

Abstract
Flagellar assembly requires intraflagellar transport of components from the cell body to the flagellar tip for assembly. The understanding of flagellar assembly has been aided by the ease of biochemistry and the availability of mutants in the unicellular green alga, Chlamydomonas reinhardtii. In this chapter, we discuss means to identify genes involved in these processes using forward and reverse genetics. In particular, the ease and low cost of whole genome sequencing (WGS) will help to make gene identification easier and promote the understanding of this important process.
INTRODUCTION
Forward genetics allows the identification of mutants with phenotypes of interest and leads to the mechanistic understanding of biological processes. Chlamydomonas has been an outstanding model for the identification and mechanistic understanding of flagellar assembly. Flagella are used for propelling cells toward light and various chemicals (Ermilova, Nikitin, & Fernandez, 2007; Hirschberg & Stavis, 1977; Wakabayashi, Misawa, Mochiji, & Kamiya, 2011) as well as for the recognition of mating partners and the initiation of fusion between them (Goodenough, 1993; Goodenough et al., 1980; Goodenough, Adair, Collin-Osdoby, & Heuser, 1985). Flagellar are not essential for propagation of Chlamydomonas cells in the lab, but they are required for mating in the absence of exogenous cAMP (Pasquale & Goodenough, 1987). Thus, the study of flagellar assembly can be approached using null mutants as well as conditional mutants. Null mutants provide information about the role of the gene, while conditional mutants allow for the isolation and analysis of flagella to study the effect of the mutations (Iomini, Babaev-Khaimov, Sassaroli, & Piperno, 2001). Null alleles in genes required for flagellar assembly are often aflagellate. Mutants with defects in flagellar assembly fall into two broad classes. They include mutants that encode motors and proteins of the intraflagellar transport machinery (IFT) (Ishikawa & Marshall, 2011) and mutants that affect the assembly of basal bodies and transition zones that serve as templates for the doublet microtubules and sites of docking for IFT machinery (Reiter, Blacque, & Leroux, 2012).
The IFT trains assemble in the cytoplasm and dock at the basal body/transition zone that is attached to the plasma membrane (Deane, Cole, Seeley, Diener, & Rosenbaum, 2001; Williamson, Silva, Richey, & Qin, 2012). IFT trains contain at least 22 proteins (Cole et al., 1998; Follit, Xu, Keady, & Pazour, 2009; Ishikawa et al., 2014; Piperno & Mead, 1997) and transport structural proteins into flagella (Cevik et al., 2010, 2013; Qin, Diener, Geimer, Cole, & Rosenbaum, 2004). They are carried by kinesin-2 to the tip and by cytoplasmic dynein-2 to the cell body (Ishikawa & Marshall, 2011). Proteomics of isolated IFT particles have identified many of these proteins (Andersen et al., 2003; Jakobsen et al., 2011; Keller, Romijn, Zamora, Yates, & Marshall, 2005; Kilburn et al., 2007; Muller et al., 2010; Ostrowski et al., 2002; Pazour, Agrin, Leszyk, & Witman, 2005).
A collection of temperature-sensitive mutants in Chlamydomonas that assemble flagella at the permissive temperature of 21 °C, but lack flagella at the restrictive temperature of 32 °C provides an important resource for the analysis of flagellar assembly (Adams, Huang, & Luck, 1982; Engel et al., 2012; Huang, Rifkin, & Luck, 1977; Iomini et al., 2001). Since many conditional mutants have reduced but sufficient function at the permissive temperature, this collection offers the opportunity to examine IFT in assembled flagella at the permissive temperature to ask about the effects of reduced function (Iomini et al., 2001; Lux & Dutcher, 1991; Williamson et al., 2012) (Table 1).
Table 1.
Identifying Gene Needed for Flagellar Assembly
| Gene | Protein | No. Alleles | Null Phenotype | Conditional Phenotype | Method | References |
|---|---|---|---|---|---|---|
|
| ||||||
| Kinesin-2 | ||||||
|
| ||||||
| FLA3 | Kinesin associated protein (KAP) | 1 | – | Flagellar assembly | Mapping | Huang et al. (1977), Mueller, Perrone, Bower, Cole, and Porter (2005) |
| FLA8 | Kinesin 2 | 3 | No flagella | Flagellar assembly | Mapping, whole genome sequencing | Adams et al. (1982), Dutcher et al. (2012), Miller et al. (2005) |
| FLA10 | Kinesin 2 | 4 | No flagella | Flagellar assembly | Mapping, insertional mutagenesis, whole genome sequencing | Lin, Nauman, et al. (2013), Matsuura, Lefebvre, Kamiya, and Hirono (2002), Miller et al. (2005), Walther, Vashishtha, and Hall (1994) |
|
| ||||||
| Cytoplasmic dynein 2 | ||||||
|
| ||||||
| FLA14 | Light chain 8 | 2 | Short flagella | – | Insertional mutagenesis | Pazour, Wilkerson, and Witman (1998) |
| DHC1b | Dynein heavy chain | 6 | Very short flagella | Flagellar assembly, slow retrograde IFT | Insertional mutagenesis, mapping, whole genome sequencing | Engel et al. (2012), Lin, Nauman, et al. (2013), Pazour, Dickert, and Witman (1999), Porter, Bower, Knott, Byrd, and Dentler (1999) |
| D1bLIC | Light intermediate chain | 2 | Very short flagella | – | Insertional mutagenesis | Hou, Pazour, and Witman (2004), Perrone et al. (2003) |
|
| ||||||
| IFT complex A | ||||||
|
| ||||||
| FLA15 | IFT144 | 1 | – | Flagellar assembly | Mapping | Iomini et al. (2001), Iomini et al. (2009) |
| FLA17 | IFT139 | 1 | – | Flagellar assembly | Mapping | Iomini et al. (2001), Iomini et al. (2009) |
| IFT122 | 1 | Aflagellate | – | Behal et al. (2012) | ||
| IFT121 | 1 | Aflagellate | – | Insertional mutagenesis | Behal et al. (2012) | |
|
| ||||||
| IFT complex B | ||||||
|
| ||||||
| FLA11 | IFT172 | 1 | – | Flagellar assembly | Mapping | Adams et al. (1982), Pedersen et al. (2005) |
| IFT88 | 1 | Aflagellate | – | Insertional mutagenesis | Pazour et al. (2000), Pazour et al. (1998) | |
| FLA9 | IFT81 | 1 | – | Flagellar assembly | Whole genome sequencing | Adams et al. (1982), Dutcher et al. (unpublished data) |
| NG6 | IFT80 | 1 | Aflagellate | – | Whole genome sequencing | Dutcher et al. (2012) |
| IFT70 | - | Short flagella | – | RNA interference | Fan et al. (2010) | |
| IFT56/Dyf13 | 1 | Short flagella with abnormal motility | – | Mapping | Ishikawa et al. (2014) | |
| BLD1 | IFT52 | 2 | Aflagellate | – | Mapping | Brazelton, Amundsen, Silflow, and Lefebvre (2001), Deane et al. (2001) |
| IFT46 | 1 | Short flagella | – | Insertional mutagenesis | Hou et al. (2007) | |
|
| ||||||
| Basal body genes | ||||||
|
| ||||||
| BLD2 | Epsilon-tubulin | 3 | Aflagellate | – | Mapping, insertional mutagenesis | Dutcher, Morrissette, Preble, Rackley, and Stanga (2002), Goodenough and St. Clair (1975) |
| UNI1 | ND | 4 | Uniflagellar | – | Mapping, whole genome sequencing | Huang, Ramanis, Dutcher, and Luck (1982), Lin, Miller, et al. (2013) |
| UNI2 | CEP120 | 2 | Uniflagellar | – | Mapping | Dutcher et al. (2002), Piasecki, LaVoie, Tam, Lefebvre, and Silflow (2008), Piasecki and Silflow (2009) |
| UNI3 | Delta-tubulin | 1 | Uniflagellar | – | Mapping | Dutcher and Trabuco (1998) |
| BLD10 | CEP135 | 1 | Aflagellate | – | Insertional mutagenesis | Hiraki, Nakazawa, Kamiya, and Hirono (2007), Matsuura, Lefebvre, Kamiya, and Hirono (2004) |
| BLD12 | SAS6 | 2 | Aflagellate | – | Insertional mutagenesis | Nakazawa et al. (2007) |
– indicates that no strain exists in this category.
ND; Not determined
1. IDENTIFYING THE CAUSATIVE GENES USING FORWARD GENETICS
1.1 REQUIREMENT FOR BACKCROSSES
Before beginning to identify the causative gene from strains in the stock center or newly isolated, backcrosses are essential. Chlamydomonas strains are stored in continuous culture. During long-term storage, cells can acquire multiple changes and thus, it is important to perform backcrosses to a wild-type strain before characterizing any strains that have been maintained long term in storage. For example, the NG6 (CC-829) and NG30 (CC-916) strains lack flagella (McVittie, 1969). Each strain was each crossed to a wild-type parent (CC-125) and immotile progeny were crossed three successive times to CC-125 as the first round of crosses had poor viability (19% and 22%, respectively), which suggests that the strains had acquired additional changes (Dutcher et al., 2012). After four rounds of crosses, the aflagellate phenotype segregates in a 2:2 pattern in 140 and 170 tetrads, respectively, with excellent viability (>90% with four viable progeny). Strains that have been mutagenized by ultraviolet light acquired ~2500 new mutations when compared to the parental strain (Lin and Dutcher, unpublished data). The removal of some of these by backcrosses will simplify the analysis. We present three methods for identifying genes identified by forward genetics.
1.2 MAPPING
Many of the mutations have been identified to date using mapping to narrow the region of interest and then rescue to identify the gene. Molecular mapping can be performed using the highly polymorphic strains, S1D2 and S1C5. They have single nucleotide difference (SNPs) with respect to most of the lab strains at a frequency of about 1 in 80 bps (Gross, Ranum, & Lefebvre, 1988) that create restriction enzyme recognition differences that can be used for mapping (Bowers, Keller, & Dutcher, 2003; Rymarquis, Handley, Thomas, & Stern, 2005; Silflow, Kathir, & Lefebvre, 1995). Many of the repetitive region repeats (GT and CA) differ in length between lab strains and S1D2; primers flanking the repetitive regions can be used to create PCR markers for mapping with size differences. Two to three markers spread across each chromosome used with about 30 meiotic progeny is generally sufficient to identify the chromosome of interest (see Table 2 for set of mapping primers). As indicated in Table 1, there are many flagellar assembly mutants that have been identified by this approach. The availability of genome sequence data from S1D2 or S1C5 and CC-124/CC-125 will make it possible to identify many more markers if fine scale mapping is the best approach (Lin, Miller, Granas, & Dutcher, 2013; Lin, Nauman, Albee, Hsu, and Dutcher, 2013). Protocols for mating have been published previously (Dutcher, 1995).
Table 2.
PCR Primers for Mapping in Meiotic Progeny
| Marker | Temp in °C | Enzyme | Sizes in CC-124 (bp) | Sizes in CC-1952 (bp) | Primer Sequences | |
|---|---|---|---|---|---|---|
|
| ||||||
| I | Chr1-0.3 | 53 | XhoI | 175 | 150, 25 | 5′CTA AGA AGC GCT TTG TGT GGG TCG A 5′CAT CCT TG CAA CAC TGT CCA TGC |
| Chr1-4 (AGK) | 53 | NaeI | 125, 75 | 200 | 5′ATT GTT GGG GCC TGG AAA TA 5′TGA ACG TAT GGT CCA GCG TA |
|
| Chr1-7.55 | 53 | BamHI | 175 | 150, 25 | 5′AGG TTT GGG CTT TCC CCA ACC GCG G 5′GGA GCT ATG AGG GCT GAG GTT GTA |
|
| II | Chr2-1.7 | 53 | – | 600 | 700, 800 | 5′GGA GCT GGA CCT GTC CTA CA 5′GTA CAG GCC GGC ATT CTC |
| Chr2-5.11 (GSP) | 58 | PstI | 140 | 175 | 5′GGG GTA GGC GGG TAG CAG GCC TGC A 5′CAC AGC GCC TAC CAC AGT ATT CCT |
|
| III | Chr3-0.35 | 58 | XbaI | 100,125 | 225 | 5′GCT TGA GGG CGG TTT GGA GAG T 5′AGC AAT CGT GCG CAT TCC TCA G |
| Chr3-1.1 | 53 | – | 300 | 200, 100 | 5′GTA TCC GGC TTG GTA CTG TTT GGA 5′AGG TGG ACC CCT CCG ACA AGG T |
|
| Chr3-3.7 (DMAT) | 48 | – | 300 | 250, 50 | 5′GGA CAT TCG TGT GGA GTG AA 5′GGG CAC GTC TGA CAG TAA CA |
|
| Chr3-6.0 (TUA1) | 53 | MspI | 150, 50 | 200 | 5′GGC CAT GAG TTG CTT CTT TC 5′AGG AAA CGC AGT CAA GGG TA |
|
| Chr3-7.9 (PHOT) | 51 | DdeI | 250, 75, 50 | 250, 125 | 5′TGC AGT TTT GCAGTT TGG AG 5′CTG CCG TCC ATG TTC CTT AT |
|
| IV | Chr4-0.5 (TUA2) | 55 | HaeIII | 350 | 200, 150 | 5′CAC AAA CAC AAG TAC CGT TGG T 5′AAG GTC AGG GGG AAG ATC C |
| Chr4-3.39 | 53 | – | 600 | 350 | 5′AGG GAG TAC GGC AAC ACC AG 5′GTC CCA AGT TCC TGC ATC C |
|
| V | Chr5-0.59 | 53 | PstI | 150 | 125, 25 | 5′AAT CCG CCA GCC GCT TCC CAG CTG C 5′GGT GCA CAA GGA GAT GAA CGT G |
| Chr5-1.9 (ALDO) | 53 | – | 300 | 700 | 5′GTT GGA GTT CCA TCG TCG TT 5′GTG GCT CCT CTC TGA ACT GC |
|
| Chr5-2.56 | 53 | – | 450 | >500 | 5′GTA GCT GTG AAA GGC CAA CG 5′GGT GAA TAC TAG CGG AGC ACA |
|
| Chr5-3.1 | 51 | – | Larger | 5′TCC GTA CAG GTG GAG TAG GG 5′TTT AGG CCA CAC ACA CAT GG |
||
| VI | MTA/MTD | 53 | MTA: 187 MTD: 124 |
– | MTA: 5′TCT CCA TGG GGT GTA TCA TC 5′CGT TTC AGC TCA CCA ACC MTD: 5′TTT GGA GTC CTC TCG TCA AG 5′GCA TGG CCT CTT AAT CAG AC |
|
| Chr6-3.41 (PP1) | 51 | MspI | 150, 50 | 200 | 5′GCG ACG ATT ATG TTT GCT GA 5′GTG TCA CCC CGA TTC AGT TT |
|
| Chr6-8.77 | 60 | HindIII | 75, 25 | 100 | 5′TCC AAC GAC TCG GCG CGT ACC ACA A 5′GCA TCT TCG TGG GCT ACC AGC A |
|
| VII | Chr7-0.5 | 57 | HindIII | 150, 100 | 250 | 5′CAT GTG GAG TGA TAT TTG TGA GCA TG 5′CAG TTA GCA GTT GGC CCA CGT CA |
| Chr7-2.1 | 53 | – | 350 | 250 | 5′ACC TGC CTC CTT GGC ATC AAG C 5′GCG ACA AAT TGA TTG GTG GCT ATA A |
|
| Chr7-6.1 | 53 | PstI | 200 | 175, 25 | 5′CTG CAC TTG TCA AAT TGG AGA TGC T 5′TAG CAA TAA GAC GGC CCA GAT GTT |
|
| VIII | Chr8-3.63 | 55 | SalI | 150 × 2 | 300 | 5′TCT CTC ACG CTC TCA CAC TCT TCC 5′GCG TTT GCT ACC AGA ACA AAA AGG |
| Chr8-4.79 | 53 | – | 650 | 400 | 5′GCA TCA TGC GAG GTG TGT TA 5′ATC ATC ATC TGG CGC TTA GG |
|
| IX | Chr9 0.29 | 53 | – | 600 | 450 | 5′CCT CTA ACT CCG CCA GTG CT 5′GTC AAG ATG GAC CTG CTG CT |
| Chr9-2.28 (TRX) | 51 | ScrFI | 200 | 150, 50 | 5′TTC CGT CCT CAC CAA AGT TC 5′CCA GCA GAA TTG ACA GCA AA |
|
| Chr9-3.24 | 51 | HaeIII | Single | Double | 5′TTT AGC TGA GGC GAG GT 5′AAA ATG GGA TGC GCA GTT AG |
|
| Chr9-4.4 | 53 | – | 500 | 650 | 5′CGG GAA GAT AGA GGC GTT AG 5′AAT TAG GCG TGC TGG GTA AA |
|
| X | Chr10-0.3 | 53 | – | 400 | 275 | 5′GAA CGA GGC AGA GCT GAA CT 5′GGC GGC TAG GTA TGT GTG AT |
| Chr10-1.71 | 53 | – | 300 | 350 | 5′AGT GCT CAT CTT CAG TAG CGG CTT 5′GTC CAT CAG GTA CGA CAG GGT TTC |
|
| Ch10-3.65 | 53 | EcoRI | 185 | 160, 25 | 5′CTC ATT TGG GGG TTT TGC TAA CGA A 5′GTC AGC ACT AGT TAC GCA GGG GAG |
|
| Chr10-3.82 | 53 | PstI | 175 | 100, 75 | 5′TAT TGG AGT GAT CGA GTG CCC TGC A 5′CGC CCA TGT AAC CAT AAG TCA |
|
| Chr10-4.94 | 55 | SalI | 275 | 150, 125 | 5′CAG TTG CAC AAA CCG ACA AAG AAA 5′CTC GGT ACA CCC TGC CAT TCT G |
|
| XI | Chr11-0.5 | 53 | – | 400 | 250 | 5′GCT ATC GCA GGT TCC AGA AG 5′GAG CGG CCT ATT TCA GCA TA |
| Chr11-3.59 | 50 | MspI | 200,175, 25 | 400 | 5′CCC CGA TTT CGA AGT TGA TA 5′ATA AGT GCG AGG GGG AGA CT |
|
| XII | Chr12-3.63 | 53 | – | 500 | 300 | 5′ACG TTG TTG AAG TGC AAT GG 5′CGG CGG ATG AAG ATG ACT |
| Chr12-3.72 (LC3) | 55 | HaeIII | 475 | Larger | 5′TGC ACA ACA CAA CAA GCA GA 5′ACA GGC GAG GAC ACA GCT AC |
|
| Chr12-5.9 | 53 | – | 250 | 200 | 5′ACT AGA GGG CCG AGA CGG AAT GTA 5′CTG CTG GGC GTA CCT GTA AAT GTT |
|
| Chr12-8.03 (LAO) | 60 | HaeIII | 150, 75 | 225 | 5′TCT TAC AGC GAC CGG ATT TC 5′GAC ACC ACA TCAGGG CTA CA |
|
| XIII | Chr13-1.25 | 53 | – | 500 | 425 | 5′GAC AAT GTG CCA ACA GCA AG 5′AGG GAC TGG ACC AGC ACA G |
| Chr13-2.09 | 56 | PstI | 225 | 150, 75 | 5′TAC CCC CTT CCT TGA ATC CAC TGA C 5′TCC ACA ACA CCA TAG TTA CCC CAA C |
|
| Chr13-4.05 | 53 | – | 500 | 700 | 5′AGC AAT GGG TGG TGT TCA GT 5′AGG GAT CGT TCT AGG GCT GT |
|
| Chr13-4.54 | 53 | TseI | 100, 50 | 175 | 5′CAC ACC CCA ATT CGT TGA GT 5′GCC TAT CCA GAC GCA AGC TA |
|
| XIV | Chr14-1.62 | 53 | – | 300 | 500 | 5′GAG AGC GAG TGC TTG GAA AT 5′GTC ATT GAG GCC GTG AAG AT |
| Chr14-1.84 (ASB) | 50 | RsaI | 300, 25 | 200, 100, 25 | 5′GGG CTA GGC CCT TTA CCT C 5′AGC TCC CGC ACT GTT TGT TGT AG |
|
| Chr14-3.69 | 53 | HindIII | 200 | 175 | 5′AGT AAT GTA TGC ATC TTG CGG AAG C 5′TGT GAA GCG AGT CAG TAG AAA CCG |
|
| XV | Chr15-1.25 | 53 | HindIII | 50, 25 | 75 | 5′ TCG TTG AGC CAA AGC ATC CGC AAG C 5′CAG CAA CAT TGA GGC TAT CCT GGT |
| XVI | Chr16-0.61 | 53 | – | 175 | 225 | 5′CTG ACA AGG GTC AGA TGG AGG AGT 5′TGT ACG GCT GTT TTG AGT ACA CGG |
| Chr16-3.04 (RAA2) | 61 | – | 650 | 500 | 5′ATT GAC CAC TGC GGC GCT AGC G 5′TAG TAG GGC CAT CCG TGG CTC TCG |
|
| Chr16-7.1 | 53 | – | 250 | 150 | 5′TGC AGA CTT GCA TTC GTA CAG AGG 5′CGT TGG AGC TGA CAG CAG TCC C |
|
| Chr16-7.25 | 58 | SalI | 150, 100 | 200 | 5′GAG CCA GTG TGA GCC TTG TGT G 5′CAG TAA GGA TGG GAT GGG TGT GAG |
|
| XVII | Chr17-0.57H | 53 | HindIII | 200 | 175, 25 | 5′GCA CAG CTG AAG CGCAAA AGG AAG C 5′CGT TTC TCG AAC TCA GCC ACT GT |
| Chr17-0.57 | 53 | – | 175 | 200 | 5′TGC GAG GAG GTA TCG AAG AAA GAG 5′CCG TCT ACC GGC ACT ACG AGT C |
|
| Chr17-2.23 (LC5) | 53 | MspI | 150, 50 | 200 | 5′GGA CGG TGG GTA TGC ATT AG 5′GCT GTC ACT ACG TGG TCT CG |
|
| Chr17-4.0 | 53 | – | 300 | 275 | 5′ATA TTA CGC CTC TCC GAC AAC AGC 5′CAG CTT CTT TGT GCG CTT GTA CTT |
|
| Chr17-6.8 | 53 | EcoRV | 125 | 100, 25 | 5′CAT CGA GCT GCT TGG AGG CCA GAT A 5′CGC TAT ACA CCA CAT AGC GTC GAG |
|
1.3 INSERTIONAL MUTAGENESIS
A second approach has been to use insertional mutagenesis (Tam & Lefebvre, 1993). DNA encoding a selectable phenotype is transformed into cells where it integrates randomly. Flanking DNA can be identified using a variety of techniques that include Southern blot analysis or PCR-based approaches (Pazour & Witman, 2000). Insertional mutagenesis can create several problems. First, it is often associated with deletions that may remove large pieces of adjacent DNA. For example, the bld12 mutation is associated with a deletion of 40 kb of genomic DNA (Nakazawa, Hiraki, Kamiya, & Hirono, 2007) and the bld2-6 mutation has an additional of deletion of about 8 kb (Esparza et al., 2013). In these examples, a single gene rescued the phenotype of interest, but this need not necessarily be the case. Second, the use of carrier DNA or the selectable marker itself may create additional mutations (Shimogawara, Fujiwara, Grossman, & Usuda, 1998). A recent library of insertional mutants that has been sequenced will provide an alternative approach (Zhang et al., 2014).
1.4 WHOLE GENOME SEQUENCING
Provides a new means to identify mutations. The genome of Chlamydomonas is only 125 Mb and it is possible to put multiple mutants in a single lane of an Illumina HiSeq machine to reduce the cost of obtaining 40–50× coverage. We describe the methods involved in using this approach.
1. Preparation of a Chlamydomonas genomic DNA library for sequencing
The genomic DNA preparation protocol is modified from our previous publications (Dutcher et al., 2012; Lin, Kwan, & Dutcher, 2010).
Wash approximately 5 × 108 cells in 1 mL TEN buffer (150 mM NaCl, 10 mM EDTA (Ethylenediaminetetraacetic acid) pH8.0, 10 mM Tris–HCl pH8.0) and resuspend in 300 μL chilled water. Add 600 μL of SDS-EB buffer (2% SDS, 100 mM Tris–HCl pH8.0, 400 mM NaCl, 40 mM EDTA pH8.0) and 100 μL 20% SDS to the cells and incubate the sample at room temperature (RT) for 15 min to ensure complete cell lysis.
Separate the 1 mL of sample into two 1.5 mL Eppendorf tubes and add 0.5 mL phenol/chloroform (1:1) into each tube. Vortex each tube briefly and centrifuge the tubes at 12,000 g for 10 at 4 °C to separate DNA/RNA from proteins and other organic materials. Transfer the clear DNA/RNA supernatant to new tubes. Repeat DNA extraction with an equal volume (0.5 mL per tube) phenol/chloroform (1:1) and then an equal volume of chloroform. Transfer the clear supernatant into new tubes and precipitate DNA with 0.5 mL of isopropanol on ice for 15 min and centrifuge at 4 °C for 10 min. Remove the supernatant from tubes and wash the pellets with 70% ethanol at 4 °C for 5 min. All DNA extraction/precipitation steps should be performed on ice/4 °C to reduce degradation of genomic DNA. Dry pelleted DNA using Savant SpeedVac for 5 min and resuspend in 200 μL of TE (10 mM Tris–HCl pH 8.0, 1 mM EDTA, pH 8.0) with 5 μg/mL RNase A each. DNA from both tubes can be combined at this step.
Treat DNA with RNase A at 37 °C for 1 h. Repeat DNA extraction with equal volume (400 μL) of phenol/chloroform (1:1) and chloroform. Precipitate DNA with 1/10 volume of 3 M NaOAc (pH 5.2) and equal volume of iso-proponal. Wash DNA with 70% ethanol. All DNA extraction/precipitation steps should be performed on ice/4 °C to reduce degradation of genomic DNA.
Dry the resultant genomic DNA in SpeedVac for 5 min and resuspend DNA with 40 μL of TE.
Determine concentration of DNA by spectrophotometry at 260 nm. Final concentration of each sample usually is ~200 ng/μL and a 260/280 ratio of 1.80–1.95. Visualize DNA by running ~2 μL of isolated DNA on a 0.8% agarose gel. A clean genomic DNA preparation would result in one single band with large molecular weight. Smears at the low molecular weights indicate RNA contamination or DNA degradation.
Before construction of library for Illumina sequencing, analyze the quality and quantity of genomic DNA by an Agilent 2100 Bioanalyzer (Agilent Technologies). Shear ~1 μg of DNA with Covaris miniTUBEs with AFA (Covaris) to ~200 bp fragments according to Manufacturer’s protocol. Purify the sheared DNA with Agencourt AMPure XP beads (Beckman Coulter) according to manufacturer’s protocol and elute DNA with 50 μL of water.
To perform end repair of the fragmented DNA, mix 1 mM ATP, 0.4 mM dNTP mix, 15 units of T4 DNA polymerase, 5 units of Klenow DNA polymerase, 50 units of T4 polynucleotide kinase, 50 mM Tris–HCl, 10 mM MgCl2, and 50 μL of sheared DNA in a single tube and adjust the final volume to 100 μL. Incubate the sample at 20 °C for 30 min. Purify the sample with AMPure XP beads and elute DNA with 10 μL of water.
To perform A-tailing to the 3′ end of the DNA fragment, mix 1× Klenow buffer, 0.2 mM dATP, 15 units of Klenow exo−, and 10 μL end-repaired DNA in a single tube and adjust the final volume to 20 μL. Incubate the sample at 37 °C for 30 min and heat inactivate the enzyme at 75 °C for 20 min.
To ligate sequencing adapters to the DNA fragments, mix 1× Rapid ligase buffer, 3 μM adapter mix, 240 units of Rapid T4 DNA ligase, 20 μL of A-tailing DNA in a single tube and adjust the final volume to 50 μL. Incubate the sample at room temperature for 15 min. Purify the sample with AMPure XP beads and elute DNA with 30 μL of water.
To enrich DNA template and to introduce unique indexed primer to each sample, mix 1× VeraSeq, 0.75 μM pair-ended primer, 0.75 μM indexed primer, 15 μL of adapter-ligated DNA in a PCR tube and adjust the volume to 50 μL. Amplify DNA using the following conditions: 30 s at 98 °C; 8–10 cycles of (10 s at 98 °C, 30 s at 64 °C, 30 s at 72 °C); and 5 min at 72 °C. Purify the sample with AMPure XP beads and elute DNAwith 30 μL of water.
Validate the library with an Agilent 2100 Bioanalyzer. The final product should have a distinct peak at 200–400 bp. The library is then subjected to Illumina whole genome sequencing.
2. Whole genome sequencing and variant calling
While the output of whole genome sequencing reads varies dependent on the sequencing system, the current Illumina HiSeq 2500 model we used in Rapid Run mode can generate ~300 million reads (~150 million pair-ended reads). It allows us to combine up to five individually indexed strains into one single flow cell for sequencing. About 60 million 101 bp reads can be generated from each strain, which converts to ~50× coverage for the ~125 Mb Chlamydomonas genome. If more than five strains need to be sequenced, all individually indexed genomic DNA libraries can be combined and run in two or three flow cells. The sequencing reads of the same strain from individual flow cells can later be combined for analysis.
-
a
Demultiplex sequencing reads to assign reads to individual strains.
-
b
Align reads to Chlamydomonas genome.
Download Chlamydomonas genome assembly sequence (version v5.3.1; ftp://ftp.jgi-psf.org/pub/compgen/phytozome/v9.0/Creinhardtii/assembly/Creinhardtii_236.fa.gz) from Phytozome and index it by Novoindex (Novocraft.com)
Align reads to the Chlamydomonas genome assembly sequence with Novoalign (v2.08 or later, Novocraft.com; options: o SAM –r random –l 30 –e 100 –I 230 140 –H –c 12 –h 90 120) and the output. SAM (Sequence Alignment/Map) file is converted to BAM (Binary Sequence Alignment/MAP) file by SAMtools (version 0.1.18 or later; H. Li et al., 2009). If an individual strain is sequenced in multiple flow cells, generate BAM files from individual flow cells and then merge the BAM files by SAMtools. The percentage of total reads that aligned to the reference genome is usually 90–97% if the sequenced strain was generated in 137c background, the same strain background as the reference strain (CC-503). In the highly polymorphic strain, S1C5, only ~67% of reads can be aligned to the genome using our alignment parameters (Lin, Miller, et al., 2013).
Sort the resultant BAM file by SAMtools. Mark PCR duplication reads by Picard MarkDuplicates (http://picard.sourceforge.net/; options: REMOVE_DUPLICATES = True ASSUME_SORTED = True VALIDATION_STRINGENCY = LENIENT). The output file is in BAM format.
Sort the resultant BAM file by SAMtools. Perform SNP and short indel calling by SAMtools mpileup (options: ugf) and BCFtools view (options: bvcg). The output is in binary call format (BCF), which is then converted into VCF (version 4.1) with a read depth cutoff of 999 (varFilter –D999).
-
c
Eliminate nonrelevant SNPs/indels by comparing to our SNP/indel library at the Website (http://stormo.wustl.edu/SNPlibrary/).
The SNP/indel library was generated using the same pipeline described above and it includes 2,557,197 SNPs/indels from 16 different Chlamydomonas strains (Lin, Miller, et al., 2013). Among these 16 strains, four of them are wild-type: CC-124, CC-125, isoloP (CC-4402), and isoloM (CC-4403). All of these wild-type strains were originated from the 137c background. Among the 11 mutant strains generated by either chemical or UV-mutagenesis, five have flagellar assembly defects (fla18, fla24, fla9, uni1, ift80), four have motility defects (ida3, pf23, pf7, pf8), and one has a mating defect (imp3). One additional mutant strain, cnk10, which is supersensitivity to Taxol, was generated by insertional mutagenesis of the CC-125 strain (Lin, James, and Dutcher, unpublished data). All of these mutant strains, with the exception of uni1, were generated in or backcrossed multiple times to the 137c background. This library also include the highly polymorphic strain S1C5 (CC-1952) that is frequently used in meiotic mapping was also included in this library (Lin, Miller, et al., 2013).
On the SNP/indel library Website, users are allowed to either find unique SNPs/indels in one of the 16 strains we have sequenced or upload their own VCF file generated by the pipeline described here. Users can choose to eliminate SNPs/indels from one or more strains in the library. The output VCF file is in the same format as the input VCF file, with elimination of common changes found in our library.
-
d
Annotate variants by SnpEff.
In general, while the SNP/indel library is powerful enough to remove 80–99% nonrelevant changes from a given strain, we usually observe 700–2000 variants in the output VCF files. These variants represent unique changes found in both coding and noncoding regions. Some of these variants may have low quality scores due to SNP/indel found in some but not all aligned reads. To facilitate identification of the causative mutation from these variants in a given strain, we opt for SnpEff, a variant annotation and effect prediction tool (Cingolani et al., 2012).
Download the latest version of SnpEff from http://snpeff.sourceforge.net/ and unzip the files under a directory/path/to/snpEff/.
Add a new folder data/under the directory/path/to/snpEff/.
Add a new folder chlamy_v5/under the directory/path/to/snpEff/data/.
Download the Chlamydomonas genomic DNA FASTA sequences from ftp://ftp.jgipsf.org/pub/compgen/phytozome/v9.0/Creinhardtii/assembly/Creinhardtii_236.fa.gz to the folder/path/to/snpEff/data/chlamy_v5/and rename the file sequences.fa.gz.
Download the gene annotation file from ftp://ftp.jgipsf.org/pub/compgen/phytozome/v9.0/Creinhardtii/annotation/Creinhardtii_236_gene.gff3.gz to the folder/path/to/snpEff/data/chlamy_v5/and rename the file genes.gff.gz.
Download the Chlamydomonas protein FASTA sequences from ftp://ftp.jgipsf.org/pub/compgen/phytozome/v9.0/Creinhardtii/annotation/Creinhardtii_236_protein.fa.gz to the folder/path/to/snpEff/data/chlamy_v5/and rename the file protein.fa.gz.
Modify the snpEff.config file under the folder/path/to/snpEff/using txt editor program such as Notepad ++ by adding the following lines under the section “Databases: Not from ENSEMBL”:
# Chlamydomonas reinhardtii chlamy_v5.genome: Chlamydomonas reinhard-tiichlamy_v5.reference: ftp://ftp.jgipsf.org/pub/compgen/nternal/v9.0/Creinhardtii/annotation/
To create the chlamy_v5 database, cd/path/to/snpEff/and run java –jar snpEff.jar build –gff3 –v chlamy_v5.
To run snpEff, move the output VCF file (e.g. strain_name.vcf) from step c, which eliminate nonrelevant variants from our SNP/indel library, to/path/to/snpEff/, and run
java –jar snpEff.jar –v chlamy_v5 strain_name.vcf > strain_name.eff.vcf. The output vcf file is further filtered by SnpSift: type strain_name.eff.vcf | java –jar SnpSift.jar filter “filters”. The commonly used filters include CHROM=“chromosome_x” (x can be a number between 1 and 17, if the mutation has been previously mapped to a specific chromosome); QUAL >= 100 (the Phred quality of a specific base calling has to be greater or equal to 100); EFF [*].IMPACT = HIGH | EFF[*].IMPACT = MODERATE (changes include nonsense, missense mutations, frame shift, and in frame deletion); isHom(Gen [0]) (all reads are consistent and show non-reference reading). When analyzing the output file, we filter out variants that result in synonymous changes and variants with low quality scores. The quality scores are Phred quality scores. A Phred quality score greater than 30 indicates that the probability of incorrect base call is less than 0.001 (10−30/10). In practice, we usually eliminate variants with quality scores lower than 100. In more than 20 mutant strains we analyzed to date, all except one have quality scores greater than 100. The one exception has a quality score of 41 and is covered by only three reads.
3. Identification of the causative mutation
While successful variant calling depends on the quality of sequencing reads and the length of sequencing reads, the program that aligns reads to the reference genome, and the variant calling program, the filtered output file may contain 10–500 variants. Identifying the causative change among these variants may require one or more additional approaches as described below.
Mapping the mutant. Map the mutation to a chromosome or a region on a chromosome. This can be achieved by standard meiotic mapping (Dutcher, 1995; Lin et al., 2010; Lin, Nauman, Albee, Hsu, and Dutcher, 2013) as described in Section 2.2 or through the whole-genome sequencing of bulk mutant progeny isolated from a cross between the mutation strain and the highly polymorphic strain S1C5 (Alford et al., 2013). Our sequencing results indicated that while S1C5 bears ~2.3 million variants, the mutant strain pf27 contains ~860,000 changes. The pooling of multiple meiotic progeny from a cross for whole genome sequencing is one means to identify the region where the gene lies. The sequencing of 14 pooled pf27 progeny from a cross between pf27 and S1C5 showed about 1.4 million changes in the pooled strains. It is expected that regions that are linked to the pf27 mutation will have fewer variants obtained from the S1C5 strain background. While it is less obvious in the sequence from a single meiotic strain, the chromosomal region is easily identified in a pool of progeny that share the mutant phenotype. As shown in Figure 1B, a small number of variants are observed in the pool between 6 and 8.5 Mb on chromosome 12. This region is in agreement with meiotic mapping results, which indicate the pf27 mutation is between 6 and 8 Mb.
Multiple alleles. Whole-genome sequencing of multiple mutant alleles of the same gene is another approach. Two or more mutant alleles in one gene, identified or characterized by linkage or complementation tests, can each be subjected to whole genome sequencing. Gene IDs, which contain variants from each mutant, can be cross referenced to identify the affected gene. This was successful for identifying a suppressor of the fla24 mutation with three alleles (unpublished results, Lin, Ferkol, and Dutcher).
Trancriptional upregulation. Comparison to genes that show transcriptional upregulation following flagellar deflagellation is a third approach (see Section 4 for details). Many flagellar genes, which include IFT proteins, radial spoke proteins, central pair proteins, motor proteins, and FAPs (flagellar associated protein), are transcriptionally upregulated at least 2.5-fold within the first hour following deflagellation (Albee et al. 2013). A total of 1850 genes show >2.5-fold upregulation, and they are listed in Table S3 of Albee et al. 2013. If one works on a mutant that has a flagellar assembly defect or motility defect, cross-referencing of gene IDs carrying variants in a mutant to the list of 1850 genes may pinpoint the causative mutation without the necessity of meiotic mapping.
FIGURE 1. Bulk segregant mapping.

The distribution of polymorphisms found in the pf27 parental strain (blue), in 14 pf27 progeny from a cross between pf27 and S1C5 (yellow (light gray in print versions)), and in S1C5 (green (dark gray in print versions)) on two chromosomes compared with the reference genome (v5.3) (Alford et al., 2013). The number of changes is binned over 100 kb intervals along the length of each chromosome and displayed on the y-axis. The position along in the chromosome in Mb is displayed on the x-axis. The pf27 mutation maps to chromosome 12 as indicated by the orange (light gray in print versions) line; these have no increase in the number of polymorphisms. Conventional mapping puts pf27 within the region marked by the black line. Chromosome 1 is representative of the pattern that is observed for the other chromosomes. The number of polymorphisms observed in the 14 pooled meiotic segregants is between the numbers observed in the two parents. From this analysis, we are able to map the centromeres on all of the chromosomes. Centromeres are indicated by a decrease in the number of unique polymorphisms and are marked by the pink (gray in print versions) arrows.
For example, the WGS from fla24 mutant contains 72,298 variants (Lin, Nauman, et al., 2013). A comparison to 15 other strains in our SNP library found 1417 unique SNPs in this strain. SnpEff 3.6 calls 79 unique changes in coding regions. Only seven variants remain when variants with a Phred quality score lower than 100 were filtered out. Cross-referencing to the list of 1850 genes indicated that only one gene, Cre06.g250300, is upregulated following deflagellation. This is consistent with mapping results that show that fla24 maps to chromosome six near the mating-type locus. The FLA24 gene encodes the cytoplasmic dynein heavy chain gene DHC1b. We were able to verify the change on DHC1b is the causative mutation by reversion/suppressor analysis as described below (Lin, Nauman, et al., 2013).
4. Verification of the causative mutation
-
Designing markers and segregation analysis. Once a small collection of potential causative changes have been found, primers to amplify PCR products around these changes provide a fast means to rule out potential candidates with a few meiotic segregants.
Generate 5–10 meiotic segregants with the phenotype of interest by mating to a wild-type lab strain. These can be obtained by random spores or by tetrad analysis (Dutcher, 1995).
Using the NEBcutter2 program (http://tools.neb.com/NEBcutter2/), paste ~300–400 bps of the wild-type sequence and the same ~300–400 bps of the mutant sequence into the Website to look for a restriction enzyme recognition site that is present in one and not the other sequence. The mutation should be in the middle of the chosen sequence (Figure 2(A) and (B)) as illustrated for the fla24 mutation. Sequence around the T to C SNP (marked in red) show that the restriction enzymes AlwNI would cut the wild-type sequence twice and the mutant sequence only once. NciI would cut the mutant sequence once but would not cut the wild-type sequence (Figure 2(B)).
Primers are designed using a variety of programs (Primer3 or IDT design). PCR products are synthesized and cut with the enzyme. In Figure 2(C), the digestion of wild-type and mutant fla24 PCR products with AlwNI are shown.
Prepare crude DNA from the two parental strains and the meiotic segregants. In a PCR tube, mix 2 μL of 10× Vent buffer (200 mM Tris–HCl, 100 mM (NH4)2SO4, 100 mM KCl, 20 mM MgSO4, 1% Triton X-100, pH 8.8), 1 μL of 20 mg/mL Proteinase K, 7 μL of water, and 10 μL of cells in liquid culture medium. Incubate cells at 58 °C for 30 min to remove proteins from cells and 95 °C for 15 min to inactivate the enzyme. Perform quick spin of the PCR tube and use 1 μL of the supernatant in PCR with the designed primers.
If this is the causative change, then all of the 5–10 meiotic segregants will show the same digestion pattern as the mutant parent. If the change is unlinked, then only one-half the progeny on average will show the mutant digestion pattern.
Not all changes generate an alteration in a restriction enzyme recognition site. The dCAPS (derived cleaved amplified polymorphic sequence) program is very useful for introducing a restriction enzyme recognition site change (Neff, Turk, & Kalishman, 2002). The Website can be found at http://helix.wustl.edu/dcaps/dcaps.html.
Paste 60 bps of sequence from the wild-type and potential mutant change. The program will not accept spaces. Place the mutant change near one of the ends. Enter one for the number of mismatches allowed and run the program. Potential primers that are ~60 bases long will be generated along with the enzyme that will cut this “mutated” fragment. You need to generate the second primer about 200–300 bps away by a different primer design program. As an example, the fla10-1 does not change or create a restriction enzyme recognition site, but with one mismatch at the 3′ end of the forward primer, the dCAPs program introduces a site in wild-type but not in fla10-1 (Figure 2(D)).
-
Reversion analysis. Point mutations are excellent candidates for reversion analysis with the postulate that changing the point mutation back to the original amino acid (true reversion) or to another amino acid (pseudor-eversion) serves as evidence that the variant is likely to be causative.
If the phenotype is flagellar assembly or lack of motility, it is straightforward to screen for restoration of the wild-type swimming phenotype. This procedure can be performed with mutagenesis to increase the frequency or simply by looking for spontaneous mutations. Mutagenesis in Chlamydomonas is easily performed with ultraviolet light. A total of 750 μJ produces about 50% lethality.
This strategy has been successful for seven published flagellar assembly strains (Table 3) as well as many flagellar motility mutant (Huang, Piperno, Ramanis, & Luck, 1981). The fla10-14 mutation provides a useful example (Miller et al., 2005). The mutant had two changes in the gene relative to the wild-type strain, which are E24K and L196F. The fla10-14 strain was mutagenized and swimming cells were identified after enrichment at 32 °C. In the nine strains that regained the ability to swim, no changes in the L196F alteration were observed, which suggested that it was a neutral change rather than the causative change. One revertant was isolated that restored the lysine at position 24 back to glutamic acid; this is referred to as a true revertant. Several pseudorevertants that changed other amino acids were isolated. These include one strain that had changed the lysine at position 25 to a glutamic acid and six strains that changed other positions in kinesin-2 (G331S, N329H, and N329T) (Table 3).
Pseudorevertants may be useful for understanding folding and function of the proteins as well.
Plasmid and BAC rescue. Perform Chlamydomonas transformation as described in Section 3.3. Instead of spreading cells on paromomycin plates, separate 50 mL of cells in R + 100 mM mannitol into 48 large tubes, which contain 20 mL of R each. For mutants that have assembly defects, recipient cells are likely to be found at the bottom of the tubes while the swimmers are found on the top of the tubes. Transformation of plasmid or BAC DNA that contains the wild-type gene is expected to restore wild-type swimming phenotype. Swimmers in the tubes at the restrictive temperature (for rescue of temperature-sensitive mutants) or at 25 °C (for null mutants) are enriched by transferring the top 5 mL into new tubes every 2 days. After about five to seven rounds of enrichment, it is expected over half of the tubes are enriched for swimmers. Dilute the cells so that about 200 cells from each tube that was enriched for swimmers are spread onto TAP (TRIS acetate phosphate) plates and pick 10 colonies from each plates to perform crude DNA extraction, PCR, and digestion. The positive colonies are expected to contain both wild-type and mutant DNA alleles present by PCR. The rescued colonies are then backcrossed to wild-type cells to show that the rescued phenotype cosegregates with the transgene DNA.
FIGURE 2. Mapping and dCAPS design.

(A). Genomic sequence around wild-type and fla24 mutation. The red nucleotide indicates the mutant SNP (T to C). (B) NEBcutter (Vincze, Posfai, & Roberts, 2003) was used to cut the wild-type and the fla24 sequence. The wild-type sequence is cut twice by AlwNI and the fla24 mutant sequence is cut only once by AlwNI and by NciI. Enzymes that cut an odd number of times in the combined wild-type and mutant sequence are desirable (C). PCR products cut by AlwNI. Wild-type and the true revertant D11 are cut twice by AlwNI. (Reproduced from (Lin, Nauman, et al., 2013).) (D) The change in fla10-1, which has an N to K change, does not have a restriction enzyme site difference between wild-type and the mutant. A dCAPS (derived cleaved amplified polymorphic sequence) marker was designed (Neff et al., 2002). The primers (in orange (light gray in print versions)) introduce a nucleotide change that generates a restriction enzyme recognition site. The forward primer (in orange (light gray in print versions)) introduces a G (underlined in magenta (light gray in print versions)) that creates a restriction site (in blue (black in print versions)) in the wild-type sequence but not in the mutant sequence. The mutant nucleotide is shown in red (light gray in print versions).
Table 3.
Reversion Analysis of Flagellar Assembly Mutants
| Mutant | Change | Number of true revertants | Number of pseudoreverants | References |
|---|---|---|---|---|
|
| ||||
| fla8-2 | E21K | 3 | 4 M313V | Miller et al. (2005) |
| 17 M313I | ||||
| 8 G314S | ||||
| fla8-1 | F55S | 6 | 6 M313I | Miller et al. (2005) |
| 3 G314S | ||||
| fla10-14 | E24K | 1 | 1 N329T | Miller et al. (2005) |
| L196F | 1 N329H | |||
| 1 K25E | ||||
| 4 G314S | ||||
| pf4 | One base pair deletion to generate frameshift | 0 | AGAAGAA insertion upstream to restore reading frame | Elam, Sale, and Wirschell (2009) |
| fla8-3 | V12E | 1 | 2 E12G | Dutcher et al. (2012) |
| 1 E12I | ||||
| 1 E12L | ||||
| 1 unknown | ||||
| bld2-6 | I163N | 0 | 2 N163S | Esparza et al. (2013) |
| 2 S144G | ||||
| fla18 | E24G | 8 | None | Lin, Nauman, et al. (2013) |
| fla24 | L3234P | 6 | 2 P3234R | Lin, Nauman, et al. (2013) |
| 1 P3234S | ||||
| 7 unknown | ||||
1.5 ADDITIONAL MUTANTS
Nonconditional flagellar assembly mutants are easy to obtain. After insertional mutagenesis, about 1–5% of the colonies have a flagellar assembly defect (Tam & Lefebvre, 1993). In addition, the Chlamydomonas Center (http://chlamycollection.org/) maintains five stumpy (very short) or aflagellate strains from the McVittie mutagenesis (McVittie, 1972) and five aflagellate strains from the Goodengough mating screen (Goodenough, Hwang, & Martin, 1976; Goodenough & St. Clair, 1975) for which the causative gene has not been identified. There are also seven temperature-sensitive flagellar assembly mutant strains for which the genes have not been identified. One has a retrograde IFT defect (FLA2) and two that have anterograde IFT defects (FLA27 and FLA28). Four of them (FLA4, FLA5, FLA12, FLA21) do not show defects in anterograde or retrograde IFT (Iomini et al., 2001).
2. IDENTIFYING GENE FUNCTION USING REVERSE GENETICS
Reverse genetics approaches start with a gene of interest and use some method to reduce the level of the protein/transcript. RNA interference and artificial micro-RNAs have been used to study flagellar genes (Fan et al., 2010; Heuser, Dymek, Lin, Smith, & Nicastro, 2012; Iomini, Li, Mo, Dutcher, & Piperno, 2006). In most cases, RNA interference has used hairpins constructed from the whole gene or from exons. This method presents two major problems and is now rarely used. Whole genes or even a single exon have the possibility of off-target effects since many short fragments are generated by the Dicer complex. Second, these genes are often silenced and the phenotypes are lost (Iomini et al., 2006). The alternative method is to use artificial microRNA (amiRNA) (Molnar et al., 2009). This vector uses an endogenous microRNA from Chlamydomonas that have been engineered to accept 21 bp sequences. In either case, it is necessary to identify multiple independent strains since insertion of the RNAi or amiRNA vector can cause phenotypes.
2.1 DESIGNING AN ARTIFICIAL microRNA (amiRNA)
The Website (http://wmd3.weigelworld.org/cgi-bin/webapp.cgi) allows the identification of 21 bp targets for many organisms including Chlamydomonas.
Enter the target gene in FASTA format in the Designer window
Select the genome release you want to use
Specify the number of targets
Enter your email. Your results are emailed to you
Hit submit
When the output is returned by email, select targets. In our hands, only the “green” targets have been successful
Past the sequence into the Oligo window and select the pChlamiRNA2 and RNA3 vector
Order the two primers for each amiRNA
2.2 MAKING AN amiRNA CONSTRUCT (MOLNAR ET AL., 2009)
Resuspend primers in distilled water to a final concentration of 100 μM each. Mix 10 μL each with 20 μL of 2× annealing buffer (20 mM Tris pH 8.0, 2 mM EDTA, 100 mM NaCl), put in boiling water for 5 min and cool down gradually overnight in the same water bath.
Clean up the annealed oligonucleotides using Qiagen PCR purification kit and elute the DNA with 40 μL of water. To perform phosphorylation on the purified annealed oligos, mix 1 μl of the eluted DNA with 10 units of T4 polynucleotide kinase, 1× T4 DNA ligase buffer and adjust the final volume to 10 μL with water. Incubate the sample at 37 °C for 30 min and heat inactivate the enzyme at 65 °C for 20 min.
To prepare the pChlamiRNA3 plasmid for cloning, digest 1 μg of the plasmid DNA with SpeI in a reaction volume of 20 μL at 37 °C for 30 min, add 1 unit of Shrimp Alkaline Phosphatase into the mix and incubate at 37 °C for another 30 min. Gel purify the DNA with gel purification kit of your choice.
Set up ligation with 1:100 dilution of the purified oligos, ~20 ng of the purified vector, 200 units of T4 DNA ligase, and 1× T4 DNA ligase buffer in a final volume of 10 μL and incubate at room temperature for 10 min. Transform 1 μL of the ligated product to bacterial competent cells and select on LB + ampicillin plates.
Identify the positive clones that contain the annealed DNA by PCR with the following set of primers: AmiRNAprecfor (5′-GGTGTTGGGTCGGTGTTTTTG-3′) and Spacerrev (5′-TAGCGCTGATCACCACCACCC-3′). Extract plasmid DNA from two of the positive clones and perform Sanger sequencing with the AmiRNAprecfor primer. Align the sequence with the forward oligo sequence to make sure they are perfectly aligned.
2.3 TRANSFORMATION WITH THE amiRNA CONSTRUCT
Innoculate Chlamydomonas cells in 100 mL of liquid R (rich) or TAP medium under continuous light for 2 days until the cells reach the density of 2–5 × 106 cells/mL.
Prepare autolysin by resuspending 5- to 7-day-old wild-type plus and minus cells in 10 mL of nitrogen-free medium, respectively, for 2 h at room temperature. Mix the wild-type plus and minus cells to mate at room temperature for 15 min. Spin down the cells and filter the supernatant through 0.22 nm filter to sterilize the autolysin and to remove Chlamydomonas cells. Alternatively, one can use a strain that carries one of the mutations that lacks or has reduced cell wall, which negates the need for autolysin.
Resuspend the recipient Chlamydomonas cells in filtered autolysin to remove cell wall for 30 min at room temperature. Chill the cells for 10 min on ice before centrifugation at 1000 g for 5 min at 4 °C. Resuspend cells gently in R (or TAP) + 100 mM mannitol to a final concentration of ~4 × 108 cells/mL. Transfer 250 μL of cells into an electroporation cuvette (4 mm gap, Bio-Rad), add 1 μg of the microRNA plasmid DNA into the cell, incubate the cuvette in a 16 °C water bath for 5 min. Perform one pulse electroporation in a Bio-Rad Gene Pulser II with the following settings: 1.12 kV, 25 μF, and 400 Ω. Incubate the cuvette at room temperature for 10 min before transferring the cells to 50 ml R (or TAP) + 100 mM mannitol liquid medium. Incubate the cells overnight at 25 °C with continuous illumination.
The next morning, resuspend cells in 1 mL of 25% cornstarch in R (or TAP) medium and spread onto 5 TAP + 10 μg/mL paromomycin plates. In these TAP plates, the concentration of glacial acetic acid is 0.75 ml/L instead of 1 ml/L. The reduction of acetic acid in the medium confers more robust resistance to paromomycin. Colonies usually appear 5–7 days at 25 °C after spreading.
To identify colonies that contain the microRNA construct, prepare crude DNA extract and PCR with the AmiRNAprecfor and Spacerrev primers for individual colonies.
2.4 ANALYZING THE amiRNA STRAINS
Once the positive Chlamydomonas colonies that contain the amiRNA plasmid have been identified, screen them for a flagellar phenotype. It is expected that multiple colonies should bear the same phenotype.
If an antibody is available, the colonies can be screen by immunoblot for the presence of the protein (Heuser, Dymek, et al., 2012)
Grow about 108 cells from each colony and extract total RNA from these cells with Qiagen Rneasy Mini Kit. Treat RNA with 1 unit of Rnase-free DNAse I at 37 °C for 30 min to remove genomic DNA and inactive the enzyme by adding 1/10 volume of 25 mM EDTA and incubate at 65 °C for 10 min. Measure the RNA concentration.
Use 1 μg of DNAse I-treated RNA in a 20 μL of reverse transcription reaction that contains 200 ng random primers (Invitrogen), 1× RT buffer (Invitrogen), 5 mM DTT (dithiothreitol), 0.5 mM dNTP (deoxynucleotide triphosphate), 20 unit of RNaseOUT (Invitrogen), and 100 unit of SuperScript III reverse transcriptase (Invitrogen). Perform the reaction according to manufacturer’s recommendation.
For real-time PCR, dilute cDNA 1:10 and use 2 μL in a 20 μL of SYBR Green-PCR reaction which contains 1× iTAQ universal SYBR Green Supermix (Biorad) and 500 nM of each primer. Carry out the reactions using a Bio-Rad iCycler iQ (or equivalent machine) under the following conditions, 95 °C 3 min, 40 cycles of (95 °C 10 s and 57 °C 45 s), followed by the melting curve program. Normalize the transcript levels of individual genes by an internal control, CRY1, which encodes the ribosomal protein S14. The primer sequences that amplify the CRY1 gene are: CRY1-F (5′ GAAACTATTTCGCGCGTCAC 3′) and CRY1-R (5′ CAGTGATGCCCAGCTCCT 3′).
3. IDENTIFYING GENES USING THE RNA TRANSCRIPTOME
Many of the known ciliary components are upregulated during ciliogenesis in Chlamydomonas. Previous methods to look at transcript levels focused on cDNA libraries (Lefebvre & Rosenbaum, 1986), genes found by genomic comparisons (Li et al., 2004) or proteomics (Pazour et al., 2005), or used an incomplete version of the Chlamydomonas genome (Stolc, Samanta, Tongprasit, & Marshall, 2005). In addition, these studies focused on genes with increased fold change at 30 min and this single time point may also yield an incomplete list of ciliogenesis genes. A more complete picture of the genes required for ciliogenesis, was obtained by comparing RNAseq at multiple time points (3, 10, 30, and 60 min) after deflagellation with the predeflagellation transcript levels. A total of 1850 genes showed an increased fold change of at least 2.5 at one or more of the time points.
Illumina sequencing of mRNA isolated from predeflagellation, 3, 10, 30 and 60 min into ciliogenesis produced a total of 99.4 million 36-mer single-end reads, for an average of 19.9 million reads per time point sample. This equates to 3.58 Gb or a 32-fold coverage of the 125 Mb Chlamydomonas genome. TopHat (Trapnell, Pachter, & Salzberg, 2009) was used to compute expression levels of 18,507 Augustus gene models. Expression values calculated by Cufflinks are reported in terms of fragments per kilobase transcribed per million reads mapped (FPKM) (Trapnell et al., 2010). In five independent sets of RNAseq sequencing (pre-shock, 3, 10, 30 and 60 min), 96% of RNAseq reads align to the v5.3.1 Chlamydomonas genome assembly. Only genes that have three FPKMs during at least one of the time points are included. By these criteria, 10,813 genes are expressed. Any gene with a time point to predeflagellation expression value ratio of 2.5 or greater is considered an upregulated gene; 1850 genes were predicted to be upregulated at one or more time points. There are 4392 genes downregulated, and 4548 genes that do not alter their expression level during ciliogenesis (Albee et al., 2013).
A set of principal regulation profiles for genes that are upregulated during the first 60 min of cilia regeneration in Chlamydomonas was generated. Sixteen principal regulation profiles are identified and then grouped into five main patterns (Figure 3(A)). The Arch group, containing patterns Arch1 and Arch2, shows increased expression at three min, a peak expression at 10 min, and then decreasing expression at 30 and 60 min (Figure 3(A)). Arch2 is the most common pattern with 343 genes while Arch1 is the second most common pattern with 336 genes. 37% of genes display the Arch pattern.
FIGURE 3. Profile clustering identifies 16 principal upregulation expression profiles organized into five pattern groups.
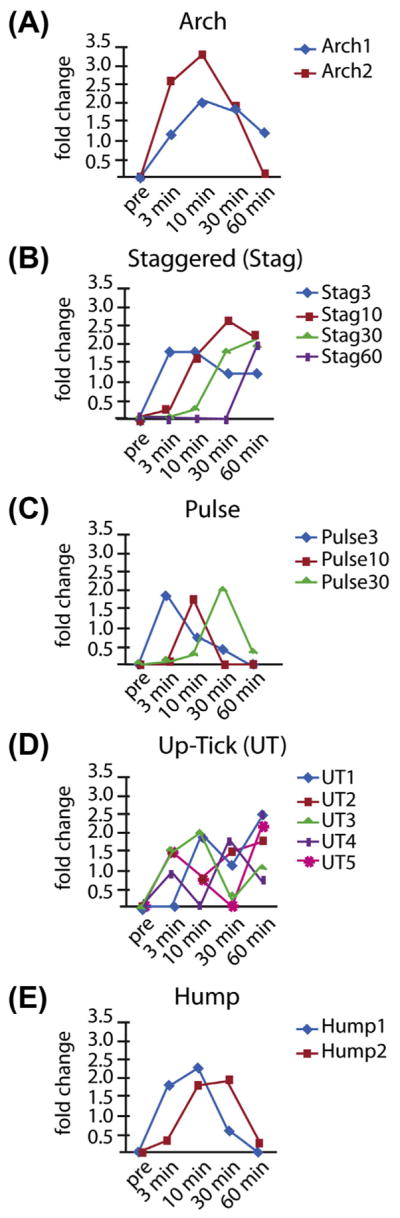
(A) The Arch pattern shows increased expression at 3 min, a peak expression at 10 min, and then decreasing expression. This profile accounts for 37% of the profiles. The Arch1 pattern shows expression that peaks at 10 or 30 min, but is still up at 60 min. The Arch2 pattern is similar to Arch1 except that the genes are not upregulated at the 60 min time point. (B) The staggered (Stag) pattern shows genes with a burst of expression at the 3, 10, 30, or 60 min time points. (C) The pulse pattern shows the upregulation at a single time point and accounts for 11% (N = 196) of the upregulated genes. (D) The Up-Tick (UT) pattern describes genes that show increased expression at one time point followed by downregulation at another time point followed by upregulation at a third time point. UT patterns make up 3% (N = 51) of upregulated genes. (E) Hump patterns make up 16% (N = 290) and are profiles that are pulse-like, but significant upregulation is sustained over two consecutive time points. 0.5% (N = 9) show profiles that are outliers in that their profiles are not adequately similar to any principal expression profile found in this analysis (Albee et al., 2013).
Of the IFT genes and associated motors, 90% of the genes fell into the Arch category. Axonemal dyneins, radial spoke, and central pair genes display more varied expression patterns with 60%, 78%, and 84% in the Arch category, respectively. Overall, structural ciliogenesis genes are more likely to have Arch expression patterns.
Many assembly factors for axonemal dynein arms have been identified over the last few years in humans and in model organisms. There are 11 factors that are not structural components of the dynein arms, but rather cytoplasmic factors that are needed for their assembly or transport. Three of them, D1 (Duquesnoy et al., 2009; Freshour, Yokoyama, & Mitchell, 2007), DNAAF2 (Omran et al., 2008), and DNAAF3 (Mitchison et al., 2012) are postulated to be involved in folding the dynein heavy chains. The others, with the exception of FBB11 and ODA16, share the property with DNAAF1-DNAAF3 that the proteins appear to localized to the cytoplasm and not the cilia. These include HEATR2 (Horani et al., 2012), LRRC6 (Horani et al., 2013; Kott et al., 2012), ZMYND10 (Albee et al., 2013; Zariwala et al., 2013), Reptin/RuvBL2 and DYX1C1 (Chandrasekar, Vesterlund, Hultenby, Tapia-Paez, & Kere, 2013; Tarkar et al., 2013). There is evidence that LRRC6 and Reptin interact (Zhao et al., 2013) as do LRRC6 and ZMYND10 (Zariwala et al., 2013). DYX1C1 interacts with DNAAF2 (Tarkar et al., 2013). The 10 of the 11 preassembly and adaptor factors show a similar pattern of expression following deflagellation in Chlamydomonas reinhardtii. These genes are among the earliest to show increased transcript levels (at 3 and 10 min after deflagellation) (Albee et al., 2013). Thirty-two genes with that pattern have human orthologs, and 27 are expressed in airway epithelial cells (Hoh, Stowe, Turk, & Stearns, 2012). These are likely to be a strong set of candidate genes for assembly/transport factors.
4. IDENTIFYING GENES USING GENETIC INTERACTIONS
Several mutants in the same process provide a way to bootstrap through a genetic network and identify other genes in the same pathway or in a different pathway that affect the pathway of interest. This approach relies on using synthetic interactions between different mutations. On can use the isolation of suppressors or the identification of synthetic interactions. Both of these approaches have been used extensively in yeast (Dixon et al., 2008), but have been underemployed in the study of flagellar assembly. Below are examples of suppressor analysis and synthetic interactions.
4.1 EXAMPLES OF SUPPRESSORS
Delta-tubulin is only found in organisms that build triplet microtubules and when deleted results in basal bodies that assembly primarily doublet microtubules (Dutcher & Trabuco, 1998). The deletion of delta-tubulin produces cells that assemble zero, one or two flagella. Suppressors that restore near wild-type numbers of flagella can be isolated and the majority map to the TUA2 locus, which encodes alpha-2-tubulin. Amino acids D205 or A208, which are nearly invariant residues in alpha-tubulin, were altered in 12 sequenced alleles. The suppressors partially restore the assembly of triplet microtubules. The tua2 mutations on their own render cells colchicine supersensitive (Fromherz, Giddings, Gomez-Ospina, & Dutcher, 2004). Thus, alterations in alpha-tubulin can substitute for the lack of delta-tubulin. Another example of a useful suppressor is seen in the suppression of the ift46 mutant, which reveals that IFT46 is required for IFT and specifically for the transport of outer dynein arms (Hou et al., 2007)
4.2 EXAMPLES OF SYNTHETIC INTERACTIONS
The kinesin-2 subunit encoded by the FLA10 gene shows a synthetic lethal phenotype with one specific allele at the APM1 gene, which encodes HSP40 (Silflow, Sun, Haas, Foley, & Lefebvre, 2011). The apm1-122 allele, which confers resistance to the plant herbicides amiprophos-methyl and oryzalin, in combination with three different fla10 alleles shows cold-sensitive cell cycle arrest. The null fla10 allele together with apm1-122 does not show arrest. This allele-specific phenotype suggests perhaps a titration of HSP40 protein by mutant kinesin-2 protein at the low temperature (Lux & Dutcher, 1991).
IFT144 and IFT139 are likely to interact with each other in Complex A of the IFT train based on several genetic interactions. Double mutants display synthetic flagellar assembly defects at the permissive temperature, and transgenes confer two-copy suppression (Iomini, Li, Esparza, & Dutcher, 2009).
4.3 PROTOCOL FOR SUPPRESSOR SCREEN
1. Mutagenesis
Chemicals and ultraviolet light can be used to isolate suppressors. A dose that gives about 50% lethality is used in most cases.
Ultraviolet light. The Stratagene Crosslinker works well for mutagenesis as discussed in Section 3.4. Alternatively, a GE 30W germicidal UV lamp (G3T08) can be used at a distance of 15 cm for 60 s. Use lawns of cells on solid medium after 2 days of growth. The cells must be able to go through a round of cell division after irradiation. After irradiation, leave cells in the dark for at least 5 h to avoid blue light activated repair of the DNA damage.
MNNG treatment. Approximately 108 cells are suspended in liquid medium and methylnitronitrosoguandine is added to a final concentration of 25 μg/mL. Incubate cells for 30 min. Pellet cells by centrifugation and wash three times with medium. Retain washes for proper disposal.
Ethylmethanesulfonate (EMS) treatment. Approximately 108 cells are suspended in liquid medium and EMS is added to a final concentration of 20 μg/mL. Incubate cells for 40 min. Pellet cells by centrifugation and wash with sterilized 1% sodium thiosulfate, which inactivates the EMS.
Methylmethanesulfonate (MMS) treatment. Approximately 108 cells are suspended in liquid medium and MMS is added to a final concentration of 1 μL/mL. Incubate cells for 120 min. Pellet cells by centrifugation and wash with medium.
2. Isolation schemes
Following mutagenesis, suppressors can be isolated by enrichment as described for the transformation and reversion experiments in Sections 3.3 and 2.4, respectively. Alternatively, cell can be selected for the ability to mate. This screen does not require that assembled flagella be motile and can be less restrictive. Cells are prepared for mating (Dutcher, 1995). After removal of nitrogen for 5 h, cells are mixed with a 10-fold excess of wild-type cells of the opposite mating-type for 1.5 h. Mating mixtures are plated to TAP plates and incubated at 21 °C for 18 h and then wrapped in foil for 5 days. The plates are exposed to chloroform vapors for 60 s to kill unmated cells as well as diploid cells and then allowed to grow. Due to the thick wall on zygotes, they are resistant to chloroform vapors and will form colonies. These colonies can be picked and the progeny analyzed by plating for single colonies. Single colonies are picked and phenotypes can be assayed.
3. Identification of intragenic versus extragenic suppressors
Once suppressed isolates are obtained from single colonies, they can be analyzed by PCR for the presence of the original mutation as described in Section 2.4. They can be crossed to a wild-type parent of the opposite mating-type and a minimum of 15 tetrads or 30 random spores should be analyzed. If only parental ditype tetrads are recovered, the suppressor mutation is likely to be intrangenic or tightly linked (less than 3.3 cM or 330 kb away) to the original flagellar mutation. Isolates that produce tetratype and/or nonparental ditype tetrads are considered extragenic as the original flagellar phenotype is recovered. To confirm that the suppression is caused by a single mutational hit, the suppressed strain should crossed to the original mutation. Suppression should be observed in one-half of the progeny in random spores and only parental ditypes should be recovered. Recovery of tetratype and nonparental ditype tetrads indicate that suppression requires more than one mutation.
4.4 PROTOCOLS FOR EPISTASIS AND SYNTHETIC INTERACTIONS
In both Saccharomyces cerevisiae and Schizosaccharomyces pombe, genome wide synthetic interactions have been performed (Dixon et al., 2008). These experiments have revealed conserved pathways. However, no similar method is available for Chlamydomonas. If a genome wide deletion collection becomes available, this type of approach will be possible. At this time, each double mutant must be constructed by mating (Dutcher, 1995), and phenotypes analyzed. A synthetic phenotype is defined as a phenotype not observed in either of parental strains. If both of the mutants are null alleles, then it suggests that the two genes act in different pathways. If the mutants are not null alleles, it can suggest that the loss of a pathway may have a more severe phenotype.
5. SUMMARY
There are many other approaches that can be taken to understand flagellar assembly. Identifying protein–protein interactions using yeast two hybrid analysis (Ahmed, Gao, Lucker, Cole, & Mitchell, 2008; Behal et al., 2012; Lucker et al., 2005; Lucker, Miller, Dziedzic, Blackmarr, & Cole, 2010), bacterial expression (Lucker et al., 2010; Taschner, Bhogaraju, & Lorentzen, 2012; Taschner, Bhogaraju, Vetter, Morawetz, & Lorentzen, 2011), and co-immunoprecipitation (Wang, Fan, Williamson, & Qin, 2009) are important avenues. Because flagella are well conserved organelles, comparative genomics has identified many key proteins (Avidor-Reiss et al., 2004; Li et al., 2004; Li, Zhang, Dutcher, & Stormo, 2005; Nishimura et al., 2005). Proteomics of isolated flagella and substructures as well as basal bodies have also been important in building the parts list for flagella and basal bodies (Andersen et al., 2003; Jakobsen et al., 2011; Keller et al., 2005; Kilburn et al., 2007; Muller et al., 2010; Ostrowski et al., 2002; Pazour et al., 2005). These approaches have been useful for building the parts list for flagella and basal bodies.
There are also many tools for studying the function and role of these proteins. Analysis of IFT in wild-type and mutants can be achieved by TIRF (total internal reflection fluorescence) or DIC (differential interference contrast) microscopy (Engel et al., 2012; Iomini et al., 2001). Electron micrograph tomography has been instrumental in understanding the structures involved (Heuser, Barber, et al., 2012; S. Li, Fernandez, Marshall, & Agard, 2012; O’Toole, Giddings, & Dutcher, 2007; Pigino et al., 2009). Dikaryons have provided an important tool in studying flagellar motility but can be used more extensively to study flagellar assembly (Dutcher, 2014). When Chlamydomonas cells mate, the flagella adhere to each other, which leads within 10 min to cell fusion and formation of a single cell with four flagella and two nuclei, which is known as a dikaryon. They allow the analysis of temporal and spatial localization patterns of proteins during flagellar assembly. These tools will lead to further understanding of how flagella are assembled and the assembly is regulated.
Acknowledgments
This work was supported by a grant to SKD from the National Institute of General Medical Sciences. The Children’s Discovery Institute of Washington University supported sequencing.
References
- Adams GM, Huang B, Luck DJ. Temperature-sensitive, assembly-defective flagella mutants of Chlamydomonas reinhardtii. Genetics. 1982;100(4):579–586. doi: 10.1093/genetics/100.4.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed NT, Gao C, Lucker BF, Cole DG, Mitchell DR. ODA16 aids axonemal outer row dynein assembly through an interaction with the intraflagellar transport machinery. Journal of Cell Biology. 2008;183(2):313–322. doi: 10.1083/jcb.200802025. http://dx.doi.org/10.1083/jcb.200802025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albee AJ, Kwan AL, Lin H, Granas D, Stormo GD, Dutcher SK. Identification of cilia genes that affect cell-cycle progression using whole-genome transcriptome analysis in Chlamydomonas reinhardtti. G3 (Bethesda) 2013;3(6):979–991. doi: 10.1534/g3.113.006338. http://dx.doi.org/10.1534/g3.113.006338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alford LM, Mattheyses AL, Hunter EL, Lin H, Dutcher SK, Sale WS. The Chlamydomonas mutant pf27 reveals novel features of ciliary radial spoke assembly. Cytoskeleton (Hoboken) 2013;70:804–818. doi: 10.1002/cm.21144. http://dx.doi.org/10.1002/cm.21144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen JS, Wilkinson CJ, Mayor T, Mortensen P, Nigg EA, Mann M. Proteomic characterization of the human centrosome by protein correlation profiling. Nature. 2003;426(6966):570–574. doi: 10.1038/nature02166. http://dx.doi.org/10.1038/nature02166. nature02166 [pii] [DOI] [PubMed] [Google Scholar]
- Avidor-Reiss T, Maer AM, Koundakjian E, Polyanovsky A, Keil T, Subramaniam S, et al. Decoding cilia function: defining specialized genes required for compartmentalized cilia biogenesis. Cell. 2004;117(4):527–539. doi: 10.1016/s0092-8674(04)00412-x. [DOI] [PubMed] [Google Scholar]
- Behal RH, Miller MS, Qin H, Lucker BF, Jones A, Cole DG. Subunit interactions and organization of the Chlamydomonas reinhardtii intraflagellar transport complex A proteins. Journal of Biological Chemistry. 2012;287(15):11689–11703. doi: 10.1074/jbc.M111.287102. http://dx.doi.org/10.1074/jbc.M111.287102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers AK, Keller JA, Dutcher SK. Molecular markers for rapidly identifying candidate genes in Chlamydomonas reinhardtii. Ery1 and ery2 encode chloroplast ribosomal proteins. Genetics. 2003;164(4):1345–1353. doi: 10.1093/genetics/164.4.1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brazelton WJ, Amundsen CD, Silflow CD, Lefebvre PA. The bld1 mutation identifies the Chlamydomonas osm-6 homolog as a gene required for flagellar assembly. Current Biology. 2001;11(20):1591–1594. doi: 10.1016/s0960-9822(01)00485-7. [DOI] [PubMed] [Google Scholar]
- Cevik S, Hori Y, Kaplan OI, Kida K, Toivenon T, Foley-Fisher C, et al. Joubert syndrome Arl13b functions at ciliary membranes and stabilizes protein transport in Caenorhabditis elegans. Journal of Cell Biology. 2010;188(6):953–969. doi: 10.1083/jcb.200908133. http://dx.doi.org/10.1083/jcb.200908133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cevik S, Sanders AA, Van Wijk E, Boldt K, Clarke L, van Reeuwijk J, et al. Active transport and diffusion barriers restrict Joubert syndrome-associated ARL13B/ARL-13 to an Inv-like ciliary membrane subdomain. PLoS Genetics. 2013;9(12):e1003977. doi: 10.1371/journal.pgen.1003977. http://dx.doi.org/10.1371/journal.pgen.1003977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandrasekar G, Vesterlund L, Hultenby K, Tapia-Paez I, Kere J. The zebrafish orthologue of the dyslexia candidate gene DYX1C1 is essential for cilia growth and function. PLoS One. 2013;8(5):e63123. doi: 10.1371/journal.pone.0063123. http://dx.doi.org/10.1371/journal.pone.0063123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cingolani P, Platts A, Wang le L, Coon M, Nguyen T, Wang L, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012;6(2):80–92. doi: 10.4161/fly.19695. http://dx.doi.org/10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole DG, Diener DR, Himelblau AL, Beech PL, Fuster JC, Rosenbaum JL. Chlamydomonas kinesin-II-dependent intraflagellar transport (IFT): IFT particles contain proteins required for ciliay assembly in Caenorhabditis elegans sensory neurons. Journal of Cell Biology. 1998;141(4):993–1008. doi: 10.1083/jcb.141.4.993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deane JA, Cole DG, Seeley ES, Diener DR, Rosenbaum JL. Localization of intraflagellar transport protein IFT52 identifies basal body transitional fibers as the docking site for IFT particles. Current Biology. 2001;11(20):1586–1590. doi: 10.1016/s0960-9822(01)00484-5. [DOI] [PubMed] [Google Scholar]
- Dixon SJ, Fedyshyn Y, Koh JL, Prasad TS, Chahwan C, Chua G, Boone C. Significant conservation of synthetic lethal genetic interaction networks between distantly related eukaryotes. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(43):16653–16658. doi: 10.1073/pnas.0806261105. http://dx.doi.org/10.1073/pnas.0806261105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duquesnoy P, Escudier E, Vincensini L, Freshour J, Bridoux AM, Coste A, Amselem S. Loss-of-function mutations in the human ortholog of Chlamydomonas reinhardtii ODA7 disrupt dynein arm assembly and cause primary ciliary dyskinesia. American Journal of Human Genetics. 2009;85(6):890–896. doi: 10.1016/j.ajhg.2009.11.008. http://dx.doi.org/10.1016/j.ajhg.2009.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutcher SK. Mating and tetrad analysis in Chlamydomonas reinhardtii. Methods in Cell Biology. 1995;47:531–540. doi: 10.1016/s0091-679x(08)60857-2. [DOI] [PubMed] [Google Scholar]
- Dutcher SK. The awesome power of dikaryons for studying flagella and basal bodies in Chlamydomonas reinhardtii. Cytoskeleton (Hoboken) 2014;71(2):79–94. doi: 10.1002/cm.21157. http://dx.doi.org/10.1002/cm.21157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutcher SK, Li L, Lin H, Meyer L, Giddings TH, Jr, Kwan AL, et al. Whole-genome sequencing to identify mutants and polymorphisms in Chlamydomonas reinhardtii. G3 (Bethesda) 2012;2(1):15–22. doi: 10.1534/g3.111.000919. http://dx.doi.org/10.1534/g3.111.000919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutcher SK, Morrissette NS, Preble AM, Rackley C, Stanga J. Epsilon-tubulin is an essential component of the centriole. Molecular Biology of the Cell. 2002;13(11):3859–3869. doi: 10.1091/mbc.E02-04-0205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutcher SK, Trabuco EC. The UNI3 gene is required for assembly of basal bodies of Chlamydomonas and encodes delta-tubulin, a new member of the tubulin superfamily. Molecular Biology of the Cell. 1998;9(6):1293–1308. doi: 10.1091/mbc.9.6.1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elam CA, Sale WS, Wirschell M. The regulation of dynein-driven microtubule sliding in Chlamydomonas flagella by axonemal kinases and phosphatases. Methods in Cell Biology. 2009;92:133–151. doi: 10.1016/S0091-679X(08)92009-4. http://dx.doi.org/10.1016/S0091-679X(08)92009-4. [DOI] [PubMed] [Google Scholar]
- Engel BD, Ishikawa H, Wemmer KA, Geimer S, Wakabayashi K, Hirono M, et al. The role of retrograde intraflagellar transport in flagellar assembly, maintenance, and function. Journal of Cell Biology. 2012;199(1):151–167. doi: 10.1083/jcb.201206068. http://dx.doi.org/10.1083/jcb.201206068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ermilova EV, Nikitin MM, Fernandez E. Chemotaxis to ammonium/methylammonium in Chlamydomonas reinhardtii: the role of transport systems for ammonium/methylammonium. Planta. 2007;226(5):1323–1332. doi: 10.1007/s00425-007-0568-1. http://dx.doi.org/10.1007/s00425-007-0568-1. [DOI] [PubMed] [Google Scholar]
- Esparza JM, O’Toole E, Li L, Giddings TH, Jr, Kozak B, Albee AJ, et al. Katanin localization requires triplet microtubules in Chlamydomonas reinhardtii. PLoS One. 2013;8(1):e53940. doi: 10.1371/journal.pone.0053940. http://dx.doi.org/10.1371/journal.pone.0053940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan ZC, Behal RH, Geimer S, Wang Z, Williamson SM, Zhang H, et al. Chlamydomonas IFT70/CrDYF-1 is a core component of IFT particle complex B and is required for flagellar assembly. Molecular Biology of the Cell. 2010;21(15):2696–2706. doi: 10.1091/mbc.E10-03-0191. http://dx.doi.org/10.1091/mbc.E10-03-0191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Follit JA, Xu F, Keady BT, Pazour GJ. Characterization of mouse IFT complex B. Cell Motility and Cytoskeleton. 2009;66(8):457–468. doi: 10.1002/cm.20346. http://dx.doi.org/10.1002/cm.20346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freshour J, Yokoyama R, Mitchell DR. Chlamydomonas flagellar outer row dynein assembly protein ODA7 interacts with both outer row and I1 inner row dyneins. Journal of Biological Chemistry. 2007;282(8):5404–5412. doi: 10.1074/jbc.M607509200. http://dx.doi.org/10.1074/jbc.M607509200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fromherz S, Giddings TH, Jr, Gomez-Ospina N, Dutcher SK. Mutations in alpha-tubulin promote basal body maturation and flagellar assembly in the absence of delta-tubulin. Journal of Cell Science. 2004;117(Pt 2):303–314. doi: 10.1242/jcs.00859. [DOI] [PubMed] [Google Scholar]
- Goodenough UW. Tipping of flagellar agglutinins by gametes of Chlamydomonas reinhardtii. Cell Motility and Cytoskeleton. 1993;25(2):179–189. doi: 10.1002/cm.970250207. [DOI] [PubMed] [Google Scholar]
- Goodenough UW, Adair WS, Caligor E, Forest CL, Hoffman JL, Mesland DA, et al. Membrane-membrane and membrane-ligand interactions in Chlamydomonas mating. Society of General Physiologists Series. 1980;34:131–152. [PubMed] [Google Scholar]
- Goodenough UW, Adair WS, Collin-Osdoby P, Heuser JE. Structure of the Chlamydomonas agglutinin and related flagellar surface proteins in vitro and in situ. Journal of Cell Biology. 1985;101(3):924–941. doi: 10.1083/jcb.101.3.924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodenough UW, Hwang C, Martin H. Isolation and genetic analysis of mutant strains of Chlamydomonas reinhardtii defective in gametic differentiation. Genetics. 1976;82(2):169–186. doi: 10.1093/genetics/82.2.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodenough UW, St Clair HS. BALD-2: a mutation affecting the formation of doublet and triplet sets of microtubules in Chlamydomonas reinhardtii. Journal of Cell Biology. 1975;66:480–491. doi: 10.1083/jcb.66.3.480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodenough UW, StClair HS. BALD-2: a mutation affecting the formation of doublet and triplet sets of microtubules in Chlamydomonas reinhardtii. Journal of Cell Biology. 1975;66(3):480–491. doi: 10.1083/jcb.66.3.480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross CH, Ranum LP, Lefebvre PA. Extensive restriction fragment length polymorphisms in a new isolate of Chlamydomonas reinhardtii. Current Genetics. 1988;13(6):503–508. doi: 10.1007/BF02427756. [DOI] [PubMed] [Google Scholar]
- Heuser T, Barber CF, Lin J, Krell J, Rebesco M, Porter ME, et al. Cryoelectron tomography reveals doublet-specific structures and unique interactions in the I1 dynein. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(30):E2067–E2076. doi: 10.1073/pnas.1120690109. http://dx.doi.org/10.1073/pnas.1120690109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuser T, Dymek EE, Lin J, Smith EF, Nicastro D. The CSC connects three major axonemal complexes involved in dynein regulation. Molecular Biology of the Cell. 2012;23(16):3143–3155. doi: 10.1091/mbc.E12-05-0357. http://dx.doi.org/10.1091/mbc.E12-05-0357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiraki M, Nakazawa Y, Kamiya R, Hirono M. Bld10p constitutes the cartwheel-spoke tip and stabilizes the 9-fold symmetry of the centriole. Current Biology. 2007;17(20):1778–1783. doi: 10.1016/j.cub.2007.09.021. S0960-9822(07)01980-X, [pii] [DOI] [PubMed] [Google Scholar]
- Hirschberg R, Stavis R. Phototaxis mutants of Chlamydomonas reinhardtii. Journal of Bacteriology. 1977;129(2):803–808. doi: 10.1128/jb.129.2.803-808.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoh RA, Stowe TR, Turk E, Stearns T. Transcriptional program of ciliated epithelial cells reveals new cilium and centrosome components and links to human disease. PLoS One. 2012;7(12):e52166. doi: 10.1371/journal.pone.0052166. http://dx.doi.org/10.1371/journal.pone.0052166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horani A, Druley TE, Zariwala MA, Patel AC, Levinson BT, Van Arendonk LG, et al. Whole-exome capture and sequencing identifies HEATR2 mutation as a cause of primary ciliary dyskinesia. American Journal of Human Genetics. 2012;91(4):685–693. doi: 10.1016/j.ajhg.2012.08.022. http://dx.doi.org/10.1016/j.ajhg.2012.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horani A, Ferkol TW, Shoseyov D, Wasserman MG, Oren YS, Kerem B, et al. LRRC6 mutation causes primary ciliary dyskinesia with dynein arm defects. PLoS One. 2013;8(3):e59436. doi: 10.1371/journal.pone.0059436. http://dx.doi.org/10.1371/journal.pone.0059436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou Y, Pazour GJ, Witman GB. A dynein light intermediate chain, D1bLIC, is required for retrograde intraflagellar transport. Molecular Biology of the Cell. 2004;15(10):4382–4394. doi: 10.1091/mbc.E04-05-0377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou Y, Qin H, Follit JA, Pazour GJ, Rosenbaum JL, Witman GB. Functional analysis of an individual IFT protein: IFT46 is required for transport of outer dynein arms into flagella. Journal of Cell Biology. 2007;176(5):653–665. doi: 10.1083/jcb.200608041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang B, Piperno G, Ramanis Z, Luck DJ. Radial spokes of Chlamydomonas flagella: genetic analysis of assembly and function. Journal of Cell Biology. 1981;88(1):80–88. doi: 10.1083/jcb.88.1.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang B, Ramanis Z, Dutcher SK, Luck DJ. Uniflagellar mutants of Chlamydomonas: evidence for the role of basal bodies in transmission of positional information. Cell. 1982;29(3):745–753. doi: 10.1016/0092-8674(82)90436-6. [DOI] [PubMed] [Google Scholar]
- Huang B, Rifkin MR, Luck DJ. Temperature-sensitive mutations affecting flagellar assembly and function in Chlamydomonas reinhardtii. Journal of Cell Biology. 1977;72(1):67–85. doi: 10.1083/jcb.72.1.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iomini C, Babaev-Khaimov V, Sassaroli M, Piperno G. Protein particles in Chlamydomonas flagella undergo a transport cycle consisting of four phases. Journal of Cell Biology. 2001;153(1):13–24. doi: 10.1083/jcb.153.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iomini C, Li L, Esparza JM, Dutcher SK. Retrograde intraflagellar transport mutants identify complex A proteins with multiple genetic interactions in Chlamydomonas reinhardtii. Genetics. 2009;183(3):885–896. doi: 10.1534/genetics.109.101915. genetics.109.101915, [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iomini C, Li L, Mo W, Dutcher SK, Piperno G. Two flagellar genes, AGG2 and AGG3, mediate orientation to light in Chlamydomonas. Current Biology. 2006;16(11):1147–1153. doi: 10.1016/j.cub.2006.04.035. [DOI] [PubMed] [Google Scholar]
- Ishikawa H, Ide T, Yagi T, Jiang X, Hirono M, Sasaki H, Marshall WF. TTC26/DYF13 is an intraflagellar transport protein required for transport of motility-related proteins into flagella. Elife. 2014;3:e01566. doi: 10.7554/eLife.01566. http://dx.doi.org/10.7554/eLife.01566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa H, Marshall WF. Ciliogenesis: building the cell’s antenna. Nature Reviews Molecular Cell Biology. 2011;12(4):222–234. doi: 10.1038/nrm3085. http://dx.doi.org/10.1038/nrm3085. [DOI] [PubMed] [Google Scholar]
- Jakobsen L, Vanselow K, Skogs M, Toyoda Y, Lundberg E, Poser I, et al. Novel asymmetrically localizing components of human centrosomes identified by complementary proteomics methods. EMBO Journal. 2011;30(8):1520–1535. doi: 10.1038/emboj.2011.63. http://dx.doi.org/10.1038/emboj.2011.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller LC, Romijn EP, Zamora I, Yates JR, 3rd, Marshall WF. Proteomic analysis of isolated Chlamydomonas centrioles reveals orthologs of ciliary-disease genes. Current Biology. 2005;15(12):1090–1098. doi: 10.1016/j.cub.2005.05.024. http://dx.doi.org/10.1016/j.cub.2005.05.024. [DOI] [PubMed] [Google Scholar]
- Kilburn CL, Pearson CG, Romijn EP, Meehl JB, Giddings TH, Jr, Culver BP, et al. New Tetrahymena basal body protein components identify basal body domain structure. Journal of Cell Biology. 2007;178(6):905–912. doi: 10.1083/jcb.200703109. http://dx.doi.org/10.1083/jcb.200703109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kott E, Duquesnoy P, Copin B, Legendre M, Dastot-Le Moal F, Montantin G, et al. Loss-of-function mutations in LRRC6, a gene essential for proper axonemal assembly of inner and outer dynein arms, cause primary ciliary dyskinesia. American Journal of Human Genetics. 2012;91(5):958–964. doi: 10.1016/j.ajhg.2012.10.003. http://dx.doi.org/10.1016/j.ajhg.2012.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefebvre PA, Rosenbaum JL. Regulation of the synthesis and assembly of ciliary and flagellar proteins during regeneration. Annual Review of Cell Biology. 1986;2:517–546. doi: 10.1146/annurev.cb.02.110186.002505. http://dx.doi.org/10.1146/annurev.cb.02.110186.002505. [DOI] [PubMed] [Google Scholar]
- Li S, Fernandez JJ, Marshall WF, Agard DA. Three-dimensional structure of basal body triplet revealed by electron cryo-tomography. EMBO Journal. 2012;31(3):552–562. doi: 10.1038/emboj.2011.460. http://dx.doi.org/10.1038/emboj.2011.460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JB, Gerdes JM, Haycraft CJ, Fan Y, Teslovich TM, May-Simera H, et al. Comparative genomics identifies a flagellar and basal body proteome that includes the BBS5 human disease gene. Cell. 2004;117(4):541–552. doi: 10.1016/s0092-8674(04)00450-7. [DOI] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N Genome Project data processing. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25(16):2078–2079. doi: 10.1093/bioinformatics/btp352. http://dx.doi.org/10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin H, Kwan AL, Dutcher SK. Synthesizing and salvaging NAD: lessons learned from Chlamydomonas reinhardtii. PLoS Genetics. 2010;6(9):e1001105. doi: 10.1371/journal.pgen.1001105. http://dx.doi.org/10.1371/journal.pgen.1001105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin H, Miller ML, Granas DM, Dutcher SK. Whole genome sequencing identifies a deletion in protein phosphatase 2A that affects its stability and localization in Chlamydomonas reinhardtii. PLoS Genetics. 2013;9(9):e1003841. doi: 10.1371/journal.pgen.1003841. http://dx.doi.org/10.1371/journal.pgen.1003841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin H, Nauman NP, Albee AJ, Hsu S, Dutcher SK. New mutations in flagellar motors identified from whole genome sequencing in Chlamydomonas reinhardtii. Cilia. 2013;2:10. doi: 10.1186/2046-2530-2-14. 1186/2046-2530-1182-1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JB, Zhang M, Dutcher SK, Stormo GD. Procom: a web-based tool to compare multiple eukaryotic proteomes. Bioinformatics. 2005;21(8):1693–1694. doi: 10.1093/bioinformatics/bti161. http://dx.doi.org/10.1093/bioinformatics/bti161. [DOI] [PubMed] [Google Scholar]
- Lucker BF, Behal RH, Qin H, Siron LC, Taggart WD, Rosenbaum JL, et al. Characterization of the intraflagellar transport complex B core: direct interaction of the IFT81 and IFT74/72 subunits. Journal of Biological Chemistry. 2005;280(30):27688–27696. doi: 10.1074/jbc.M505062200. [DOI] [PubMed] [Google Scholar]
- Lucker BF, Miller MS, Dziedzic SA, Blackmarr PT, Cole DG. Direct interactions of intraflagellar transport complex B proteins IFT88, IFT52, and IFT46. Journal of Biological Chemistry. 2010;285(28):21508–21518. doi: 10.1074/jbc.M110.106997. http://dx.doi.org/10.1074/jbc.M110.106997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lux FG, 3rd, Dutcher SK. Genetic interactions at the FLA10 locus: suppressors and synthetic phenotypes that affect the cell cycle and flagellar function in Chlamydomonas reinhardtii. Genetics. 1991;128(3):549–561. doi: 10.1093/genetics/128.3.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuura K, Lefebvre PA, Kamiya R, Hirono M. Kinesin-II is not essential for mitosis and cell growth in Chlamydomonas. Cell Motility and Cytoskeleton. 2002;52(4):195–201. doi: 10.1002/cm.10051. [DOI] [PubMed] [Google Scholar]
- Matsuura K, Lefebvre PA, Kamiya R, Hirono M. Bld10p, a novel protein essential for basal body assembly in Chlamydomonas: localization to the cartwheel, the first ninefold symmetrical structure appearing during assembly. Journal of Cell Biology. 2004;165(5):663–671. doi: 10.1083/jcb.200402022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McVittie A. Studies on flagella-less, stumpy, and short flagellum mutants of Chlamydomonas reinhardii. Proceedings of the Royal Society of London B: Biological Sciences. 1969;173(30):59–60. doi: 10.1098/rspb.1969.0036. [DOI] [PubMed] [Google Scholar]
- McVittie A. Flagellum mutants of Chlamydomonas reinhardii. Journal of General Microbiology. 1972;71(3):525–540. doi: 10.1099/00221287-71-3-525. [DOI] [PubMed] [Google Scholar]
- Miller MS, Esparza JM, Lippa AM, Lux FG, 3rd, Cole DG, Dutcher SK. Mutant kinesin-2 motor subunits increase chromosome loss. Molecular Biological Cell. 2005;16(8):3810–3820. doi: 10.1091/mbc.E05-05-0404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchison HM, Schmidts M, Loges NT, Freshour J, Dritsoula A, Hirst RA, et al. Mutations in axonemal dynein assembly factor DNAAF3 cause primary ciliary dyskinesia. Nature Genetics. 2012;44(4):381–389. doi: 10.1038/ng.1106. http://dx.doi.org/10.1038/ng.1106. S381–S382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molnar A, Bassett A, Thuenemann E, Schwach F, Karkare S, Ossowski S, et al. Highly specific gene silencing by artificial microRNAs in the unicellular alga Chlamydomonas reinhardtii. Plant Journal. 2009;58(1):165–174. doi: 10.1111/j.1365-313X.2008.03767.x. http://dx.doi.org/10.1111/j.1365-313X.2008.03767.x. [DOI] [PubMed] [Google Scholar]
- Mueller J, Perrone CA, Bower R, Cole DG, Porter ME. The FLA3 KAP subunit is required for localization of kinesin-2 to the site of flagellar assembly and processive anterograde intraflagellar transport. Molecular Biological Cell. 2005;16(3):1341–1354. doi: 10.1091/mbc.E04-10-0931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller H, Schmidt D, Steinbrink S, Mirgorodskaya E, Lehmann V, Habermann K, et al. Proteomic and functional analysis of the mitotic Drosophila centrosome. EMBO Journal. 2010;29(19):3344–3357. doi: 10.1038/emboj.2010.210. http://dx.doi.org/10.1038/emboj.2010.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakazawa Y, Hiraki M, Kamiya R, Hirono M. SAS-6 is a cartwheel protein that establishes the 9-fold symmetry of the centriole. Current Biology. 2007;17(24):2169–2174. doi: 10.1016/j.cub.2007.11.046. S0960-9822(07)02285-3, [pii] [DOI] [PubMed] [Google Scholar]
- Neff MM, Turk E, Kalishman M. Web-based primer design for single nucleotide polymorphism analysis. Trends in Genetics. 2002;18(12):613–615. doi: 10.1016/s0168-9525(02)02820-2. [DOI] [PubMed] [Google Scholar]
- Nishimura DY, Swiderski RE, Searby CC, Berg EM, Ferguson AL, Hennekam R, et al. Comparative genomics and gene expression analysis identifies BBS9, a new Bardet-Biedl syndrome gene. American Journal of Human Genetics. 2005;77(6):1021–1033. doi: 10.1086/498323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Toole ET, Giddings TH, Jr, Dutcher SK. Understanding microtubule organizing centers by comparing mutant and wild-type structures with electron tomography. Methods in Cell Biology. 2007;79:125–143. doi: 10.1016/S0091-679X(06)79005-7. S0091-679X(06)79005-7, [pii] [DOI] [PubMed] [Google Scholar]
- Omran H, Kobayashi D, Olbrich H, Tsukahara T, Loges NT, Hagiwara H, et al. Ktu/PF13 is required for cytoplasmic pre-assembly of axonemal dyneins. Nature. 2008;456(7222):611–616. doi: 10.1038/nature07471. http://dx.doi.org/10.1038/nature07471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrowski LE, Blackburn K, Radde KM, Moyer MB, Schlatzer DM, Moseley A, et al. A proteomic analysis of human cilia: identification of novel components. Molecular Cell Proteomics. 2002;1(6):451–465. doi: 10.1074/mcp.m200037-mcp200. [DOI] [PubMed] [Google Scholar]
- Pasquale SM, Goodenough UW. Cyclic AMP functions as a primary sexual signal in gametes of Chlamydomonas reinhardtii. Journal of Cell Biology. 1987;105(5):2279–2292. doi: 10.1083/jcb.105.5.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pazour GJ, Agrin N, Leszyk J, Witman GB. Proteomic analysis of a eukary-otic cilium. Journal of Cell Biology. 2005;170(1):103–113. doi: 10.1083/jcb.200504008. http://dx.doi.org/10.1083/jcb.200504008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pazour GJ, Dickert BL, Vucica Y, Seeley ES, Rosenbaum JL, Witman GB, et al. Chlamydomonas IFT88 and its mouse homologue, polycystic kidney disease gene tg737, are required for assembly of cilia and flagella. Journal of Cell Biology. 2000;151(3):709–718. doi: 10.1083/jcb.151.3.709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pazour GJ, Dickert BL, Witman GB. The DHC1b (DHC2) isoform of cytoplasmic dynein is required for flagellar assembly. Journal of Cell Biology. 1999;144(3):473–481. doi: 10.1083/jcb.144.3.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pazour GJ, Wilkerson CG, Witman GB. A dynein light chain is essential for the retrograde particle movement of intraflagellar transport (IFT) Journal of Cell Biology. 1998;141(4):979–992. doi: 10.1083/jcb.141.4.979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pazour GJ, Witman GB. Forward and reverse genetic analysis of microtubule motors in Chlamydomonas. Methods. 2000;22(4):285–298. doi: 10.1006/meth.2000.1081. http://dx.doi.org/10.1006/meth.2000.1081. [DOI] [PubMed] [Google Scholar]
- Pedersen LB, Miller MS, Geimer S, Leitch JM, Rosenbaum JL, Cole DG. Chlamydomonas IFT172 is encoded by FLA11, interacts with CrEB1, and regulates IFT at the flagellar tip. Current Biology. 2005;15(3):262–266. doi: 10.1016/j.cub.2005.01.037. [DOI] [PubMed] [Google Scholar]
- Perrone CA, Tritschler D, Taulman P, Bower R, Yoder BK, Porter ME. A novel dynein light intermediate chain colocalizes with the retrograde motor for intraflagellar transport at sites of axoneme assembly in Chlamydomonas and Mammalian cells. Molecular Biology of the Cell. 2003;14(5):2041–2056. doi: 10.1091/mbc.E02-10-0682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piasecki BP, LaVoie M, Tam LW, Lefebvre PA, Silflow CD. The Uni2 phosphoprotein is a cell cycle regulated component of the basal body maturation pathway in Chlamydomonas reinhardtii. Molecular Biology of the Cell. 2008;19(1):262–273. doi: 10.1091/mbc.E07-08-0798. http://dx.doi.org/10.1091/mbc.E07-08-0798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piasecki BP, Silflow CD. The UNI1 and UNI2 genes function in the transition of triplet to doublet microtubules between the centriole and cilium in Chlamydomonas. Molecular Biology of the Cell. 2009;20(1):368–378. doi: 10.1091/mbc.E08-09-0900. http://dx.doi.org/10.1091/mbc.E08-09-0900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pigino G, Geimer S, Lanzavecchia S, Paccagnini E, Cantele F, Diener DR, et al. Electron-tomographic analysis of intraflagellar transport particle trains in situ. Journal of Cell Biology. 2009;187(1):135–148. doi: 10.1083/jcb.200905103. http://dx.doi.org/10.1083/jcb.200905103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piperno G, Mead K. Transport of a novel complex in the cytoplasmic matrix of Chlamydomonas flagella. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(9):4457–4462. doi: 10.1073/pnas.94.9.4457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter ME, Bower R, Knott JA, Byrd P, Dentler W. Cytoplasmic dynein heavy chain 1b is required for flagellar assembly in Chlamydomonas. Molecular Biology of the Cell. 1999;10(3):693–712. doi: 10.1091/mbc.10.3.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin H, Diener DR, Geimer S, Cole DG, Rosenbaum JL. Intraflagellar transport (IFT) cargo: IFT transports flagellar precursors to the tip and turnover products to the cell body. Journal of Cell Biology. 2004;164(2):255–266. doi: 10.1083/jcb.200308132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiter JF, Blacque OE, Leroux MR. The base of the cilium: roles for transition fibres and the transition zone in ciliary formation, maintenance and compartmentalization. EMBO Reports. 2012;13(7):608–618. doi: 10.1038/embor.2012.73. http://dx.doi.org/10.1038/embor.2012.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rymarquis LA, Handley JM, Thomas M, Stern DB. Beyond complementation. Map-based cloning in Chlamydomonas reinhardtii. Plant Physiology. 2005;137(2):557–566. doi: 10.1104/pp.104.054221. pp.104.054221, [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimogawara K, Fujiwara S, Grossman A, Usuda H. High-efficiency transformation of Chlamydomonas reinhardtii by electroporation. Genetics. 1998;148(4):1821–1828. doi: 10.1093/genetics/148.4.1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silflow CD, Kathir P, Lefebvre PA. Molecular mapping of genes for flagellar proteins in Chlamydomonas. Methods in Cell Biology. 1995;47:525–530. doi: 10.1016/s0091-679x(08)60856-0. [DOI] [PubMed] [Google Scholar]
- Silflow CD, Sun X, Haas NA, Foley JW, Lefebvre PA. The Hsp70 and Hsp40 chaperones influence microtubule stability in Chlamydomonas. Genetics. 2011;189(4):1249–1260. doi: 10.1534/genetics.111.133587. http://dx.doi.org/10.1534/genetics.111.133587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolc V, Samanta MP, Tongprasit W, Marshall WF. Genome-wide transcriptional analysis of flagellar regeneration in Chlamydomonas reinhardtii identifies orthologs of ciliary disease genes. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(10):3703–3707. doi: 10.1073/pnas.0408358102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam LW, Lefebvre PA. Cloning of flagellar genes in Chlamydomonas reinhardtii by DNA insertional mutagenesis. Genetics. 1993;135(2):375–384. doi: 10.1093/genetics/135.2.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarkar A, Loges NT, Slagle CE, Francis R, Dougherty GW, Tamayo JV, et al. DYX1C1 is required for axonemal dynein assembly and ciliary motility. Nature Genetics. 2013;45(9):995–1003. doi: 10.1038/ng.2707. http://dx.doi.org/10.1038/ng.2707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taschner M, Bhogaraju S, Lorentzen E. Architecture and function of IFT complex proteins in ciliogenesis. Differentiation. 2012;83(2):S12–S22. doi: 10.1016/j.diff.2011.11.001. http://dx.doi.org/10.1016/j.diff.2011.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taschner M, Bhogaraju S, Vetter M, Morawetz M, Lorentzen E. Biochemical mapping of interactions within the intraflagellar transport (IFT) B core complex: IFT52 binds directly to four other IFT-B subunits. Journal of Biological Chemistry. 2011;286(30):26344–26352. doi: 10.1074/jbc.M111.254920. http://dx.doi.org/10.1074/jbc.M111.254920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25(9):1105–1111. doi: 10.1093/bioinformatics/btp120. http://dx.doi.org/10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature Biotechnology. 2010;28(5):511–515. doi: 10.1038/nbt.1621. http://dx.doi.org/10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincze T, Posfai J, Roberts RJ. NEBcutter: a program to cleave DNA with restriction enzymes. Nucleic Acids Research. 2003;31(13):3688–3691. doi: 10.1093/nar/gkg526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakabayashi K, Misawa Y, Mochiji S, Kamiya R. Reduction-oxidation poise regulates the sign of phototaxis in Chlamydomonas reinhardtii. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(27):11280–11284. doi: 10.1073/pnas.1100592108. http://dx.doi.org/10.1073/pnas.1100592108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walther Z, Vashishtha M, Hall JL. The Chlamydomonas FLA10 gene encodes a novel kinesin-homologous protein. Journal of Cell Biology. 1994;126(1):175–188. doi: 10.1083/jcb.126.1.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Fan ZC, Williamson SM, Qin H. Intraflagellar transport (IFT) protein IFT25 is a phosphoprotein component of IFT complex B and physically interacts with IFT27 in Chlamydomonas. PLoS One. 2009;4(5):e5384. doi: 10.1371/journal.pone.0005384. http://dx.doi.org/10.1371/journal.pone.0005384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson SM, Silva DA, Richey E, Qin H. Probing the role of IFT particle complex A and B in flagellar entry and exit of IFT-dynein in Chlamydomonas. Protoplasma. 2012;249(3):851–856. doi: 10.1007/s00709-011-0311-4. http://dx.doi.org/10.1007/s00709-011-0311-4. [DOI] [PubMed] [Google Scholar]
- Zariwala MA, Gee HY, Kurkowiak M, Al-Mutairi DA, Leigh MW, Hurd TW, et al. ZMYND10 is mutated in primary ciliary dyskinesia and interacts with LRRC6. American Journal of Human Genetics. 2013;93(2):336–345. doi: 10.1016/j.ajhg.2013.06.007. http://dx.doi.org/10.1016/j.ajhg.2013.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, Patena W, Armbruster U, Gang SS, Blum SR, Jonikas MC. High-throughput genotyping of green algal mutants reveals random distribution of mutagenic insertion sites and endonucleolytic cleavage of transforming DNA. Plant Cell. 2014;26(4):1398–1409. doi: 10.1105/tpc.114.124099. http://dx.doi.org/10.1105/tpc.114.124099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L, Yuan S, Cao Y, Kallakuri S, Li Y, Kishimoto N, et al. Reptin/Ruvbl2 is a Lrrc6/Seahorse interactor essential for cilia motility. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(31):12697–12702. doi: 10.1073/pnas.1300968110. http://dx.doi.org/10.1073/pnas.1300968110. [DOI] [PMC free article] [PubMed] [Google Scholar]