

Summary
Rhodococcus equi is a multi-host, facultative intracellular bacterial pathogen that primarily causes pneumonia in foals less than six months in age and immunocompromised people. Previous studies determined that the major virulence determinant of R. equi is the surface bound virulence associated protein A (VapA). The presence of VapA inhibits the maturation of R. equi-containing phagosomes and promotes intracellular bacterial survival, as determined by the inability of vapA deletion mutants to replicate in host macrophages. While the mechanism of action of VapA remains elusive, we show that soluble recombinant VapA32-189 both rescues the intramacrophage replication defect of a wild type R. equi strain lacking the vapA gene and enhances the persistence of nonpathogenic Escherichia coli in macrophages. During macrophage infection, VapA was observed at both the bacterial surface and at the membrane of the host-derived R. equi containing vacuole, thus providing an opportunity for VapA to interact with host constituents and promote alterations in phagolysosomal function. In support of the observed host membrane binding activity of VapA, we also found that rVapA32-189 interacted specifically with liposomes containing phosphatidic acid in vitro. Collectively, these data demonstrate a lipid binding property of VapA, which may be required for its function during intracellular infection.
Keywords: R. equi, host-pathogen interactions, macrophage, phosphatidic acid, protein:lipid interaction
Abbreviated Summary
Virulence associated protein A (VapA), a key virulence determinant of the intracellular pathogen Rhodococcus equi, was observed to alter macrophage killing capacity and localize to eukaryotic membranes during infection. Direct binding of recombinant VapA protein to liposomes containing phosphatidic acid was demonstrated.
Introduction
The Gram-positive coccobacillus Rhodococcus equi is a causative agent of pyogranulomatous pneumonia in foals less than six months in age and in immunocompromised people (Giguere et al., 2011; Vazquez-Boland et al., 2013; Weinstock and Brown, 2002). Cattle and swine are also occasional hosts but disease presentation in these species is different and typically manifests as respiratory lymph node abscessation and sub-maxillary lymphadenitis respectively (Flynn et al., 2001; Komijn et al., 2007; Valero-Rello et al., 2015). Delayed type hypersensitivity testing of horses has determined that exposure to R. equi is widespread. Disease however, is either sporadic or endemic depending on the horse farm (Barton and Hughes, 1984; Takai, 1997). While a significant portion of subclinical disease resolves naturally, the unrestrained use of antibiotics on some horse farms has led to the emergence of antimicrobial resistant strains of the bacterium, complicating treatment during disease progression (Giguere, 2017). Additionally, the rise in human immunocompromised populations, whether it be from chemotherapy or HIV, has allowed the bacterium to become a relevant opportunistic pathogen of humans (Weinstock and Brown, 2002).
R. equi exposure of foals typically occurs through inhalation of aerosolized bacteria from contaminated soil, wherein the bacterium is introduced to the lower airway (Muscatello et al., 2006). Delivery of the bacterium to the lungs allows alveolar macrophages to recognize and phagocytose the bacterium. Thereafter, the macrophage will attempt to kill the bacterium through a variety of antimicrobial mechanisms, including the production of reactive oxygen and nitrogen species, activation of hydrolytic enzymes, generation of an acidified vacuolar compartment, and depletion of essential nutrients (Flannagan et al., 2009). Despite these tactics, R. equi is able to survive and replicate within macrophages of susceptible hosts, designating the bacterium a facultative intracellular pathogen (Hondalus and Mosser, 1994).
Initial studies indicated that there are both virulent and avirulent strains of R. equi, differing in the possession of a virulence-associated plasmid. (Giguere et al., 1999). Subsequent work has shown that the type of virulence plasmid carried by disease-causing strains of R. equi is host specific; wherein pVapA is the plasmid type carried by R. equi isolates infecting equine and is the most studied of the virulence plasmid types (Ocampo-Sosa et al., 2007). pVapB and pVapN comprise the remainder of the sequenced virulence plasmids and are carried by R. equi strains infecting swine and bovine species respectively (Valero-Rello et al., 2015). Human infection can arise from R. equi carrying any of the aforementioned virulence associated plasmids, but the most commonly detected pathogen is R. equi harboring the pVapB type virulence plasmid (Ocampo-Sosa et al., 2007). The ability of multiple R. equi strains harboring different virulence plasmids to cause disease in immunocompromised humans confirms the opportunistic capacity of this bacterium. Sequence analysis of these virulence plasmids revealed the presence of four common regions known as the conjugation, replication, unknown function and the virulence or pathogenicity island (PAI) regions (Letek et al., 2008; Valero-Rello et al., 2015). Within the PAI region of these plasmids, members of a novel gene family, the vap (virulence associated protein) family reside. Deletion of the plasmid pathogenicity island renders the bacterium incapable of replication in macrophages (Coulson et al., 2010). Further experimentation on the pVapA type virulence plasmid has shown that, of the twenty open reading frames present in the PAI, only three are required for intramacrophage replication: virR, virS, and vapA (Coulson et al., 2015; Jain et al., 2003; Valero-Rello et al., 2015).
virR and virS encode for transcriptional regulators crucial for the expression of vapA as well as a number of other genes found on both the virulence-associated plasmid and the R. equi chromosome. The vapA gene produces the highly immunogenic protein, virulence associated protein A (VapA) (Coulson et al., 2015; Jain et al., 2003; Kakuda et al., 2015; Takai et al., 1991). There are five other open reading frames located in the pVapA PAI encoding for proteins with a high degree of identity to vapA: vapG, vapH, vapC, vapD, and vapE (Letek et al., 2008). Targeted deletion studies of each of these vap genes have determined that deletion of vapA alone attenuates the bacterium’s ability to replicate within macrophages (Coulson et al., 2010; Jain et al., 2003; Wang et al., 2014). Therefore, VapA activity does not appear to be shared among these related Vap homologs even though many of these Vap-family proteins appear to be expressed during R. equi infection (Byrne et al., 2001; Hooper-McGrevy et al., 2003; Jacks et al., 2007). While the mechanism of action of VapA has yet to be discerned, it has been determined that VapA is localized on the surface of R. equi, giving it the capability to directly interact with the host (Takai et al., 1992). Studies have shown that virulent R. equi reside within an enlarged neutral compartment in the macrophage and that the presence of vapA inhibits the acidification of the R. equi containing vacuole (RCV), thereby supporting the intracellular survival of the bacterium (Fernandez-Mora et al., 2005; Toyooka et al., 2005; von Bargen et al., 2009).
Direct visualization of R. equi after phagocytic uptake has determined that the recruitment of early trafficking markers to the bacteria-laden phagosome is unaffected. The early endosomal markers Rab5 and early endosomal antigen 1 (EEA1) are gained and lost by the RCV in a timely manner. Late endosomal markers lysobisphosphatidic acid, Rab7, and lysosomal-associated membrane proteins (LAMP) 1 and 2 increase subsequently, suggesting that maturation of these endosomal compartments to late endosomes proceeds unhindered by the presence of the bacterium (Fernandez-Mora et al., 2005). The two aberrant phenotypes associated with R. equi infection of macrophages are the lack of acidification of, and replication within, an enlarged phagosomal compartment (Fernandez-Mora et al., 2005; Toyooka et al., 2005; von Bargen et al., 2009).
Sequence characterization of the Vap proteins suggests that they can be broken into three domains, designated as the signal sequence, disordered, and conserved domains (Letek et al., 2008). Molecular work on the Vap proteins has determined the protein structure of the conserved domain of VapG, VapD, and VapB; VapB is a virulence associated protein located in the PAI of pVapB (Geerds et al., 2014; Makrai et al., 2002; Okoko et al., 2015; Takai et al., 1996; Valero-Rello et al., 2015; Whittingham et al., 2014). Solved structures resolved an eight stranded anti-parallel β-barrel connected by an α-helix for each of the preceding proteins, although the solved structures could not provide much insight into the function of these Vaps (Geerds et al., 2014; Okoko et al., 2015; Whittingham et al., 2014). Attempts to crystallize VapA have been unsuccessful, but it is presumed that the protein obtains a similar structure to the other Vaps due to high sequence homology. More recent work on endocytosed recombinant VapA has determined that the protein does not directly inhibit acidification but can reduce hydrolytic capacity and cause endolysosomal swelling of normal rat kidney cells; however, a mechanism for how VapA alters cellular trafficking remains to be determined (Rofe et al., 2016).
This study further characterizes VapA, the major virulence determinant of Rhodococcus equi isolates carrying pVapA. Experiments focused on localizing the protein during both macrophage infection and in vitro expression in yeast show a distinct membrane-binding activity of VapA, which may be critical for its ability to alter endolysosomal traffic in host cells during infection. Additionally, the ability of rVapA to bind to liposomes of varying lipid composition was analyzed, and provides evidence that membrane phosphatidic acid is a ligand for VapA.
Results
Recombinant VapA complements the intramacrophage replication defect of an R. equi strain lacking vapA
In order to survive and replicate intracellularly, R. equi harboring the pVapA-type virulence plasmid requires only the activity of vapA, and two transcriptional regulators, virR and virS, all residing within the plasmid-borne pathogenicity island (Coulson et al., 2015). Since VapA appears to be the only pVapA-encoded Vap family protein essential for the growth of R. equi in macrophages (Jain et al., 2003), and considering a recent study that showed that VapA could directly alter lysosomal activity when endocytosed by the normal rat kidney cell line (Rofe et al., 2016), we assessed whether macrophages pre-treated with purified recombinant VapA lacking the putative N-terminal signal sequence, rVapA32-189, could rescue the intramacrophage replication defect of R. equi ΔvapA. It is important to note that this fragment of the VapA protein is known to be functional, as mature VapA lacks the first 31 N-terminal amino acids (Tan et al., 1995). In these experiments, bacterial intracellular growth in the J774A.1 macrophage cell line was followed over time by traditional lysis and plating of infected monolayer lysates. As anticipated, intracellular bacterial loads of wild type R. equi (103S) increased ~17 fold over 48 hr. This was in contrast to bacteria lacking the pVapA-type virulence plasmid (103P−), whose growth was reduced 10-fold over the same time frame (Figures 1A and 1B). Similar to 103P−, the wild type R. equi strain lacking vapA (ΔvapA) was unable to replicate intracellularly, although intracellular bacterial loads persisted over the course of the infection (Figures 1A and 1B). Strikingly, the addition of rVapA32-189 restored the ability of the ΔvapA strain to replicate in a dose-dependent manner, wherein macrophages pre-treated with 100nM of rVapA32-189 could replicate to levels comparable to that of the wild type strain (Figures 1A and 1B). Thus, exogenous addition of soluble recombinant VapA could compensate for the loss of the bacterially-encoded VapA protein during R. equi intramacrophage growth.
Figure 1. Recombinant VapA32-189 and VapK232-202 complement the replication defect of R. equi ΔvapA.

Murine J774A.1 macrophage monolayers were incubated overnight with the indicated concentration of recombinant Vap protein (dashed lines with open symbols) or media (solid lines with closed symbols), then infected with the indicated R. equi strain. Each experimental condition was performed in triplicate per infection; n = 3. Symbols denote the mean number of bacteria observed and the bars denote the mean bacterial fold change. Error bars represent the standard deviation using a two-way analysis of variation (ANOVA) by way of the Holm-Sidak test. (A,C) Letters to the right of each curve denote statistical significance; the same letter signifies no statistical difference, while different letters signify statistical difference at P ≤ 0.05. (B,D) ns = not significant; (*) = P < 0.05; (**) = P < 0.01; (***) = P < 0.001.
To assess the specificity of rVapA32-189 activity, other recombinant Vap-family proteins lacking their putative signal sequences were tested in their ability to rescue the intramacrophage replication defect of ΔvapA. Of the five other functional vap open reading frames (vapG, vapH, vapE, vapC and vapD) located in the PAI of pVapA, VapG displays the highest sequence identity to VapA at 48%, and is known to have high mRNA expression levels during R. equi infection of foals (Jacks et al., 2007; Letek et al., 2008; Okoko et al., 2015; Valero-Rello et al., 2015). Despite these characteristics, purified recombinant VapG27-172 was unable to rescue the replication defect of ΔvapA bacteria (Figures 1C and 1D).
Isolates of R. equi that harbor a pVapB-type plasmid contain six distinct vap genes (vapJ, vapK1, vapL, vapK2, vapM, and vapB) (Letek et al., 2008; Valero-Rello et al., 2015). Because of the high sequence identity (78%) between the VapB and VapA proteins, it has been proposed that the VapB protein in R. equi strains harboring pVapB would be functionally equivalent to VapA during pathogenesis (Letek et al., 2008). Despite this, a recent report speculates that VapK1/K2, with 59% identity to VapA, is the functional equivalent of VapA in pVapB-carrying R. equi strains (Letek et al., 2008; Valero-Rello et al., 2015). Interestingly, in support of the latter, we found that while the addition of rVapB35-197 was unable to reverse the intracellular replication defect of ΔvapA, rVapK232-202 addition resulted in a 10-fold increase of ΔvapA bacterial loads 48 hours post-infection (Figures 1C and 1D). Importantly, this increase was in-line with the overall magnitude of intracellular replication displayed by that of the wild type pVapB-carrying R. equi strain 33705 (Figures 1C and 1D), although the overall extent of bacterial replication was reduced when compared to the growth displayed by the ΔvapA strain in the presence of rVapA32-189. To assess whether the lack of effect of rVapG and rVapB could be explained by improper folding of the recombinant proteins, all of the recombinant Vap proteins used in these studies were subjected to Proteinase K digestion and analyzed by gel electrophoresis, as previously performed by Geerds and coworkers, who described the presence of a properly-folded ~12 kDa core domain of VapA and VapB that was resistant to proteolytic degradation (Geerds et al., 2014). Upon exposure to Proteinase K digestion, we detected this Proteinase K-resistant core domain in each of the rVaps used in this study; this protease-resistant fragment was lost upon the denaturation of the protein in SDS (Supplemental Figure 1). These data suggest that each of the Vap proteins tested was appropriately folded and that the inability of rVapB35-197 or rVapG27-271 to rescue the intracellular growth defect of a R. equi ΔvapA strain was likely not due to improperly folded recombinant protein. Taken together, these data show that exogenous addition of either rVapA32-189 or rVapK232-202 can reverse the ΔvapA replication defect in macrophages, whereas related Vap-family proteins do not share this property. Surprisingly, VapA does not need to be directly produced by R. equi for its activity, likely because this virulence factor directly alters macrophage physiology.
rVapA promotes the intramacrophage persistence of nonpathogenic Escherichia coli
It is known that R. equi expressing vapA has the ability to both neutralize the pH of the RCV during infection and to inhibit the degradative capacity of host macrophages (Fernandez-Mora et al., 2005; Toyooka et al., 2005; von Bargen et al., 2009). Because exogenous addition of rVapA32-189 was able to reverse the intramacrophage replication defect of the ΔvapA strain, we hypothesized that rVapA32-189 could directly alter the killing capacity of macrophages. Thus, we questioned whether the effect was broad in scope, and therefore, we assessed the impact of the presence of rVapA32-189 on the intracellular survival of a nonpathogenic E. coli strain. As expected, the nonpathogenic E. coli strain was unable to replicate or persist in J774A.1 cells, with intracellular bacterial loads reduced by ~80% at 12 hours post infection (Figures 2A and 2B). Upon preincubation of macrophages with rVapA32-189, however, the survival of E. coli was greatly enhanced and bacterial numbers were only reduced by 40% at 12 hours post infection (Figures 2A and 2B). In contrast, macrophages incubated with rVapG27-271 were not significantly different from untreated macrophages in their ability to kill E. coli under these conditions (Figures 2A and 2B). In order to support the intracellular persistence of a nonpathogenic bacterium, it is likely that rVapA32-189 directly alters host phagolysosomal function.
Figure 2. Recombinant VapA32-189 enhances intracellular persistence of nonpathogenic E. coli in J774A.1 cells.

J774A.1 macrophage monolayers were incubated overnight with either media (triangles) or 150 nM recombinant Vap protein (VapA, circles; VapG, crosses), and infected with E. coli. Symbols delineate the mean bacterial number and bars denote the mean percent survival. The error bars represent the standard deviation calculated using a two-way ANOVA via the Holm-Sidak method. Each experimental condition was performed in triplicate per infection; n = 3. (A) Statistical significance is expressed as letters to the right of the curve, with same letters defining a lack of significant difference, and different letters defining significance with P ≤ 0.05. (B) Percent of viable E. coli cells detected, as compared to 1 hour post infection (HPI) is shown; ns = not significant; (***) = P < 0.001.
Localization of VapA during R. equi infection shows the protein at the bacterial surface and the RCV membrane
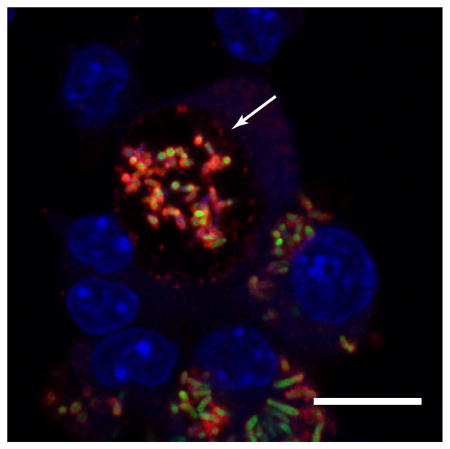
Because rVapA32-189 appeared to be able to directly modulate the activity of host phagolysosomal compartments, we sought to localize R. equi-produced VapA over the course of an infection. For these studies, we utilized murine bone marrow-derived macrophages (BMDMs) as the host for R. equi. Both wild type 103S and ΔvapA R. equi strains harboring a GFP expression plasmid were utilized to locate bacteria during infection of BMDM monolayers, and intracellular bacterial loads were measured via direct visualization and enumeration. As expected, the 103S-GFP wild type strain was capable of replicating intracellularly over 72 hr, and the ΔvapA-GFP strain was cleared from these macrophages over the same timeframe (Figures 3A and 3B). As previously observed, supplementation with exogenous rVapA32-189 restored the ability of the ΔvapA-GFP strain to survive and replicate intracellularly (Figures 3A and 3B). Of note, these results (Figure 3B) likely underestimate the total number of intracellular bacteria because of the clumping nature of R. equi and the fact that macrophages containing more than 10 organisms were quantified as containing only 10 bacteria. Therefore, the number of macrophages containing ten or more bacteria was also followed over time. After 72 hr, 35 of 200 macrophages infected with 103S harbored ≥10 intracellular bacteria, compared to 1 macrophage infected with the ΔvapA strain. In the presence of exogenous rVapA32-189, almost 80 of 200 macrophages assessed contained 10 or more ΔvapA bacteria, thus directly confirming the ability of rVapA32-189 to support the intracellular replication of ΔvapA R. equi (Figure 3C).
Figure 3. VapA associates with the RCV membrane during infection.

Murine bone marrow-derived macrophages (BMDMs) were infected with R. equi 103S or ΔvapA strains harboring the GFP expression plasmid, pGFPmut2. Where indicated, 100 nM rVapA was added to the BMDM monolayer the night before the infection. R. equi (GFP, green), BMDM nucleus (DAPI, blue), and VapA (anti-VapA, red) were observed. (A) Representative confocal images of infection, bar = 5 μ, inset bar = 1 μ. Arrows indicate VapA detected at the RCV membrane. (B) Bacterial numbers per 200 macrophages were quantified by direct visualization at the indicated time points. (C) Macrophages containing ten or more bacteria, discerned via direct visualization. (B,C) Bars indicate the mean number of quantified bacteria or macrophages, while the error bars represent the standard deviation calculated by a two-way ANOVA using the Holm-Sidak method. n = 3; ns = not significant and (***) = P ≤ 0.001.
Visualization of monolayers infected with 103S R. equi showed the presence of VapA at the bacterial surface throughout the course of infection (Figure 3A). After 48 hours, however, in addition to remaining on the surface of the bacterium, we found that VapA was also observed to accumulate on the membrane of the R. equi-containing vacuole (RCV) (Figure 3A, arrows and insets). In ΔvapA-infected monolayers supplemented with rVapA32-189, the recombinant protein displayed a highly punctate staining pattern at 1 hour post-infection, reminiscent of vesicular packaging upon endocytic or pinocytotic uptake (Figure 3A, T1). After 48 hr, rVapA32-189 appears to accumulate around and within RCVs, suggesting that not only is rVapA32-189 eventually trafficked to bacteria-laden compartments within these cells, but that its activity is needed within (or on) the RCV to support the replication of the R. equi ΔvapA strain. Taken together, these images indicate that VapA likely functions in the lumen of the RCV, and that VapA may begin to associate with the membrane of bacteria-laden host vacuoles by 48 hr after infection.
VapA localizes to the yeast plasma membrane
Because of the discovery that VapA appears to localize to the limiting membrane of mature RCVs during R. equi infection, we decided to exploit a model eukaryotic system to aid in characterizing the cellular localization of the various Vap-family proteins. The budding yeast Saccharomyces cerevisiae is a robust model system for essential eukaryotic processes and has often been used to study the biochemical activities of secreted bacterial effector proteins that alter eukaryotic physiology (Botstein and Fink, 2011; Siggers and Lesser, 2008). N-terminal GFP fusions to full-length VapA, VapA32-189, VapG27-172, VapB35-197, and VapK232-202 were constructed in the yeast GFP expression vector pGO35 (Burd and Emr, 1998; Odorizzi et al., 1998), and protein expression of each of these constructs in yeast was examined via immunoblot (Figure 4A). Each GFP-Vap construct, with the exception of GFP-VapG27-172, generated the expected ~44kDa band upon exposure with the polyclonal and cross-reactive anti-VapA antibody. Subsequently, the localization of each GFP-Vap fusion (other than GFP-VapG) within S. cerevisiae was examined. Expression of GFP alone showed a diffuse cytosolic staining, as expected (Figure 4B). However, expression of either GFP-tagged VapA construct (VapA or VapA32-189) resulted in a striking localization of GFP to the yeast plasma membrane. In fact, plasma membrane localization of GFP was observed in 100% of the GFP-VapA expressing yeast cells (Figure 4C). In contrast, yeast expressing either GFP-VapK232-202 or GFP-VapB35-197, showed a marked decrease in the amount of GFP localized to the plasma membrane (Figure 4B). Only ~40% of the GFP-VapK232-202 expressing yeast cells showed GFP at the plasma membrane, while ~23% of GFP-VapB35-197 expressing yeast cells displayed plasma membrane-localized GFP (Figure 4C). Cumulatively, VapA membrane localization in both BMDM infection and upon expression in yeast suggest that VapA can either directly bind to cellular lipids or a conserved membrane-bound receptor. That GFP-VapK232-202 can also moderately localize to membranes in yeast may be relevant in its capacity to rescue the intramacrophage growth impairment of the vapA deletion mutant.
Figure 4. GFP-VapA binds to the yeast plasma membrane upon expression in vivo.

(A) Saccharomyces cerevisiae strain BY4742 harboring either GFP (vector), GFP-VapA, GFP-VapA32-189, GFP-VapG27-172, GFP-VapB35-197, or GFP-VapK232-202 plasmid constructs were grown overnight and proteins were extracted from equal amounts of cell pellets, separated via SDS-PAGE, and probed with polyclonal anti-VapA antisera. (B) Cells from (A), with the exception of the strain harboring GFP-VapG27-172, were visualized for GFP localization via fluorescence microscopy. Bar = 5 μ. (C) Quantification of GFP localization from yeast strains imaged in (A); Cells with clear accumulation of GFP-Vap protein at the plasma membrane was counted as “plasma membrane”, diffuse GFP staining of the cytoplasm with no plasma membrane localization was counted as “cytoplasm;” all other morphologies were counted as “other.” 100 yeast cells were counted per experimental condition, bars denote the mean percent of cells displaying a particular Vap localization pattern. Error bars represent the standard deviation using a two-way analysis of variation (ANOVA) by way of the Holm-Sidak test, and significance pertaining to the difference in plasma membrane localization of GFP-Vap proteins is indicated. n = 3; ns = not significant and (***) = P ≤ 0.001.
rVapA binds liposomes containing phosphatidic acid
As VapA was seen to interact with both the yeast plasma membrane and the phagosomal membrane containing R. equi, we examined the possibility that rVapA32-189 could interact directly with phospholipid bilayers. Therefore, liposomes of varying lipid concentrations were constructed, and the ability of rVapA32-189 to bind to these membranes was investigated. We chose to explore rVapA32-189’s interaction with four major phospholipid constituents of the eukaryotic plasma membrane: phoshphatidylcholine (PC), phosphaditylethanolamine (PE), phosphatidylserine (PS), and phosphatidic acid (PA). At a near-physioloigcal pH of 7.4, interactions of rVapA32-189 with liposomes containing either 100% PC or supplemented with 20% PE, PS, or PA were not detected, as measured by liposome co-floatation (Figure 5A). In contrast, rVapA32-189 was found to bind to liposomes containing 20% PA when incubations were performed at a pH of 5.5; a minor interaction with PS-containing liposomes was also observed (Figure 5B). Furthermore, rVapA32-189 bound PA-containing liposomes in a concentration-dependent manner (Figure 5C). Thus, rVapA32-189 appears to bind to some negatively-charged lipids under acidic conditions, with a preference for membranes containing phosphatidic acid. Finally, we assayed each of the other closely related Vap proteins isolated in this study (VapG27-172, VapB35-197, and VapK232-202) for their ability to interact with membranes containing phosphatidic acid. Results showed that none of these Vaps could appreciably interact with these liposomes (Figure 5D). Therefore, the interaction of VapA with phosphatidic acid appears to be unique among the Vap-family proteins tested.
Figure 5. rVapA binds phosphatidic acid containing liposomes.

Liposomes of indicated compositions (PA, 20% phosphatidic acid; PC, 100% phosphatidylcholine; PE; 20% phosphatidylethanolamine; PS, 20% phosphatidylserine) were generated as in Experimental Procedures. Liposomes were incubated with recombinant Vap protein at a pH of either (A) 7.4 or (B) 5.5, and liposomes were isolated by flotation (Experimental Procedures). Equal fractions representing 10% of the total reaction (T) and either 900 nmol (A,B) or 1 μmol (C,D) total floated (F) liposomes were separated via SDS-PAGE and immunoblotted for VapA. (C) Liposomes containing increasing amounts of PA were assayed for VapA binding, as above in (5B). (D) Liposomes containing 20% PA were incubated with 1 μg indicated recombinant Vap protein, and assayed for binding, as above in (5B). All recombinant Vap proteins tested cross-react with VapA antiserum.
R. equi resides in LAMP1-negative vesicles in the presence of VapA
Because the existence of VapA at the phagosomal membrane could impact host phagolysosomal trafficking, we followed the presence of the late endosomal/lysosomal marker LAMP1 (lysosomal-associated membrane protein 1) during the course of macrophage infection (Flannagan et al., 2009; Vieira et al., 2002). It has been reported that RCVs associate with LAMP1 up to twenty-four hours post infection, and it has been presumed that wild type R. equi remain in a late endosome-like compartment by inhibiting the fusion of bacterial-containing vesicles with degradative lysosomes (Fernandez-Mora et al., 2005). Using the GFP-expressing 103S and immunofluorescence staining for LAMP1, we similarly found R. equi-containing vesicles to be associated with LAMP1 up to twenty-four hours post infection, and noted that the vesicular membranes were closely associated with individual bacteria (Figure 6, 103S, T24). We extended these observations and at 48h post infection, when bacterial loads were high, we noted that the RCV became enlarged and LAMP1 was no longer associated with R. equi-containing vacuoles (Figure 6, 103S, T48). Interestingly, LAMP1 appeared to be excluded from VapA-positive membranes (Figure 6, 103S, insets), which continued until at least 72 h post infection (Figure 6B). Furthermore, there was a noticeable increase in LAMP1 staining of macrophages harboring replicating bacteria than the surrounding uninfected cells.
Figure 6. The presence of VapA prevents the accumulation of LAMP1 within the RCV 48 and 72 h post infection.

(A) BMDMs were infected with either R. equi 103S or ΔvapA strains, and 100 nM rVapA32-189 was added the night before the infection, where indicated. At indicated time points, cells were fixed, immunostained, and visualized via confocal microscopy. R. equi (GFP, green), BMDM nucleus (DAPI, blue), VapA (anti-VapA, red), and murine LAMP1 (anti-LAMP1, purple) were observed. n = 3, bar = 5 μ, inset bar = 1 μ. (B) Representative confocal image of 103S infection of BMDMs at T72, performed and stained as in (A). A large RCV containing replicating bacteria and RCV membrane-associated VapA (arrow) is shown in comparison to macrophages containing strongly LAMP1-positive compartments surrounding bacteria that lack detectable VapA at the RCV membrane (asterisks), bar = 5 μ.
In contrast, RCVs containing ΔvapA bacteria were found to be associated with LAMP1 throughout the course of infection, suggesting delivery of these mutant R. equi to the degradative lysosome and a lack of intracellular replication (Figure 6, ΔvapA). Contrastingly, in the presence of 100nM rVapA32-189, ΔvapA bacteria were found to be located in both LAMP1-positive and -negative compartments at 48–72 hours post infection (Figure 6, ΔvapA+rVapA, insets). Although the RCV was never found to be enlarged under these conditions, as in the wild type infections, intracellular bacterial loads increased in spite of some overlap with LAMP1-containing vesicles. Taken together, these data suggest that the presence of VapA at the RCV membrane is associated with altered phagolysosomal maturation or altered endolysosomal trafficking pathways in macrophages, and that LAMP1 displacement away from VapA-producing R. equi occurs beyond twenty-four hours post infection.
Discussion
Herein, we show that exogenous addition of rVapA32-189 protein to macrophage monolayers both reversed the intracellular growth defect of R. equi ΔvapA bacteria and supported the persistence of nonpathogenic E. coli, highlighting the ability of the major virulence determinant of R. equi, VapA, to broadly inhibit the killing capacity of macrophages (Figures 1 and 2). Our work determined that soluble rVapA32-189 at 100 nm (1.7 μg ml−1) was sufficient to restore intramacrophage replication of ΔvapA to wild type levels. Our findings are supported by those of another group who very recently described the rescue of ΔvapA growth in macrophages in the presence of 100 ug ml−1 soluble rVapA (Sangkanjanavanich et al., 2017). The group also reported that addition of rVapA allowed for the intracellular growth of an avirulent plasmid-cured strain of R. equi, 103P−. The latter was a surprising result, given it is well-established that expression of wild type levels of VapA alone by virulence plasmid-free R. equi is not sufficient to promote intramacrophage replication (Giguere et al., Coulson et al 2015), although Sangkanjanavanich and colleagues used at least 50-fold more rVapA than we found is required to support the intracellular replication of ΔvapA R. equi in this study. Interestingly, we also found that a related Vap protein encoded on the pVapB-type plasmid carried by some R. equi strains (e.g. 33705), rVapK232-202, showed a similar capacity to restore the growth of ΔvapA bacteria in macrophages, albeit to a lesser degree (Fig. 1). No other recombinant Vap protein tested shared this activity, suggesting that the ability to alter macrophage antimicrobial capabilities is specific for VapA and its functional homologs (like VapK2) across R. equi strains harboring different virulence plasmids.
To better understand VapA’s mechanism of action, we sought to observe the localization of VapA during extended R. equi macrophage infection. While previous in vitro experimentation has determined that VapA is located on the surface of the bacterium (Takai et al., 1992), to our knowledge, this is the first time in which this localization has been confirmed during macrophage infection. Strikingly, at 48 hours post-infection, VapA was no longer solely confined to the bacterial surface, but also appeared at the membrane of the RCV (Figure 3A). The mechanism by which VapA is delivered to the RCV membrane is unknown, but reasoning suggests that active secretion of VapA from intracellular R. equi, release of VapA from shedding of the outer cell envelope from intracellularly replicating bacteria, degradation of some bacteria within the RCV, or delivery via an interaction between the bacterium and phagosomal membrane during initial phagocytic uptake are all viable possibilities. Byrne and coworkers previously described the secretion of VapC, VapD, and VapE into R. equi culture supernatant, but could not detect VapA via immunoblot (Byrne et al., 2001). While this demonstrates that R.equi does not secrete VapA across the outer lipid envelope into the supernatant during routine laboratory culture, it may be that signals within the intramacrophage environment triggers release of the protein from the bacterial surface. Some intracellular pathogens harbor type three (T3SS) and type 4 (T4SS) secretion systems allowing for bacterial effectors to be secreted into the cytoplasm of their host cell (Deng et al., 2017). While genes encoding for a T3SS or classical T4SS are not present in R. equi, the R. equi virulence plasmid harbors genes required for the conjugal transfer of this plasmid (Tripathi et al., 2012). It is unlikely that these genes are responsible for the secretion of VapA during infection, however, as this region is expendable for the proper localization of VapA on the surface of R. equi (Coulson et al 2010). There is genetic evidence in R. equi for the presence of a type seven secretion system (T7SS; ESX) similar to Mycobacterium tuberculosis (Letek et al., 2010). T7SS have been identified in a variety of Gram-positive organisms, where they participate in a wide number of functions including virulence and permitting the transport of substrates across the inner membrane and outer lipid envelope, or mycomembrane; however, these T7SS have not been shown to deliver substrates into the host cytoplasm. What role, if any, the T7SS may play in R. equi pathogenesis remains unknown at present.
This study provides the first experimental evidence showing VapA deposition on the membrane of the RCV during macrophage infection. VapA was not observed outside of the RCV (e.g. in the host cytosol) during macrophage infection with wild type R. equi. As a first step to better understand how the presence of VapA at the limiting membrane of the RCV might potentially affect phagolysosomal trafficking, we assessed the locations of both native VapA and of lysosomal-associated membrane protein 1 (LAMP1), found on late endosomes and lysosomes, over the course of macrophage infection (Figure 6). Previously published work has established that LAMP1 associates with RCVs containing virulent R. equi during the 24 hours following uptake (Fernandez-Mora et al., 2005). By extending this assessment further, however, we made a surprising observation: R. equi-containing vacuoles appear to exist in a mixed population during infection. Notably, enlarged RCVs with VapA at the limiting membrane were devoid of LAMP1, while RCVs that lacked VapA at the membrane were LAMP1 positive (Figure 6). This finding suggests that VapA’s presence at the RCV membrane is important in aiding the bacteria in avoiding destruction by the lysosomal compartment. While LAMP1 is initially gained, the RCV containing actively replicating bacteria can either displace, or avoid further accumulation of, LAMP1-containing endolysosomal vesicles (Figure 6). Thus, there appears to be a late RCV stage that warrants further characterization, but such is beyond the scope of this work.
VapA localization to eukaryotic membranes was not restricted to macrophages, as expression of GFP-VapA in S. cerevisiae (VapA or VapA32-189) localized strongly to the plasma membrane (Figures 4B and C). Likewise, GFP-VapK232-202 displayed plasma membrane localization in yeast, albeit to a lesser extent, and this reduction might account for the decreased ability of exogenous rVapK232-202 to rescue the ΔvapA mutant and restore its replicative capacity in macrophages, at least as compared to rVapA32-189 (Figures 4B, 4C, and 1C). GFP-VapB35-197 showed even less yeast plasma membrane localization than rVapK232-202 and was predominantly localized to the cytoplasm. That the two Vap proteins (rVapA32-189 and rVapK232-202) with the capacity to reverse the intramacrophage replication defect of ΔvapA displayed localization to the yeast plasma membrane (Figure 4B and 4C) prompted us to postulate that VapA might possess lipid binding properties. Thus, we found that rVapA32-189 bound to synthetic liposomes containing phosphatidic acid (PA) (Figures 5B–5D), and to a much lesser extent, phosphatidylserine (PS) (Figure 5B). Interestingly, membrane PA concentrations in yeast is highest at the outer mitochondrial and plasma membranes, wherein both of these organelles also contain a high percentage of phosphatidylethanolamine (PE). Unique to the plasma membrane, however, is a high amount of PS along with PA and PE (Zinser et al., 1991). Additionally, the more acidic environment (pH 5.5) used in these assays allowed rVapA32-189 to bind PA as compared to neutral conditions (pH 7.4) (Figures 5A and 5B). This increased interaction at a lower pH could be accounted for because of the lower charge PA holds in this environment (Putta et al., 2016; Shin and Loewen, 2011). The fact that we found GFP-VapA able to bind the yeast plasma membrane in a near-neutral cytosolic pH may identify additional unknown lipid or protein ligands for VapA, which are required to increase the affinity of VapA for PA under these conditions.
PA is an anionic glycerophospholipid that induces the formation of negative curvature in membranes (Kooijman et al., 2005), and can participate in cellular signaling via both electrostatic and hydrophobic interactions with protein ligands (Lemmon, 2008). During phagocytosis, phospholipase D actively generates PA in the phagocytic cup and post-phagocytic vesicles, which is required to properly deform the plasma membrane in order to generate the limiting membrane of the phagocytic vesicle (Corrotte et al., 2006). This phagosomal pool of PA would provide VapA on the bacterial surface its host binding target upon phagocytic uptake. That we observed rVapA32-189 binding to PA-containing liposomes only in an acidic environment (Figures 5A and 5B) was interesting. Lack of lysosomal acidification has been well documented during R. equi infections using Lysotracker dye, which responds to a pH of 6.5 or lower by a shift in fluorescence intensity (Fernandez-Mora et al., 2005; Toyooka et al., 2005; von Bargen et al., 2009). Therefore, the VapA:PA interaction may occur during an initial acidification step during infection that is later hindered by the replicating bacterium. Support of this theory comes from the ability of R. equi to withstand acidic conditions and the upregulation of VapA during the bacterium’s acid response (Benoit et al., 2001; Kakuda et al., 2014). Previous measurements of phagosomal acidification during macrophage infections with R. equi have shown that while colocalization between the bacterium and Lysotracker dye is low, Lysotracker-positive vesicles are still detected early during the infection (Fernandez-Mora et al., 2005; Toyooka et al., 2005; von Bargen et al., 2009). These acidic vesicles may fuse with the nascent R. equi-containing phagosome, thus providing the trigger for VapA binding. Recently, it was discerned that rVapA supplemented normal rat kidney cells had high colocalization between Lysotracker dye and lysosomes but that the lysosomes had a lower hydrolytic capacity (Rofe et al., 2016). Taken together, we can speculate that VapA is not directly altering phagosomal/lysosmal pH, but rather still-undescribed mechanisms downstream of VapA activity are required for R. equi to limit RCV acidification.
Signaling mediated by PA is becoming better understood as an important means for cellular communication, and is implicated in the regulation of critical cellular events including vesicular trafficking, actin polymerization, and respiratory burst (Corrotte et al., 2006; Fang et al., 2001; Frondorf et al., 2010; Lim et al., 2003; Schlam et al., 2013; Wang et al., 2006). Phosphatidic acid is able to mediate the Akt-mTOR-S6K signaling cascade at each of the pathway components and may be a way in which the bacterium promotes intramacrophage growth (Fang et al., 2001; Frondorf et al., 2010; Lim et al., 2003; Wang et al., 2006). Macrophages grown in PA-supplemented media activated the Akt-dependent signaling cascade, thus leading to the production of proinflammatory cytokines, nitric oxide, and prostaglandin E2. This response was abrogated in mutant Akt lines, wherein it was presumed that PA indirectly affects Akt localization, and therefore activity, through its ability to modulate phosphoinositide 3-kinase (Lim et al., 2003). Notably, macrophages infected with either the wild type or plasmid-cured strain of R. equi have very similar cytokine production profiles, suggesting that Akt is likely not the primary regulatory target of VapA (Giguere and Prescott, 1998). While PA has an indirect effect on Akt, the lipid has been found to directly interact with mTOR via the FRB domain (Fang et al., 2001; Frondorf et al., 2010). mTOR works in conjunction with the V-type ATPase on lysosomal membranes to sense available nutrients inside the phagosome (Flinn et al., 2010; Zoncu et al., 2011). Thereafter, mTOR mediates downstream signaling events to alter protein synthesis and autophagic pathways (Flinn et al., 2010). Lastly, experiments studying phagocyte chemotaxis have shown that PA stimulates S6K to promote cellular migration through a Transwell plate, and therefore likely promotes the migration of phagocytes into infected tissues (Frondorf et al., 2010). Accordingly, the VapA:PA interaction could have drastic effects on a number of PA-dependent cellular physiologies, wherein modulation of the mTOR or S6K proteins later in the Akt-mTOR-S6K signaling cascade is one such example.
While we found that rVapA32-189 interacted with PA-containing liposomes, neither of the pVapB encoded Vaps tested, rVapB35-197 or rVapK232-202, bound PA liposomes to levels observed with rVapA32-189 (Figure 5D). R. equi isolates carrying alternate virulence plasmids differ in their disease presentation and host species tropism; wherein equine isolates, carrying exclusively pVapA, present with pyogranulomatous pneumonia and swine isolates, carrying primarily pVapB, show submaxillary lymphadenitis (Giguere et al., 2011; Komijn et al., 2007; Makrai et al., 2005; Takai et al., 1996). It has been determined, however, that R. equi equipped with either the pVapA or pVapB type virulence plasmids can replicate within macrophages of many species; showing that interspecies macrophage differences are not responsible for the plasmid-specific species tropism observed (Willingham-Lane et al., 2016). It has been presumed that the Vaps responsible for supporting the intramacrophage growth of differing R. equi isolates would employ similar molecular mechanisms during pathogenesis. Here, we have shown that while rVapK232-202 supplementation rescues the intramacrophage growth phenotype of ΔvapA R. equi and that GFP-VapK232-202 to some extent localizes to the yeast plasma membrane, the protein was not detected to interact with PA (Figures 1C, 1D, and 6D). While it is likely that the core structures of the Vap proteins are conserved (Geerds et al., 2014; Okoko et al., 2015; Whittingham et al., 2014), it is possible that rVapK232-202 cooperates with a different lipid in the eukaryotic membrane. Alternatively, rVapK232-202 may lack the capacity to interact with PA, but contains a shared function with VapA that has yet to be determined. Future work is required to elucidate or eliminate the possibility of lipid binding by rVapK2. It should be noted that the bulk of research on R. equi pathogenesis has focused on pVapA carrying isolates, as these are typical of strains derived from both diseased horses and humans. It is therefore unknown if the R. equi-containing vacuole generated by strains harboring the pVapB-type virulence plasmid follows the same phagolysosomal maturation process as those strains harboring the pVapA-type plasmid (Fernandez-Mora et al., 2005; Toyooka et al., 2005; von Bargen et al., 2009). This represents a gap in the knowledge regarding macrophage infection with R. equi and should be considered when addressing R. equi virulence in the future. In closing, the identification of VapA:host membrane and VapA:PA interactions define a new understanding of VapA:host molecular interactions that should provide insight into the activity of VapA during R. equi pathogenesis. Future work will identify the region of VapA responsible for PA binding, and may help answer the question of why the other highly homologous Vap proteins of R. equi are insufficient to support intramacrophage replication in the absence of the VapA protein.
Experimental Procedures
Bacterial and yeast cells
Bacterial and yeast strains used in these studies are listed in Table 1. Escherichia coli was grown in Luria-Bertani (LB) broth at 37°C with antibiotics where appropriate. Selective antibiotics used for E. coli were carbenicillin or hygromycin at 50 μg ml−1 or 180 μg ml−1, respectively. R. equi was grown in Brain Heart Infusion (BHI) broth at 37°C or 30°C and supplemented with either 80 μg ml−1 apramycin or 180 μg ml−1 hygromycin, when appropriate.
Table 1.
Bacterial and yeast strains
| Bacteria | Description | Source |
|---|---|---|
| R. equi | ||
| 103S | Wild type strain isolated from pneumonic foal (~80kb pVapA type virulence plasmid) | (Giguere et al., 1999) |
| 103P− | Plasmid cured 103+ R. equi variant | (Jain et al., 2003) |
| ΔvapA | 103S R. equi variant with vapA deletion; AprR | (Jain et al., 2003) |
| 103S/pGFPmut2 | 103S harboring pGFPmut2; HygR | (Burton et al., 2015) |
| ΔvapA/pGFPmut2 | ΔvapA harboring pGFPmut2; HygR and AprR | This Study |
| 33705 | Wild type strain isolated from pig lymph node (~80kb pVapB type virulence plasmid) | (Takai et al., 1985) |
| E. coli | ||
| pTRC His-GFP | Common lab strain of E. coli carrying GFP expression vector | A. Medlock (UGA) |
| S. cerevisiae | ||
| BY4742 | MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 | GE Dharmacon |
Saccharomyces cerevisiae strain BY4742 cells were grown on yeast extract peptone dextrose (YPD) agar or broth at 30°C. BY4742 cells transformed with a plasmid that conferred auxotrophic selection through uracil were grown at 30° C and either plated on or in complete supplemental mixture (CSM) lacking uracil.
Recombinant Vap purification
Plasmids utilized and constructed in this study are shown in Table S1. Vap proteins were cloned into the pHis-Parallel1 overexpression plasmid (Sheffield et al., 1999) as follows: Vap open reading frames were amplified from the respective virulence plasmid templates (103S plasmid for vapA and vapG; 33705 plasmid for vapB and vapK2) using the primers listed in Table S2. These primers only amplify the part of the vap gene that is responsible for the disordered and conserved domains of the proteins, and lack the putative N-terminal signal sequences; accession numbers for proteins are CAQ30407 (VapA), CAQ30394.1 (VapG), CAQ30336.1 (VapK2), and CAQ30339.1 (VapB) (Letek et al., 2008). Amplicons were digested using the appropriate restriction enzymes underlined in Table S2 and cloned into pHis-Parallel1, which had been digested with the same enzymes. Resultant plasmids, pVapALMW, pVapGLMW, pVapBLMW, and pVapK2LMW, were confirmed by sequencing at the Georgia Genomics Facility (University of Georgia).
Plasmids expressing N-terminal hexahistidine fusions to Vap proteins were electroporated into the E. coli Tuner™ strain (EMD Millipore). Acquired transformants were grown to an OD600 = 0.8–1.0 at 37 °C in LB supplemented with carbenicillin. His-Vap expression was induced with 1 mM isopropyl β-D-1-thiogalactopyranoside and cells were grown for another 4 hr at 37 °C with shaking. Cells were harvested and suspended in lysis buffer (1 ml 0.5 g−1 wet weight pellet; 50 mM sodium phosphate pH 7.0, 300 mM NaCl, 10 mM imidazole, 1 mg ml−1 lysozyme, 1 mM β-mercaptoethanol, and EDTA-free protease inhibitor cocktail (Thermo Scientific)). Cell mixture was incubated for 1 hr at room temperature with gentle agitation, and sonicated to disrupt cells (6 × 30 s).
Lysed cells were clarified via centrifugation (19000 × g, 15 min, 4 °C), and passed over Ni-NTA resin, which had been pre-equilibrated with lysis buffer lacking protease inhibitors, lysozyme, and β-mercaptoethanol. The resin was washed once each with 10 column volumes wash buffer (50 mM sodium phosphate pH 7.0 and 300 mM NaCl) containing increasing amounts of imidazole (20 mM, 40 mM, 60 mM). Protein was eluted with wash buffer containing 500 mM imidazole. Eluted protein was dialyzed into 50 mM potassium phosphate pH 7.4, 150 mM KCl, and 10% (v/v) glycerol, and stored at −80 °C.
Proteinase K digestion of recombinant Vap proteins
Vap proteins were digested by Proteinase K (Novagen) in a 500:1 (protein:enzyme) mass ratio as described by Geerds and colleagues (Geerds et al., 2014). Briefly, Proteinase K and Vap proteins were incubated either with or without 1% SDS at 37°C for 75 min. Digestion was halted by addition of 2 mM phenylmethanesulfonyl-fluoride (PMSF) and resulting products were run on an SDS-PAGE gel.
Construction of yeast expressing GFP tagged Vaps
To generate GFP-VapA yeast expression constructs, primer pairs GFP-VapA SS F and GFP-VapA R or GFP-VapA NSS F and GFP-VapA R were used to amplify the open reading frames from103S corresponding to VapA or VapA32-189 respectively. The remaining vap genes were amplified using their respective GFP-Vap(X) NSS F and GFP-Vap(X) R primers from either the 103S or 33705S plasmid; wherein a region containing vapL-M from 33705 was initially amplified in order to PCR vapK2. The resultant PCR products from these primers (Table S2) provided sequence homology to the pGO35 vector, which allows for the constitutive expression of an N-terminally GFP tagged protein of interest (a gift from Alexey Merz, UW Seattle) (Burd and Emr, 1998; Odorizzi et al., 1998). pGO35 was digested with BglII, and co-transformed with the corresponding amplicons via standard lithium acetate methods (Gietz and Woods, 2002). The resultant plasmids, pGFP-VapA32-189, pGFP-VapG27-172, pGFP-VapB35-197, and pGFP-VapK232-202 express GFP-tagged Vap proteins without their N-terminal signal sequences. In contrast, pGFP-VapA will express full length GFP-VapA. Imaging of these strains was completed via fluorescence microscopy.
SDS-PAGE and western blotting
Pure protein samples were boiled for 8 min in 1 X loading dye consisting of 50 mM Tris-Cl (pH6.8), 2 % SDS, 0.1 % (w/v) bromophenol blue, 10% glycerol, and 100 mM DTT. For extracting protein from whole yeast cells, approximately 1 X109 cells were suspended in 200 μl lysis buffer (0.1 M NaOH, 0.05 M EDTA, 2 % SDS, and 2 % β-mercaptoethanol) before boiling for 10 min. Afterwards, 5 μl of 5 M glacial acetic acid was added to the yeast lysates before vortexing for 30 s and boiling samples for an additional 10 min. Yeast samples then had loading dye added to a 1 X concentration.
Prepped samples were run on an SDS-PAGE gel in 1 X Tris-Glycine SDS running buffer (Novex®). Transfer of the samples from the SDS-PAGE gel to a nitrocellulose membrane was done in a 1.4% (w/v) glycine, 0.3% (w/v) Tris Base, and 20% methanol buffer. Thereafter, the nitrocellulose membrane was blocked using the SuperBlock® T20 (TBS) blocking buffer (Thermo Scientific) for either an hour at room temperature or overnight at 4°C with agitation. Primary polyclonal rabbit αVapA was diluted 1:1,000 in blocking buffer. Primary antibodies were then incubated with the membrane for either 1 hour at room temperature or overnight at 4°C with agitation. Afterwards, while rocking, the membrane was washed 4 X for 10 minutes each with a f.05% (v/v) Tween 20 and 1X PBS solution. Thereafter, secondary goat αrabbit antibody, conjugated to horseradish peroxidase (HRP), was diluted 1:20,000 in blocking buffer and incubated with the membrane as done with the primary antibody. Next, the membrane was washed as done previously. To expose the western blot, enhanced chemiluminescent HRP substrate and peroxide buffer, both from Thermo Scientific, were mixed and added to the membrane and allowed to incubate at room temperature for 5 minutes.
Electroporation of R. equi
An overnight 100 ml culture of ΔvapA R. equi in BHI broth was diluted to an OD600 of 0.4 with fresh medium, and grown at 30°C until it reached an OD600 of 0.8–1.0. Bacteria were harvested via centrifugation (3600 × g, 10 min, 4°C), the pellet was washed with 50 ml of cold sterile dH2O, and centrifuged for an additional 30 min. This wash step was repeated and additional time, then cells were suspended in 4 ml cold, sterile dH2O water containing 5% glycerol. Approximately 200 ng pGFPmut2 DNA was added to 400 μl of washed bacteria and mixed by gentle pipetting. The bacteria/DNA mixture was then placed in a pre-chilled 2 mm electroporation cuvette (Bio-Rad) and electroporated (2500 V, 25 μF, 1000 Ω). Immediately following electroporation, 1 ml of filter sterilized BHI supplemented with 0.5 M sucrose was added to the cuvette, collected, and incubated for one hour at 30°C. Following incubation, bacteria were harvested via centrifugation, 400μl of the supernatant was discarded, and the bacterial pellet was suspended in the remaining supernatant. Aliquots of the resuspension were then plated to appropriate selective media, and incubated at 30°C for two days.
Macrophage growth conditions and isolation
J774A.1 macrophages (ATCC) were grown at 37°C with 5% CO2 in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% heat inactivated Fetal Bovine Serum (FBS) and 2 mM glutamine (complete medium).
Bone marrow-derived macrophages (BMDM) were isolated from femurs and tibias of BALB/c mice (Jackson Laboratories). In order to isolate BMDM precursors, femurs and tibias were dissected from the mouse and flushed with cold cation-deficient 1X PBS containing 100 units ml−1 penicillin and 10 mg ml−1 streptomycin (P/S). Cells retrieved from the bone marrow were centrifuged (260 × g, 10 min, 4°C), washed once with complete media containing P/S, then suspended in complete media supplemented with P/S and 10% supernatant from Colony Stimulating Factor-1 (CSF-1) producing L929 cells (24 ml per mouse). These precursor cells were incubated in 6-well non-tissue culture treated plates (37°C, 5% CO2; 4ml suspended cells per well) for 3 days. After incubation, 4 ml complete media containing 10% L929 cell supernatant (no P/S) were added to each well and incubation continued. On days five and six, all of the media on the cells was removed and replaced with complete media supplemented with 10% CSF-1 containing L929 cell supernatant. Following a 2 hr incubation with the fresh media on day six, the media was removed from the wells and replaced with cold 1X PBS, incubated at 4 °C for 10 min, and macrophages were harvested. Macrophages obtained were either used immediately in assays, or suspended in 90% FBS with 10% DMSO at 5X106 cells per ml and frozen in liquid nitrogen. For assays, cells were maintained in DMEM supplemented with 10% FBS, 2 mM glutamine, and 10% L929 cell supernatant containing CSF-1.
Macrophage infection assays
1 × 105 macrophages were placed on 13 mm-diameter glass coverslips or in 24-well tissue culture plates. When applicable, filter sterilized recombinant protein was suspended in macrophage media at a concentration of 50–150 nM and incubated with the monolayer overnight. The next morning, monolayers were washed with complete media. Bacteria, suspended in PBS, were added to the monolayer at a multiplicity of infection of 5:1 (J774A.1) or 7:1 (BMDM) during R. equi infections or 10:1 during E. coli infections. After 1 hr, the monolayer was washed three times with warm DMEM without supplementation. The monolayers were maintained in complete media supplemented with 20–40 μg ml−1 amikacin in order to kill any remaining extracellular bacteria; while one plate was lysed for bacterial quantification at T1.
To quantify R. equi loads in macrophages, cells were lysed (37 °C, 5% CO2, 20 min) with 500μl of sterile dH2O. For E. coli infections, 500 μl 0.5% (v/v) Triton X-100 was added to macrophages, and the incubation time was reduced to 5 min. The resultant lysates containing bacteria were collected, serially diluted in PBS, and plated on BHI (R. equi) or LB (E. coli) agar and incubated at 37°C for 24 to 48 hr. For later time points, the monolayer was washed three times with DMEM, as before, and the lysis procedure was performed.
Immunofluorescence assays
Infected macrophage monolayers on 13mm diameter coverslips in 24-well plates were fixed with 4% (v/v) paraformaldehyde in PBS (22 °C, 20 min, in the dark). Coverslips were washed four times with PBS supplemented with 5% FBS, and fixed cells were permeabilized with 350 μl of PBS containing 0.1% Triton X-100 (3 min). 250μl anti-VapA polyclonal antibody (diluted 1:1600 in PBS with 5% FBS and 0.1% (v/v) Triton X-100) was incubated on the coverslips for one hour in the dark at 22 °C. Coverslips were washed four times in PBS supplemented with 5% FBS and 0.1% (v/v) Triton X-100. Afterwards, 250 μl goat anti-rabbit antibody conjugated to fluorescein isothiocyanate (FITC) (1:500 in PBS with 5% FBS and 0.1 % (v/v) Triton X-100, Thermo Scientific), was incubated with the fixed monolayer and washed, as above. 250μl of an α-LAMP1 mouse monoclonal conjugated to Alexa Fluor 647 (diluted 1:500 in PBS with 5% FBS and 0.1% (v/v) Triton X-100) was used to stain monolayers for LAMP1 as described for the aforementioned primary and secondary antibodies. Washing was completed as done previously. A final wash with with PBS was immediately performed before the coverslips were mounted on microscope slides using ProLong Gold antifade reagent with 4′,6-diamidino-2-phenylindole (DAPI) (ThermoFisher Scientific), and were allowed to dry for 24 hours before visualization via confocal microscopy.
Liposome production
Stock 16:0–18:1 phospholipids (Avanti Polar Lipids) were dissolved in chloroform and mixed in glass tubes to provide a final amount of 2 μmol total lipid. For quantitation, 0.5% (mol/mol) rhodamine-phosphatidylethanolamine (Rh-PE) was added to all mixtures. Lipid mixtures were composed of 1-palmitoyl-2-oleoyl-sn-glyceryo-3-phosphocholine (POPC) and either 1-palmitoyl-2-oleoyl-sn-glyceryo-3-phosphate (POPA), 1-palmitoyl-2-oleoyl-sn-glyceryo-3-phosphoethanolamine (POPE), or 1-palmitoyl-2-oleoyl-sn-glyceryo-3-phospho-L-serine (POPS) were added to the indicated percentage. Chloroform was removed via a stream of argon gas, and complete solvent removal was obtained under vacuum for 1 hr. Dried lipids were suspended in 1 ml buffer containing either 20 mM 4-2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) pH 7.4 or 50 mM 2-ethanesulfonic (MES) and 3-(N-morpholino)propanesulfonic (MOPS) acid pH 5.5, depending on the pH desired, 10% (v/v) glycerol, and 150 mM NaCl. The suspended lipids were placed in a 37 °C water bath and intermittently vortexed at high speed for 60 min. After 10 freeze-thaw cycles in liquid nitrogen, suspended lipids were passaged 11 times through a Mini-Extruder (Avanti Polar Lipids) fitted with a 1 μm polycarbonate membrane, which had been previously equilibrated on a heating block set to 37 °C.
Liposome quantification was determined with the use of a Rh-PE standard curve in RB150 (20 mM HEPES pH 7.4 or 50mM MES pH 5.5, 150 mM NaCl, 10% (v/v) glycerol) containing 1% (v/v) Thesit; liposomes of interest were also serially diluted in this same buffer. Fluorescence of the suspensions was determined by loading 20 μl each suspension into a 384-well, low-volume, black microplate (Corning) and a computer-controlled multimode plate reader (λex = 540 nm, λem = 585 nm; BioTek). Liposomes were stored at 4°C until the time of assay.
Liposome floatation
50 μl reactions containing 500 μM total lipids and 1 μg of recombinant protein of interest were incubated for 1 hr in a 37°C water bath. Afterwards, 5 μl of the reaction was collected as a control before adding 50μl of 80% filter-sterilized Histodenz (Sigma Aldrich) in the appropriate pH RB150 buffer. This mixture was inverted to mix, and placed into a 7 X 20 mm polycarbonate centrifuge tube (Beckman Coulter). Sampled were then overlaid with 75μl of filter sterilized 30% Histodenz in RB150 and 75μl of RB150 media. Samples were centrifuged (96,000 × g for 2 hours at 4°C) before collecting 40μl of the floated liposomes. To analyze the samples, the harvested liposome concentrations were determined via Rh-PE fluorescence, equivalent amounts of liposomes were separated via SDS-PAGE, and immunoblotted for Vap proteins.
Confocal Microscopy
Fluorescence images were captured on a Nikon A1R confocal microscope system (UGA College of Veterinary Medicine Cytometry Core Facility) using a 100X 1.45NA (Nikon) objective, equipped with an S-P 50 mW multiline Ar laser for imaging GFP and Alexa 488™, a Coherent Sapphire 561nm 20 mW laser for imaging dsRed, and a Coherent Cube 640nm 40mW laser for imaging Alexa 647™. Images were captured using NIS Elements software and processed with the Fiji (ImageJ v. 1.48) software package (Schindelin et al., 2012; Schneider et al., 2012).
Statistical Analysis
Sigma Plot version 11.2.0.5 (Systat Software, San Jose, CA) and GraphPad Prism version 6.0b (GraphPad Software, Inc., La Jolla, CA) were used for statistical analysis. Bacterial quantification was evaluated via a two-way analysis of variation (ANOVA) using the Holm-Sidak method, where a P value of ≤ 0.05 was considered significant.
Supplementary Material
Acknowledgments
The authors would like to thank Drs. Vihbay Tripathi and Jennifer Willingham-Lane for their input and discussions regarding this work, as well as Dr. Alexey Merz for providing pGO35. We also thank Leanna Ritson for technical expertise and Dr. Amy Medlock for providing the E. coli strain harboring pTRCHis-GFP. V.J.S. is supported by a grant from National Institute of Allergy and Infectious Diseases (R01-AI100913). The authors have no conflicts of interest to declare.
Footnotes
Author Contributions
LMW has made major contributions to design of the study, data acquisition/analysis, and manuscript preparation. EMC and TLB have made key contributions to data acquisition. Both MKH and VJS have substantial contributions to study conceptualization and writing of the manuscript.
References
- Barton MD, Hughes KL. Ecology of Rhodococcus equi. Vet Microbiol. 1984;9:65–76. doi: 10.1016/0378-1135(84)90079-8. [DOI] [PubMed] [Google Scholar]
- Benoit S, Benachour A, Taouji S, Auffray Y, Hartke A. Induction of vap genes encoded by the virulence plasmid of Rhodococcus equi during acid tolerance response. Res Microbiol. 2001;152:439–449. doi: 10.1016/s0923-2508(01)01217-7. [DOI] [PubMed] [Google Scholar]
- Botstein D, Fink GR. Yeast: an experimental organism for 21st Century biology. Genetics. 2011;189:695–704. doi: 10.1534/genetics.111.130765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burd CG, Emr SD. Phosphatidylinositol(3)-phosphate signaling mediated by specific binding to RING FYVE domains. Mol Cell. 1998;2:157–162. doi: 10.1016/s1097-2765(00)80125-2. [DOI] [PubMed] [Google Scholar]
- Burton AJ, Giguere S, Berghaus LJ, Hondalus MK, Arnold RD. Efficacy of liposomal gentamicin against Rhodococcus equi in a mouse infection model and colocalization with R. equi in equine alveolar macrophages. Vet Microbiol. 2015;176:292–300. doi: 10.1016/j.vetmic.2015.01.015. [DOI] [PubMed] [Google Scholar]
- Byrne BA, Prescott JF, Palmer GH, Takai S, Nicholson VM, Alperin DC, Hines SA. Virulence plasmid of Rhodococcus equi contains inducible gene family encoding secreted proteins. Infect Immun. 2001;69:650–656. doi: 10.1128/IAI.69.2.650-656.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrotte M, Chasserot-Golaz S, Huang P, Du G, Ktistakis NT, Frohman MA, Vitale N, Bader MF, Grant NJ. Dynamics and function of phospholipase D and phosphatidic acid during phagocytosis. Traffic. 2006;7:365–377. doi: 10.1111/j.1600-0854.2006.00389.x. [DOI] [PubMed] [Google Scholar]
- Coulson GB, Agarwal S, Hondalus MK. Characterization of the role of the pathogenicity island and vapG in the virulence of the intracellular actinomycete pathogen Rhodococcus equi. Infect Immun. 2010;78:3323–3334. doi: 10.1128/IAI.00081-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulson GB, Miranda-CasoLuengo AA, Miranda-CasoLuengo R, Wang X, Oliver J, Willingham-Lane JM, Meijer WG, Hondalus MK. Transcriptome reprogramming by plasmid-encoded transcriptional regulators is required for host niche adaption of a macrophage pathogen. Infect Immun. 2015;83:3137–3145. doi: 10.1128/IAI.00230-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng W, Marshall NC, Rowland JL, McCoy JM, Worrall LJ, Santos AS, Strynadka NCJ, Finlay BB. Assembly, structure, function and regulation of type III secretion systems. Nat Rev Microbiol. 2017;15:323–337. doi: 10.1038/nrmicro.2017.20. [DOI] [PubMed] [Google Scholar]
- Fang Y, Vilella-Bach M, Bachmann R, Flanigan A, Chen J. Phosphatidic acid-mediated mitogenic activation of mTOR signaling. Science. 2001;294:1942–1945. doi: 10.1126/science.1066015. [DOI] [PubMed] [Google Scholar]
- Fernandez-Mora E, Polidori M, Luhrmann A, Schaible UE, Haas A. Maturation of Rhodococcus equi-containing vacuoles is arrested after completion of the early endosome stage. Traffic. 2005;6:635–653. doi: 10.1111/j.1600-0854.2005.00304.x. [DOI] [PubMed] [Google Scholar]
- Flannagan RS, Cosio G, Grinstein S. Antimicrobial mechanisms of phagocytes and bacterial evasion strategies. Nat Rev Microbiol. 2009;7:355–366. doi: 10.1038/nrmicro2128. [DOI] [PubMed] [Google Scholar]
- Flinn RJ, Yan Y, Goswami S, Parker PJ, Backer JM. The late endosome is essential for mTORC1 signaling. Mol Biol Cell. 2010;21:833–841. doi: 10.1091/mbc.E09-09-0756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn O, Quigley F, Costello E, O’Grady D, Gogarty A, Mc Guirk J, Takai S. Virulence-associated protein characterisation of Rhodococcus equi isolated from bovine lymph nodes. Vet Microbiol. 2001;78:221–228. doi: 10.1016/s0378-1135(00)00297-2. [DOI] [PubMed] [Google Scholar]
- Frondorf K, Henkels KM, Frohman MA, Gomez-Cambronero J. Phosphatidic acid is a leukocyte chemoattractant that acts through S6 kinase signaling. J Biol Chem. 2010;285:15837–15847. doi: 10.1074/jbc.M109.070524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geerds C, Wohlmann J, Haas A, Niemann HH. Structure of Rhodococcus equi virulence-associated protein B (VapB) reveals an eight-stranded antiparallel beta-barrel consisting of two Greek-key motifs. Acta Crystallogr F Struct Biol Commun. 2014;70:866–871. doi: 10.1107/S2053230X14009911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gietz RD, Woods RA. Transformation of yeast by lithium acetate/single-stranded carrier DNA/polyethylene glycol method. Methods Enzymol. 2002;350:87–96. doi: 10.1016/s0076-6879(02)50957-5. [DOI] [PubMed] [Google Scholar]
- Giguere S. Treatment of Infections Caused by Rhodococcus equi. Vet Clin North Am Equine Pract. 2017 doi: 10.1016/j.cveq.2016.11.002. [DOI] [PubMed] [Google Scholar]
- Giguere S, Cohen ND, Chaffin MK, Hines SA, Hondalus MK, Prescott JF, Slovis NM. Rhodococcus equi: clinical manifestations, virulence, and immunity. J Vet Intern Med. 2011;25:1221–1230. doi: 10.1111/j.1939-1676.2011.00804.x. [DOI] [PubMed] [Google Scholar]
- Giguere S, Hondalus MK, Yager JA, Darrah P, Mosser DM, Prescott JF. Role of the 85-kilobase plasmid and plasmid-encoded virulence-associated protein A in intracellular survival and virulence of Rhodococcus equi. Infect Immun. 1999;67:3548–3557. doi: 10.1128/iai.67.7.3548-3557.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giguere S, Prescott JF. Cytokine induction in murine macrophages infected with virulent and avirulent Rhodococcus equi. Infect Immun. 1998;66:1848–1854. doi: 10.1128/iai.66.5.1848-1854.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hondalus MK, Mosser DM. Survival and replication of Rhodococcus equi in macrophages. Infect Immun. 1994;62:4167–4175. doi: 10.1128/iai.62.10.4167-4175.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooper-McGrevy KE, Wilkie BN, Prescott JF. Immunoglobulin G subisotype responses of pneumonic and healthy, exposed foals and adult horses to Rhodococcus equi virulence-associated proteins. Clin Diagn Lab Immunol. 2003;10:345–351. doi: 10.1128/CDLI.10.3.345-351.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacks S, Giguere S, Prescott JF. In vivo expression of and cell-mediated immune responses to the plasmid-encoded virulence-associated proteins of Rhodococcus equi in foals. Clin Vaccine Immunol. 2007;14:369–374. doi: 10.1128/CVI.00448-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain S, Bloom BR, Hondalus MK. Deletion of vapA encoding Virulence Associated Protein A attenuates the intracellular actinomycete Rhodococcus equi. Mol Microbiol. 2003;50:115–128. doi: 10.1046/j.1365-2958.2003.03689.x. [DOI] [PubMed] [Google Scholar]
- Kakuda T, Hirota T, Takeuchi T, Hagiuda H, Miyazaki S, Takai S. VirS, an OmpR/PhoB subfamily response regulator, is required for activation of vapA gene expression in Rhodococcus equi. BMC Microbiol. 2014;14:243. doi: 10.1186/s12866-014-0243-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakuda T, Miyazaki S, Hagiuda H, Takai S. Transcriptional regulation by VirR and VirS of members of the Rhodococcus equi virulence-associated protein multigene family. Microbiol Immunol. 2015;59:495–499. doi: 10.1111/1348-0421.12277. [DOI] [PubMed] [Google Scholar]
- Komijn RE, Wisselink HJ, Rijsman VM, Stockhofe-Zurwieden N, Bakker D, van Zijderveld FG, Eger T, Wagenaar JA, Putirulan FF, Urlings BA. Granulomatous lesions in lymph nodes of slaughter pigs bacteriologically negative for Mycobacterium avium subsp. avium and positive for Rhodococcus equi. Vet Microbiol. 2007;120:352–357. doi: 10.1016/j.vetmic.2006.10.031. [DOI] [PubMed] [Google Scholar]
- Kooijman EE, Chupin V, Fuller NL, Kozlov MM, de Kruijff B, Burger KN, Rand PR. Spontaneous curvature of phosphatidic acid and lysophosphatidic acid. Biochemistry. 2005;44:2097–2102. doi: 10.1021/bi0478502. [DOI] [PubMed] [Google Scholar]
- Lemmon MA. Membrane recognition by phospholipid-binding domains. Nat Rev Mol Cell Biol. 2008;9:99–111. doi: 10.1038/nrm2328. [DOI] [PubMed] [Google Scholar]
- Letek M, Gonzalez P, Macarthur I, Rodriguez H, Freeman TC, Valero-Rello A, Blanco M, Buckley T, Cherevach I, Fahey R, et al. The genome of a pathogenic rhodococcus: cooptive virulence underpinned by key gene acquisitions. PLoS Genet. 2010;6:e1001145. doi: 10.1371/journal.pgen.1001145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letek M, Ocampo-Sosa AA, Sanders M, Fogarty U, Buckley T, Leadon DP, Gonzalez P, Scortti M, Meijer WG, Parkhill J, et al. Evolution of the Rhodococcus equi vap pathogenicity island seen through comparison of host-associated vapA and vapB virulence plasmids. J Bacteriol. 2008;190:5797–5805. doi: 10.1128/JB.00468-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim HK, Choi YA, Park W, Lee T, Ryu SH, Kim SY, Kim JR, Kim JH, Baek SH. Phosphatidic acid regulates systemic inflammatory responses by modulating the Akt-mammalian target of rapamycin-p70 S6 kinase 1 pathway. J Biol Chem. 2003;278:45117–45127. doi: 10.1074/jbc.M303789200. [DOI] [PubMed] [Google Scholar]
- Makrai L, Takai S, Tamura M, Tsukamoto A, Sekimoto R, Sasaki Y, Kakuda T, Tsubaki S, Varga J, Fodor L, et al. Characterization of virulence plasmid types in Rhodococcus equi isolates from foals, pigs, humans and soil in Hungary. Vet Microbiol. 2002;88:377–384. doi: 10.1016/s0378-1135(02)00157-8. [DOI] [PubMed] [Google Scholar]
- Makrai L, Takayama S, Denes B, Hajtos I, Sasaki Y, Kakuda T, Tsubaki S, Major A, Fodor L, Varga J, et al. Characterization of virulence plasmids and serotyping of rhodococcus equi isolates from submaxillary lymph nodes of pigs in Hungary. J Clin Microbiol. 2005;43:1246–1250. doi: 10.1128/JCM.43.3.1246-1250.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muscatello G, Anderson GA, Gilkerson JR, Browning GF. Associations between the ecology of virulent Rhodococcus equi and the epidemiology of R. equi pneumonia on Australian thoroughbred farms. Appl Environ Microbiol. 2006;72:6152–6160. doi: 10.1128/AEM.00495-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ocampo-Sosa AA, Lewis DA, Navas J, Quigley F, Callejo R, Scortti M, Leadon DP, Fogarty U, Vazquez-Boland JA. Molecular epidemiology of Rhodococcus equi based on traA, vapA, and vapB virulence plasmid markers. J Infect Dis. 2007;196:763–769. doi: 10.1086/519688. [DOI] [PubMed] [Google Scholar]
- Odorizzi G, Babst M, Emr SD. Fab1p PtdIns(3)P 5-kinase function essential for protein sorting in the multivesicular body. Cell. 1998;95:847–858. doi: 10.1016/s0092-8674(00)81707-9. [DOI] [PubMed] [Google Scholar]
- Okoko T, Blagova EV, Whittingham JL, Dover LG, Wilkinson AJ. Structural characterisation of the virulence-associated protein VapG from the horse pathogen Rhodococcus equi. Vet Microbiol. 2015;179:42–52. doi: 10.1016/j.vetmic.2015.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putta P, Rankenberg J, Korver RA, van Wijk R, Munnik T, Testerink C, Kooijman EE. Phosphatidic acid binding proteins display differential binding as a function of membrane curvature stress and chemical properties. Biochim Biophys Acta. 2016;1858:2709–2716. doi: 10.1016/j.bbamem.2016.07.014. [DOI] [PubMed] [Google Scholar]
- Rofe AP, Davis LJ, Whittingham JL, Latimer-Bowman EC, Wilkinson AJ, Pryor PR. The Rhodococcus equi virulence protein VapA disrupts endolysosome function and stimulates lysosome biogenesis. Microbiologyopen. 2016 doi: 10.1002/mbo3.416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sangkanjanavanich N, Kawai M, Kakuda T, Takai S. Rescue of an intracellular avirulent Rhodococcus equi replication defect by the extracellular addition of virulence-associated protein A. J Vet Med Sci. 2017 doi: 10.1292/jvms.17-0350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlam D, Bohdanowicz M, Chatgilialoglu A, Steinberg BE, Ueyama T, Du G, Grinstein S, Fairn GD. Diacylglycerol kinases terminate diacylglycerol signaling during the respiratory burst leading to heterogeneous phagosomal NADPH oxidase activation. J Biol Chem. 2013;288:23090–23104. doi: 10.1074/jbc.M113.457606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheffield P, Garrard S, Derewenda Z. Overcoming expression and purification problems of RhoGDI using a family of “parallel” expression vectors. Protein Expr Purif. 1999;15:34–39. doi: 10.1006/prep.1998.1003. [DOI] [PubMed] [Google Scholar]
- Shin JJ, Loewen CJ. Putting the pH into phosphatidic acid signaling. BMC Biol. 2011;9:85. doi: 10.1186/1741-7007-9-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siggers KA, Lesser CF. The Yeast Saccharomyces cerevisiae: a versatile model system for the identification and characterization of bacterial virulence proteins. Cell Host Microbe. 2008;4:8–15. doi: 10.1016/j.chom.2008.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takai S. Epidemiology of Rhodococcus equi infections: a review. Vet Microbiol. 1997;56:167–176. doi: 10.1016/s0378-1135(97)00085-0. [DOI] [PubMed] [Google Scholar]
- Takai S, Fukunaga N, Ochiai S, Imai Y, Sasaki Y, Tsubaki S, Sekizaki T. Identification of intermediately virulent Rhodococcus equi isolates from pigs. J Clin Microbiol. 1996;34:1034–1037. doi: 10.1128/jcm.34.4.1034-1037.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takai S, Iie M, Watanabe Y, Tsubaki S, Sekizaki T. Virulence-associated 15- to 17-kilodalton antigens in Rhodococcus equi: temperature-dependent expression and location of the antigens. Infect Immun. 1992;60:2995–2997. doi: 10.1128/iai.60.7.2995-2997.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takai S, Koike K, Ohbushi S, Izumi C, Tsubaki S. Identification of 15- to 17-kilodalton antigens associated with virulent Rhodococcus equi. J Clin Microbiol. 1991;29:439–443. doi: 10.1128/jcm.29.3.439-443.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takai S, Michizoe T, Matsumura K, Nagai M, Sato H, Tsubaki S. Correlation of in vitro properties of Rhodococcus (Corynebacterium) equi with virulence for mice. Microbiol Immunol. 1985;29:1175–1184. doi: 10.1111/j.1348-0421.1985.tb00907.x. [DOI] [PubMed] [Google Scholar]
- Tan C, Prescott JF, Patterson MC, Nicholson VM. Molecular characterization of a lipid-modified virulence-associated protein of Rhodococcus equi and its potential in protective immunity. Can J Vet Res. 1995;59:51–59. [PMC free article] [PubMed] [Google Scholar]
- Toyooka K, Takai S, Kirikae T. Rhodococcus equi can survive a phagolysosomal environment in macrophages by suppressing acidification of the phagolysosome. J Med Microbiol. 2005;54:1007–1015. doi: 10.1099/jmm.0.46086-0. [DOI] [PubMed] [Google Scholar]
- Tripathi VN, Harding WC, Willingham-Lane JM, Hondalus MK. Conjugal transfer of a virulence plasmid in the opportunistic intracellular actinomycete Rhodococcus equi. J Bacteriol. 2012;194:6790–6801. doi: 10.1128/JB.01210-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valero-Rello A, Hapeshi A, Anastasi E, Alvarez S, Scortti M, Meijer WG, MacArthur I, Vazquez-Boland JA. An Invertron-Like Linear Plasmid Mediates Intracellular Survival and Virulence in Bovine Isolates of Rhodococcus equi. Infect Immun. 2015;83:2725–2737. doi: 10.1128/IAI.00376-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazquez-Boland JA, Giguere S, Hapeshi A, MacArthur I, Anastasi E, Valero-Rello A. Rhodococcus equi: the many facets of a pathogenic actinomycete. Vet Microbiol. 2013;167:9–33. doi: 10.1016/j.vetmic.2013.06.016. [DOI] [PubMed] [Google Scholar]
- Vieira OV, Botelho RJ, Grinstein S. Phagosome maturation: aging gracefully. Biochem J. 2002;366:689–704. doi: 10.1042/BJ20020691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Bargen K, Polidori M, Becken U, Huth G, Prescott JF, Haas A. Rhodococcus equi virulence-associated protein A is required for diversion of phagosome biogenesis but not for cytotoxicity. Infect Immun. 2009;77:5676–5681. doi: 10.1128/IAI.00856-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Coulson GB, Miranda-Casoluengo AA, Miranda-Casoluengo R, Hondalus MK, Meijer WG. IcgA is a virulence factor of Rhodococcus equi that modulates intracellular growth. Infect Immun. 2014;82:1793–1800. doi: 10.1128/IAI.01670-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Devaiah SP, Zhang W, Welti R. Signaling functions of phosphatidic acid. Prog Lipid Res. 2006;45:250–278. doi: 10.1016/j.plipres.2006.01.005. [DOI] [PubMed] [Google Scholar]
- Weinstock DM, Brown AE. Rhodococcus equi: an emerging pathogen. Clin Infect Dis. 2002;34:1379–1385. doi: 10.1086/340259. [DOI] [PubMed] [Google Scholar]
- Whittingham JL, Blagova EV, Finn CE, Luo H, Miranda-CasoLuengo R, Turkenburg JP, Leech AP, Walton PH, Murzin AG, Meijer WG, et al. Structure of the virulence-associated protein VapD from the intracellular pathogen Rhodococcus equi. Acta Crystallogr D Biol Crystallogr. 2014;70:2139–2151. doi: 10.1107/S1399004714012632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willingham-Lane JM, Berghaus LJ, Giguere S, Hondalus MK. Influence of Plasmid Type on the Replication of Rhodococcus equi in Host Macrophages. mSphere. 2016;1 doi: 10.1128/mSphere.00186-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinser E, Sperka-Gottlieb CD, Fasch EV, Kohlwein SD, Paltauf F, Daum G. Phospholipid synthesis and lipid composition of subcellular membranes in the unicellular eukaryote Saccharomyces cerevisiae. J Bacteriol. 1991;173:2026–2034. doi: 10.1128/jb.173.6.2026-2034.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoncu R, Bar-Peled L, Efeyan A, Wang S, Sancak Y, Sabatini DM. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Science. 2011;334:678–683. doi: 10.1126/science.1207056. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.