

Abstract
Ischemic preconditioning (IPC) represents a potential therapy against cerebral ischemia. While our group has previously shown IPC to induce neuroprotection through various pathways, the role of astrocytes in supporting IPC-induced neuroprotection has not been extensively studied. Astrocyte-derived lactate has gained attention as a potential soluble mediator through which astrocytes could impart ischemic tolerance to neurons. Therefore, the goal of this study was to determine if i) IPC-treatment of astrocytes alone could transfer ischemic tolerance to neurons; ii) if IPC-treatment of astrocytes increases lactate production; and if iii) exogenous lactate administration to neurons could induce neuroprotection against lethal ischemia in vitro. For this purpose, a co-culture system was used and modified from a previous method. This system allows astrocytes and neurons to be separated by a physical barrier, while allowing secreted substances from either cell type to interact with each other. Oxygen-glucose deprivation was used as a model of cerebral ischemia and IPC in cultured rodent astrocytes and neurons. Neurons incubated with IPC-treated astrocytes were significantly protected against lethal ischemic injury compared to neurons incubated with sham-treated astrocytes. In addition, IPC-treatment of astrocytes significantly increased lactate secretion into the extracellular media. Finally, exogenous lactate administration can significantly attenuate cell death in neuronal cultures following exposure to lethal OGD. Our results suggest that IPC-treatment of astrocytes alone can transfer ischemic tolerance to neurons. In addition, the ability of IPC to increase lactate production in astrocytes suggest that lactate could represent a neuroprotective agent to protect neurons against lethal ischemic injury.
Keywords: glial cells, anoxia, glycolysis, stroke, cardiac arrest, lactate shuttle
Cerebrovascular disease, primarily “stroke,” is the fourth leading cause of death in the United States. Many of the individuals who succumb to stroke survive, yet require long-term rehabilitation and often have a poor prognosis (Kernan et al., 2014). Thus, the development of novel prophylactic therapies that target individuals at high-risk of developing stroke (i.e. patients with hypertension, diabetes, or history of smoking) could lead to improvements in survival and neurological outcomes following such a devastating condition.
One such therapy could be represented by ischemic preconditioning (IPC). IPC is an intrinsic neuroprotective mechanism whereby a brief, sublethal ischemic exposure protects against a subsequent lethal ischemic insult. Our group has previously shown IPC to induce cytoprotection in both astrocytes (Narayanan et al., 2015) and neurons (Kim et al., 2010; Thompson et al., 2013; Morris-Blanco et al., 2014), and shown to provide neuroprotection in in vivo rodent models of global cerebral ischemia (Della-Morte et al., 2009). However, the field of IPC has focused on neuroprotection from a predominantly “neuro-centric” point of view; as a result, the role of astrocytes in mediating neuronal ischemic tolerance is not been well understood.
Astrocytes have been shown to serve many neuroprotective functions. Astrocytes can supply neurons with antioxidants, recycle neurotransmitters, and modulate synaptic transmission (Ullian et al., 2001) (reviewed in (Trendelenburg and Dirnagl, 2005)). Rat retinal ganglion cells cultured in the presence of astrocytes or astrocyte-conditioned media have been shown to have increased neurite outgrowth and increased density of synaptic connections (Risher et al., 2014). A more recent study showed that neurons and astrocytes exchange mitochondria and that this exchange is neuroprotective (Hayakawa et al., 2016). These results suggest that astrocytes can profoundly impact neuronal functioning at the synaptic level. As glutamate-excitotoxicity mediates much of the pathogenesis in cerebral ischemia (Camacho and Massieu, 2006), astrocytes can perhaps induce neuroprotection by modulating synaptic activity in the face of excess glutamate release. Indeed, glutamate uptake by astrocytes has been suggested as a principal function of these cells, and under basal conditions is thought to be a key mechanism in coupling neuronal activity to astrocyte glucose uptake from the cerebral vasculature (Howarth, 2014).
While many of these functions have ample support, a more controversial role of astrocytes is the supplying of neurons with bioenergetic substrates, particularly lactate. This theorized function has received considerable attention for many years, attributed to the astrocyte-neuronal lactate shuttle (ANLS). While controversial to some, the ANLS theory has focused on the bioenergetic properties of lactate and the compartmentalization of glycolysis and oxidative phosphorylation in the brain (Kasischke et al., 2004; Aubert and Costalat, 2005). Lactate has traditionally been relegated as a waste-product of glycolysis. However, lactate has since been shown to have a dynamic role between astrocytes and neurons. The current model of lactate shuttling consists of astrocyte production of lactate through glycolysis or glycogenolysis; the lactate is then exported via the astrocyte-specific monocarboxylate transporter 1 (MCT1). In addition, astrocytes express an isoform of lactate dehydrogenase (LDH), LDH5, which preferentially converts glycolysis-derived pyruvate into lactate. Lactate can then be exported out of astrocytes and taken up by neurons, which occurs by a different neuronal monocarboxylate transporter, MCT2. Finally, neurons express a different isoform of LDH (LDH1) which facilitates conversion of lactate to pyruvate for subsequent entry into the Krebs cycle and oxidative phosphorylation (reviewed in (Pellerin and Magistretti, 2012)). While lactate generation can occur even during periods of low glucose and oxygen availability (ischemia), the neuroprotective role of lactate in the context of ischemic preconditioning has not been well investigated.
Therefore, our hypothesis was that IPC-treatment of astrocytes can transfer protection to neurons, and that a potential factor that mediates this protection is the soluble transport of lactate.
Methods
Animal Use
All animal protocols were approved by the Animal Care and Use Committee of the University of Miami (assurance number: A-3224-01). All experiments were conducted in accordance to ARRIVE guidelines. 16-17 day-pregnant Sprague-Dawley rats were purchased from Charles Rivers Laboratories and housed in a temperature controlled environment with 12 hr light -12 h dark cycle and ad libitum food and water.
Materials
Minimum Essential Medium (MEM), Hanks Balanced Salt Solution (HBSS), Fetal Bovine Serum (FBS), Propidium iodide, and NucBlue Hoechst 33342 nuclear stain were purchased from Life Technologies (Grand Island, NY). All other reagents were purchased from Sigma (St. Louis, MO) unless otherwise noted.
Preparation of post-natal astrocyte cultures
Cortex from postnatal Sprague Dawley rat pups (P2-P4) was harvested, followed by digestion with 0.25% trypsin and DNase. Following tituration and filtration through a 70 μM filter, the resulting filtrate was centrifuged at 200 g for 5 minutes to remove trypsin and DNase. The pelleted cells were then resuspended in Astrocyte Media, consisting of MEM base supplemented with 20 mM glucose, 1% GlutaMAX, 1% Penicillin/Streptomycin, and 10% FBS and plated in 100 mm cell culture dishes at a density of 2 hemispheres per plate. Complete media changes were performed every 2-3 days until cultures reached 70% confluency 5-7 days following the initial plating. Astrocyte cultures were then passaged and plating in appropriate culture vessels at a density of 50,000 cells/cm2, and allowed to reach full confluency and maintained for an additional 6-7 days. Passages P0-P3 were used for experiments.
Preparation of neuronal cultures
Cortical tissue from embryonic pups was isolated and digested with 0.25 % Trypsin and 1% DNase, triturated, and filtered through a 70 μM mesh filter to remove any undigested tissue debris. Neurons were plated in neuronal plating media (MEM supplemented with 5% FBS, 1% Glutamax, and 20 mM Glucose) at a density of 2 hemispheres for each 24 well plate, with each well containing a 12 mm poly-d lysine coated coverslip. Cultures were maintained 10-14 days with neuronal maintenance media (MEM supplemented with 1% Glutamax and 20 mM Glucose) with ½ media exchanges occurring every 3-4 days. Proliferation of non-neuronal cells was inhibited by the addition of cytosine arabinoside (5 uM final) 72 hours after the initial plating.
Astrocyte-Neuronal co-culture model
A modified co-culture system was adapted from a previously described protocol(Kaech and Banker, 2006). Astrocytes were prepared in 100 mm dishes containing paraffin pedestals, and were cultured until fully confluent. In parallel, neuronal cultures were prepared by culturing neurons on poly-D-lysine-coated 12 mm coverslips. Following treatment of astrocyte cultures with a preconditioning paradigm, astrocyte cultures were then placed in neuronal maintenance media. Neuronal coverslips were placed on top of the astrocyte cultures, such that the paraffin dots suspended the neuronal coverslips above the astrocytes without direct cell-to-cell contact, while still allowing both cell types to be bathed in the media. The coverslips were then incubated with the astrocytes for 48 hours. Following this incubation, the neuronal coverslips were removed and placed in a new 24 well culture dish, and were subjected to lethal OGD (3 hrs). 48 hours following lethal OGD, the neuronal coverslips were then assayed for cell death through immunofluorescence with propidium iodide and NucBlue nuclear stain.
Oxygen Glucose Deprivation
To mimic sublethal cerebral ischemia, astrocyte cultures were exposed to oxygen and glucose deprivation (OGD) as previously described (Kim et al., 2010) for 2 hr. Through empirical testing, 2 hr was determined to be a sublethal duration of OGD that induced the highest degree of protection to astrocytes following a lethal OGD insult (6 hours), thus this is considered IPC. 6 hour time point was chosen as both a significant amount of cell death occurred along with minimal cell detachment. Neuronal cultures were placed on the astrocyte cultures immediately after IPC (see Fig. 1) and thus, they were exposed to the conditioning media for 48 hrs. Similarly, for lethal OGD treatment of neurons for in vitro neuroprotection studies, neuronal cultures were exposed to 3 hours of lethal OGD. To induce OGD, cells were washed two times with glucose-free HBSS (CaCl2. 2H2O 1.26 mM, KCl 5.37 mM, KH2PO4 0.44 mM, MgCl2 0.49 mM, MgSO4·7H2O 0.41 mM, NaCl 136.9 mM, NaHCO3 4.17 mM, Na2HPO4·7H2O 0.34 mM, sucrose 15 mM, pH 7.4) and exposed to an oxygen-free environment (90% nitrogen, 5% hydrogen, and 5% CO2, 37°C) using a COY anaerobic chamber (COY Laboratory Products Inc, Lake Charter Township, MI). OGD was terminated by placing the cells back into normal aerobic media and returning the cultures to a5% CO2, 37°C incubator.
Figure 1. Schematic of modified segregated astrocyte-neuronal co-culture system.
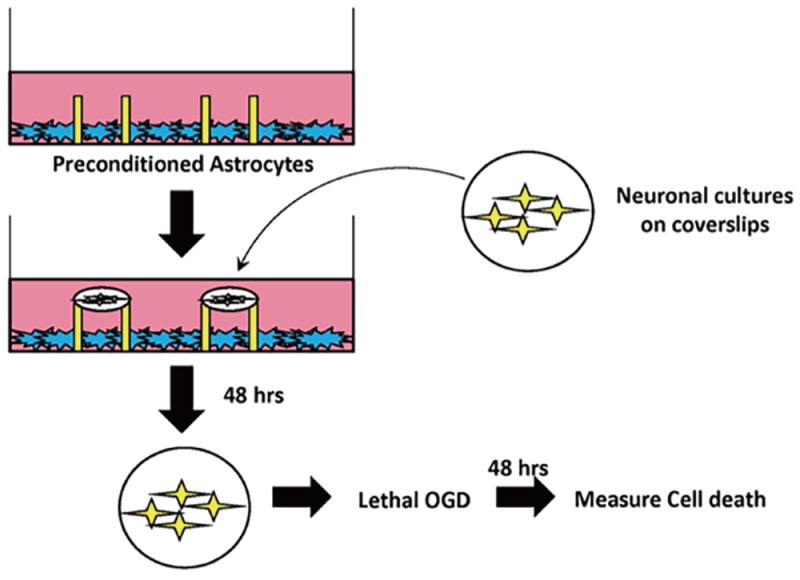
Confluent astrocytes were plated in a cell culture dish containing paraffin pedestals. Following exposure to IPC or Sham preconditioning, neurons (10-14 DIV) were placed on top of the pedestals. The paraffin pedestals allowed for physical separation of neurons and astrocytes but free exchange of soluble mediates. Neurons were incubated with IPC- or Sham-preconditioned astrocytes for 48 hours. After 48 hours, coverslips were removed and placed in a new cell culture dish, and were exposed to lethal OGD (3 hr). 48 hours following termination of lethal OGD, cell death was assayed in neuronal cultures with LDH release assay.
Cell Death Assay
In addition, neuronal cell death was determined by propidium iodide staining. Following removal from the modified astrocyte co-culture system and 48 hours following exposure to lethal OGD, neuronal coverslips were incubated in culture medium supplemented with 2 μg/ml propidium iodide for 15 minutes before imaging. Micrographs of the neuronal culture propidium iodide staining were captured (1) before preconditioning or sham treatment; (2) 48 hours after lethal ischemia; and (3) 24 hours after N-methyl-D-aspartate (NMDA) treatment (500 μmol/l) to determine maximum neuronal cell death. Fluorescence images were obtained using a SPOT CCD camera and were digitized using SPOT advanced software (Sterling Heights, MI, USA). Cell death was quantified as fraction of propidium iodide positive cells (number of propidium iodide-positive cells/Hoechst 3324-positive cells) for at least 3 fields per treatment group for each independent experiment.
Alternatively, cell death was assessed in neuronal cultures by measuring lactate dehydrogenase release after sham or lethal OGD using a commercially available kit (Roche, Nutley, NJ, USA). Maximum neuronal lactate dehydrogenase release was determined by exposing cultures to 500 μmol/l NMDA for 24 hours. Lactate dehydrogenase was measured at 340 nm absorbance using a microplate reader (Bio-Tek Instrument, Winooski, VT, USA). Values are expressed relative to maximal NDMA-induced neuronal lactate dehydrogenase release.
Lactate assay
Lactate was measured from the extracellular media of astrocyte cultures using a calorimetric assay according to manufacturer’s protocol (Eton Biosciences, Lactate Assay Kit I catalogue# 120001100A), which measures lactate concentration at an absorbance of 490 nm. Extracellular media from astrocyte cultures were collected 0, 1, 24, or 48 hours following termination of ischemic preconditioning, sham preconditioning, or 1 μM oligomycin treatment for lactate measurements. Media was collected and centrifuged through a 0.22 um filter to clear the media of cellular debris. Media was then diluted 1:5 and added to the manufacturer’s developing solution. 30 minutes following development, the assay reaction was stopped with 0.5 M acetic acid and lactate concentration was measured at 490 nm. Absorbencies were correlated with lactate concentration based on a standard curve (0.05 – 3.2 mM) that were performed concurrently with the samples to be measured.
Polarography
Mitochondrial respiration studies were conducted as previously described(Dave et al., 2008). In brief, naïve rat astrocytes were trypsinized and resuspended to a concentration of 2×106/mL of complete astrocyte media. The ratio of State III/State IV respiration was measured using a Clark-type oxygen electrode. State III was induced by the addition of astrocyte cells to the polarographic chamber, and basal respiration was allowed to occur for at least 1 minute. To produce State IV respiration, oligomycin (1 uM final) was added to the chamber to inhibit coupled-respiration, as previously described. This ratio represented the respiratory control index (RCI), an established measure of mitochondrial coupling.
Statistical Analysis
All data are expressed as mean ± SEM. Statistical analysis between two groups was performed using the unpaired Student’s t-test. Statistical analysis between more than two groups was performed using a one-way ANOVA with Bonferroni multiple comparison post hoc test. P < 0.05 was considered statistically significant.
Results
IPC treatment of astrocytes transfers ischemic tolerance to neurons
For our first experiment, we wanted to investigate the ability of astrocytes to transfer neuroprotection to neurons. For these experiments, we developed a modified co-culture system (Figure 1) that allows neurons and astrocytes to be physically separated through the use of paraffin pedestals, thus suspending neuronal coverslips above a confluent layer of astrocytes.
In the next set of experiments, we preconditioned astrocytes and then immediately after returning astrocytes to regular media, we suspended neuronal coverslips as depicted in Figure 1. From Figure 2, our results indicate that neurons that were incubated with astrocytes treated with IPC were significantly protected from lethal OGD compared to neurons incubated with sham-preconditioned astrocytes, as determined by the fraction of propidium iodide-positive cells. The use of the co-culture model from Figure 1 and results from Figure 2 suggest that IPC-treatment of astrocytes alone can transfer protection to neurons against lethal ischemic injury through the exchange of soluble mediators.
Figure 2. IPC-treatment of astrocytes can transfer ischemic tolerance to neurons.
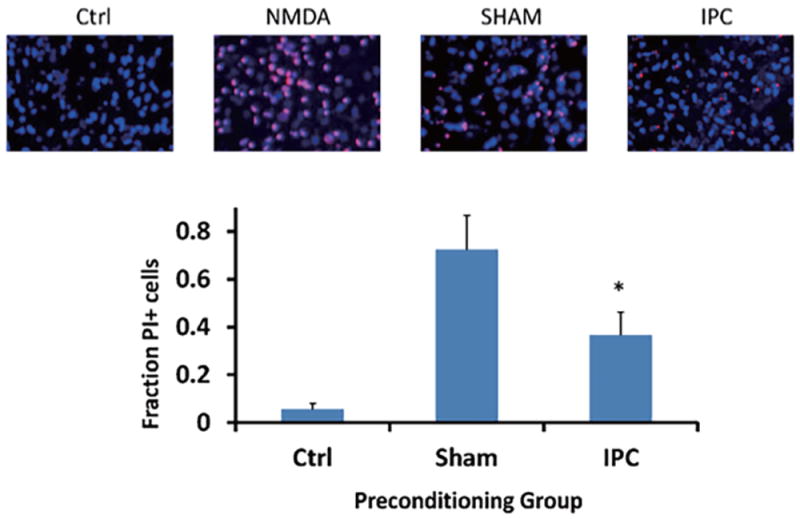
Neurons were subjected to lethal OGD following incubation with either IPC- or Sham-treated astrocytes. Panels above are representative images of propidium iodide and Hoechst 3324 staining of neuronal coverslips 48 hours following lethal OGD exposure. Ctrl: naïve neurons. NMDA: 500 μM N-Methyl-D-aspartic acid treatment (maximal cell death). Bar graph indicates quantification of cell death of neuronal cultures based on fraction of propidium iodide-positive cells compared to all Hoechst 3324-labeled cells. Sham: neurons incubated with sham-preconditioned astrocytes. IPC: neurons incubated with IPC-treated astrocytes. PI: propidium iodide. n = 5, *p<0.05.
Lactate production following IPC
While there is a plethora of soluble factors that astrocytes and neurons can freely exchange with each other, previous studies have suggested an important role of lactate in neuronal bioenergetics and neuronal synaptic transmission. Therefore, to determine if lactate could be one of the soluble factors responsible for the transference of protection observed in Figure 2, we next investigated whether IPC-treatment of astrocytes could increase astrocyte lactate production. We measured lactate concentrations in extracellular media 0, 1, 24, and 48 hours following IPC or Sham treatment. Lactate concentration was normalized to total protein content of each sampled well of astrocyte cultures. As a positive control, oligomycin (1 μM final) was added to astrocyte cultures to inhibit oxidative phosphorylation and to ensure that the assay could detect increases in lactate production in vitro.
From the results in Figure 3A, lactate production was significantly decreased in the IPC-treatment group immediately following termination of treatment compared to Sham and oligomycin-treated astrocyte cultures (Figure 3A; IPC: 0.17±0.03; Sham: 1.092±0.08; oligomycin: 2.011±0.1, n = 4, p<0.005). As expected, oligomycin significantly increased lactate production compared to both Sham and IPC-treated cultures (p<0.005). However, at 1 hr post-IPC treatment, IPC-treated astrocytes had a significant increase in lactate production compared with either sham or oligomycin-treated groups (Figure 3B; IPC: 0.98±0.14; Sham: 0.70±0.15; Oligomycin: 0.63±0.08, n = 4, p<0.01). Finally, lactate production in both IPC and Sham-treated astrocyte cultures were not significantly different at the 24 and 48 hour time points (Figure 3C and 3D). However, oligomycin-treated astrocyte cultures significantly increased lactate production at these time points relative to the preconditioned groups. The time course of lactate production is summarized in the Figure 3E. To ensure that the positive control of oligomycin was sufficient to inhibit oxidative phosphorylation in astrocyte cultures, we performed polarographic experiments to measure basal respiration of astrocyte cultures and respiration in the presence of 1 μM oligomycin. From Figure 3F, 1 μM oligomycin was sufficient to inhibit astrocyte respiration, therefore supporting its use as a positive control to inhibit oxidative phosphorylation and increase lactate production.
Figure 3. IPC increases lactate production in astrocyte cultures.
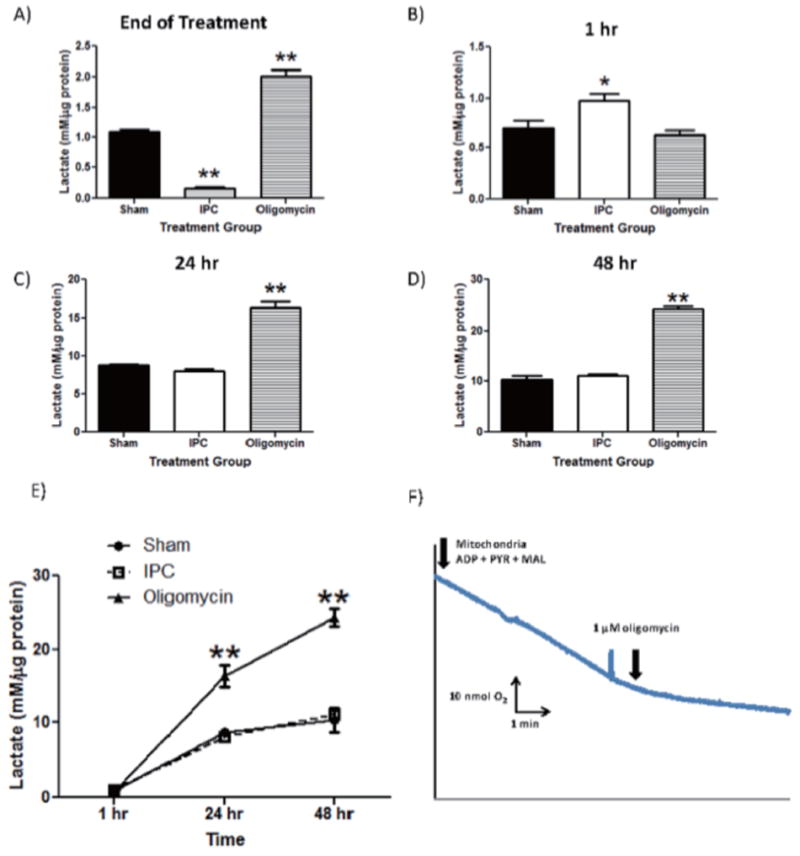
Lactate concentrations were measured and normalized to protein content in each sampled well. Extracellular lactate concentrations were measured by harvesting astrocyte culture media immediately following termination of preconditioning treatment and 1, 24, 48 hours following treatment (Panels 3A-3D). Oligomycin was used as a positive control to inhibit OXPHOS and promote lactate production through glycolysis. E) Compiled time course of lactate production from astrocyte cultures from each treatment group. F) Polarographic measurement of astrocyte respiration in the presence and absence of oligomycin (1 uM final). n = 5, ** p<0.005, *p<0.05.
Exogenous administration of lactate to neurons
Based on the previous findings, IPC-treatment of astrocytes was able to transfer ischemic tolerance to neurons, and IPC-treatment of astrocytes increased lactate production. These results suggest that lactate could represent one soluble compound that could mediate astrocyte-transference of ischemic tolerance in the context of IPC. To determine if lactate could represent a neuroprotective compound, we treated neuronal cultures with 5 mM lactate for 48 hours prior to exposure to lethal OGD. 48 hours following termination of lethal OGD, neurons treated with lactate had a significant reduction in cell death compared to control neurons, as determined by LDH release assay (Figure 4). To determine if this effect is specifically due to lactate, we incubated neurons with lactate in the presence of a 100 μM Alpha-cyano-4-hydroxycinnamate (4-CN). Previous studies have indicated that this concentration of 4-CN can preferentially inhibit MCT-2, the monocarboxylate transporter located on neurons and responsible for lactate uptake. When 4-CN was administered with lactate, the protection of neurons against lethal OGD was mitigated and was no longer significantly different compared to untreated neurons. Finally, 4-CN alone had no significant effect on neuronal cell death following lethal OGD. These results suggest that exogenous lactate administration can induce ischemic tolerance to neurons in vitro.
Figure 4. Exogenous lactate administration decreases neuronal cell death following lethal OGD.
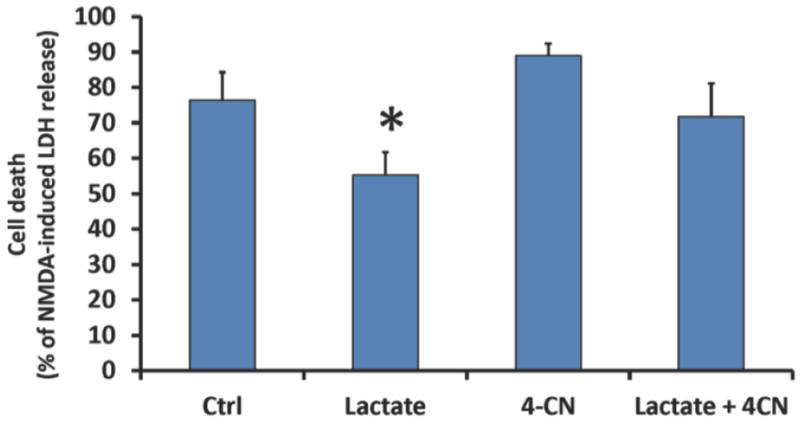
Neurons were treated with 5 mM lactate, the MCT inhibitor 4-CN (100 μM final), or a combination of lactate and 4-CN for 48 hours prior to lethal OGD. 48 hours following lethal OGD, cell death was measured with LDH-release assay and normalized to NMDA-induced LDH release. MCT: monocarboxylate transporter. 4-CN: Alpha-cyano-4-hydroxycinnamate. n = 4-5, *p<0.05.
Discussion
The role of astrocytes in mediating IPC-induced neuroprotection in neurons is not well understood, despite astrocytes serving many well-documented functions that are important for normal neuronal functioning. Our current study provides evidence that IPC-treatment of astrocytes can transfer ischemic tolerance to neurons through soluble mediators. In addition, IPC was found to increase lactate production in astrocytes, suggesting that lactate plays a role in mediating this protection. Finally, neuroprotection induced by exogenous lactate administration may indicate that lactate could serve as a neuroprotective agent. Mechanisms that could stimulate lactate production from astrocytes could also serve as means to induce ischemic tolerance in neurons.
Our current co-culture system includes a slight modification from a previous method (Kaech and Banker, 2006) (Figure 1). This model allows us to manipulate astrocytes independent of neurons, while still allowing the two cell types to freely exchange soluble mediators. Incubating neurons with IPC-treated astrocytes induced protection against a subsequent lethal ischemic insult when compared to neurons incubated with sham-treated astrocytes (Figure 2). Astrocytes have previously been documented to secrete several soluble factors that could modulate neuronal function. Perhaps most well documented are factors belonging to the family of proteins known as glypicans and thrombospondins that modulate synaptogenesis and synaptic transmission in neurons (Kucukdereli et al., 2011). Indeed, incubation of neurons with astrocytes or astrocyte conditioned media induced significant increases in synaptic connections and neurite outgrowth. As cerebral ischemia consists of an excitotoxicity injury mediated by deranged glutametergic synaptic transmission, modulation of neuronal synapses by astrocyte function could represent an important therapeutic strategy to combat cerebral ischemic injury.
One possible consequence of exposure of astrocytes to brief periods of ischemia could be an increase in lactate production. In general, most cells adapt to ischemia by increasing glycolysis and lactate production in an effort to sustain metabolism. Astrocytes are more capable of this function than neurons due to their glycogen content (Swanson and Choi, 1993) and expression of certain isoforms of lactate dehydrogenase (LDH) (Bittar et al., 1996) that favor the conversion of pyruvate to lactate. In addition, astrocytes express MCT isoforms that favor the efflux of lactate, while neurons express the necessary receptors and enzymatic machinery to uptake and convert lactate back to pyruvate (Debernardi et al., 2003). Therefore, we measured the lactate concentration in the extracellular media in astrocyte cultures following IPC. We found that 1 hour following termination of IPC, astrocytes were able to significantly increase lactate production compared to sham or OXPHOS inhibition via oligomycin treatment (Figure 3B). Interestingly, immediately following IPC treatment, there was a significant decrease in lactate production compared to sham or oligomyicn-treated groups (Figure 3A).
While we expected brief periods of OGD to increase lactate production based on previous studies (Marrif and Juurlink, 1999), our results appear to contradict the ability of astrocytes to significantly upregulate glycolysis during periods of ischemia. Following replenishment of glucose in IPC-treated astrocyte cultures, astrocytes appeared to recover their ability to produce lactate, as determined by Figure 3B. It is possible that under periods of OGD in our astrocyte cultures, astrocytes are readily producing and consuming lactate, which we detect as an observed decrease in extracellular media lactate when compared to Sham or oligomycin-treated groups. When glucose is replenished in the media following IPC treatment, astrocytes can perhaps utilize the glucose for metabolic needs, sparing the consumption of lactate for use by neurons and resulting in the observed increased concentration of lactate in the extracellular media after 1 hr of IPC treatment (Figure 3B). At 24 and 48 hours, we did not observe any increase in lactate production compared to sham or IPC, although both were significantly different compared to 0 and 1 hr time points for both treatment groups. This increase in lactate during the first hour following IPC-treatment in astrocytes suggests a temporal profile of how ischemic tolerance is transferred from astrocytes to neurons by lactate. Further investigation of the temporal release of lactate and other soluble mediators between astrocytes and neurons could provide a wealth of information into the mechanisms of IPC-induced transference of ischemic tolerance.
To corroborate these findings, we wished to determine if exogenous lactate administration could induce neuroprotection. Following 48 hours of lactate treatment, neurons were significantly protected against lethal ischemic injury (Figure 4). In addition, these effects appeared to be specific to lactate as this protection was inhibited by the MCT2 blocker 4-CN. These findings suggest that lactate may be an important neuroprotective agent, and could represent an important soluble factor that could be transferred between astrocytes and neurons to induce ischemic tolerance. While the role of lactate in neuronal metabolism and the astrocyte-neuronal lactate shuttle remains controversial, it is possible that lactate may serve bioenergetic-independent roles to induce neuronal ischemic tolerance through maintenance of synaptic connections. Future studies will need to define the temporal profile of lactate release and actual concentration over the 48 hrs following IPC. We also expect that other factors released by astrocytes, or other cell types in our cultures, are required for neuronal ischemic tolerance.
It is important to note that astrocytes are just one subtype of glial cells. Like astrocytes, other glial cells play significant roles in neuronal metabolism. For example, oligodendrocytes provide pivotal support to axons in the form of metabolites and in electrical conduction (Annunziato et al., 2013). Microglial cells are highly responsive to injury and infection and have been implicated in ischemic preconditioning (Annunziato et al., 2013). In the current study, oligodendrocytes are not present in our in vitro preparation. However, microglia expression is present in our culture model and is probably playing a role in neuronal ischemic tolerance in our model. Our in vitro model is an excellent model to test the potential role of microglia in IPC and is a goal of our future studies.
In conclusion, our current investigation serves to better understand the role of astrocytes in ischemic preconditioning-mediated neuroprotection. The results suggest that lactate may induce neuroprotection and could be a soluble factor that can be transferred between astrocytes and neurons to induce ischemic tolerance. Future therapies that could selectively stimulate lactate production from astrocytes by either OXPHOS inhibition (such as the use of oligomycin) could represent an important neuroprotective therapy.
Acknowledgments
None.
Sources of Funding
This work was supported by grants from the National Institutes of Health, national institute of neurological disease and stroke (NINDS) NS45676, NS054147, NS34773, NS097658, F31 NS080344.
References
- Annunziato L, Boscia F, Pignataro G. Ionic transporter activity in astrocytes, microglia, and oligodendrocytes during brain ischemia. J Cereb Blood Flow Metab. 2013;33:969–982. doi: 10.1038/jcbfm.2013.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aubert A, Costalat R. Interaction between astrocytes and neurons studied using a mathematical model of compartmentalized energy metabolism. J Cereb Blood Flow Metab. 2005;25:1476–1490. doi: 10.1038/sj.jcbfm.9600144. [DOI] [PubMed] [Google Scholar]
- Bittar PG, Charnay Y, Pellerin L, Bouras C, Magistretti PJ. Selective distribution of lactate dehydrogenase isoenzymes in neurons and astrocytes of human brain. J Cereb Blood Flow Metab. 1996;16:1079–1089. doi: 10.1097/00004647-199611000-00001. [DOI] [PubMed] [Google Scholar]
- Camacho A, Massieu L. Role of glutamate transporters in the clearance and release of glutamate during ischemia and its relation to neuronal death. Archives of medical research. 2006;37:11–18. doi: 10.1016/j.arcmed.2005.05.014. [DOI] [PubMed] [Google Scholar]
- Dave KR, DeFazio RA, Raval AP, Torraco A, Saul I, Barrientos A, Perez-Pinzon MA. Ischemic preconditioning targets the respiration of synaptic mitochondria via protein kinase C epsilon. J Neurosci. 2008;28:4172–4182. doi: 10.1523/JNEUROSCI.5471-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debernardi R, Pierre K, Lengacher S, Magistretti PJ, Pellerin L. Cell-specific expression pattern of monocarboxylate transporters in astrocytes and neurons observed in different mouse brain cortical cell cultures. Journal of neuroscience research. 2003;73:141–155. doi: 10.1002/jnr.10660. [DOI] [PubMed] [Google Scholar]
- Della-Morte D, Dave KR, DeFazio RA, Bao YC, Raval AP, Perez-Pinzon MA. Resveratrol pretreatment protects rat brain from cerebral ischemic damage via a sirtuin 1-uncoupling protein 2 pathway. Neuroscience. 2009;159:993–1002. doi: 10.1016/j.neuroscience.2009.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayakawa K, Esposito E, Wang X, Terasaki Y, Liu Y, Xing C, Ji X, Lo EH. Transfer of mitochondria from astrocytes to neurons after stroke. Nature. 2016;535:551–555. doi: 10.1038/nature18928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howarth C. The contribution of astrocytes to the regulation of cerebral blood flow. Frontiers in neuroscience. 2014;8:103. doi: 10.3389/fnins.2014.00103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaech S, Banker G. Culturing hippocampal neurons. Nature protocols. 2006;1:2406–2415. doi: 10.1038/nprot.2006.356. [DOI] [PubMed] [Google Scholar]
- Kasischke KA, Vishwasrao HD, Fisher PJ, Zipfel WR, Webb WW. Neural activity triggers neuronal oxidative metabolism followed by astrocytic glycolysis. Science. 2004;305:99–103. doi: 10.1126/science.1096485. [DOI] [PubMed] [Google Scholar]
- Kernan WN, et al. Guidelines for the prevention of stroke in patients with stroke and transient ischemic attack: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2014;45:2160–2236. doi: 10.1161/STR.0000000000000024. [DOI] [PubMed] [Google Scholar]
- Kim EJ, Raval AP, Hirsch N, Perez-Pinzon MA. Ischemic preconditioning mediates cyclooxygenase-2 expression via nuclear factor-kappa B activation in mixed cortical neuronal cultures. Translational stroke research. 2010;1:40–47. doi: 10.1007/s12975-009-0006-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kucukdereli H, Allen NJ, Lee AT, Feng A, Ozlu MI, Conatser LM, Chakraborty C, Workman G, Weaver M, Sage EH, Barres BA, Eroglu C. Control of excitatory CNS synaptogenesis by astrocyte-secreted proteins Hevin and SPARC. Proc Natl Acad Sci U S A. 2011;108:E440–449. doi: 10.1073/pnas.1104977108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrif H, Juurlink BH. Astrocytes respond to hypoxia by increasing glycolytic capacity. Journal of neuroscience research. 1999;57:255–260. doi: 10.1002/(SICI)1097-4547(19990715)57:2<255::AID-JNR11>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Morris-Blanco KC, Cohan CH, Neumann JT, Sick TJ, Perez-Pinzon MA. Protein kinase C epsilon regulates mitochondrial pools of Nampt and NAD following resveratrol and ischemic preconditioning in the rat cortex. J Cereb Blood Flow Metab. 2014;34:1024–1032. doi: 10.1038/jcbfm.2014.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan SV, Dave KR, Saul I, Perez-Pinzon MA. Resveratrol Preconditioning Protects Against Cerebral Ischemic Injury via Nuclear Erythroid 2-Related Factor 2. Stroke. 2015 doi: 10.1161/STROKEAHA.115.008921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellerin L, Magistretti PJ. Sweet sixteen for ANLS. J Cereb Blood Flow Metab. 2012;32:1152–1166. doi: 10.1038/jcbfm.2011.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risher WC, Patel S, Kim IH, Uezu A, Bhagat S, Wilton DK, Pilaz LJ, Singh Alvarado J, Calhan OY, Silver DL, Stevens B, Calakos N, Soderling SH, Eroglu C. Astrocytes refine cortical connectivity at dendritic spines. eLife. 2014;3 doi: 10.7554/eLife.04047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson RA, Choi DW. Glial glycogen stores affect neuronal survival during glucose deprivation in vitro. J Cereb Blood Flow Metab. 1993;13:162–169. doi: 10.1038/jcbfm.1993.19. [DOI] [PubMed] [Google Scholar]
- Thompson JW, Dave KR, Saul I, Narayanan SV, Perez-Pinzon MA. Epsilon PKC increases brain mitochondrial SIRT1 protein levels via heat shock protein 90 following ischemic preconditioning in rats. PloS one. 2013;8:e75753. doi: 10.1371/journal.pone.0075753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trendelenburg G, Dirnagl U. Neuroprotective role of astrocytes in cerebral ischemia: focus on ischemic preconditioning. Glia. 2005;50:307–320. doi: 10.1002/glia.20204. [DOI] [PubMed] [Google Scholar]
- Ullian EM, Sapperstein SK, Christopherson KS, Barres BA. Control of synapse number by glia. Science. 2001;291:657–661. doi: 10.1126/science.291.5504.657. [DOI] [PubMed] [Google Scholar]