

Abstract
Purpose of Review
As a subset of the organism-wide reaction to severe infection, the host vascular response has received increasing attention in recent years. The transformation that small blood vessels undergo to facilitate the clearance of pathogens may become harmful to the host if it occurs too broadly or if it is sustained too long. Adverse clinical manifestations of leaky and inflamed blood vessels include edema impairing the function of critical organs and circulatory shock.
Recent findings
suggest that this host vascular response may be both measurable and potentially targetable. Tie2 is a receptor tyrosine kinase heavily enriched in the vascular endothelium whose tonic signaling actively maintains vascular quiescence. When Tie2 becomes inactivated, important molecular brakes are released in the endothelium that in turn, potentiate inflammation and vascular leakage. The ligands of Tie2, Angiopoietin-1 and -2, regulate its activation status. Genetic and molecular studies spanning thousands of human subjects link Tie2 and imbalance of the Angiopoietins to major adverse clinical events arising from bacterial sepsis, other severe infections, and even acute sterile inflammation.
Summary
The Tie2 signaling axis may constitute a molecular switch in systemic inflammation that can be measured and manipulated to target the host vascular response therapeutically.
Keywords: angiopoietin, Tek, vascular permeability
INTRODUCTION
The inflammatory response to infections and sterile stressors has been intensely investigated at least since the 1940s, with reports of host-derived “pyrogens” later becoming leukocyte-secreted factors in tissue culture that were subsequently cloned in the 1980s as interleukin-2 (IL-2), interleukin-1 (IL1), and tumor necrosis factor alpha (TNFα) (1-10). Neutralizing proteins against each of these host factors have been part of the clinical armamentarium since the late 1990s. For individuals with rheumatoid arthritis and other autoimmune conditions, these drugs have been transformative. Yet the full promise of untangling the host inflammatory response has not been realized. Infections such as bacterial sepsis, influenza, and falciparum malaria continue to claim millions of lives around the world yearly despite common access to highly effective antimicrobials in most situations. Among other factors, the rapidity, severity, and redundancy of the “pyrogenic” attack have thwarted therapeutic efforts to tamp host-driven inflammation.
This review focuses on a comparatively late-developing feature of severe inflammation that is both more proximate to adverse clinical manifestations and more directly contributory—the host vascular response. Often taught as “rubor, calor, dolor, tumor,” the hyperermia, warmth, and swelling that develop at sites of tissue injury relate directly to microvascular changes. When this response occurs locally, humoral and cellular mediators of immunity are efficiently delivered to the injured site to clear pathogens. If the same response is not constrained however, the host can develop shock and edema of critical organs culminating in death. A signaling axis in the endothelium of blood vessels, anchored by the receptor tyrosine kinase Tie2, has an important role in regulating the molecular, cellular, and physical responses of blood vessels to the inflammatory milieu, that in turn, may impact the host vascular response.
Members of the Tie2 Axis
The cloning of Tie2 was reported in 1992 from a cDNA library of blood vessel tunica interna (11). Tie2 is a transmembrane tyrosine kinase whose expression is highly enriched in the endothelium. Germline deletion is lethal in utero around E10.5 with knockout (KO) embryos exhibiting edema, hemorrhages, and an underdeveloped cardiovascular system (12). The hearts show fewer myocardial trabeculations and retraction of the endocardium from the myocardium. Vascular patterning is less complex, suggesting a defect in developmental vessel remodeling. Results from conditional mice in which TIE2 is deleted at mid-gestation or beyond exhibit gross subcutaneous edema, supporting the notion of Tie2-dependent vascular stabilization mechanisms independent of developmental cardiovascular patterning defects (13, 14). Activating mutations in human TIE2 are strongly associated with venous malformations (15, 16), affirming an important role for this pathway in mammalian vascular development.
The ligands of Tie2, Angiopoietin-1 (Angpt-1) and Angiopoietin-2 (Angpt-2), are highly homologous to each other, but exert different effects on Tie2 (Fig 1) (17, 18) Angpt-1 is a canonical agonist of Tie2. Angpt-1 KO mice largely phenocopy Tie2 KO. Angpt-1 is made and secreted from peri-endothelial cells and platelet α-granules (19). Angpt-1 uses a carboxy-terminal fibrinogen-like domain to bind the 2nd of 3 extracellular immunoglobulin domains on Tie2 (20). The oligomerization state of Angpt-1 may be important for its ability to “cluster” Tie2 monomers to promote cross-phosphorylation in the intracellular domain of Tie2. A central coiled-coil domain enables Angpt-1 to dimerize, and an amino-terminal superclustering domain promotes higher-order multimers (21).
Figure 1. Tie2 signaling is finely tuned.

Tie2 expression is heavily enriched in the endothelium. Its 2nd immunoglobulin (Ig) domain ligates Angpts. Angpt-1 or -2 bind Tie2 via a C-terminal fibrinogen domain, and quarternary structural differences driven by the N-terminal superclustering (SCD) or coiled-coil (CCD) domains may account for the distinct actions of Angpt-1 as agonist vs. Angpt-2 as antagonist on Tie2. Angpt-1 is more highly oligomerized than Angpt-2 in vivo, and clustering of Tie2 monomers may be important for phosphorylation in its kinase domain. Tie1 is an orphan receptor in the endothelium that optimizes Tie2 signaling during inflammation. VE-PTP is a transmembrane tyrosine phosphatase that inhibits Tie2 signaling. Other abbreviations: EGF = epidermal growth factor repeat; FNIII = type 3 fibronectin domain. Adapted from (20).
Angpt-2 competitively inhibits Angpt-1-Tie2 binding and Tie2 activation in endothelial cells (18). This antagonist role may relate to the observation that Angpt-2 primarily forms a dimer in solution rather than higher-order multimers despite possessing its own coiled-coil and superclustering domain; specific amino acid differences between Angpt-1 and Angpt-2’s fibrinogen-like domain have also been cited for this differential response (21, 22). Angpt-2 transgenic mice share a vascular disruption phenotype with Angpt-1 and Tie2 KO mice whereas Angpt-2 deleted mice survive gestation exhibiting lymphatic defects beyond the current scope of discussion (18, 23). Angpt-2 is primarily made and secreted by the endothelium from pre-formed stores in Weibel-Palade bodies (24). Angpt-2 may therefore act in a paracrine, autocrine, or even “intracrine” fashion on Tie2 (25). Although Angpt-2 can, in certain contexts, activate rather than inhibit Tie2 signaling (26), results spanning a decade of studies from numerous laboratories consistently implicate Angpt-2 as a Tie2 antagonist during inflammation (27-36). Two other ligands of Tie2—Angpt-3 in mice and Angpt-4 in humans—are less-well studied.
VE-PTP (vascular endothelial protein tyrosine phosphatase) is a transmembrane tyrosine phosphatase that physically associates with and inhibits Tie2 signaling (37). VE-PTP also de-phosphorylates the junctional effector molecule VE-cadherin and the angiogenic receptor of VEGF (VEGFR2, Kdr). The interaction of VE-PTP with VE-cadherin maintains the endothelial barrier, but this association rapidly disassembles after the endothelium is exposed to VEGF (38). During angiogenesis when VEGF expression is high, the interaction of VE-PTP with VEGFR2 helps to organize the vectorial growth of new blood vessel sprouts (39). The net effect of VE-PTP activity on endothelial barrier function is in part determined by the degree of Tie2 expression. When Tie2 expression is artificially suppressed by RNAi, VE-PTP inhibition induces vascular hyperpermeability, presumably by leaving VEGFR2 and/or VE-cadherin in junction-destabilizing phosphorylated states (40). However, when Tie2 expression is intact, VE-PTP inhibition fortifies barrier function in a Tie2-requiring fashion.
Angiopoietins may also physically interact with integrins expressed in both endothelial cells and non-endothelial cells, specifically α5β1, α2β1, and αvβ5 (41-45). The Angpt-integrin interaction is of low affinity compared to nanomolar binding of Tie2, but in contexts of reduced Tie2 expression, this alternative signaling pathway may become functionally more important (43, 44). The role of these and perhaps additional integrins in Angpt-associated vascular inflammation requires further study.
Finally, a paralog of Tie2, Tie1 was cloned before Tie2 and yet remains an orphan receptor (46). Understanding Tie1’s role in biology, physiology and disease has also been more challenging than unraveling Tie2’s actions. For example, the global KO for Tie1 does not phenocopy Tie2 KO; instead, these mice die at E13.5 without the cardiac defects of Tie2 KO (47). More detailed studies using hypomorphic Tie1 alleles reveal a lymphatic defect in mice with low Tie1 protein (48). Tie1 associates with Tie2, but crystallographic data suggest that the Angpts are unlikely to bind Tie1 directly (20, 49). This receptor interaction has been described as inhibitory to Tie2 signaling: through heterodimerization or by inhibiting surface presentation of Tie2 (49, 50). But in vivo results suggest the opposite—when Tie1 is conditionally deleted from the endothelium of mice, Tie2 is less potently activated by Angpt-1 (36). As discussed below, acute inflammation leads to rapid Tie1 cleavage, which may contribute to Tie2 phosphorylation (35, 36).
Tie2 Signaling during Quiescence and Inflammation
Several features distinguish Tie2 signaling from other vascular receptor tyrosine kinases—the existence of multiple endogenous ligands with differential effects at the receptor; the membrane-bound paralog Tie1 that modulates signaling, the relocalization of activated Tie2 to junctions where it participates in dynamic signaling complexes, and the inhibitory effect of another membrane protein that is often in physical association with Tie2, VE-PTP. Together, these facets of Tie2 signaling are consistent with the notion that the degree of Tie2 activation can be finely tuned by distinct combinations of protein-protein interactions.
The high degree of Tie2 activation in quiescent, non-angiogenic blood vessels is also unique among this larger family of RTKs (51), suggesting one or more roles in vascular maintenance. Experimental gain-of-function with genetic or transient Angpt-1 overexpression demonstrates a remarkable vascular barrier defense phenotype (52, 53). With Angpt-1 stimulation, endothelial cells form a tightened barrier that is able to resist leakage as signaled by diverse ligands ranging from host proteins thrombin (54) and VEGF (52, 53) to bacterial lipopolysaccharides and anthrax lethal toxin (55-57). In animal models, this anti-leak effect of Tie2 stimulation protects organ function—prominently reducing edema in the lung but other organs, too—and enhances survival of animals subjected to endotoxemic shock, bacterial sepsis, anthrax, and malaria (56-61).
That Tie2 stimulation can promote broad-ranging anti-leakiness implicates the activation of central and conserved cellular effectors of barrier function that are downstream of many permeability mediators (reviewed in (62)). Indeed, barrier defense induced by Angpt-1 activating Tie2 is achieved by signaling the reorganization of the actin cytoskeleton and the accumulation of VE-cadherin at inter-endothelial junctions. Angpt-1 signals via PI3K and through NADPH oxidases to activate the Rho family GTPase Rac1 (55, 63). Rac1 is stabilized in its GTP-bound active form by the scaffolding protein IQGAP1 to signal the inhibition of another GTPase, RhoA, via the GTPase signaling transducer p190RhoGAP (Fig 2) (54, 55). These events unfold rapidly (~30 min) after Angpt-1 application to activate myosin-actin interactions that distribute actin into a “cortical” arrangement at the periphery of the cell. This rearrangement toward a spread morphology may reflect relaxation of tension within the endothelial cell or, conversely, development of pulling forces in the peri-junctional region; this remains to be determined. Angpt-1 stimulation favors the junctional accumulation of VE-cadherin. This may relate to transcriptional and post-translational actions (27, 31, 54, 55, 57, 64-70). Activated Tie2 is also thought to inhibit the inflammatory transcription factor NFκB via a signaling intermediate ABIN2 (71).
Figure 2. Tie2 signaling prevents vascular leakage.

Activated Tie2 migrates to cell junctions, remodels the cytoskeleton, and augments junctional accumulation of VE-cadherin. Compartmentalized intracellular signaling at intercellular junction may be important. Tie2 signals a complex of NADPH oxidase, its regulatory subunit p47phox, and the small GTPase Rac1. Rac1 is stabilized in its active GTP-bound form by IQGAP1. Rac1 itself signals the reorganization of the actin cytoskeleton toward a spread morphology, and by acting via the regulatory protein p190RhoGAP, inhibits the GTPase RhoA. Interaction of cortical actin via catenins (not depicted for ease of illustration) stabilizes VE-cadherin at the junction. VE-cadherin is essential for effective barrier formation in several microvascular beds.
During inflammation, a series of changes collaborate to attenuate Tie2 signaling (Fig 3). Pre-formed Angpt-2 protein is rapidly released from Weibel-Palade bodies. Tie1 is cleaved. Tie2 is cleaved, albeit more slowly than Tie1. Tie2 and Angpt-1 expression decline through mechanisms requiring further study—for example, reduced microvascular flow may itself downregulate Tie2 expression (72, 73). Angpt-2 production is induced by the de-repressed transcription factor Foxo1, which is otherwise held in an inactivated and phosphorylated state as long as Tie2 signals to PI3-kinase and Akt (33, 74). During inflammation, the fall in Tie2 signaling enables Foxo1 to relocate to the nucleus and transcribe ANGPT2 (33). Thus a pernicious feedback loop is activated. While Angpt-2 is not required for endothelial barrier disruption or endothelial inflammation, it potentiates both processes (30-32, 75).
Figure 3. Effects of Tie2 signaling in quiescence vs. vascular inflammation.
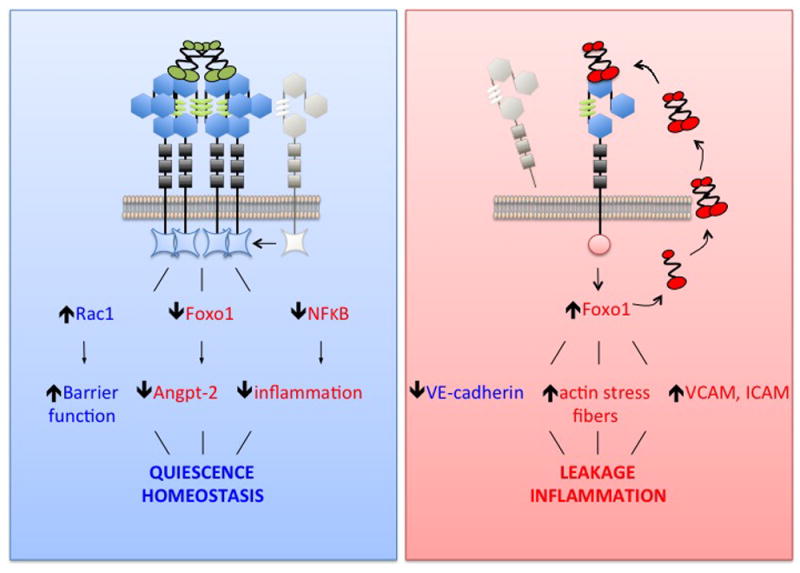
During quiescence, Tie2 is highly phosphorylated and actively maintains homeostasis. One example of active maintenance of homeostasis is the inhibition of inflammatory transcription factor NFκB via the signaling protein ABIN2. During inflammatory stress, several changes collaborate to switch off Tie2 signaling: reduced Tie2 expression, reduced surface Tie2, reduced surface Tie1 by proteolysis, reduced extracellular Angpt-1, and increased extracellular Angpt-2 that itself arises from at least two distinct mechanisms—rapid release of pre-formed Angpt-2 protein from Weibel-Palade bodies and de novo biosynthesis as dephosphorylated Tie2 is no longer able to repress Foxo1. Induction of Angpt-2 potentiates vascular inflammation and exacerbates vascular leakage
Angpt-2’s pivotal role in the phenotypic shift of inflamed blood vessels is emphasized by striking results from multiple laboratories in which endothelial-specific genetic, RNAi, or antibody-induced Angpt-2 neutralization enhances survival in experimental infections, protects organ function, defends barrier function, reduces inflammation, and thwarts long-term pathological vascular remodeling (34, 35, 76). A recent study has gone one step further by developing an antibody that clusters Angpt-2 monomers, thereby converting Angpt-2 into a Tie2 activator (75). Perhaps not surprisingly, this clustering antibody was found to be superior to an Angpt-2 neutralizing ability in terms of enhancing Tie2 phosphorylation in vivo. The clustering antibody more effectively protected organ function and enhanced survival in experimental sepsis.
Why the Tie2 axis would undergo these several changes that collectively attenuate signaling is unclear. Not only would such changes promote inflammation and leakage, but reduced Tie2 signaling may also influence the behavior of peri-endothelial cells in ways that would be deleterious to blood vessel function—e.g., (A) activated Tie2 induces the production of nitric oxide by the endothelium, which helps normal blood vessels maintain their vasomotor reactivity and (B) signaling via Tie2 appears to promote closer contacts between the endothelium and pericytes, which in turn may physically stabilize microvessels and even contribute to barrier function (76, 77).
Perhaps the shift in Tie2 signaling during inflammation relates to an adaptive function to facilitate local resolution of infection. A recent study has proposed that genetic variants that may lower TIE2 gene expression are actually highly prevalent in the general population (70). These variants were associated with a greater risk of developing acute respiratory distress syndrome in the intensive care unit. This study in humans echoes another genetics study conducted in outbred mice exposed to Ebola virus which showed that mortality was linked to subspecies allelic differences at Tie2 (78). Together, these studies suggest that vascular leakage and inflammation in a given host may arise at the nexus of genetic susceptibility and environmental exposure. Certain animals (or humans) may be genetically predisposed to Tie2 axis suppression, but only blossom a measurable phenotype under the stress of inflammation. Consistent with this two-hit model, Tie2 heterozygous mice appear normal with no excess leakage or inflammation at baseline, but develop more severe leakage and greater mortality during severe infection and inflammatory stress (57, 70, 79, 80).
The links between the Tie2 signaling axis and human disease are further buttressed by genetic studies implicating polymorphisms of ANGPT2 in acute lung injury (81, 82), and even more conclusively by blood-based studies spanning thousands of subjects with sepsis, sterile inflammation (e.g., from major trauma or surgery), malaria, and other acute inflammatory settings. The first such study linked early elevation of circulating Angpt-2 to pulmonary vascular leakage and impaired blood oxygenation during adult sepsis (27). And a second early study demonstrated that a fall in circulating Angpt-1, an increase in Angpt-2, or an elevated ratio of Angpt-2/Angpt-1 at the time of hospital admission was associated with mortality in pediatric septic shock (83). Peripheral levels of the Angiopoietins assayed within the first hour of hospitalization may even predict the future development of shock and death in the intensive care unit (31). These remarkable findings have been confirmed and advanced by numerous groups around the world (reviewed in (84)). The body of evidence supports the proposition that circulating Angiopoietin levels may enable operator-independent quantitative risk assessment. One of the important reasons that clinical trials in critical care are difficult is the wide heterogeneity; early determination of Angiopoietin levels may facilitate enrichment of future trial populations for a vascular-dominant pathophysiology. And because Angpt-2 expression is itself and indirect readout of Tie2 activity (Fig 3), serial measures of peripheral Angpt-2 may enable investigators to monitor Tie2 signaling over time in individual patients. Such applications could accelerate drug development for adjuvant therapies that optimize the host vascular response.
CONCLUSIONS
The Tie receptors and Angpts were identified in the 1990s and initially implicated in vascular inflammation in the early 2000s. Recent studies have advanced our understanding of this signaling axis in the contexts of infection and sterile inflammation. Angpt-1 protects organs from edema and enhances survival in a growing list of infections. Angpt-2, on the other hand, potentiates leakage and inflammation such that maneuvers to reduce Angpt-2 action are protective for the organism. Whether Tie2 hyperstimulation—e.g., via exogenous Angpt-1-like therapy such as an agonistic antibody—or normalization by Angpt-2 inhibition confers a better efficacy/safety profile requires further study. These questions will become better addressed as Tie2 and its ligands are manipulated in the context of inflammation using various tissue-specific genetic strategies. Nonetheless, the Tie2 axis is poised for medical translation. Not only can its signaling be manipulated with modern biologics, but the status of the Tie2 axis can be rapidly assessed, enabling future trials to focus on individuals with measurably deranged Tie2 signaling while minimizing dilution from clinical heterogeneity. Efforts toward individualization may be further facilitated by insights into the genetics of Tie2 axis members. Understanding the molecular control of the host vascular response may open new avenues to predict, track, and treat a spectrum of acute diseases.
Key Points.
The host vascular response to severe infections and systemic inflammation may contribute to adverse outcomes across a spectrum of diseases
The Tie2 receptor actively signals vascular quiescence
Inflammation signals a series of changes that attenuate Tie2 signaling, which in turn potentiates leakage and inflammation
Results from genetic, molecular, and cellular studies collectively implicate Tie2 signaling as a measurable and modifiable feature of the host vascular response
Translational efforts to assess signaling and target Tie2 may improve outcomes in diseases of vascular leakage and inflammation
Acknowledgments
For space considerations, the author was unable to cite numerous contributions to this topic.
Financial Support and sponsorship: This work is supported by the National Institutes of Health grants R01-HL093234, R01-HL125275, and R41-GM121153.
Footnotes
Conflicts of Interest: SMP is listed as an inventor on patent applications filed by Beth Israel Deaconess Medical Center on Angiopoietins. SMP is a consultant to Eunoia, which is developing therapies targeted to the vasculature.
BIBLIOGRAPHY
- 1.Kolb WP, Granger GA. Lymphocyte in vitro cytotoxicity: characterization of human lymphotoxin. Proc Natl Acad Sci U S A. 1968;61(4):1250–5. doi: 10.1073/pnas.61.4.1250. Epub 1968/12/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Williams TW, Granger GA. Lymphocyte in vitro cytotoxicity: lymphotoxins of several mammalian species. Nature. 1968;219(5158):1076–7. doi: 10.1038/2191076a0. Epub 1968/09/07. [DOI] [PubMed] [Google Scholar]
- 3.Granger GA, Williams TW. Lymphocyte cytotoxicity in vitro: activation and release of a cytotoxic factor. Nature. 1968;218(5148):1253–4. doi: 10.1038/2181253a0. Epub 1968/06/29. [DOI] [PubMed] [Google Scholar]
- 4.Ruddle NH, Waksman BH. Cytotoxicity mediated by soluble antigen and lymphocytes in delayed hypersensitivity. 3. Analysis of mechanism. J Exp Med. 1968;128(6):1267–79. doi: 10.1084/jem.128.6.1267. Epub 1968/12/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ruddle NH, Waksman BH. Cytotoxicity mediated by soluble antigen and lymphocytes in delayed hypersensitivity. II. Correlation of the in vitro response with skin reactivity. J Exp Med. 1968;128(6):1255–65. doi: 10.1084/jem.128.6.1255. Epub 1968/12/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ruddle NH, Waksman BH. Cytotoxicity mediated by soluble antigen and lymphocytes in delayed hypersensitivity. I. Characterization of the phenomenon. J Exp Med. 1968;128(6):1237–54. doi: 10.1084/jem.128.6.1237. Epub 1968/12/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pennica D, Nedwin GE, Hayflick JS, Seeburg PH, Derynck R, Palladino MA, et al. Human tumour necrosis factor: precursor structure, expression and homology to lymphotoxin. Nature. 1984;312(5996):724–9. doi: 10.1038/312724a0. Epub 1984/12/20. [DOI] [PubMed] [Google Scholar]
- 8.Auron PE, Webb AC, Rosenwasser LJ, Mucci SF, Rich A, Wolff SM, et al. Nucleotide sequence of human monocyte interleukin 1 precursor cDNA. Proc Natl Acad Sci U S A. 1984;81(24):7907–11. doi: 10.1073/pnas.81.24.7907. Epub 1984/12/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Taniguchi T, Matsui H, Fujita T, Takaoka C, Kashima N, Yoshimoto R, et al. Structure and expression of a cloned cDNA for human interleukin-2. Nature. 1983;302(5906):305–10. doi: 10.1038/302305a0. Epub 1983/03/24. [DOI] [PubMed] [Google Scholar]
- 10.Bennett IL, Jr, Beeson PB. Studies on the pathogenesis of fever. I. The effect of injection of extracts and suspensions of uninfected rabbit tissues upon the body temperature of normal rabbits. J Exp Med. 1953;98(5):477–92. doi: 10.1084/jem.98.5.477. Epub 1953/11/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dumont DJ, Yamaguchi TP, Conlon RA, Rossant J, Breitman ML. tek, a novel tyrosine kinase gene located on mouse chromosome 4, is expressed in endothelial cells and their presumptive precursors. Oncogene. 1992;7(8):1471–80. Epub 1992/08/01. [PubMed] [Google Scholar]
- 12.Dumont DJ, Gradwohl G, Fong GH, Puri MC, Gertsenstein M, Auerbach A, et al. Dominant-negative and targeted null mutations in the endothelial receptor tyrosine kinase, tek, reveal a critical role in vasculogenesis of the embryo. Genes Dev. 1994;8(16):1897–909. doi: 10.1101/gad.8.16.1897. Epub 1994/08/15. [DOI] [PubMed] [Google Scholar]
- 13.Souma T, Tompson SW, Thomson BR, Siggs OM, Kizhatil K, Yamaguchi S, et al. Angiopoietin receptor TEK mutations underlie primary congenital glaucoma with variable expressivity. J Clin Invest. 2016;126(7):2575–87. doi: 10.1172/JCI85830. Epub 2016/06/09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thomson BR, Heinen S, Jeansson M, Ghosh AK, Fatima A, Sung HK, et al. A lymphatic defect causes ocular hypertension and glaucoma in mice. J Clin Invest. 2014;124(10):4320–4. doi: 10.1172/JCI77162. Epub 2014/09/10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vikkula M, Boon LM, Carraway KL, 3rd, Calvert JT, Diamonti AJ, Goumnerov B, et al. Vascular dysmorphogenesis caused by an activating mutation in the receptor tyrosine kinase TIE2. Cell. 1996;87(7):1181–90. doi: 10.1016/s0092-8674(00)81814-0. Epub 1996/12/27. [DOI] [PubMed] [Google Scholar]
- 16.Limaye N, Wouters V, Uebelhoer M, Tuominen M, Wirkkala R, Mulliken JB, et al. Somatic mutations in angiopoietin receptor gene TEK cause solitary and multiple sporadic venous malformations. Nature genetics. 2009;41(1):118–24. doi: 10.1038/ng.272. Epub 2008/12/17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Suri C, Jones PF, Patan S, Bartunkova S, Maisonpierre PC, Davis S, et al. Requisite role of angiopoietin-1, a ligand for the TIE2 receptor, during embryonic angiogenesis. Cell. 1996;87(7):1171–80. doi: 10.1016/s0092-8674(00)81813-9. Epub 1996/12/27. [DOI] [PubMed] [Google Scholar]
- 18.Maisonpierre PC, Suri C, Jones PF, Bartunkova S, Wiegand SJ, Radziejewski C, et al. Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science. 1997;277(5322):55–60. doi: 10.1126/science.277.5322.55. Epub 1997/07/04. [DOI] [PubMed] [Google Scholar]
- 19.Li JJ, Huang YQ, Basch R, Karpatkin S. Thrombin induces the release of angiopoietin-1 from platelets. Thrombosis and haemostasis. 2001;85(2):204–6. Epub 2001/03/15. [PubMed] [Google Scholar]
- 20.Barton WA, Tzvetkova-Robev D, Miranda EP, Kolev MV, Rajashankar KR, Himanen JP, et al. Crystal structures of the Tie2 receptor ectodomain and the angiopoietin-2-Tie2 complex. Nature structural & molecular biology. 2006;13(6):524–32. doi: 10.1038/nsmb1101. Epub 2006/05/30. [DOI] [PubMed] [Google Scholar]
- 21.Davis S, Papadopoulos N, Aldrich TH, Maisonpierre PC, Huang T, Kovac L, et al. Angiopoietins have distinct modular domains essential for receptor binding, dimerization and superclustering. Nature structural biology. 2003;10(1):38–44. doi: 10.1038/nsb880. Epub 2002/12/07. [DOI] [PubMed] [Google Scholar]
- 22.Yu X, Seegar TC, Dalton AC, Tzvetkova-Robev D, Goldgur Y, Rajashankar KR, et al. Structural basis for angiopoietin-1-mediated signaling initiation. Proc Natl Acad Sci U S A. 2013;110(18):7205–10. doi: 10.1073/pnas.1216890110. Epub 2013/04/18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gale NW, Thurston G, Hackett SF, Renard R, Wang Q, McClain J, et al. Angiopoietin-2 is required for postnatal angiogenesis and lymphatic patterning, and only the latter role is rescued by Angiopoietin-1. Dev Cell. 2002;3(3):411–23. doi: 10.1016/s1534-5807(02)00217-4. Epub 2002/10/04. [DOI] [PubMed] [Google Scholar]
- 24.Fiedler U, Scharpfenecker M, Koidl S, Hegen A, Grunow V, Schmidt JM, et al. The Tie-2 ligand angiopoietin-2 is stored in and rapidly released upon stimulation from endothelial cell Weibel-Palade bodies. Blood. 2004;103(11):4150–6. doi: 10.1182/blood-2003-10-3685. Epub 2004/02/21. [DOI] [PubMed] [Google Scholar]
- 25.Scharpfenecker M, Fiedler U, Reiss Y, Augustin HG. The Tie-2 ligand angiopoietin-2 destabilizes quiescent endothelium through an internal autocrine loop mechanism. J Cell Sci. 2005;118(Pt 4):771–80. doi: 10.1242/jcs.01653. Epub 2005/02/03. [DOI] [PubMed] [Google Scholar]
- 26.Yuan HT, Khankin EV, Karumanchi SA, Parikh SM. Angiopoietin 2 is a partial agonist/antagonist of Tie2 signaling in the endothelium. Mol Cell Biol. 2009;29(8):2011–22. doi: 10.1128/MCB.01472-08. Epub 2009/02/19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Parikh SM, Mammoto T, Schultz A, Yuan HT, Christiani D, Karumanchi SA, et al. Excess circulating angiopoietin-2 may contribute to pulmonary vascular leak in sepsis in humans. PLoS Med. 2006;3(3):e46. doi: 10.1371/journal.pmed.0030046. Epub 2006/01/19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fiedler U, Reiss Y, Scharpfenecker M, Grunow V, Koidl S, Thurston G, et al. Angiopoietin-2 sensitizes endothelial cells to TNF-alpha and has a crucial role in the induction of inflammation. Nat Med. 2006;12(2):235–9. doi: 10.1038/nm1351. Epub 2006/02/08. [DOI] [PubMed] [Google Scholar]
- 29.Bhandari V, Choo-Wing R, Lee CG, Zhu Z, Nedrelow JH, Chupp GL, et al. Hyperoxia causes angiopoietin 2-mediated acute lung injury and necrotic cell death. Nat Med. 2006;12(11):1286–93. doi: 10.1038/nm1494. Epub 2006/11/07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ziegler T, Horstkotte J, Schwab C, Pfetsch V, Weinmann K, Dietzel S, et al. Angiopoietin 2 mediates microvascular and hemodynamic alterations in sepsis. J Clin Invest. 2013;123(8):3436–45. doi: 10.1172/JCI66549. Epub 2013/07/19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.David S, Mukherjee A, Ghosh CC, Yano M, Khankin EV, Wenger JB, et al. Angiopoietin-2 may contribute to multiple organ dysfunction and death in sepsis*. Crit Care Med. 2012;40(11):3034–41. doi: 10.1097/CCM.0b013e31825fdc31. Epub 2012/08/15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stiehl T, Thamm K, Kaufmann J, Schaeper U, Kirsch T, Haller H, et al. Lung-targeted RNA interference against angiopoietin-2 ameliorates multiple organ dysfunction and death in sepsis. Crit Care Med. 2014;42(10):e654–62. doi: 10.1097/CCM.0000000000000524. Epub 2014/08/02. [DOI] [PubMed] [Google Scholar]
- 33.Ghosh CC, Thamm K, Berghelli AV, Schrimpf C, Maski MR, Abid T, et al. Drug Repurposing Screen Identifies Foxo1-Dependent Angiopoietin-2 Regulation in Sepsis. Crit Care Med. 2015;43(7):e230–40. doi: 10.1097/CCM.0000000000000993. Epub 2015/04/10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tabruyn SP, Colton K, Morisada T, Fuxe J, Wiegand SJ, Thurston G, et al. Angiopoietin-2-driven vascular remodeling in airway inflammation. Am J Pathol. 2010;177(6):3233–43. doi: 10.2353/ajpath.2010.100059. Epub 2010/10/19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim M, Allen B, Korhonen EA, Nitschke M, Yang HW, Baluk P, et al. Opposing actions of angiopoietin-2 on Tie2 signaling and FOXO1 activation. J Clin Invest. 2016;126(9):3511–25. doi: 10.1172/JCI84871. Epub 2016/08/23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Korhonen EA, Lampinen A, Giri H, Anisimov A, Kim M, Allen B, et al. Tie1 controls angiopoietin function in vascular remodeling and inflammation. J Clin Invest. 2016;126(9):3495–510. doi: 10.1172/JCI84923. Epub 2016/08/23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fachinger G, Deutsch U, Risau W. Functional interaction of vascular endothelial-protein-tyrosine phosphatase with the angiopoietin receptor Tie-2. Oncogene. 1999;18(43):5948–53. doi: 10.1038/sj.onc.1202992. Epub 1999/11/11. [DOI] [PubMed] [Google Scholar]
- 38.Nottebaum AF, Cagna G, Winderlich M, Gamp AC, Linnepe R, Polaschegg C, et al. VE-PTP maintains the endothelial barrier via plakoglobin and becomes dissociated from VE-cadherin by leukocytes and by VEGF. J Exp Med. 2008;205(12):2929–45. doi: 10.1084/jem.20080406. Epub 2008/11/19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hayashi M, Majumdar A, Li X, Adler J, Sun Z, Vertuani S, et al. VE-PTP regulates VEGFR2 activity in stalk cells to establish endothelial cell polarity and lumen formation. Nature communications. 2013;4:1672. doi: 10.1038/ncomms2683. Epub 2013/04/12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Frye M, Dierkes M, Kuppers V, Vockel M, Tomm J, Zeuschner D, et al. Interfering with VE-PTP stabilizes endothelial junctions in vivo via Tie-2 in the absence of VE-cadherin. J Exp Med. 2015;212(13):2267–87. doi: 10.1084/jem.20150718. Epub 2015/12/09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Carlson TR, Feng Y, Maisonpierre PC, Mrksich M, Morla AO. Direct cell adhesion to the angiopoietins mediated by integrins. J Biol Chem. 2001;276(28):26516–25. doi: 10.1074/jbc.M100282200. Epub 2001/05/11. [DOI] [PubMed] [Google Scholar]
- 42.Dallabrida SM, Ismail N, Oberle JR, Himes BE, Rupnick MA. Angiopoietin-1 promotes cardiac and skeletal myocyte survival through integrins. Circ Res. 2005;96(4):e8–24. doi: 10.1161/01.RES.0000158285.57191.60. Epub 2005/02/05. [DOI] [PubMed] [Google Scholar]
- 43.Felcht M, Luck R, Schering A, Seidel P, Srivastava K, Hu J, et al. Angiopoietin-2 differentially regulates angiogenesis through TIE2 and integrin signaling. J Clin Invest. 2012 doi: 10.1172/JCI58832. Epub 2012/05/16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hakanpaa L, Sipila T, Leppanen VM, Gautam P, Nurmi H, Jacquemet G, et al. Endothelial destabilization by angiopoietin-2 via integrin beta1 activation. Nature communications. 2015;6:5962. doi: 10.1038/ncomms6962. Epub 2015/01/31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee J, Kim KE, Choi DK, Jang JY, Jung JJ, Kiyonari H, et al. Angiopoietin-1 guides directional angiogenesis through integrin alphavbeta5 signaling for recovery of ischemic retinopathy. Sci Transl Med. 2013;5(203):203ra127. doi: 10.1126/scitranslmed.3006666. Epub 2013/09/21. [DOI] [PubMed] [Google Scholar]
- 46.Partanen J, Armstrong E, Makela TP, Korhonen J, Sandberg M, Renkonen R, et al. A novel endothelial cell surface receptor tyrosine kinase with extracellular epidermal growth factor homology domains. Mol Cell Biol. 1992;12(4):1698–707. doi: 10.1128/mcb.12.4.1698. Epub 1992/04/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Puri MC, Rossant J, Alitalo K, Bernstein A, Partanen J. The receptor tyrosine kinase TIE is required for integrity and survival of vascular endothelial cells. EMBO J. 1995;14(23):5884–91. doi: 10.1002/j.1460-2075.1995.tb00276.x. Epub 1995/12/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.D’Amico G, Korhonen EA, Anisimov A, Zarkada G, Holopainen T, Hagerling R, et al. Tie1 deletion inhibits tumor growth and improves angiopoietin antagonist therapy. J Clin Invest. 2014;124(2):824–34. doi: 10.1172/JCI68897. Epub 2014/01/17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Seegar TC, Eller B, Tzvetkova-Robev D, Kolev MV, Henderson SC, Nikolov DB, et al. Tie1-Tie2 interactions mediate functional differences between angiopoietin ligands. Mol Cell. 2010;37(5):643–55. doi: 10.1016/j.molcel.2010.02.007. Epub 2010/03/17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Savant S, La Porta S, Budnik A, Busch K, Hu J, Tisch N, et al. The Orphan Receptor Tie1 Controls Angiogenesis and Vascular Remodeling by Differentially Regulating Tie2 in Tip and Stalk Cells. Cell reports. 2015;12(11):1761–73. doi: 10.1016/j.celrep.2015.08.024. Epub 2015/09/08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wong AL, Haroon ZA, Werner S, Dewhirst MW, Greenberg CS, Peters KG. Tie2 expression and phosphorylation in angiogenic and quiescent adult tissues. Circ Res. 1997;81(4):567–74. doi: 10.1161/01.res.81.4.567. Epub 1997/10/07. [DOI] [PubMed] [Google Scholar]
- 52.Thurston G, Suri C, Smith K, McClain J, Sato TN, Yancopoulos GD, et al. Leakage-resistant blood vessels in mice transgenically overexpressing angiopoietin-1. Science. 1999;286(5449):2511–4. doi: 10.1126/science.286.5449.2511. Epub 2000/01/05. [DOI] [PubMed] [Google Scholar]
- 53.Thurston G, Rudge JS, Ioffe E, Zhou H, Ross L, Croll SD, et al. Angiopoietin-1 protects the adult vasculature against plasma leakage. Nat Med. 2000;6(4):460–3. doi: 10.1038/74725. Epub 2000/03/31. [DOI] [PubMed] [Google Scholar]
- 54.David S, Ghosh CC, Mukherjee A, Parikh SM. Angiopoietin-1 Requires IQ Domain GTPase-Activating Protein 1 to Activate Rac1 and Promote Endothelial Barrier Defense. Arterioscler Thromb Vasc Biol. 2011 doi: 10.1161/ATVBAHA.111.233189. Epub 2011/09/03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mammoto T, Parikh SM, Mammoto A, Gallagher D, Chan B, Mostoslavsky G, et al. Angiopoietin-1 requires p190 RhoGAP to protect against vascular leakage in vivo. J Biol Chem. 2007;282(33):23910–8. doi: 10.1074/jbc.M702169200. Epub 2007/06/15. [DOI] [PubMed] [Google Scholar]
- 56.Witzenbichler B, Westermann D, Knueppel S, Schultheiss HP, Tschope C. Protective role of angiopoietin-1 in endotoxic shock. Circulation. 2005;111(1):97–105. doi: 10.1161/01.CIR.0000151287.08202.8E. Epub 2004/12/22. [DOI] [PubMed] [Google Scholar]
- 57.Ghosh CC, Mukherjee A, David S, Knaus UG, Stearns-Kurosawa DJ, Kurosawa S, et al. Impaired function of the Tie-2 receptor contributes to vascular leakage and lethality in anthrax. Proc Natl Acad Sci U S A. 2012 doi: 10.1073/pnas.1120755109. Epub 2012/06/06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.David S, Ghosh CC, Kumpers P, Shushakova N, Van Slyke P, Khankin EV, et al. Effects of a synthetic PEG-ylated Tie-2 agonist peptide on endotoxemic lung injury and mortality. Am J Physiol Lung Cell Mol Physiol. 2011;300(6):L851–62. doi: 10.1152/ajplung.00459.2010. Epub 2011/03/23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.David S, Park JK, Meurs M, Zijlstra JG, Koenecke C, Schrimpf C, et al. Acute administration of recombinant Angiopoietin-1 ameliorates multiple-organ dysfunction syndrome and improves survival in murine sepsis. Cytokine. 2011;55(2):251–9. doi: 10.1016/j.cyto.2011.04.005. Epub 2011/05/03. [DOI] [PubMed] [Google Scholar]
- 60.Kumpers P, Gueler F, David S, Van Slyke P, Dumont DJ, Park JK, et al. The synthetic Tie2 agonist peptide vasculotide protects against vascular leakage and reduces mortality in murine abdominal sepsis. Crit Care. 2011;15(5):R261. doi: 10.1186/cc10523. Epub 2011/11/02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Higgins SJ, Purcell LA, Silver KL, Tran V, Crowley V, Hawkes M, et al. Dysregulation of angiopoietin-1 plays a mechanistic role in the pathogenesis of cerebral malaria. Sci Transl Med. 2016;8(358):358ra128. doi: 10.1126/scitranslmed.aaf6812. Epub 2016/09/30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Milam KE, Parikh SM. The angiopoietin-Tie2 signaling axis in the vascular leakage of systemic inflammation. Tissue barriers. 2015;3(1-2):e957508. doi: 10.4161/21688362.2014.957508. Epub 2015/04/04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ghosh CC, Mukherjee A, David S, Milam KE, Hunter JT, Parikh SM. Angiopoietin-1 requires oxidant signaling through p47phox to promote endothelial barrier defense. PLoS One. 2015;10(3):e0119577. doi: 10.1371/journal.pone.0119577. Epub 2015/03/12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bentley K, Franco CA, Philippides A, Blanco R, Dierkes M, Gebala V, et al. The role of differential VE-cadherin dynamics in cell rearrangement during angiogenesis. Nat Cell Biol. 2014;16(4):309–21. doi: 10.1038/ncb2926. Epub 2014/03/25. [DOI] [PubMed] [Google Scholar]
- 65.Broermann A, Winderlich M, Block H, Frye M, Rossaint J, Zarbock A, et al. Dissociation of VE-PTP from VE-cadherin is required for leukocyte extravasation and for VEGF-induced vascular permeability in vivo. J Exp Med. 2011;208(12):2393–401. doi: 10.1084/jem.20110525. Epub 2011/10/26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Corada M, Mariotti M, Thurston G, Smith K, Kunkel R, Brockhaus M, et al. Vascular endothelial-cadherin is an important determinant of microvascular integrity in vivo. Proc Natl Acad Sci U S A. 1999;96(17):9815–20. doi: 10.1073/pnas.96.17.9815. Epub 1999/08/18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gavard J, Patel V, Gutkind JS. Angiopoietin-1 prevents VEGF-induced endothelial permeability by sequestering Src through mDia. Dev Cell. 2008;14(1):25–36. doi: 10.1016/j.devcel.2007.10.019. Epub 2008/01/16. [DOI] [PubMed] [Google Scholar]
- 68.Sun Z, Li X, Massena S, Kutschera S, Padhan N, Gualandi L, et al. VEGFR2 induces c-Src signaling and vascular permeability in vivo via the adaptor protein TSAd. J Exp Med. 2012;209(7):1363–77. doi: 10.1084/jem.20111343. Epub 2012/06/13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zheng W, Nurmi H, Appak S, Sabine A, Bovay E, Korhonen EA, et al. Angiopoietin 2 regulates the transformation and integrity of lymphatic endothelial cell junctions. Genes Dev. 2014;28(14):1592–603. doi: 10.1101/gad.237677.114. Epub 2014/07/18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ghosh CC, David S, Zhang R, Berghelli A, Milam K, Higgins SJ, et al. Gene control of tyrosine kinase TIE2 and vascular manifestations of infections. Proc Natl Acad Sci U S A. 2016;113(9):2472–7. doi: 10.1073/pnas.1519467113. Epub 2016/02/18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tadros A, Hughes DP, Dunmore BJ, Brindle NP. ABIN-2 protects endothelial cells from death and has a role in the antiapoptotic effect of angiopoietin-1. Blood. 2003;102(13):4407–9. doi: 10.1182/blood-2003-05-1602. Epub 2003/08/23. [DOI] [PubMed] [Google Scholar]
- 72.Kurniati NF, Jongman RM, Vom Hagen F, Spokes KC, Moser J, Regan ER, et al. The flow dependency of Tie2 expression in endotoxemia. Intensive Care Med. 2013;39(7):1262–71. doi: 10.1007/s00134-013-2899-7. Epub 2013/04/09. [DOI] [PubMed] [Google Scholar]
- 73.Li R, Zijlstra JG, Kamps JA, van Meurs M, Molema G. Abrupt Reflow Enhances Cytokine Induced Pro-Inflammatory Activation of Endothelial Cells During Simulated Shock and Resuscitation. Shock. 2014 doi: 10.1097/SHK.0000000000000223. Epub 2014/07/23. [DOI] [PubMed] [Google Scholar]
- 74.Daly C, Wong V, Burova E, Wei Y, Zabski S, Griffiths J, et al. Angiopoietin-1 modulates endothelial cell function and gene expression via the transcription factor FKHR (FOXO1) Genes Dev. 2004;18(9):1060–71. doi: 10.1101/gad.1189704. Epub 2004/05/11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Han S, Lee SJ, Kim KE, Lee HS, Oh N, Park I, et al. Amelioration of sepsis by TIE2 activation-induced vascular protection. Sci Transl Med. 2016 doi: 10.1126/scitranslmed.aad9260. accepted. [DOI] [PubMed] [Google Scholar]
- 76.Fuxe J, Tabruyn S, Colton K, Zaid H, Adams A, Baluk P, et al. Pericyte requirement for anti-leak action of angiopoietin-1 and vascular remodeling in sustained inflammation. Am J Pathol. 2011;178(6):2897–909. doi: 10.1016/j.ajpath.2011.02.008. Epub 2011/05/10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Fukuhara S, Sako K, Minami T, Noda K, Kim HZ, Kodama T, et al. Differential function of Tie2 at cell-cell contacts and cell-substratum contacts regulated by angiopoietin-1. Nat Cell Biol. 2008;10(5):513–26. doi: 10.1038/ncb1714. Epub 2008/04/22. [DOI] [PubMed] [Google Scholar]
- 78.Rasmussen AL, Okumura A, Ferris MT, Green R, Feldmann F, Kelly SM, et al. Host genetic diversity enables Ebola hemorrhagic fever pathogenesis and resistance. Science. 2014;346(6212):987–91. doi: 10.1126/science.1259595. Epub 2014/11/02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kugathasan L, Ray JB, Deng Y, Rezaei E, Dumont DJ, Stewart DJ. The angiopietin-1-Tie2 pathway prevents rather than promotes pulmonary arterial hypertension in transgenic mice. J Exp Med. 2009;206(10):2221–34. doi: 10.1084/jem.20090389. Epub 2009/09/10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.McCarter SD, Mei SH, Lai PF, Zhang QW, Parker CH, Suen RS, et al. Cell-based angiopoietin-1 gene therapy for acute lung injury. Am J Respir Crit Care Med. 2007;175(10):1014–26. doi: 10.1164/rccm.200609-1370OC. Epub 2007/02/27. [DOI] [PubMed] [Google Scholar]
- 81.Meyer NJ, Li M, Feng R, Bradfield J, Gallop R, Bellamy S, et al. ANGPT2 Genetic Variant is Associated with Trauma-Associated Acute Lung Injury and Altered Plasma Angiopoietin-2 Isoform Ratio. Am J Respir Crit Care Med. 2011 doi: 10.1164/rccm.201005-0701OC. Epub 2011/01/25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Su L, Zhai R, Sheu CC, Gallagher DC, Gong MN, Tejera P, et al. Genetic variants in the angiopoietin-2 gene are associated with increased risk of ARDS. Intensive Care Med. 2009;35(6):1024–30. doi: 10.1007/s00134-009-1413-8. Epub 2009/03/10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Giuliano JS, Jr, Lahni PM, Harmon K, Wong HR, Doughty LA, Carcillo JA, et al. Admission angiopoietin levels in children with septic shock. Shock. 2007;28(6):650–4. Epub 2007/12/20. [PMC free article] [PubMed] [Google Scholar]
- 84.Parikh SM. Dysregulation of the angiopoietin-Tie-2 axis in sepsis and ARDS. Virulence. 2013;4(6) doi: 10.4161/viru.24906. Epub 2013/05/09. [DOI] [PMC free article] [PubMed] [Google Scholar]