

Abstract
Background
ELND005 (scyllo-Inositol; cyclohexane-1,2,3,4,5,6-hexol) has been evaluated as a potential disease-modifying treatment for Alzheimer’s disease (AD). Individuals with Down syndrome (DS) have an increased risk for developing AD dementia.
Objective
To evaluate the safety and tolerability of ELND005 and to determine its pharmacokinetics (PK) and relationship between PK parameters, safety outcome measures, and exploratory efficacy outcome measures in young adults with DS without dementia.
Methods
This was a prospective, randomized, double-blind, placebo-controlled, parallel-group, three-arm, multicenter Phase 2 study of the safety and pharmacokinetics of ELND005 administered orally for 4 weeks (ClinicalTrials.gov NCT01791725). Participants who met study eligibility criteria were randomly assigned in a 2:1:1 ratio to receive ELND005 at either 250 mg twice daily (BID) or 250 mg once daily (QD) or matching placebo for 4 weeks.
Results
There were no apparent treatment group-related trends on cognitive or behavioral measures and there were no SAEs and no deaths in the study. Overall, mean changes from baseline in clinical laboratory parameters, vital sign measurements, electrocardiogram (ECG) results, and other physical findings were unremarkable. ELND005 accumulation averaged approximately 2-fold with QD dosing, and 3- to 4-fold with BID dosing.
Conclusion
Overall, treatment of adults with DS with ELND005 at both doses was well tolerated, achieved measurable blood levels and demonstrated no safety findings. Further studies will be needed to test efficacy.
Keywords: Alzheimer’s disease, Down syndrome, Dementia, Amyloid, Myo-inositol
INTRODUCTION
Down syndrome (DS), due to trisomy 21, is one of the most common genetic disorders, with a prevalence rate of approximately 12.6 per 10,000, or 1 in 792 live births in the United States [1]. Persons with DS exhibit a variety of well-characterized physical and behavioral conditions during development that persist throughout life including phenotypic features [2, 3] as well as impaired cognitive function ranging from mild to profound in severity [4, 5]. Of particular interest, triplication of chromosome 21 results in life-long overproduction of amyloid-β and consistently high levels of brain myo-inositol. Overproduction of amyloid-β leads to the early development of AD pathology in DS [6]. Individuals with DS develop the same biomarker changes as in sporadic AD [7, 8]. Amyloid-β oligomers are thought to lead to neuronal toxicity and synaptic dysfunction even before the appearance of amyloid plaques [9]. Another contributory factor to dementia in DS is thought to involve abnormalities in myo-inositol metabolism as the serum sodium myo-inositol co-transporter gene is localized to chromosome 21 [10]. High levels of brain myo-inositol, in both children and adults with DS correlates with the severity of cognitive dysfunction [11, 12]. Elevated myo-inositol levels are also thought to be associated with abnormal neuronal signaling mechanisms leading to abnormal synaptic activity and cortical network dysfunction [13].
ELND005 (scyllo-Inositol), an endogenous myo-inositol isomer, is a novel compound in clinical development that could reduce amyloid toxicity and regulate myo-inositiol levels to improve function in patients with DS. In preclinical studies, ELND005 has shown amyloid anti-aggregation effects in vitro, protective effects on oligomer-induced neuronal toxicity, and positive effects on learning in animal models of AD [14-16]. ELND005 has also shown both amyloid and myo-inositol lowering effects in cerebrospinal fluid (CSF) and brain respectively in patients with AD [17]. In a previous phase II study of ELND005 in sporadic AD [17], there were four treatment arms: placebo or ELND005 (250, 1,000, or 2,000 mg) administered orally BID. Based on the 48-week safety review, patients in the 1,000 and 2,000 mg groups were withdrawn. Specifically, at the higher doses, there was an increased risk of respiratory infections. The incidence of withdrawals due to AE was higher in the 1,000 mg (16.9%) and 2,000 mg (13.2%) groups than in the 250 mg (10.2%) and placebo (9.6%) groups. The incidence of SAEs was also higher in the ELND005 groups compared with placebo (23.1%, 22.5%, 21.6%, and 13.3%, in the 2,000 mg, 1,000 mg, 250 mg, and placebo groups, respectively). The incidence of respiratory tract infections was higher in the 1,000 mg and 2,000 mg groups than in the placebo and 250 mg groups. Therefore, the 250 mg qd and 250 mg bid dose were selected due to their acceptable long-term safety and beneficial trends on cognition in mild AD within the general population [17]. In the current study, a chest x-ray was performed at screening to evaluate for any potential respiratory infections.
In addition, at the 250-mg bid dose of ELND005 in Phase II AD studies, there was an approximately 40% reduction in brain myo-inositol levels in participants who underwent brain magnetic resonance spectroscopy (MRS) [17]. The phosphoinositol (PI) cascade in neurons is an important signaling pathway in response to stimulation by neurotransmitters and growth factors. Despite the fact, that neuronal myo-inositol might enhance synaptic activity, the compound has not be tested in DS.
The mechanism of action of ELND005 directly affects both β-amyloid clearance and myo-inositol regulation to improve cognitive function. This study is a 4-week randomized, double-blind, placebo-controlled, safety and pharmacokinetic (PK) study of oral ELND005 in people with DS, ages 18-45, without dementia. Exploratory cognitive assessments were also included in this study to evaluate the ability of the people with DS to complete the tests and support their utility as cognitive measures in future trials involving people with DS.
There is a dearth of drugs approved for DS, with no consensus on optimal cognitive outcome measures for clinical trials. Thus far, the main approach to improving cognitive function in people with DS has been to evaluate drugs that have been in clinical trials for the treatment of AD. Cholinesterase inhibitors and memantine have not been successful in consistently improving cognitive function in either demented or non-demented people with DS [18-22]. Other studies of potential therapeutics, such as vitamin E, have not been successful in DS [23]. Hoffman La-Roche conducted a phase II trial of the GABAA α5 antagonist, RG1662 to improve cognition in people with DS (NCT02024789). That study did not meet its primary endpoints on improvement in cognition and function. AC Immune has recently launched a phase Ib placebo-controlled, multicenter study to evaluate the anti-amyloid-β vaccine ACI-24 in 24 adults with DS (NCT02738450). This current study aims to understand the safety and tolerability of ELND-005 in adults with DS.
MATERIALS AND METHODS
Participants
This study was approved by the human subjects review committees of the University of California at San Diego, Massachusetts General Hospital and the University of California at Irvine. Treatment groups were similar with regard to demographic and baseline characteristics (Table 1). All procedures involving experiments on human subjects were carried out in accord with the Helsinki Declaration of 1975. All participants signed informed consent in person, with the participation of a legally authorized representative. The overall mean age was 27.6 years (SD = 6.49 years), and the majority (90.9%) of participants were Caucasian. The modified intent to treat (mITT) analysis set included a total of 22 participants, 14 males and 8 females; 1 additional participant did not complete the study and was included only in the safety analysis (Figure 1). Both the placebo and the ELND005 - QD groups had an equal number of males and females, but there were more males (nine (75.0%) participants) than females (three (25.0%) participants) in the ELND005 - BID group. Overall, participants had a mean IQ score of 54.3 (median: 52.0). The mean IQ score in the ELND005 - QD group (63.3) was higher than in the ELND005 – BID (50.6) or placebo (55.8) group. Participants were exposed to study drug for a mean of 27.7 days (median: 29 days). Mean and median exposure to ELND005 in the BID group were 29.3 and 29.0 days, respectively, and in the QD group were 23.0 and 28.0 days, respectively; mean and median exposure to placebo were 28.3 and 29.0 days, respectively.
Table 1.
Demographic and Baseline Characteristics (mITT Set)
| Placebo (n = 6) |
ELND005 250 mg BID (n = 12) |
ELND005 250 mg QDa (n = 4) |
Total N = 22a |
|
|---|---|---|---|---|
| Gender (n (%)) | ||||
| Male | 3 (50.0) | 9 (75.0) | 2 (50.0) | 14 (63.6) |
| Female | 3 (50.0) | 3 (25.0) | 2 (50.0) | 8 (36.4) |
| Race (n (%)) | ||||
| Asian | 0 | 1 (8.3) | 0 | 1 (4.5) |
| Black or African American | 0 | 1 (8.3) | 0 | 1 (4.5) |
| White | 6 (100) | 10 (83.3) | 4 (100) | 20 (90.9) |
| Ethnicity (n (%)) | ||||
| Hispanic or Latino | 1 (16.7) | 1 (8.3) | 0 | 2 (9.1) |
| Not Hispanic or Latino | 5 (83.3) | 11 (91.7) | 4 (100) | 20 (90.9) |
| Age (Years) | ||||
| Mean (SD) | 30.0 (5.93) | 26.3 (7.43) | 27.8 (4.03) | 27.6 (6.49) |
| Median | 32.0 | 25.0 | 29.0 | 28.0 |
| Minimum, Maximum | 19, 35 | 18, 40 | 22, 31 | 18, 40 |
| IQ (K-BIT)Score | ||||
| Mean (SD) | 55.8 (6.65) | 50.6 (13.81) | 63.3 (19.62) | 54.3 (13.75) |
| Median | 54.0 | 46.0 | 66.5 | 52.0 |
| Minimum, Maximum | 48, 68 | 40, 79 | 40, 80 | 40, 80 |
Abbreviations: BID = twice daily; IQ = intelligence quotient; mITT = modified intent to treat; QD = once daily.to treat; QD = once daily.
The mITT Set does not include 1 participant, who was randomized to the ELND005 – QD group, but did not complete the study. This participant was an 18-year-old female with an IQ score of 67.
Figure 1. Participant Flow.
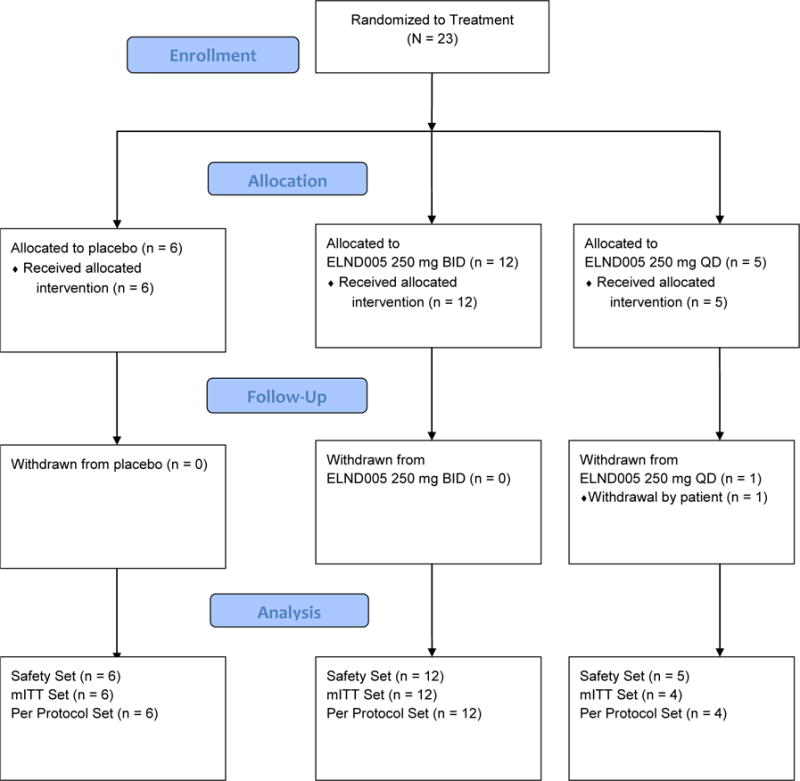
Abbreviations: BID = twice daily, mITT = modified intent to treat, QD = once daily
Study Procedures
Data was collected from participants from September 2013 to June 2014. The Screening/Baseline Period was up to 2 weeks in duration when participants underwent standard clinical tests for eligibility. Participants were assessed using the Kaufman Brief Intelligence Test – Second Edition (KBIT-II) and the Columbia-Suicide Severity Rating Scale (C-SSRS), in order to determine suitability for study and then underwent poster-anterior and lateral chest X-rays, ECG, and brain magnetic resonance imaging (MRI) at screening. Chest X-rays were included in the protocol as the incidence of respiratory tract infections was higher in the 1,000 mg and 2,000 mg groups than in the placebo and 250 mg groups in the prior AD study [17]. All efficacy instruments were administered to obtain baseline scores, and blood samples were obtained for baseline ELND005 plasma concentrations.
Participants who met study eligibility criteria were randomly assigned in a 2:1:1 ratio to receive ELND005 250 mg twice daily (BID), or ELND005 250 mg once daily (QD), or matching placebo for 4 weeks. The first dose of study drug was administered at the Baseline visit (Day 1). See Table 2 for schedule of events.
Table 2.
Schedule of Events
| Procedure | Screen/Baseline Period | Treatment Period | Unsch Visit | Follow-up Period Day 56 (± 5) |
|||||
|---|---|---|---|---|---|---|---|---|---|
| Screen | Baseline | ||||||||
| Visit | 1 | 2 | 3 | 4 | 5 | 6 | |||
| Day | Week -2 to Day -1 | 0 | 7 (± 2) |
14 (± 3) |
21 (± 3) |
28(± 3) (ET/EOS) |
|||
| Clinic Visit (C) or Telephone (T) Contact | C | C | T | C | T | C | C | C | |
| Obtain Informed Consent | X | ||||||||
| Eligibility Criteria | X | X | |||||||
| PA and Lateral Chest X- rays | X | ||||||||
| Brain MRI | X7 | X | |||||||
| Clinical Laboratory Tests | X | X | X | ||||||
| Urine Pregnancy Test | X | X | X | ||||||
| Blood Samples for ELND005 Concentrations | X | X | X | X | X | ||||
| Medical and Medication History | X | X | |||||||
| Physical and Neurological Examinations | X | X | X | ||||||
| 12 lead ECG | X | X | X | ||||||
| Vital Signs, Height, and Weight | X | X | X | X | X | X | |||
| Clinic Visit (C) or Telephone (T) Contact | C | C | T | C | T | C | C | C | |
| K-BIT | X | ||||||||
| C-SSRS | X | X | X | X | X | X | X | X | |
| CDR System, RADD-2, VABS-II, NPI. Block Design (WAIS-IV) | X | X | |||||||
| Treatment Randomization | X | ||||||||
| Dispense Study Drug | X | X | |||||||
| Concomitant Medication Review | X | X | X | X | X | X | X | X | |
| Drug Accountability | X | X | X | ||||||
| Study Drug Dosing | → | ||||||||
| Adverse Event Assessment16 |
X | X | X | X | X | X | X | X | |
Abbreviations: C = clinic visit; CDR = Cognitive Drug Research; C-SSRS = Columbia- Suicide Severity Rating Scale;; ECG = electrocardiogram; EOS = end of study; ET = early termination; MRI = magnetic resonance imaging; NPI = Neuropsychiatric Inventory, PA = posteroanterior; RADD-2 = Rapid Assessment for Developmental Disabilities, Second Edition; Vineland Adaptive Behavior Scales, Second Edition (VABS-II); Wechsler Adult Intelligence Scale IV (WAIS-IV);T = telephone contact; Unsch Visit = unscheduled visit.
During the 4-week Treatment Period, scheduled visits occurred on Days 14 and 28, and at the Early Termination (ET) visit, if applicable. Telephone contact/visits were conducted on Days 7 and 21. Concomitant medication review, Adverse Events assessment, and C-SSRS suicidality assessment were conducted at every participant visit or telephone contact; PK blood draws and additional safety assessments were performed on Days 14 and 28. Exploratory Efficacy instruments were administered on the Day 28 (or ET) visit.
Cognitive, Functional and Behavioral Efficacy Instruments
The Rapid Assessment for Developmental Disabilities – Second Edition (RADD-2) test was developed from the low-difficulty items from published intelligence tests [24] for evaluation of individuals with intellectual disabilities and developmental disabilities.
The Cognitive Drug Research (CDR) System is a computerized platform for testing cognition, including memory and attention that can be completed in approximately 20 minutes and is validated in 60 languages [25],[26].
The Block Design subtest of the Wechsler Adult Intelligence Scale IV (WAIS-IV) [27].
The Vineland Adaptive Behavior Scales, Second Edition (VABS-II) assessment tool is in the form of an interview-based questionnaire of functioning [28].
The Neuropsychiatric Inventory (NPI) [29] is a behavioral measure that assesses psychopathology in dementia patients.
A Safety Follow-Up visit was scheduled at 4 weeks after study completion or discontinuation of study drug for any reason; participants were evaluated for suicidality and a PK blood sample was collected.
Blood Collection
Peripheral venous blood samples were collected; assays for the concentration of ELND005 in plasma samples were performed at inVentiv Health Clinical Lab, Inc. (Princeton, NJ) and determined by a validated gas chromatography/tandem mass spectrometry (GC-MS/MS) method. The assay ranges were 0.4 to 80 μg/mL in plasma.
Blood samples were also collected at Baseline (Visit 2) and Day 28 End-of-Study (EOS) for ELND005 plasma levels. Samples were collected before dosing (time 0), and at 1.5 (±0.5), 3 (±0.5), and 4.5 (±0.5) hours after dosing. In addition, participants were encouraged to provide optional blood samples at 6 (±1) and 8 (±1) hours after dosing. At Day 14, one blood sample was collected, either before or after the first dose of study drug.
Neuroimaging
Brain MRI scans were performed at screening and included fluid-attenuated-inversion-recovery (FLAIR) and gradient echo sequences for the assessment of potential amyloid-related imaging abnormalities (ARIAs). All MRI scans were sent to a blinded central MRI processing and reading center for analysis, central safety review documented presence or absence of cerebral vasogenic edema (or ARIA-E) and incident microhemorrhages (or ARIA-H) [30].
Statistical Analysis
Approximately 24 participants were planned to be randomly assigned in a 2:1:1 ratio to ELND005 250 mg BID (12 participants), ELND005 250 mg QD (6 participants), or placebo (6 participants), respectively. Sample size was not determined based on statistical powering for efficacy endpoints and was considered acceptable for this type of safety and PK study. All study endpoints were summarized by treatment group using descriptive statistics. Categorical results (e.g., gender, race) were reported using frequency and percent. Continuous variables (e.g., age, weight) were reported using the number of participants, mean, SD, median, minimum, and maximum. Due to the small size of the study, only descriptive summary analyses were performed. No formal statistical testing was conducted, and therefore, no p-values reported. Because no statistical analyses were performed, there were no adjustments for covariates.
RESULTS
A total of 23 participants were randomly assigned and received treatment: 12 participants, ELND005 - BID; 5 participants, ELND005 - QD; and 6 participants, placebo. Participants were enrolled at 3 study sites in the United States. Of the 23 participants randomized, 22 (95.7%) completed the study per the protocol. One participant (4.3% overall; ELND005 - QD group) discontinued the study after a single dose of drug for the primary reason of “withdrawal by participant.” There were no major protocol deviations reported in this study.
Treatment Compliance
Overall, mean dosing compliance was 93.7% (SD = 20.31%). Mean (SD) dosing compliance was higher in the placebo (97.3% (1.75%)) and ELND005 - BID (98.0% (10.53%)) groups than in the ELND005 - QD group (79.0% (40.19%)), although the latter group also had more inter-participant variability in compliance rate than the other two groups. One participant in the ELND005 - QD group discontinued the study after a single dose of study drug, for the primary reason of “withdrawal by participant.”
Safety Evaluation
Twenty-three participants were exposed to study drug for a mean of 27.7 days (median, 29 days). Mean exposure to ELND005 in the BID group was 29.3 days, QD group 23.0 days, and placebo 28.3 days. All participants enrolled in the treatment arms received at least one dose of study drug and were included in analyses. ELND005 was well tolerated with no newly identified safety risks. Treatment-emergent AEs (TEAE) were experienced by more participants in the treatment groups than in the placebo group: five (41.7%) participants in the ELND005 250 mg BID group, two (40.0%) participants in the ELND005 250 mg QD group, and no participants in the placebo group (Table 3).
Table 3.
Overview of Adverse Events
| Placebo (n = 6) n (%) |
ELND005 250 mg BID (n = 12) n (%) |
ELND005 250 mg QD (n = 5) n (%) |
|
|---|---|---|---|
| Participants with | |||
| Any TEAE | 0 | 5 (41.7) | 2 (40.0) |
| Any drug-related TEAE | 0 | 1 (8.3) | 0 |
| Serious AEs | 0 | 0 | 0 |
| Serious drug-related AEs | 0 | 0 | 0 |
| Participants withdrawn due to an AE | |||
| TEAE | 0 | 0 | 1 (20.0) |
| Drug-related TEAE | 0 | 0 | 0 |
| TEAEs by maximum intensity | |||
| Mild | 0 | 5 (41.7) | 2 (40.0) |
| Moderate | 0 | 0 | 0 |
| Severe | 0 | 0 | 0 |
| Life threatening | 0 | 0 | 0 |
| Fatal | 0 | 0 | 0 |
| Total | 0 | 5 (41.7) | 2 (40.0) |
| Drug-related TEAEs by maximum intensity | |||
| Mild | 0 | 1 (8.3) | 0 |
| Moderate | 0 | 0 | 0 |
| Severe | 0 | 0 | 0 |
| Life threatening | 0 | 0 | 0 |
| Fatal | 0 | 0 | 0 |
| Total | 0 | 1 (8.3) | 0 |
| Deaths | 0 | 0 | 0 |
Abbreviations: AE = adverse event; BID = twice daily; QD = once daily; TEAE = treatment-emergent adverse event.
Note: A participant with more than one event in the same category is counted only once in that category. A participant may appear in multiple categories. TEAEs from the same category are counted as reported and may appear multiple times in any category.
All reported preferred terms were experienced by no more than a single participant in a given treatment group (Table 4). Of the total 12 TEAEs that were reported, 7 (58%) resolved (after 1 to 29 days; median duration 5 days) and 4 (33%) were ongoing at the end of the study; outcome was not reported for 1 TEAE. All TEAEs were mild in intensity. One participant in the study experienced TEAEs considered related to study drug. A mild TEAE of oral disorder and a mild TEAE of resting tremor were both assessed as possibly related to study drug. No action was taken as a result of either event. The event of oral disorder resolved after a duration of 29 days, and the event of resting tremor resolved after a duration of 3 days. All reported AEs were experienced by no more than a single participant in a given treatment group.
Table 4.
Summary of Treatment-Emergent Adverse Events by Preferred Term
| Adverse Event Preferred Term | Placebo (n = 6) n (%) |
ELND005 250 mg BID (n = 12) n (%) |
ELND005 250 mg QD (n = 5) n (%) |
TEAE Outcome (Duration) |
|---|---|---|---|---|
| Participants with at least one TEAE | 0 | 5 (41.7) | 2 (40.0) | |
| Bradycardia | 0 | 1 (8.3) | 0 | (unknown) |
| Diarrhoea | 0 | 1 (8.3) | 0 | Resolved (1 day) |
| Oral disorder | 0 | 1 (8.3) | 0 | Resolved (29 days) |
| Pseudofolliculitis barbae | 0 | 1 (8.3) | 0 | Ongoing |
| Resting tremor | 0 | 1 (8.3) | 0 | Resolved (3 days) |
| Restless legs syndrome | 0 | 1 (8.3) | 0 | Ongoing |
| Seasonal allergy | 0 | 1 (8.3) | 0 | 2 episodes, both resolved (5 days, 7 days) |
| Catheter site pain | 0 | 0 | 1 (20.0)a | Resolved (5 days) |
| Muscle strain | 0 | 0 | 1 (20.0) | Ongoing |
| Electrocardiogram abnormal | 0 | 0 | 1 (20.0) | Ongoing |
| Anger | 0 | 0 | 1 (20.0)a | Resolved (1 day) |
Abbreviations: BID = twice daily; QD = once daily; TEAE = treatment-emergent adverse event.
Note: A participant with more than one event in the same category is counted only once in that category. A participant may appear in multiple categories.
These 2 events were reported by participant (001-007) who withdrew from the study on Day 7.
Overall, mean changes from baseline in clinical laboratory parameters, vital sign measurements, ECG results, chest x-rays, and other physical findings were transient and unremarkable. There were no apparent treatment group-related trends. One participant (ELND005 - BID group) was reported to have a TEAE associated with vital sign measurements (bradycardia) and one participant (ELND005 - QD group) experienced a TEAE of abnormal ECG. Both events were assessed as unrelated to study drug. One participant discontinued the study after a single dose of ELND005 due to a mild TEAE of anger, which was unrelated to study drug. There were no SAEs and no deaths in the study. There were no safety signals noted in laboratory tests, ECGs or chest X-rays for the duration of the study.
Pharmacokinetic and Pharmacodynamic Assessment
Plasma levels were calculated based on peripheral venous blood samples that were collected from indwelling catheters or by direct venipuncture into collection tubes. Baseline (Visit 2) and Day 28 (EOS) blood samples for ELND005 plasma levels were collected before dosing (time 0), and at 1.5 (±0.5), 3 (±0.5), and 4.5 (±0.5) hours after dosing. In addition, patients were encouraged to provide blood samples at 6 (±1) and 8 (±1) hours after dosing. At Day 14, one blood sample was collected (either before or after the first dose of study drug on Day 14).
On the first day of dosing with ELND005 (Baseline Visit; “Day 0”), the plasma concentration profile of the ELND005 - QD group (N=5) and the BID group (N=12) overlapped, and both increased gradually during the first few hours after dosing. Mean maximum (peak) serum concentration (Cmax) was 1.68 to 2.61 μg/mL, and mean Time to maximum (peak) serum concentration (Tmax) was 2.7 to 3.2 hours post-dose on Day 0 (Figure 2). Volume of distribution (VoD) was calculated to be 69.36 L. The area under the concentration versus time curve (AUCtau) was 17.58 μg hr/ml.
Figure 2. Mean (SD) Plasma Concentration of ELND005 (μg/mL) versus Time.
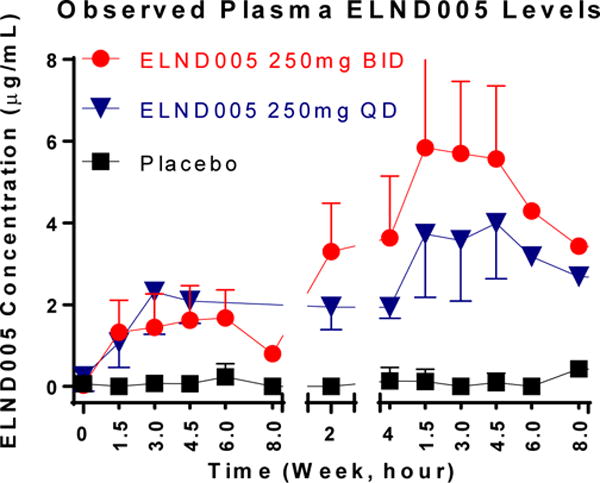
Plasma levels were calculated based on peripheral venous blood samples that were collected from indwelling catheters or by direct venipuncture into collection tubes. Baseline (Visit 2) and Day 28 (EOS) blood samples for ELND005 plasma levels were collected before dosing (time 0), and at 1.5 (±0.5), 3 (±0.5), and 4.5 (±0.5) hours after dosing. In addition, patients were encouraged to provide blood samples at 6 (±1) and 8 (±1) hours after dosing. At Day 14, one blood sample was collected (either before or after the first dose of study drug on Day 14).
By Day 14, mean ELND005 plasma concentration in the BID group was approximately 40% higher than in the QD group, and at Day 28, the plasma concentration profiles of the two treatment groups were clearly separated from each other. Mean Cmax was 4.48 and 6.33 mg/mL, and mean Tmax was 6.6 and 2.5 hours post-dose, for the QD and BID groups, respectively, on Day 28. Apparent steady state was reached by week 4. ELND005 accumulation averaged approximately 2-fold with QD dosing, and 3- to 4-fold with BID dosing (Figure 2).
At the Day 56 Safety Follow-up Visit (4 weeks after last dose of study drug), the average plasma concentrations of ELND005 had significantly decreased and were near the lower limit of quantitation (<2X LLOQ) in both ELND005 treatment groups. Concentration-time profiles of ELND005 at apparent terminal phase could not be reasonably characterized due to limited samples at the later time points of each regimen.
Exploratory Efficacy Results
All efficacy analyses were performed on the mITT Set (total of 22 participants). WAIS Block design scores, VABS-II, RADD-2 and CDR scores were similar across the treatment arms and placebo (Table 5). RADD-2 scores were similar across the treatment arms and placebo. Mean (SD) change from baseline to Week 4 was higher in the ELND005 - QD (6.3 (4.6)) group than the ELND005 - BID (0.4 (4.3)) or placebo (0.7 (2.3)) group. Overall, there was wide variability in CDR scores across groups; this is not unexpected, given the small number of participants per group.
Table 5a.
Summary of the Block Design (WAIS-IV) Total Score (mITT Population)
| Score | Placebo (N=6) |
ELND005 250 mg BID (N=12) |
ELND005 250 mg QD (N=4) |
|---|---|---|---|
|
| |||
| Baseline | |||
| Mean (SD) | 14.7 (6.0) | 15.6 (11.8) | 19.0 (17.9) |
| Median | 16.0 | 11.0 | 13.0 |
| Range | 8.0–24.0 | 0.0–37.0 | 6.0–44.0 |
|
| |||
| Week 4 | |||
| Mean (SD) | 14.7 (5.5) | 16.5 (10.4) | 21.3 (20.8) |
| Median | 14.0 | 20.0 | 14.0 |
| Range | 8.0–24.0 | 0.0 – 29.0 | 6.0–51.0 |
|
| |||
| Week 4 CFB | |||
| Mean (SD) | 0.0 (4.4) | 0.9 (6.8) | 2.3 (11.1) |
| Median | 0.0 | 0.0 | 3.5 |
| Range | −8.0-4.0 | −13.0 – 16.0 | −12.0 – 14.0 |
CFB = Change from baseline, SD= Standard Deviation
Total NPI score at baseline was higher in the ELND005 - BID group (mean (SD) 8.6 (14.7)) than either the ELND005 - QD group (1.8 (1.5)) or the placebo group (2.2 (3.1)). At Week 4, the scores were more similar across all groups (mean (SD) 2.3 (3.6), placebo group; 2.4 (3.4), ELND005 - BID group; and 4.5 (5.2), ELND005 - QD group), noting that the score decreased in the BID group but increased in the QD group (Table 6). The most common neuropsychiatric symptoms at baseline were agitation/aggression (8/18 participants), aberrant motor behavior (5/18), and disinhibition (4/18). Participants with baseline neuropsychiatric symptoms in the NPI assessment showed a numerical trend favoring the 250-mg BID dose. Among the subset of participants who had non-zero scores on the NPI at baseline (i.e., participants with neuropsychiatric symptoms at baseline), there was a decrease in NPI total scores (i.e., improvement) at Week 4 in the ELND005 - BID group in 7 of 8 participants (Table 6). In comparison, only 1 of 3 participants in the placebo group, and 0 of 4 participants in the ELND005 - QD group, had an improved NPI total score at Week 4.
Table 6.
Change from Baseline in NPI Total Scores for Participants with Baseline NPI Score ≥1 (n=15)
| Group | Participant | K-BIT IQ Score | Total NPI Score at Baseline | Total NPI Score at Week 4 | Change from Baseline |
|---|---|---|---|---|---|
|
| |||||
| Placebo | 001–001 | 48 | 1 | 1 | 0 |
| 002–003 | 68 | 5 | 9 | 4 | |
| 001–004 | 57 | 7 | 4 | −3 | |
|
| |||||
| ELND005 250 mg QD | 001–003 | 79 | 1 | 4 | 3 |
| 001–006 | 54 | 1 | 1 | 0 | |
| 001–008 | 40 | 1 | 1 | 0 | |
| 004–005 | 80 | 4 | 12 | 8 | |
|
| |||||
| ELND005 250 mg BID | 002–001 | 40 | 1 | 0 | −1 |
| 004–006 | 48 | 2 | 0 | −2 | |
| 001–002 | 79 | 3 | 6 | 3 | |
| 004–003 | 40 | 6 | 3 | −3 | |
| 004–004 | 40 | 12 | 0 | −12 | |
| 001–009 | 70 | 13 | 8 | −5 | |
| 001–011 | 40 | 14 | 3 | −11 | |
| 004–002 | 40 | 52 | 9 | −43 | |
BID = twice daily; K-BIT = Kaufman-Brief Intelligence Test; NPI = neuropsychiatric inventory; QD = once daily; Higher scores on the NPI indicate more behavioral disturbance.
DISCUSSION
This study enrolled a total of 23 participants with DS, 18 to 45 years of age (inclusive) without dementia, to receive either ELND005 or placebo. Overall, treatment with ELND005 was well tolerated with no newly identified safety risks for ELND005. All TEAEs were experienced by participants in the ELND005 treatment groups and none in the placebo group. All reported TEAEs were experienced by no more than a single participant in a given treatment group. All TEAEs were mild. One participant discontinued the study due to a mild TEAE of anger, which was deemed unrelated to study drug. There were no SAEs and no deaths in the study. There were no apparent treatment group-related trends. One participant (ELND005 - BID group) experienced a TEAE associated with vital sign measurements (bradycardia) and 1 participant (ELND005 - QD group) experienced a TEAE of abnormal ECG. Both events were assessed as unrelated to study drug. The incidence of AEs was greater in the treatment groups as compared to the placebo group. However, the sample size is too small to draw any clear conclusions regarding this observation. There is no clear pattern of the nature of AEs associated with drug treatment and no AEs were attributed to study drug exposure.
Steady state levels of ELND005 in plasma were achieved by Week 4. ELND005 accumulation averaged approximately 2-fold with QD dosing, and 3- to 4-fold with BID dosing. The plasma concentration profiles of the ELND005 - QD group and the BID group overlapped during the first day of dosing and both increased gradually during the first few hours after dosing. However, by the Day 14 visit, mean ELND005 plasma concentration in the BID group was notably higher than in the QD group; and, at Day 28, the plasma concentration profiles of the two treatment groups were clearly separated from each other. In a prior phase I study of ELD005 in the general population of healthy adults, concentrations in plasma appeared to reach steady state by Day 5 (Liang et al, 2013). Prior PK studies of ELND005 in the general (non-DS) population demonstrated that after initial oral administration, ELND005 in plasma was rapidly absorbed reaching the maximum concentrations at 3.8 hours post-dose and apparent steady state by day 5 [31].
A dose-response effect was not observed in any of the exploratory efficacy assessments, nor was it expected given the four week duration of drug exposure. PK modeling of data from previous dose-range findings studies indicated that a dose of 250 mg QD was likely to be below the therapeutic range of the compound. An important aspect of this study was to better understand the utility of exploratory cognitive assessments in clinical trial participants with DS. In this regard, we found that all participants were able to successfully complete these assessments in a clinical trial setting. Within the functional outcomes, the total VABS-II score increased from baseline to Week 4 in the ELND005 - BID group but decreased in the ELND005 - QD and placebo groups.
The most common neuropsychiatric behavioral symptoms (NPI subscores) at baseline were agitation/aggression, aberrant motor behavior, and disinhibition. There were no apparent treatment-related findings in the behavioral outcomes of the NPI total scores when the overall treatment groups were compared. However, among the subset of participants who had non-zero scores on the NPI at baseline (i.e., participants with neuropsychiatric symptoms), there was a trend of a decrease in NPI scores (i.e. improvement) at the 250-mg BID dose. This data should be interpreted with caution since the NPI has not yet been validated in DS. Indeed, there is a great need for validation studies of cognitive and functional outcome measures for AD in DS. Specifically, cognitive outcome measures warrant development of lower floors, in order to be appropriate for individuals with DS who may be lower cognitive functioning at baseline. Lower floors allow clinical trials to be applicable to a broader range of individuals with DS.
This study was focused on safety and pharmacokinetics of ELND005 in young adults with DS; our findings indicate the treatment with ELND005 was well tolerated and no safety findings to suggest any newly identified safety risks. The study was not powered to prove differences between treatment groups. A dose-response effect was not observed in any of the exploratory efficacy assessments, nor was it expected. Although limited in sample size, this small study may provide support for future longitudinal studies in this population. Study limitations include short duration of treatment, small number of participants and potential practice effects on exploratory cognitive measures. Nonetheless, PK blood sample were reliably obtained in this population and all participants were able to complete the relatively complex study procedures.
Table 5b.
Summary of the VABS-II Total Score (mITT Population)
| VABS-II Score | Placebo (N=6) |
ELND005 250 mg BID (N=12) |
ELND005 250 mg QD (N=4) |
|---|---|---|---|
|
| |||
| Baseline | |||
| Mean (SD) | 682.3 (60.9) | 660.5 (49.1) | 690.0 (31.8) |
| Median | 687.0 | 666.0 | 691.0 |
| Range | 597.0–775.0 | 550.0–736.0 | 652.0–726.0 |
|
| |||
| Week 4 | |||
| Mean (SD) | 674.3 (48.9) | 672.6 (53.7) | 683.3 (32.0) |
| Median | 689.5 | 659.5 | 681.5 |
| Range | 594.0–716.0 | 565.0–749.0 | 650.0–720.0 |
|
| |||
| Week 4 CFB | |||
| Mean (SD) | −8.0 (26.0) | 12.1 (31.2) | −6.8 (32.7) |
| Median | 0.5 | 12.5 | 3.0 |
| Range | −59.0 – 13.0 | −25.0–82.0 | −53–20.0 |
Table 5c.
Summary of the RADD-2 Total Score (mITT Population)
| RADD Total Score (calculated) | Placebo (N=6) |
ELND005 250 mg BID (N=12) |
ELND005 250 mg QD (N=4) |
|---|---|---|---|
|
| |||
| Baseline | |||
| Mean (SD) | 62.2 (9.8) | 58.7 (10.2) | 58.0 (15.6) |
| Median | 66.5 | 61.5 | 63.0 |
| Range | 49.0–73.0 | 43.0–70.0 | 36.0–70.0 |
|
| |||
| Week 4 | |||
| Mean (SD) | 62.8 (10.3) | 59.1 (11.5) | 64.3 (13.5) |
| Median | 65.0 | 62.5 | 70.5 |
| Range | 49.0–75.0 | 35.0–72.0 | 44.0–72.0 |
|
| |||
| Week 4 CFB | |||
| Mean (SD) | 0.7 (2.3) | 0.4 (4.3) | 6.3 (4.6) |
| Median | 0.5 | 1.0 | 5.5 |
| Range | –3.0–4.0 | −8.0–8.0 | 2.0–12.0 |
Acknowledgments
We deeply thank all the participants and their families for participating in the Phase II study of scyllo-inositol in Down syndrome.
Funding: This study was funded entirely by Elan Pharmaceuticals, though all authors had access to any data they requested and all authors contributed, reviewed and agreed with this manuscripts content, language and conclusions.
Role of the Sponsor: Elan Pharmaceuticals conducted of all required nonclinical research and regulatory filings and approvals for this study. The IND is now held by Transition Therapeutics.
References
- 1.de Graaf G, Buckley F, Skotko BG. Live births, natural losses, and elective terminations with Down syndrome in Massachusetts. Genet Med. 2016;18:459–66. doi: 10.1038/gim.2016.15. [DOI] [PubMed] [Google Scholar]
- 2.Gardiner K, Herault Y, Lott I, Antonarakis S, Reeves RH, Dierssen M. Down syndrome: from understanding the neurobiology to therapy. J Neurosci. 2010;30:14943–5. doi: 10.1523/JNEUROSCI.3728-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pinter JD, Brown WE, Eliez S, Schmitt JE, Capone GT, Reiss AL. Amygdala and hippocampal volumes in children with Down syndrome: a high-resolution MRI study. Neurology. 2001;56:972–4. doi: 10.1212/wnl.56.7.972. [DOI] [PubMed] [Google Scholar]
- 4.Edgin JO, Mason GM, Allman MJ, Capone GT, Deleon I, Maslen C, Reeves RH, Sherman SL, Nadel L. Development and validation of the Arizona Cognitive Test Battery for Down syndrome. J Neurodev Disord. 2010;2:149–164. doi: 10.1007/s11689-010-9054-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lott IT, Dierssen M. Cognitive deficits and associated neurological complications in individuals with Down’s syndrome. Lancet Neurol. 2010;9:623–33. doi: 10.1016/S1474-4422(10)70112-5. [DOI] [PubMed] [Google Scholar]
- 6.Wisniewski KE, Wisniewski HM, Wen GY. Occurrence of neuropathological changes and dementia of Alzheimer’s disease in Down’s syndrome. Ann Neurol. 1985;17:278–82. doi: 10.1002/ana.410170310. [DOI] [PubMed] [Google Scholar]
- 7.Handen BL, Cohen AD, Channamalappa U, Bulova P, Cannon SA, Cohen WI, Mathis CA, Price JC, Klunk WE. Imaging brain amyloid in nondemented young adults with Down syndrome using Pittsburgh compound B. Alzheimers Dement. 2012;8:496–501. doi: 10.1016/j.jalz.2011.09.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rafii MS, Wishnek H, Brewer JB, Donohue MC, Ness S, Mobley WC, Aisen PS, Rissman RA. The down syndrome biomarker initiative (DSBI) pilot: proof of concept for deep phenotyping of Alzheimer’s disease biomarkers in down syndrome. Front Behav Neurosci. 2015;9:239–259. doi: 10.3389/fnbeh.2015.00239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Walsh DM, Selkoe DJ. A beta oligomers - a decade of discovery. J Neurochem. 2007;101:1172–84. doi: 10.1111/j.1471-4159.2006.04426.x. [DOI] [PubMed] [Google Scholar]
- 10.Berry GT, Mallee JJ, Kwon HM, Rim JS, Mulla WR, Muenke M, Spinner NB. The human osmoregulatory Na+/myo-inositol cotransporter gene (SLC5A3): molecular cloning and localization to chromosome 21. Genomics. 1995;25:507–13. doi: 10.1016/0888-7543(95)80052-n. [DOI] [PubMed] [Google Scholar]
- 11.Huang W, Alexander GE, Daly EM, Shetty HU, Krasuski JS, Rapoport SI, Schapiro MB. High brain myo-inositol levels in the predementia phase of Alzheimer’s disease in adults with Down’s syndrome: a 1H MRS study. Am J Psychiatry. 1999;156:1879–86. doi: 10.1176/ajp.156.12.1879. [DOI] [PubMed] [Google Scholar]
- 12.Shonk T, Ross BD. Role of increased cerebral myo-inositol in the dementia of Down syndrome. Magn Reson Med. 1995;33:858–61. doi: 10.1002/mrm.1910330619. [DOI] [PubMed] [Google Scholar]
- 13.Shinno H, Inagaki T, Miyaoka T, Okazaki S, Kawamukai T, Utani E, Inami Y, Horiguchi J. A decrease in N-acetylaspartate and an increase in myoinositol in the anterior cingulate gyrus are associated with behavioral and psychological symptoms in Alzheimer’s disease. J Neurol Sci. 2007;260:132–8. doi: 10.1016/j.jns.2007.04.017. [DOI] [PubMed] [Google Scholar]
- 14.Fenili D, Brown M, Rappaport R, McLaurin J. Properties of scyllo-inositol as a therapeutic treatment of AD-like pathology. J Mol Med (Berl) 2007;85:603–11. doi: 10.1007/s00109-007-0156-7. [DOI] [PubMed] [Google Scholar]
- 15.McLaurin J, Golomb R, Jurewicz A, Antel JP, Fraser PE. Inositol stereoisomers stabilize an oligomeric aggregate of Alzheimer amyloid beta peptide and inhibit abeta-induced toxicity. J Biol Chem. 2000;275:18495–502. doi: 10.1074/jbc.M906994199. [DOI] [PubMed] [Google Scholar]
- 16.Townsend M, Cleary JP, Mehta T, Hofmeister J, Lesne S, O’Hare E, Walsh DM, Selkoe DJ. Orally available compound prevents deficits in memory caused by the Alzheimer amyloid-beta oligomers. Ann Neurol. 2006;60:668–76. doi: 10.1002/ana.21051. [DOI] [PubMed] [Google Scholar]
- 17.Salloway S, Sperling R, Keren R, Porsteinsson AP, van Dyck CH, Tariot PN, Gilman S, Arnold D, Abushakra S, Hernandez C, Crans G, Liang E, Quinn G, Bairu M, Pastrak A, Cedarbaum JM, ELND005-AD201 Investigators A phase 2 randomized trial of ELND005, scyllo-inositol, in mild to moderate Alzheimer disease. Neurology. 2011;77:1253–62. doi: 10.1212/WNL.0b013e3182309fa5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mohan M, Bennett C, Carpenter PK. Rivastigmine for dementia in people with Down syndrome. Cochrane Database Syst Rev. 2009;(1):CD007658. doi: 10.1002/14651858.CD007656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mohan M, Bennett C, Carpenter PK. Memantine for dementia in people with Down syndrome. Cochrane Database Syst Rev. 2009;(1):CD007657. doi: 10.1002/14651858.CD007657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mohan M, Bennett C, Carpenter PK. Galantamine for dementia in people with Down syndrome. Cochrane Database Syst Rev. 2009;(1):CD007656. doi: 10.1002/14651858.CD007658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mohan M, Carpenter PK, Bennett C. Donepezil for dementia in people with Down syndrome. Cochrane Database Syst Rev. 2009;(1):CD007178. doi: 10.1002/14651858.CD007178.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Boada R, Hutaff-Lee C, Schrader A, Weitzenkamp D, Benke TA, Goldson EJ, Costa AC. Antagonism of NMDA receptors as a potential treatment for Down syndrome: a pilot randomized controlled trial. Transl Psychiatry. 2012;2:1–11. doi: 10.1038/tp.2012.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sano M, Aisen PS, Andrews HF, Tsai WY, Lai F, Dalton AJ, International Down Syndrome and Alzheimer’s Disease Consortium Vitamin E in aging persons with Down syndrome: A randomized, placebo-controlled clinical trial. Neurology. 2016;86:2071–6. doi: 10.1212/WNL.0000000000002714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Walsh DM, Finwall J, Touchette PE, McGregor MR, Fernandez GE, Lott IT, Sandman CA. Rapid assessment of severe cognitive impairment in individuals with developmental disabilities. J Intellect Disabil Res. 2007;51:91–100. doi: 10.1111/j.1365-2788.2006.00853.x. [DOI] [PubMed] [Google Scholar]
- 25.Edgar CJ, Wesnes KA. Cognition assessment in paediatric clinical trials. Drug Discov Today. 2008;13:79–85. doi: 10.1016/j.drudis.2007.10.006. [DOI] [PubMed] [Google Scholar]
- 26.Lloyd A, Brett D, Wesnes K. Coherence training in children with attention-deficit hyperactivity disorder: cognitive functions and behavioral changes. Altern Ther Health Med. 2010;16:34–42. [PubMed] [Google Scholar]
- 27.Wechsler D, Coalson DL, Raiford SE. Wechsler Adult Intelligence Test: Fourth Edition Technical and Interpretive Manual. San Antonio: Pearson; 2008. [Google Scholar]
- 28.Sparrow SS, Balla DA, editors. Vineland Adaptive Behavior Scales. Second. Minneapolis: Pearson; p. 2005. [Google Scholar]
- 29.Cummings JL, Mega M, Gray K, Rosenberg-Thompson S, Carusi DA, Gornbein J. The Neuropsychiatric Inventory: comprehensive assessment of psychopathology in dementia. Neurology. 1994;44:2308–14. doi: 10.1212/wnl.44.12.2308. [DOI] [PubMed] [Google Scholar]
- 30.Sperling RA, Jack CR, Jr, Black SE, Frosch MP, Greenberg SM, Hyman BT, Scheltens P, Carrillo MC, Thies W, Bednar MM, Black RS, Brashear HR, Grundman M, Siemers ER, Feldman HH, Schindler RJ. Amyloid-related imaging abnormalities in amyloid-modifying therapeutic trials: recommendations from the Alzheimer’s Association Research Roundtable Workgroup. Alzheimers Dement. 2011;7:367–85. doi: 10.1016/j.jalz.2011.05.2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liang E, Garzone P, Cedarbaum JM, Koller M, Tran T, Xu V, Ross, Jhee SS, Ereshefsky L, Pastrak A, Abushakra S. Pharmacokinetic Profile of Orally Administered Scyllo-Inositol (Elnd005) in Plasma, Cerebrospinal Fluid and Brain, and Corresponding Effect on Amyloid-Beta in Healthy Subjects. Clin Pharmacol Drug Dev. 2013;2:186–94. doi: 10.1002/cpdd.14. [DOI] [PubMed] [Google Scholar]