5 and in vivo models.6 This work benefits from a series of crystal structures of sterol 14α-demethylase (CYP51) co-crystalized with VNI and its analogs5 which resulted in the structure-based design of VFV, a compound with the broader antiprotozoan spectrum of activity. Furthermore, co-crystal structures provided an explanation of the significant difference in potency of enantiomeric forms of the chiral scaffold (cf. Fig. 1).5c In addition to compound potency, drug metabolism and pharmacokinetic properties (DMPK) have been evaluated using animal models and were found to be very favorable.6b
Fig. 1.
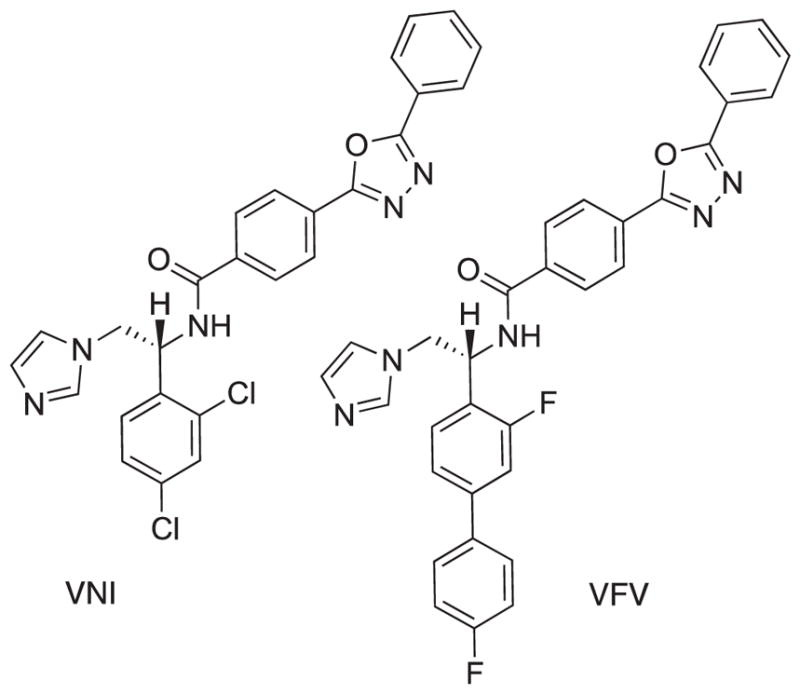
Structures of CYP51 inhibitors VNI and VFV.
In order to provide support for Structure-Activity Relationship (SAR) studies a convergent synthesis enabling structural modification at three key regions of the VNI scaffold was required; the azole iron binding heterocycle, the hydrophobic aromatic residue and the polycyclic ring system occupying the enzyme substrate entrance tunnel. In addition, selective access to the more potent (R)-configured isomer was needed. Finally, in vivo animal studies required VNI and VFV to be available in multi-gram quantities and the material to be of >98% purity.
In earlier work we developed a racemic and scalable synthesis of VNI readily allowing variation at the azole and the hydrophobic aromatic residue (Scheme 1).5b In general, azole (2) opening of aromatic epoxide 1 provides secondary alcohol 3. The latter is converted to the corresponding amine by way of simple three-step reaction sequence. Primary amine 4 is then condensed with carboxylic acid 5 using a HATU coupling. As this five-step synthetic route allows variation of all three regions of the scaffold [azole, hydrophobic (aryl) and polycyclic (carboxylic acid)] we favored its modification to an enantioselective synthetic process. We also required a process that would scale up in order to enable access to multi-gram quantities of the azole products for in vivo animal studies. To this end, we required epoxide 1 as a single enantiomer and a stereospecific (i.e. no loss of ee) interconversion of the secondary alcohol to the corresponding primary amine (3 to 4).
Scheme 1.
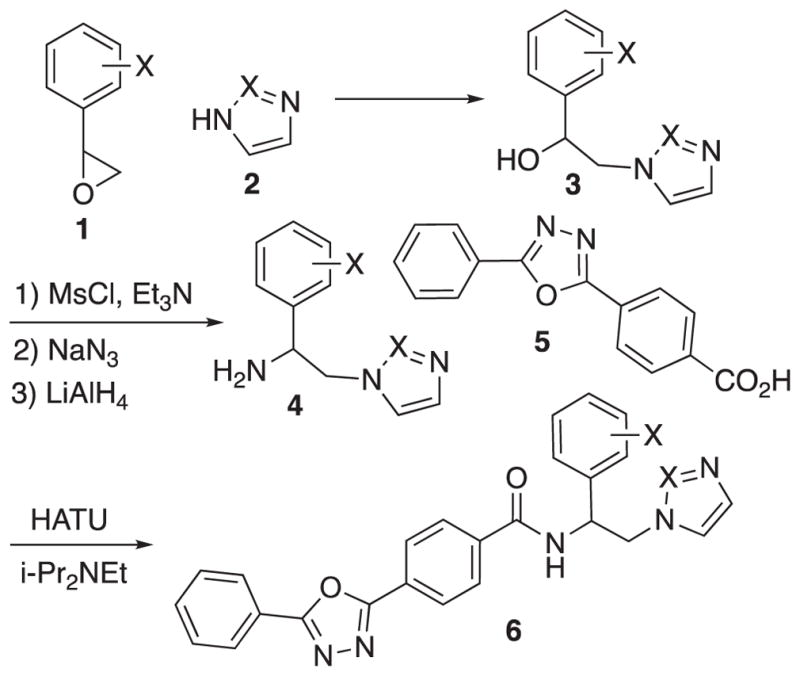
Racemic route to VNI and derivatives.5b
Single enantiomer azole agents such as VNI have been accessed by asymmetric reductions,7 (aza) Henry reactions,8 and Lipase mediated kinetic resolutions.9 We found the use of the Jacobsen hydrolytic kinetic resolution (HKR)10 of epoxide 7 to be most convenient as the reaction is conducted at high concentration (minimal solvent waste), uses low catalyst loading (5 mol%), has broad substrate scope and readily proceeds on large reaction scales (>25 g). In this way application of the HKR reaction to epoxide (±)- 7 proceeded in 43% yield on a 30 g scale to provide (S)-7 in >98% enantiomeric excess. The epoxide product was reacted with imidazole under basic conditions to afford azole alcohol 8, the product of epoxide opening at the less hindered primary carbon. Stereospecific conversion of alcohol 8 to amine 10 was achieved using a two step process. First, reaction of 8 with diphenylphosphoryl azide11 provided azide 9 followed by its reduction with lithiumaluminum hydride to give amine 10 (>98% ee). In the racemic synthesis series the conversion of alcohol 8 to azide 9 proceeded by way of an intermediate mesylate (cf. 3 to 4, Scheme 1), however this transformation occured with complete racemization when conducted with optically active alcohol (8, Scheme 2). The synthesis of (+)-VNI was completed using previously described HATU coupling of amine 5 and carboxylic acid 5.5b Using this route we prepared over 20 g of (+)-VNI of >95% purity and enantiomeric excess.
Scheme 2.

Multi-gram synthesis of (+)-VNI.
The synthesis of (+)-VFV (Scheme 3) illustrates the flexibility of our enantioselective and enabled the preparation of greater than 20 g of (+)-VFV (Scheme 3). The reaction sequence started with a Suzuki coupling of aryl bromide 11 and boronic acid 12. Aldehyde 13 was methylenated using the Corey-Chaykovsky reaction to give racemic epoxide 14. The latter was resolved on 28 g scale using the Jacobsen HKR to give (S)-14 in >98% ee. Epoxide (S)-14 was reacted with imidazole and the product, alcohol 15, converted to amine 17 by way of azide 16 without loss of optical activity. Installation of the oxadiazole proceeded using established HATU coupling conditions. Using this synthetic route 28 g of optically pure (+)-VFV was prepared.
Scheme 3.

Synthesis of (+)-VFV.
In conclusion, we have developed a robust and reliable synthetic route to azole antimicrobial agents as illustrated by multi-gram synthesis of (+)-VNI and (+)-VFV. The purity (>98%) of these products was verified by LC-MS analysis using multiple wavelengths and 1H and 13C NMR analysis of high concentration analytical solutions. The synthetic products were employed in animal studies by the Lepesheva group6 and demonstrated their effectiveness as antiprotozoan agents and are under consideration for clinical trials for Chagas disease.
Supplementary Material
Acknowledgments
This research was supported by the National Institutes of General Health (GM 067871) and the Vanderbilt Institute of Chemical Biology.
A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at https://doi.org/10.1016/j.tetlet.2017.09.070.
References
- 1.(a) Lepesheva GI, Waterman MR. Biochim Biophys Acta. 2007:467–477. doi: 10.1016/j.bbagen.2006.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lepesheva GI, Waterman MR. Curr Top Med Chem. 2011;11:2060–2071. doi: 10.2174/156802611796575902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lepesheva GI, Ott RD, Hargrove TY, et al. Chem Biol. 2007;14:1283–1293. doi: 10.1016/j.chembiol.2007.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Hargrove TY, Friggeri L, Wawrzak Z, et al. J Lipid Res. 2016;57:1552–1563. doi: 10.1194/jlr.M069229. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hu Q, Negri M, Olgen S, Harmann RW. Chem Med Chem. 2010;5:899–910. doi: 10.1002/cmdc.201000065. [DOI] [PubMed] [Google Scholar]
- 4.(a) Choi JY, Podust LM, Roush WR. Chem Rev. 2014;114:11242–11271. doi: 10.1021/cr5003134. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Emami S, Tavangar P, Keighobaki M. Eur J Med Chem. 2017;135:241–259. doi: 10.1016/j.ejmech.2017.04.044. [DOI] [PubMed] [Google Scholar]
- 5.Biochemical and In Vitro Studies: Lepesheva GI, Park H-W, Hargrove T, et al. J Biol Chem. 2010;285:1773–1780. doi: 10.1074/jbc.M109.067470.Hargrove TY, Kim K, Soeiro MNC, et al. Int J Parasitol Drugs Drug Resist. 2012;2:178–186. doi: 10.1016/j.ijpddr.2012.06.001.Friggeri L, Hargrove TY, Rachakonda G, et al. J Med Chem. 2014;57:6704–6717. doi: 10.1021/jm500739f.
- 6.In Vivo Studies: Soeiro MDC, de Souza EM, da Silva CF, et al. Antimicrob Agents Chemother. 2013;57:4151–4163. doi: 10.1128/AAC.00070-13.Lepesheva GI, Hargrove TY, Rachakonda G, et al. J Infectious Diseases. 2015;212:1439–1446. doi: 10.1093/infdis/jiv228.Villalta F, Dobish MC, Nde PN, et al. J Infectious Diseases. 2013;208:504–511. doi: 10.1093/infdis/jit042.Guedes-da-Silva FH, Bastista DG, Da Silva CF, et al. Antimicrob Agents Chemother. 2017;61:e02098–16. doi: 10.1128/AAC.02098-16.
- 7.(a) Oh K, Shimura Y, Ishikawa K, et al. Bioorg Med Chem. 2008;16:1090–1095. doi: 10.1016/j.bmc.2007.10.095. [DOI] [PubMed] [Google Scholar]; (b) Bisaha SN, Malley MF, Pudzianowski A, et al. Bioorg Med Chem Lett. 2005;15:2749–2751. doi: 10.1016/j.bmcl.2005.03.115. [DOI] [PubMed] [Google Scholar]
- 8.(a) Dobish MC, Villalta F, Waterman MR, Lepesheva GI, Johnston JN. Org Lett. 2012;14:6322–6325. doi: 10.1021/ol303092v. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Blay G, Domingo LR, Hernandez-Olmos V, Pedro JR. Chem Eur J. 2008;14:4725–4730. doi: 10.1002/chem.200800069. [DOI] [PubMed] [Google Scholar]
- 9.Mangas-Sanchez J, Busto E, Gotor-Fernandez V, Malpartida F, Gotor V. J Org Chem. 2011;76:2115–2122. doi: 10.1021/jo102459w. [DOI] [PubMed] [Google Scholar]
- 10.Tokunaga M, Larrow JF, Kakiuchi F, Jacobsen EN. Science. 1997;277:936–938. doi: 10.1126/science.277.5328.936. [DOI] [PubMed] [Google Scholar]
- 11.Thompson AS, Humphrey GR, DeMarco AM, Mathre DJ, Grabowski EJJ. J Org Chem. 1993;58:5886–5888. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.