

Abstract
Atrial fibrillation (AF) is the most common arrhythmia in the clinic. While previous studies have identified AF-associated mutations in several genes, the genetic basis for AF remains unclear. Here, we identified a novel T361S missense mutation in potassium voltage-gated channel, shal-related subfamily, member 3 (KCND3) from a Chinese Han family ancestor with lone AF. The wild-type (WT) or mutant T361S of Kv4.3 protein (encoded by KCND3) were co-expressed with the auxiliary subunit K+ channel-Interacting Protein (KChIP2) in HEK293 cells, and transient outward potassium current (Ito) were recorded using patch-clamp methods, and the surface or total protein levels of Kv4.3 were analyzed by western blot. Ito density, measured at 60 mV, for T361S was significantly higher than that for WT. Both the steady-state activation and inactivation curves showed a remarkable hyperpolarizing shift in T361S. Moreover, recovery from inactivation after a 500-ms depolarizing pulse was significantly delayed for T361S compared with that for WT. Mechanistically, the gain of function of Ito elicited by T361S was associated with the increased expression of cell surface and total cell protein of Kv4.3. The computer stimulation revealed that the T361S mutation shortened the action potential duration through an increased Itoin Human Atrial Model. In conclusion, we identified a novel T361S mutation in KCND3 associated with AF in the Chinese Han family. The T361S mutant result in the changes in channel kinetics as well as the up-regulation of Kv4.3 protein, which may be a critical driver for lone AF as observed in the patient.
Keywords: atrial fibrillation, KCND3, transient outward potassium current, patch clamp
INTRODUCTION
Atrial fibrillation (AF) is the most common sustained cardiac arrhythmia that results in serious cardiovascular outcomes such as stroke, heart failure and death [1]. The prevalence of AF is approximately 1% in the general population and markedly increases in aging populations, occurring in approximately 10% of individuals over age 80. AF is predicted to affect more than 33 million individuals worldwide [2]. AF is associated with a five-fold increased risk in the incidence of stroke and other cardiovascular mortalities and is rapidly becoming a public health challenge worldwide [3].
However, the defined etiology and pathogenesis of AF are incompletely understood. AF is usually associated with recognizable overt cardiovascular disease or precipitating illness, but more than 30% AF cases occurs in the absence of these complications (conventionally referred to as lone AF) [4, 5]. Genetic susceptibility is associated with the development of AF, especially to lone AF. Following the identification of the S140G mutation in KCNQ1 in familial AF, mutations have also been identified in genes encoding ion channels and their accessory subunits, including sodium channels (SCN5A and β subunits) and potassium channels [6–10]. Subsequent studies found that mutations in non-ion channel genes are also linked to AF, such as connexins, atrial natriuretic peptide gene NPPA, T-box transcription factor 5 (TBX5) and nuclear pore complex 155 (NUP155) [10–13]. However, given that the aforementioned gene mutations occur in low prevalence in AF patients, it is reasonable to hypothesize that additional disease genes remain to be identified.
The rapidly activating and inactivating transient outward potassium currents (Ito), mediated by the pore-forming potassium channel subunit Kv4.3 (encoded by the KCND3 gene) together with the K+ channel-interacting protein 2 (KChIP2, encoded by the KCNIP2 gene), plays a vital role in the early phase of repolarization and excitation-contraction coupling [14, 15]. Dysfunctions of Ito contribute to pathophysiological conditions, including cardiac hypoxia and arrhythmia. Previous studies revealed that hypoxia (e.g., myocardial infarction) markedly decreased the Ito densities by a Ca2+-dependent mechanism [16–18]. In addition, a mutation in KCNE3 (V17M), an accessory subunit that interacts with the Kv4.3, leads to increased Ito and has been reported to be associated with the pathogenesis of early-onset long AF [19]. Multiple mutations in KCND3 have been reported to be associated with the pathogenesis of dominantly inherited spinocerebellar ataxia (SCA) 19/22, Brugada syndrome (BrS) and sudden unexplained death syndrome [20–22]. A mutation in KCND3 has also been reported in the pathogenesis of AF in Danish cohort populations [23]. However, mutation screening of the KCND3 gene obtained from patients of Chinese Han ancestry with early onset lone AF has not been reported.
In the present study, given the genetic susceptibility of different racial groups, we pursued the identification of novel lone AF-associated mutations among individuals of Chinese Han ancestry.
RESULTS
Study cohort and mutation screening
A total of 180 patients with lone AF ranging from 16 to 39 years, without any concomitant disease, were screened for mutations in the cardiac potassium channel subunit Kv4.3, encoded by KCND3. The clinical data from the study population are shown in Table 1. A heterozygous nucleotide variant of KCND3 (c.1081A>T) leading to the substitution of a threonine residue at position 361 by a serine residue (label T361S) was identified in the proband AF patient and his father. This variant was not detected in the other family members or the 600 Chinese Han control cases (Figure 1A and 1B), and were not found in the database of Human Gene Mutation Data base (HGMD), Ensembl and HapMap. Other atrial fibrillation–associated genes encoding ion channels and their accessory subunits were sequenced in the proband patient. However, no variant was detected in the proband patient.
Table 1. Clinical characteristics of the lone AF population (n=180).
| Median age of onset, y (IQR) | 34.2(26–41) |
| Male gender, % | 72 |
| Height, cm | 171±6 |
| Weight, kg | 69±17 |
| BMI, kg/m2 | 23.6±4.3 |
| Blood Pressure, mmHg | |
| Systolic | 127±16 |
| Diastolic | 76±10 |
| AF type | |
| Paroxysmal, % | 53.7 |
| Persistent, % | 31.7 |
| Permanent, % | 8.2 |
| Unknown, % | 6.4 |
| Family history of AF | |
| First-degree relatives with AF (%) | 32 |
All numbers are reported as mean±standard deviation unless otherwise noted. IQR Interquartile range.
Figure 1. Identification of the mutant residues at position 361 in KCND3 associated with lone AF.

(A) Family pedigree showing squares that indicate male family members, circles that indicate female family members and arrows that indicate the proband patient. His father was affected by persistent AF at age 38. (B) Direct sequencing chromatograms of the KCND3 gene in the index patient and control case showing a heterozygous transition c.1081A>T resulting in replacement of threonine by serine at position 361. (C) Predicted topology schematic of Kv4.3 protein showing the position of the T361S mutation in the third extracellular loop linking segment 5 and segment 6, which is regarded as channel pore-forming. (D) Amino acid sequence alignment showing that serine at position 361 is highly conserved among multiple species. (E) The proband patient had onset of paroxysmal AF at the age of 28. ECG data: HR 102 bpm; no P-wave; QRS 98 ms. (F) In sinus rhythm, the patient had a normal ECG (ECG data: HR 90 bpm; P-wave duration 98 ms; PR 168 ms; QRS 96 ms; QTc 392 ms).
The proband was a 38-year-old male patient affected with paroxysmal AF at the age of 28 years (ECG data: HR 102 bpm; no P-wave; QRS 98 ms) (Figure 1E). The patient had normal sinus rhythm ECG (HR 90 bpm; P-wave duration 98 ms; PR 168 ms; QRS 96 ms; QTc 392 ms) (Figure 1F) and his echocardiography was normal (left ventricular ejection fraction 57%; left atrium <40 mm). His father (age 66) was affected by persistent AF at age of 38 years and had the same mutation. Other family members were normal.
Electrophysiological characterization of AF-associated T361S-Kv4.3 mutant
The residue at the mutation position is highly conserved from Homo. sapiens to Xenopus. tropicalis (Figure 1D), suggesting that it is critical for the physiological function of the Kv4.3 protein. Despites Kv4.3 subunit generates Kv currents with similar to myocardial Ito, additional subunits such as the regulatoryβcurrents and KChIPs are indispensable for the generation of cardiac Ito [24]. Thus, we recorded Ito under whole-cell voltage-clamp by transiently expressing WT or T361S-KCND3 together with KChIP2 in HEK293 cells. As shown in Figure 2A, cells expressing T361S-KCND3 had markedly increased Ito density (peak current normalized to cell capacitance, pA/pF) across the range of test potentials compared to WT-KCND3. The mutant T361S-KCND3 had an especially prominent enlargement of Ito density (807.79±80.99 pA/pF) at 60 mV compared to WT-KCND3 (557.9±40.61 pA/pF, P<0.01) (Figure 2B) by about 45%. Given that the mutation at position 361 in Kv4.3 is located in the third extracellular loop linking S5 and S6, which is regarded as channel pore-forming (Figure 1C), it was necessary to analyze kinetic characteristics. As illustrated in Figure 3B, a significant hyperpolarizing shift of the steady-state activation curve was observed in the T361S mutant channel [WT: V1/2=-13.69±1.48, k=11.35±0.77, n=15 and T361S: V1/2=-22.98±0.75 (P<0.05), k=9.67±0.39, n=17], indicating a significantly negative shift of the activation curve of the T361S-KCND3 by 9.29 mV. A 13.14 mV negative shift of the steady state inactivation was also observed for the T361S channel (WT: V1/2=-41.16±0.47, k=5.37±0.39, n=17 and T361S: V1/2=-54.30±1.25 (P<0.05), k=8.56±0.36, n=18) (Figure 3B). Additionally, Kv4.3 T361S expressed in HEK293 cells displays a slower recovery from the time-dependence of inactivation compared to the WT channel (T361S: t=81.17±10.18 ms; WT: t=27.33±2.80 ms; P<0.05, Figure 3C-3D). These results indicate that the T361S mutation results in a gain of function of Ito.
Figure 2. The mutant T361S-KCND3 prominently increases Ito density.

(A) Typical currents recorded from HEK293 cells transfected WT or T361S of Kv4.3 together with KChIP2 in response to increasing step potential. (B) Current-voltage relationship. The protocol was shown in the inset. Data was shown as means ± SEM.
Figure 3.

The mutant T361S-KCND3 alters the kinetic properties of Ito channel (A). Typical currents of voltage-dependent inactivation are shown. (B) Voltage-dependent inactivation and activation curves are plotted with Boltzmann equation. The recorded protocol was shown in the inset. (C) The curve of recovery from inactivation for WT and T361S channels is obtained using double pulse protocol. Representative recordings are shown. (D) The curve of recovery from inactivation. Peak Ito currents elicited by P2 were normalized (P2/P1) and the data is fitted with a single exponential function (X axis: time, ms; Y axis: P2/P1). Data was shown as means ± SEM.
T361S mutants increased protein expression level in cell-surface
The current density of Kv4.3 is mainly determined by its expression level on the cell surface, the single channel conductance and the open probability of Kv4.3 channel. To investigate the underlying molecular mechanism, the cell surface protein levels of the Kv4.3 subunit were measured by western blot analysis. HEK293 cells were co-transfected KChIP2 with wild type or mutant Kv4.3. As shown in Figure 4A, the T361S mutation up-regulated the expression of Kv4.3 on the cell surface compared to wild type together with KChIP2 by about 2.8 folds. Interestingly, we also found that the expression level of mutant T361S in total cell extracts dramatically increased compared to WT (Figure 4B). Therefore, the results indicate that the up-regulation of the Kv4.3 subunit in cellular surface contribute to the enlargement of Ito in T361S mutant.
Figure 4. The effect of the missense mutation of Kv4.3 on the cell-surface expression and the total expression of Kv4.3 protein.

(A) Cell surface biotinylation assays were performed in Hek293 cells co-transfected KChIP2 with wild type or mutant T361S of Kv4.3 after 48 hours. Intergrin α5 was used as loading to calibrate the cell-surface proteins. No GAPDH was detected in the biotinylated fraction (data not shown). (B) The total expression of Kv4.3 protein was extracted from HEK293 cells co-transfected KChIP2 with wild type or mutant T361S of Kv4.3. This experiment was repeated at least three times, and similar results were obtained. Data was shown as means ± SEM. *P < 0.05.
T361S shortened action potential duration through an increased Ito
Compared to WT, the increase in the surface expression of the T361S mutation is by about 2.8 folds while the peak current at 60 mV only increased by about 145%, it suggest that per molecule activity of the T361S mutant channel is much lower than of WT (Figure 2B, Figure 4A). However, though computer simulation, the effect of the T361S mutation (in the presence of KChIP2), in the model, the mutation results in a shortening of the action potential duration through an increased Ito (Figure 5).
Figure 5. Computer simulation of the effect of the T361S mutation.
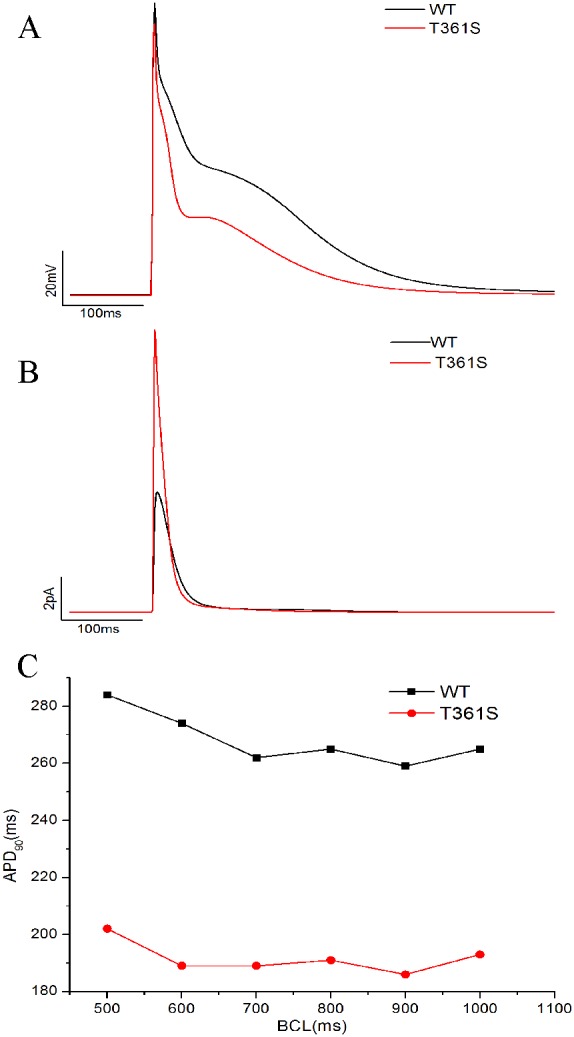
(A) Action potentials (APs) simulated at a BCL of 1000 ms. (B) The Ito current associated with the APs in (A). (C) Rate adaptation based on the model simulations. APD90 was calculated and plotted against BCL.
DISCUSSION
In the present study, we carried out a sequencing analysis of the KCND3 gene encoding the pore-forming K+ channel subunit Kv4.3 in 180 AF patients. A novel mutation, T361S, was identified in a young patient with lone AF. The missense mutation resulted in a substitution of a threonine residue at position 361 in Kv4.3 protein by a serine residue. The T361S mutation was absent in 600 control individuals and in publicly available databases. Electrophysiological studies revealed that the T361S mutation resulted in a gain of function compared to WT. In addition, the mutation facilitated the expression of Kv4.3 protein subunits on the cellular surface and resulted in a shortening of the atrial action potential in Human Atrial Model. Collectively, our findings identify a novel T361S mutation associated with lone AF and suggest that a gain of function resulting from Kv4.3 mutation may drive the pathogenesis of AF.
Ito channels are expressed in cardiomyocytes of larger mammals, and are important for early phase of repolarization and excitation-contraction coupling. Dysfunction of Ito channels in inherited or acquired cardiac diseases contributed to cardiac arrhythmias, including BrS and AF. In chronic AF patients, Ito has been reported to be reduced by down-regulation of the Kv4.3 mRNA levels [25, 26]. The efficacy of Ito blockers in terminating AF and a silico modeling suggested that reduced Ito may be a protective compensation during human chronic AF [27]. Olesen et al. first identified a gain of function mutation (A545P) in KCND3 associated with AF in Danish populations, which led to reduce action potential duration in an atrial cell model, but the mechanism underlying the enlargement of the current amplitude by the A545P mutation remains unclear [23]. Recently, a genome-wide association study (GWAS) has identified a SNP (rs12044963) in KCND3 gene, is relevant to AF susceptibility in the Japanese population [28]. Here, we observed T361S mutant dramatically increased Ito amplitudes, and up-regulated the surface and total Kv4.3 expression in heterologous expression systems. Previous studies revealed that the translation efficiency in the threonine residue mutant (tcc) increased compared to the wild serine residue (acc), and a recent report has revealed that cold-inducible RNA-binding protein (CIRP) modulates cardiac repolarization by negatively regulating the expression of the Kv4.2/Kv4.3 proteins at the post-transcriptional level [29, 30]. We speculate that the mutation may disrupt the interaction of CIRP protein with KCND3 mRNAs, increasing the translation efficiency which leads to an upregulation of protein levels of Kv4.3 subunits. Enhanced Ito can accelerate atrial repolarization and shorten the atrial effective refractory period, which may facilitate reentry leading to the occurrence and maintenance of AF. Therefore, our findings expand the spectrum of mutations in KCND3 that cause the pathogenesis of AF and various forms of cardiac arrhythmias.
Several mutations in KCND3 were identified in dominantly inherited spinocerebellar ataxia (SCA) 19/22, early-onset of persistent lone atrial fibrillation, Brugada syndrome (BrS) and a case of sudden unexplained death [20, 22, 23, 31]. Mutant residues at positions 450 and 600 (L450F and G600R) associated with BrS significantly resulted in a gain of function and increased peak current density compared to WT [21, 32]. Because the ECG changes associated with BrS can be dynamic and are often concealed, and 10% to 20% of patients with BrS have AF, it is necessary to perform drug challenge to eliminate the BrS in the proband [33]. As ajmaline are unobtainable in China, propafenone challenge is also effective to reveal BrS as previously described [34]. The proband exhibited a negative reaction following propafenone challenge (data no shown). Interestingly, the mutant residues at positions 352 and 373 (T352P and M373I) in KCND3 associated with SCA19 were also located in the third extracellular loop linking S5 and S6 [20]. However, the peak current density for the mutants was significantly reduced when compared to WT-KCND3. The results of the functional experiments were contrary to those for mutant residues at position 361 in KCND3 associated with AF. Mutant residues at positions 352 and 373 (T352P and M373I) markedly reduced cell surface expression and enhanced protein degradation [20]. These results suggested that residues located in the extracellular loop linking S5 and S6 are crucial for the integrity of function of Kv4.3 channel, and its mutation played an important and diverse role in the inherited diseases. This difference may be associated with tissue-specific gene expression, which remains to be investigated in the future.
As we used the candidate gene sequencing approach, other disease-associated genes cannot be ruled out. We limited our analysis to the KCND3 encoding regions and the possibility of mutations occurring in regions beyond encoding regions cannot be excluded. However, lone AF is usually considered as a monogenic disease. A previous report revealed that gain-of-function mutations in KCND3 are associated with AF [23], and our functional analyses point towards the identified KCND3 mutation T361S as underlying AF.
Taken together, we identified a novel T361S mutation in KCND3 associated with lone AF from a Chinese Han family, which results in a gain of function of Ito. Mechanistically, the gain of function of Ito elicited by the T361S mutation was associated with the increased expression of cell surface protein and total cell protein of Kv4.3, which may be a molecular substrate leading to lone AF observed in our patient.
MATERIALS AND METHODS
Clinical data
This study was approved by the Ethics Committee of the First Affiliated Hospital of Xiamen University. Informed consent was obtained from all patients. The patients were enrolled through GeneID, a Chinese population database [35]. Patients with early onset lone AF (i.e., without other cardiovascular diseases, essential hypertension, ischemic stroke, metabolic or pulmonary diseases, or diabetes) and onset of AF before the age of 40 years were recruited. All the participants were diagnosed as lone AF based on the ACC/AHA/ESC 2006 AF guidelines by expert cardiologists using electrocardiogram (ECG) recordings. The proband exhibited a negative reaction following propafenone challenge. The 600 Chinese Han controls were normal healthy individuals without AF or any other cardiovascular diseases.
Mutation analysis
Genomic DNA was extracted from peripheral blood samples using the Wizard Genomic DNA Purification Kit (Promega, Madison, Wisconsin, USA) according to the standard protocols. All of the exons and exon-intron boundaries of the candidate ion gene KCND3 were amplified using genomic DNA from AF and the control patients as templates by polymerase chain reaction (PCR). The PCR products for the DNA sequencing were performed by Invitrogen Company (Shanghai, China) following the standard protocols. A variant was identified as a mutation if not present in the control population and NCBI database, associated with affected family members for segregation and affected a change of conserved amino acid.
Construction of plasmids for KChIP2 and KCND3
T vectors containing cDNA encoding human KChIP2 (NM_173192.2) or human KCND3 (NM_172198.2) were purchased from Proteintech Company (Wuhan, China). The KChIP2 cDNA was amplified by polymerase chain reactions (PCR) using PCR primers containing restriction enzyme sites (forward primer: 5-ATATCTCGAGATGCGGGGCCAGGGCCGCAAGGA-3 and reverse primer: 5-GCGCGGATCCTCTCCTGGGGGCTAGATGACAT-3), digested with the restriction enzymes Xho I and BamH I, and inserted into the vector pIRES2-EGFP. The KCND3 cDNA was also amplified by PCR using PCR primers (forward primer: 5- ATATCTCGAGATGGCGGCCGGAGTTGCGGCCT-3 and reverse primer: 5-GCGCGAATTCTTACAAGGCGGAGACCTTGACA-3), digested with the restriction enzymes Xho I and EcoR I, and inserted into the vector pcDNA3.1 (-). The point mutation T361S (acc→tcc) in pcDNA3.1(-)-KCND3 was introduced using a PCR-based mutagenesis method (pcDNA3.1(-)-KCND3-T361S) as previously described [36, 37]. All of the constructed plasmids were verified by direct DNA sequencing analysis.
Cell culture and transfection
HEK293 cells were cultured in Dulbecco’s Modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum, L-glutamine (2 mM), penicillin G (100 μnits/ml) and streptomycin (10 mg/ml) in a humidified incubator with 5% CO2 at 37°C. The cells were transiently co-transfected with pIRES2-EGFP-KChIP2 and pcDNA3.1(-)-KCND3 (WT or T361S) at 70-80% confluence using GenJet™ In Vitro DNA Transfection Reagent (Signagen, Rockville, Maryland, USA) according to the manufacturer’s instructions.
Electrophysiology
For patch clamp experiments, HEK293 cells in 24-well plates were transiently co-transfected with 0.5 μg of pIRES2-EGFP-KChIP2 and 0.5 μg of pcDNA3.1 (-)-KCND3 (WT or T361S). The experiments were performed 48 hours after transfection. A similar expression level of GFP-positive cells, which successfully expressed KChIP2 proteins, was selected to record Ito. Cells were bathed in extracellular solution containing (in mM) 150 NaCl, 5 KCl, 2.2 CaCl2, 1 MgCl2, 10 HEPES, 5 glucose, pH 7.3 adjusted with HCl. Patch pipettes were filled with an internal pipette solution containing 120 K-gluconate, 24 KCl, 0.2 EGTA, 10 HEPES, pH 7.2 adjusted with KOH [18]. All reagents were purchased from the Sigma-Aldrich Company (ST.Lous, Missouri, USA). The currents were recorded at room temperature (22°C) using the whole-cell recording configuration and performed with an Axon MultiClamp 700B amplifier (Axon Instruments, Molecular Devices, Sunnyvale, California, USA). The pipette resistances ranged from 2-3 MΏ and the series resistances recorded in the whole cell configuration were compensated (85%) to minimize voltage errors. The holding potential for all pulse protocols was −80 mV, and the currents were low-pass filtered at 5 kHz with a 4-pole Bessel filter, digitized with the Digidata 1440A (Axon Instruments, Molecular Devices, Sunnyvale, California, USA) and sampled at 10 kHz. The data were acquired and analyzed using a software combination of Clamfit 10.2, Microsoft Excel, and Origin 8.5 (OriginLab, USA). The Ito densities (pA/pF) were obtained by dividing the peak current by the cell capacitance. The activation-potential and steady-state inactivation curves were all fitted with a Boltzmann equation (y= {1+exp [(V-V1/2)/k)]}−1) to determine the membrane potential for the half-maximal (in) activation V1/2, and the slope factor k. Recovery from inactivation was analyzed by fitting the data with a mono-exponential equation, y = y0+ [A1 exp(−x/τ1)], to determine the time constant τ1 for recovery from inactivation.
Total cell protein and cell surface protein of Kv4.3 for western blot analysis
Total protein lysates and plasma membrane proteins were extracted from HEK293 cells co-transfected KChIP2 with wild type or mutant Kv4.3 after 48 hours following a standard protocol as previously described [38]. Plasma membrane proteins were isolated by the Pierce Cell Surface protein Isolation Kit (Thermo Scientific, Waltham, MA, USA). The total proteins or plasma membrane proteins were subjected to western blot analysis using a polyclonal anti-Kv4.3 antibody (Alomone, Jerusalem, Israel) or GAPDH antibody (Proteintech Company, Wuhan, China).
Computer simulation
Computer simulations were performed using the Grandi et al Human Atrial Model (HAR) [39]. The model code was obtained from the Grandilab (http://elegrandi.wixsite.com/grandilab/atrial-cell-models). All simulations were performed in Matlab (MathWorks, Natick, Massachusetts, USA) using the ode15s solver. To simulate the effects of the T361S mutation, the maximum conductance (gto) and current density for the Ito component of the HAR model was modified as follows: gto was increased by a factor of 1.5 and current density was decreased by a certain factor. Based on the experimental data from measurements of WT+KChIP2 and T361S+ KChIP2 channels, the steady-state inactivation, steady-state activation curve, and I-V curve in the Grandi model are re-fitted. The simulations were performed at basic cycle lengths (BCLs) of 1000, 900, 800, 700, 600, and 500 ms. At each BCL, the action potential duration at 90% repolarization (APD90) was calculated for the 40th beat.
Statistical analysis
All data were obtained from three independent experiments and expressed as the mean±SEM. The statistical analyses were carried out using a two tailed paired or unpaired Student’s t-test between two groups. The differences between groups over a time period were analyzed by two-way ANOVA. P<0.05 was considered statistically significant.
Footnotes
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
FUNDING
This work was supported by the grants from the National Natural Science Foundation of China (No. 81170090 and 81270277), and the Natural Science Foundation of Fujian Province (No.2013D020 and 2012D037).
Author contributions
Z.R.H. and Z.G.S. conceived and designed the experiments; Y.H., J.W.Y, W.Y.X, Q.C.L., Z.P.Z. and H.B.S. performed the experiments; J.W.Y., Y.H. and Z.R.H. analyzed the data; Y.H., Z.R.H. and Z.G.S. wrote the paper.
REFERENCES
- 1.Krijthe BP, Kunst A, Benjamin EJ, Lip GY, Franco OH, Hofman A, Witteman JC, Stricker BH, Heeringa J. Projections on the number of individuals with atrial fibrillation in the european union, from 2000 to 2060. Eur Heart J. 2013(34):2746–2751. doi: 10.1093/eurheartj/eht280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chugh SS, Havmoeller R, Narayanan K, Singh D, Rienstra M, Benjamin EJ, Gillum RF, Kim YH, McAnulty JH, Jr, Zheng ZJ, Forouzanfar MH, Naghavi M, Mensah GA, et al. Worldwide epidemiology of atrial fibrillation a global burden of disease 2010 study. Circulation. 2014(129):837–847. doi: 10.1161/CIRCULATIONAHA.113.005119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wolf PA, Benjamin EJ, Belanger AJ, Kannel WB, Levy D, D’Agostino RB. Secular trends in the prevalence of atrial fibrillation: the Framingham study. Am Heart J. 1996(131):790–795. doi: 10.1016/s0002-8703(96)90288-4. [DOI] [PubMed] [Google Scholar]
- 4.Kopecky SL, Gersh BJ, McGoon MD, Whisnant JP, Holmes DR, Jr, Ilstrup DM, Frye RL. The natural history of lone atrial fibrillation. A population-based study over three decades. N Engl J Med. 1987(317):669–674. doi: 10.1056/NEJM198709103171104. [DOI] [PubMed] [Google Scholar]
- 5.Lévy S. Epidemiology and classification of atrial fibrillation. J Cardiovasc Electrophysiol. 1998(9):S78–82. [PubMed] [Google Scholar]
- 6.Chen YH, Xu SJ, Bendahhou S, Wang XL, Wang Y, Xu WY, Jin HW, Sun H, Su XY, Zhuang QN, Yang YQ, Li YB, Liu Y, et al. Kcnq1 gain-of-function mutation in familial atrial fibrillation. Science. 2003(299):251–254. doi: 10.1126/science.1077771. [DOI] [PubMed] [Google Scholar]
- 7.Tada H, Kawashiri MA, Yamagishi M, Hayashi K. Atrial fibrillation: an inherited cardiovascular disease-a commentary on genetics of atrial fibrillation: from families to genomes. J Hum Genet. 2016(61):3–4. doi: 10.1038/jhg.2015.63. [DOI] [PubMed] [Google Scholar]
- 8.Wang P, Yang Q, Wu X, Yang Y, Shi L, Wang C, Wu G, Xia Y, Yang B, Zhang R, Xu C, Cheng X, Li S, et al. Functional dominant-negative mutation of sodium channel subunit gene scn3b associated with atrial fibrillation in a chinese geneid population. Biochem Biophys Res Commun. 2010(398):98–104. doi: 10.1016/j.bbrc.2010.06.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Olesen MS, Jespersen T, Nielsen JB, Liang B, Moller DV, Hedley P, Christiansen M, Varro A, Olesen SP, Haunso S, Schmitt N, Svendsen JH. Mutations in sodium channel beta-subunit scn3b are associated with early-onset lone atrial fibrillation. Cardiovasc Res. 2011(89):786–793. doi: 10.1093/cvr/cvq348. [DOI] [PubMed] [Google Scholar]
- 10.Fatkin D, Santiago CF, Huttner IG, Lubitz SA, Ellinor PT. Genetics of atrial fibrillation: state of the art in 2017. Heart Lung Circ. 2017(26):894–901. doi: 10.1016/j.hlc.2017.04.008. [DOI] [PubMed] [Google Scholar]
- 11.Hodgson-Zingman DM, Karst ML, Zingman LV, Heublein DM, Darbar D, Herron KJ, Ballew JD, de Andrade M, Burnett JC, Jr, Olson TM. Atrial natriuretic peptide frameshift mutation in familial atrial fibrillation. N Engl J Med. 2008(359):158–165. doi: 10.1056/NEJMoa0706300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ren X, Xu C, Zhan C, Yang Y, Shi L, Wang F, Wang C, Xia Y, Yang B, Wu G, Wang P, Li X, Wang D, et al. Identification of nppa variants associated with atrial fibrillation in a Chinese geneid population. Clin Chim Acta. 2010(411):481–485. doi: 10.1016/j.cca.2009.12.019. [DOI] [PubMed] [Google Scholar]
- 13.Zhang X, Chen S, Yoo S, Chakrabarti S, Zhang T, Ke T, Oberti C, Yong SL, Fang F, Li L, de la Fuente R, Wang L, Chen Q, et al. Mutation in nuclear pore component nup155 leads to atrial fibrillation and early sudden cardiac death. Cell. 2008(135):1017–1027. doi: 10.1016/j.cell.2008.10.022. [DOI] [PubMed] [Google Scholar]
- 14.Abbott GW, Goldstein SA. Potassium channel subunits encoded by the kcne gene family: physiology and pathophysiology of the mink-related peptides (mirps) Mol Interv. 2001(1):95–107. [PubMed] [Google Scholar]
- 15.Sah R, Ramirez RJ, Oudit GY, Gidrewicz D, Trivieri MG, Zobel C, Backx PH. Regulation of cardiac excitation-contraction coupling by action potential repolarization: role of the transient outward potassium current (i(to)) J Physiol. 2003(546):5–18. doi: 10.1113/jphysiol.2002.026468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aimond F, Alvarez JL, Rauzier JM, Lorente P, Vassort G. Ionic basis of ventricular arrhythmias in remodeled rat heart during long-term myocardial infarction. Cardiovasc Res. 1999(42):402–415. doi: 10.1016/s0008-6363(99)00070-x. [DOI] [PubMed] [Google Scholar]
- 17.Guo W, Kamiya K, Toyama J. Effect of chronic hypoxia on ion channel development in cultured cardiac cells. Environ Med. 1995(39):57–60. [PubMed] [Google Scholar]
- 18.Vandier C, Delpech M, Bonnet P. Spontaneous transient outward currents and delayed rectifier K+ current: effects of hypoxia. Am J Physiol. 1998(275):L145–154. doi: 10.1152/ajplung.1998.275.1.L145. [DOI] [PubMed] [Google Scholar]
- 19.Lundby A, Ravn LS, Svendsen JH, Hauns S, Olesen SP, Schmitt N. Kcne3 mutation v17m identified in a patient with lone atrial fibrillation. Cell Physiol Biochem. 2008(21):47–54. doi: 10.1159/000113746. [DOI] [PubMed] [Google Scholar]
- 20.Duarri A, Jezierska J, Fokkens M, Meijer M, Schelhaas HJ, den Dunnen WF, van Dijk F, Verschuuren-Bemelmans C, Hageman G, van de Vlies P, Kusters B, van de Warrenburg BP, Kremer B, et al. Mutations in potassium channel kcnd3 cause spinocerebellar ataxia type 19. Ann Neurol. 2012(72):870–880. doi: 10.1002/ana.23700. [DOI] [PubMed] [Google Scholar]
- 21.Giudicessi JR, Ye D, Kritzberger CJ, Nesterenko VV, Tester DJ, Antzelevitch C, Ackerman MJ. Novel mutations in the kcnd3-encoded kv4.3 k+ channel associated with autopsy-negative sudden unexplained death. Hum Mutat. 2012(33):989–997. doi: 10.1002/humu.22058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee YC, Durr A, Majczenko K, Huang YH, Liu YC, Lien CC, Tsai PC, Ichikawa Y, Goto J, Monin ML, Li JZ, Chung MY, Mundwiller E, et al. Mutations in kcnd3 cause spinocerebellar ataxia type 22. Ann Neurol. 2012(72):859–869. doi: 10.1002/ana.23701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Olesen MS, Refsgaard L, Holst AG, Larsen AP, Grubb S, Haunso S, Svendsen JH, Olesen SP, Schmitt N, Calloe K. A novel kcnd3 gain-of-function mutation associated with early-onset of persistent lone atrial fibrillation. Cardiovasc Res. 2013(98):488–495. doi: 10.1093/cvr/cvt028. [DOI] [PubMed] [Google Scholar]
- 24.Niwa N, Nerbonne JM. Molecular determinants of cardiac transient outward potassium current (I(to)) expression and regulation. J Mol Cell Cardiol. 2010;48:12–25. doi: 10.1016/j.yjmcc.2009.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bosch RF, Zeng X, Grammer JB, Popovic K, Mewis C, Kuhlkamp V. Ionic mechanisms of electrical remodeling in human atrial fibrillation. Cardiovasc Res. 1999(44):121–131. doi: 10.1016/s0008-6363(99)00178-9. [DOI] [PubMed] [Google Scholar]
- 26.Grammer JB, Bosch RF, Kuhlkamp V, Seipel L. Molecular remodeling of Kv4.3 potassium channels in human atrial fibrillation. J Cardiovasc Electrophysiol. 2000(11):626–633. doi: 10.1111/j.1540-8167.2000.tb00024.x. [DOI] [PubMed] [Google Scholar]
- 27.Pandit SV, Berenfeld O, Anumonwo JM, Zaritski RM, Kneller J, Nattel S, Jalife J. Ionic determinants of functional reentry in a 2-D model of human atrial cells during simulated chronic atrial fibrillation. Biophys J. 2005;88:3806–3821. doi: 10.1529/biophysj.105.060459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Low SK, Takahashi A, Ebana Y, Ozaki K, Christophersen IE, Ellinor PT, AFGen Consortium. Ogishima S, Yamamoto M, Satoh M, Sasaki M, Yamaji T, Iwasaki M, et al. Identification of six new genetic loci associated with atrial fibrillation in the Japanese population. Nat Genet. 2017(49):953–958. doi: 10.1038/ng.3842. [DOI] [PubMed] [Google Scholar]
- 29.Li J, Xie D, Huang J, Lv F, Shi D, Liu Y, Lin L, Geng L, Wu Y, Liang D, Chen YH. Cold-inducible rna-binding protein regulates cardiac repolarization by targeting transient outward potassium channels. Circ Res. 2015(116):1655–1659. doi: 10.1161/CIRCRESAHA.116.306287. [DOI] [PubMed] [Google Scholar]
- 30.Sabi R, Tuller T. Modelling the efficiency of codon-trna interactions based on codon usage bias. DNA Res. 2014(21):511–526. doi: 10.1093/dnares/dsu017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Duarri A, Lin MC, Fokkens MR, Meijer M, Smeets CJ, Nibbeling EA, Boddeke E, Sinke RJ, Kampinga HH, Papazian DM, Verbeek DS. Spinocerebellar ataxia type 19/22 mutations alter heterocomplex kv4.3 channel function and gating in a dominant manner. Cell Mol Life Sci. 2015(72):3387–3399. doi: 10.1007/s00018-015-1894-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Giudicessi JR, Ye D, Tester DJ, Crotti L, Mugione A, Nesterenko VV, Albertson RM, Antzelevitch C, Schwartz PJ, Ackerman MJ. Transient outward current (i(to)) gain-of-function mutations in the kcnd3-encoded kv4.3 potassium channel and brugada syndrome. Heart Rhythm. 2011(8):1024–1032. doi: 10.1016/j.hrthm.2011.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Antzelevitch C, Brugada P, Borggrefe M, Brugada J, Brugada R, Corrado D, Gussak I, LeMarec H, Nademanee K, Perez Riera AR, Shimizu W, Schulze-Bahr E, Tan H, et al. Brugada syndrome: report of the second consensus conference: endorsed by the Heart Rhythm Society and the European Heart Rhythm Association. Circulation. 2005(111):659–670. doi: 10.1161/01.CIR.0000152479.54298.51. [DOI] [PubMed] [Google Scholar]
- 34.Zeng Z, Xie Q, Huang Y, Zhao Y, Li W, Huang Z. p.D1690N sodium voltage-gated channel α subunit 5 mutation reduced sodium current density and is associated with Brugada syndrome. Mol Med Rep. 2016(13):5216–5222. doi: 10.3892/mmr.2016.5162. [DOI] [PubMed] [Google Scholar]
- 35.Xu C, Yang Q, Xiong H, Wang L, Cai J, Wang F, Li S, Chen J, Wang C, Wang D, Xiong X, Wang P, Zhao Y, et al. Candidate pathway-based genome-wide association studies identify novel associations of genomic variants in the complement system associated with coronary artery disease. Circ Cardiovasc Genet. 2014(7):887–894. doi: 10.1161/CIRCGENETICS.114.000738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zeng Z, Zhou J, Hou Y, Liang X, Zhang Z, Xu X, Xie Q, Li W, Huang Z. Electrophysiological characteristics of a scn5a voltage sensors mutation r1629q associated with brugada syndrome. PLoS One. 2013(8):e78382. doi: 10.1371/journal.pone.0078382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huang Y, Wang Z, Liu Y, Xiong H, Zhao Y, Wu L, Yuan C, Wang L, Hou Y, Yu G, Huang Z, Xu C, Chen Q, et al. αB-crystallin interacts with Nav1.5 and regulates ubiquitination and internalization of cell surface Nav1.5. J Biol Chem. 2016(291):11030–11041. doi: 10.1074/jbc.M115.695080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chakrabarti S, Wu X, Yang Z, Wu L, Yong SL, Zhang C, Hu K, Wang QK, Chen Q. Mog1 rescues defective trafficking of na(v)1.5 mutations in brugada syndrome and sick sinus syndrome. Circ Arrhythm Electrophysiol. 2013(6):392–401. doi: 10.1161/CIRCEP.111.000206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Grandi E, Pandit SV, Voigt N, Workman AJ, Dobrev D, Jalife J, Bers DM. Human atrial action potential and Ca2+ model: sinus rhythm and chronic atrial fibrillation. Circ Res. 2011(109):1055–1066. doi: 10.1161/CIRCRESAHA.111.253955. [DOI] [PMC free article] [PubMed] [Google Scholar]