

Abstract
Purpose
The mechanism of action of CNDAC (2′-C-cyano-2′-deoxy-1-β-d-arabino-pentofuranosyl-cytosine) is unique among deoxycytidine analogs because upon incorporation into DNA it causes a single strand break which is converted to a double strand break after DNA replication. This lesion requires homologous recombination (HR) for repair. CNDAC, as the parent nucleoside, DFP10917, and as an oral prodrug, sapacitabine, are undergoing clinical trials for hematological malignancies and solid tumors. The purpose of this study is to investigate the potential of CNDAC for the therapy of ovarian cancer (OC).
Methods
Drug sensitivity was evaluated using a clonogenic survival assay. Drug combination effects were quantified by median effect analysis.
Results
OC cells lacking function of the key HR genes, BRCA1 or BRCA2, were more sensitive to CNDAC than corresponding HR proficient cells. The sensitization was associated with greater levels of DNA damage in response to CNDAC at clinically achievable concentrations, manifested as chromosomal aberrations. Three classes of CNDAC-based drug combinations were investigated. First, the PARP1 inhibitors, rucaparib and talazoparib, were selectively synergistic with CNDAC in BRCA1/2 deficient OC cells (combination index < 1) at a relatively low concentration range. Second, cisplatin and oxaliplatin had additive combination effects with CNDAC (combination index ~ 1). Finally, paclitaxel and docetaxel achieved additive cell-killing effects with CNDAC at concentration ranges of the taxanes similar for both BRCA1/2 deficient and proficient OC cells.
Conclusions
This study provides mechanistic rationales for combining CNDAC with PARP inhibitors, platinum compounds and taxanes in ovarian cancer lacking BRCA1/2 function.
Keywords: CNDAC, PARP inhibitor, Platinum compound, Taxane, Clonogenic survival, Drug combination
Introduction
Ovarian cancer (OC) ranks the fifth of all women’s cancers with very high mortality (69%) relative to breast cancer (19%) [1]. Nearly 30 out of 100 women with a BRCA1 or BRCA2 gene mutation will develop OC by the age of 70 years, compared to fewer than 1 out of 100 women in the general US population [2]. Although a small population (5–13%) of OC patients carry germline mutations in BRCA1 or BRCA2, somatic BRCA1/2 mutations [3, 4] and hypermethylation in the BRCA1 promoter region [5, 6, 7, 8] have been revealed in sporadic ovarian cancers. Loss of BRCA1/2 function causes a deficiency in homologous recombination repair of DNA double strand breaks.
Current therapies for OC include the first-line chemotherapeutics, platinum compounds and taxanes. Tumors with loss of BRCA1/2 function are sensitized to platinum compound therapy, whereas the taxanes are largely non-selective. In recent years, PARP inhibitors (PARPis) have emerged as a class of novel therapeutic agents preferentially targeting BRCA1/2-deficient cells due to synthetic lethality [9, 10, 11]. Three PARPis—olaparib, rucaparib and niraparib—have been approved by the FDA to treat patients with advanced ovarian cancer that is associated with deleterious BRCA mutations. However, BRCA1/2-mutated cancer cells are able to acquire resistance to cisplatin and PARPis by restoring BRCA1/2 functions through secondary BRCA1/ 2 mutations [12, 13, 14, 15, 16]. Therefore, there is a need to develop novel therapeutic agents and combinations to target BRCA1/2-deficient OC and overcome such resistance.
CNDAC is a nucleoside analog with a unique action mechanism of inducing single strand breaks (SSBs) after incorporation into DNA. Incorporated CNDAC is transformed into a de facto chain terminator, CNddC, which is extremely hard to remove [17, 18, 19]. The SSBs are subsequently converted into double strand breaks (DSBs) during a second S-phase. This unique mechanism of action has distinguished CNDAC among deoxycytidine analogs. We demonstrated that CNDAC-induced DNA damage is primarily repaired through the ATM-dependent homologous recombination (HR) pathway, shown by sensitization of cells deficient in either ATM, RAD51D, XRCC3 or BRCA2 [20]. In contrast, cells lacking HR function are not sensitized to cytarabine or gemcitabine [21]. Repair of CNDAC-induced damage is independent of p53 [20]. Shapiro’s group reported hypersensitivity of human colon cancer cells lacking BRCA1 or BRCA2 to CNDAC [22], confirming our findings in hamster lines [20].
Sapacitabine, the orally bioavailable prodrug of CNDAC, is undergoing clinical trials in acute myeloid leukemia and MDS with encouraging outcomes [23, 24]. It is currently in Phase III registration trial for elderly AML patients (NCT01303796). CNDAC (as DFP-10,917) is in Phase I/II trial for AML and ALL (NCT01702155), and clinical responses in treatment for refractory or relapsed AML patients has been reported [25]. In this study, we investigated response of ovarian cancer (OC) cell lines to CNDAC and explored CNDAC-based combination strategies in BRCA1/2 deficient and proficient OC cells. A corollary is that matching the mechanism of action of sapacitabine or CNDAC to the Brca1/2 deficiency in OC may selectively sensitize the tumor.
Materials and methods
Materials
The nucleoside analogue CNDAC was synthesized as described [17]. CO-338 (PF-01367338-BW, rucaparib camsylate salt) was obtained from Clovis Oncology through MTA. Talazoparib (BMN 673), cisplatin, oxaliplatin, paclitaxel, and docetaxel were purchased from Selleck Chemicals (Houston, TX).
Cell lines
The BRCA1-null ovarian carcinoma cell line, UWB1.289 (designated as “− BRCA1”) and its complemented line, UWB1.289 + Brca1 (designated as “+BRCA1”) [26] were purchased from the American Type Culture Collection (ATCC). Both lines were grown in a mixture of two basic media − 50% ATCC-formulated RPMI-1640 and 50% mammary epithelial growth medium (http://www.atcc.org/Products/All/CRL-2945.aspx#culturemethod (http://www.atcc.org/Products/All/CRL-2945.aspx#culturemethod)) supplemented with 3% fetal bovine serum (FBS). G418 was added in the culture medium for the + BRCA1 line to a final concentration of 200 μg/mL. BRCA2-mutant ovarian adenocarcinoma cell line, PEO1 (designated as “− BRCA2”) and its revertant line, PEO1 C4-2 (designated as “BRCA2 rev.”) [16] were kindly provided by Dr. Tashiyasu Taniguchi (Fred Hutchinson Cancer Research Center, Seattle, WA). Both lines were cultured in DMEM supplemented with 15% FBS and Glutamax. All cells were free of mycoplasma, as certified by Characterized Cell Line Core Facility at M. D. Anderson Cancer Center (MDACC) using the MycoAlert kit from Lonza (Switzerland). Short tandem repeat DNA fingerprinting was used to authenticate human cell lines by the Characterized Cell Line Core. All lines were used in experiments for no more than 15 passages after recovering from frozen seed stocks that were validated prior to freezing.
Immunoblotting
Whole cell lysates were loaded on 4–12% Bis-Tris gradient gels (Bio-Rad) and separated proteins were transferred onto nitrocellulose membranes. Immunoblotting images were captured by Odyssey CLx Imaging System (Li-Cor, Lincoln, NE). The following antibodies were used: BRCA1 polyclonal C-20, Santa Cruz (sc-642); BRCA1 monoclonal MS110, Calbiochem (OP92); BRCA2 polyclonal, BioVision (#3675); β-actin monoclonal AC-15, Sigma–Aldrich (A5441).
Cytogenetic analysis
Colcemid (100 ng/mL) was added to cell cultures before harvesting. Mitotic spreads were prepared as described [27]. A minimum of 50 metaphase spreads were analyzed for each sample by the T.C. Hsu Molecular Cytogenetics Core at MDACC.
Clonogenic cell survival assay
Exponentially growing cells were seeded to attach in 6-well plates 1 day before cells were exposed to a range of concentrations of drugs for 24 h. After washing into drug-free medium or medium containing a PARPi, cells were incubated for 7–8 days depending on the growth rate of the particular cell line, and colonies (containing ≥ 50 cells) in triplicate wells were stained with crystal violet after fixation. Then colonies were counted either manually under a dissecting microscope or electronically using GelCount (Oxford Optronix, Oxford, UK). IC50 values of single agents were calculated using the GraphPad Prism6 software (GraphPad Software, Inc. San Diego, CA) based on clonogenic survival rates in triplicate.
Median-effect analysis of drug combinations
Cells were treated with two agents combined in fixed molar ratios over a range of concentrations for each agent that achieve 10–90% clonogenic inhibition alone. Clonogenic survival was analyzed by the median-effect method [28, 29] using the CalcuSyn software (Biosoft, Ferguson, MO) as described [30]. The calculated combination index (CI) < 1, = 1 and > 1 indicate being synergistic, additive and antagonist, respectively.
Results
Ovarian cancer cells deficient in BRCA1 and BRCA2 are sensitive to CNDAC
To determine the impact of BRCA1 and BRCA2 on sensitivity to CNDAC, we used clonogenic assays to compare OC cells lacking BRCA1 or BRCA2 with the respective proficient cells. The BRCA1-null OC cell line, UWB1.289, was > threefold more sensitive to CNDAC (IC50 ~ 10 nM, mean of three independent experiments) than the BRCA1-complemented line, UWB1.289 + BRCA1 (mean IC50 ~ 31 nM, Fig. 1a). Similarly, the BRCA2 mutant and defective cell line, PEO1, was > fivefold more sensitive to CNDAC (IC50 ~ 10 nM, mean of two independent experiments) compared to PEO1 C4-2 line (mean IC ~ 59 nM, Fig. 2a), which has restored BRCA2 function by a secondary 50 mutation [16]. Thus, both BRCA1 and BRCA2 are required for OC cells to repair CNDAC-induced DSBs.
Fig. 1.
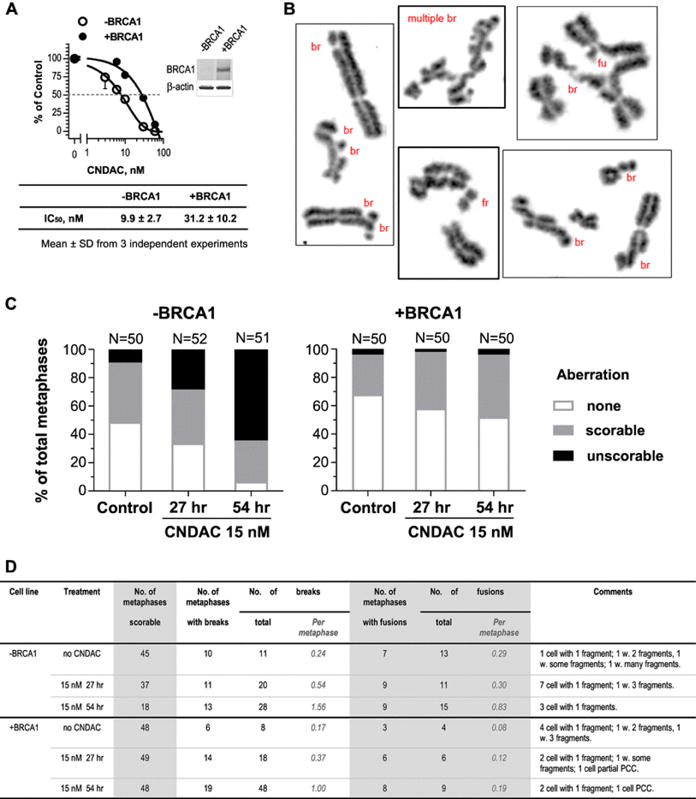
BRCA1 deficiency confers sensitivity to CNDAC in ovarian cancer cells, which is associated with greater level of chromosomal aberrations. a Clonogenic survival of UWB1.289 (BRCA1 deficient, − BRCA1) and UWB1.289 + BRCA1 (complemented, + BRCA1) cell lines in response to CNDAC (3–60 nM, 24-hour exposure). Percentage of cell survival in both cell lines is plotted as a function of drug concentration, and a representative plot is shown. IC50 values of CNDAC (Mean ± SD from three independent experiments) are listed. The insert shows the immunoblot of BRCA1 and β-actin (loading control) proteins. b–d CNDAC induces chromosomal aberrations in cells deficient and proficient in Brca1. b Representative images of chromosome abnormalities in − BRCA1 cells with many aberrations are shown. br Break, fu fusion, fr fragment. c Percentage of metaphases with chromosomal structural aberrations (including breaks, fusions and fragments) detected in − BRCA1 and + BRCA1 cell lines in response to 15 nM CNDAC for 27 and 54 h. d More details of chromosomal aberrations detected in metaphases that are scorable (with or without aberration) as in c. The number of scorable metaphases is calculated by subtracting the number of unscorable metaphases from the examined total. PCC stands for premature chromosome condensation
Fig. 2.
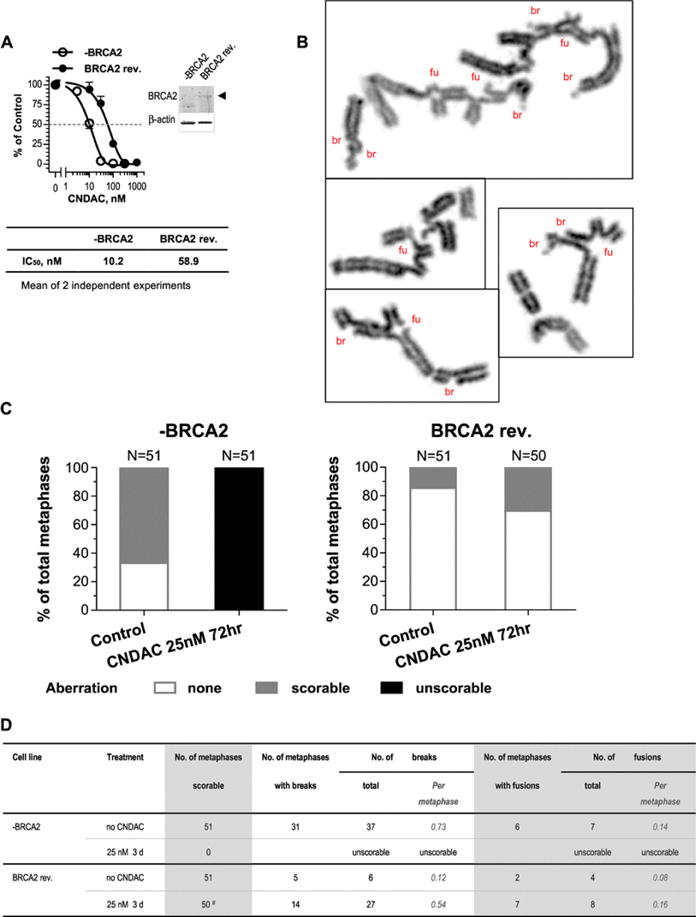
BRCA2 deficiency also confers sensitivity to CNDAC in ovarian cancer cells, which is associated with greater level of chromosomal aberrations. a Clonogenic survival of PEO1 (BRCA2 deficient, -BRCA2) and PEO1 C4-2 (BRCA2 revertant, BRCA2 rev.) cell lines after a 24-hour incubation with CNDAC (3–300 nM and 10–1000 nM, respectively). Percentage of cell survival in both cell lines is plotted as a function of drug concentration, and a representative plot is shown. IC50 values of CNDAC (mean of two independent experiments) are presented. The insert shows the immunoblot of BRCA2 and β-actin proteins. b–d CNDAC induces chromosomal aberrations in Brca2 deficient and repleted cells. b Representative images of chromosome abnormalities in − BRCA2 cells with many aberrations are shown. br Break, fu fusion. c Percentage of metaphases with chromosomal aberrations determined in − BRCA2 and BRCA2 rev. cell lines in response to 25 nM CNDAC for 72 h. d More details of chromosomal aberrations detected in metaphases that are scorable. The number of scorable metaphases is calculated by subtracting the number of unscorable metaphases from the examined total. # one metaphase with too many aberrations excluded. Those numbers are not added in the table. That particular metaphase has over 25 breaks and about 11 fusions
To find the underlying cause for the sensitization of BRCA1/2 deficient OC cells to CNDAC, we measured DNA damage at the chromosomal level (Fig. 1 b). Before CNDAC treatment, 52% (26/50) of BRCA1-null UWB1.289 cells had aberrant metaphases, including 10% (5/50) metaphases which had too many aberrations to allow accurate counting (unscorable, Fig. 1 c). Among the scorable metaphases—the sum of metaphases without aberration and with scorable aberrations (90%, 45/50,), each had average 0.24 chromosomal break and 0.29 fusion per metaphase (Fig. 1 d). The ratio of aberrant metaphases in BRCA1-complemented cells was 32% (16/50), including 4% (2/50) with unscorable aberrations (Fig. 1 c). The scorable metaphases (96%, 48/50) each had average 0.17 break and 0.08 fusion (Fig. 1 d). Thus, lack of BRCA1 led to a greater basal level of chromosomal aberrations. When exposed to 15 nM CNDAC for 27 h (approx. one cell cycle), more BRCA1-null cells had aberrant mitosis (67%, 35/52), including 29% (15/52) metaphases with severe chromosomal damage (unscorable, Fig. 1 c). Among the scorable metaphases (71%, 37/52), the average break number was 0.54 and fusion number 0.3 per metaphase (Fig. 1 d). The ratios of aberrant metaphase (94%, 48/51) and unscorable aberrant metaphase (65%, 33/51) further increased dramatically with cells exposed to CNDAC after a second cell cycle at 54 h (Fig. 1 c), demonstrating that more DSBs are formed after a second S-phase. The average numbers of breaks and fusions were 1.56 and 0.83, respectively, among those scorable metaphases (35%, 18/51, Fig. 1 b, d). In contrast, when BRCA1-repleted cells were treated with 15 nM CNDAC, the ratio of aberrant mitosis increased to 42% (21/50) at 27 h and 48% (24/50) at 54 h. The ratio of unscorable aberrant mitosis remained constant (2–4%, Fig. 1 c). The average break and fusion numbers in those scorable metaphases did increase, but far less than the BRCA1-null cells (Fig. 1 d). These results demonstrate a clear relationship between the deficiency in BRCA1 and the greater level of chromosomal damage after CNDAC, indicating that BRCA1 is essential for the repair of CNDAC-induced DSBs.
The basal level of chromosomal aberrations in BRCA2 deficient PEO1 cells (67% with aberrations) was much greater than that in BRCA2 restored PEO1 C4–2 cells (14% with aberrations, Fig. 2c). This is similar to the BRCA1 deficient situation, suggesting innate genetic instability in the absence of BRCA1/2. Remarkably, after a 72-hour incubation with 25 nM CNDAC, all metaphases in BRCA2 mutant cells presented severe chromosomal damage which could not warrant accurate counting (100% unscorable, Fig. 2b–d), whereas only 30% (15/50) metaphases in BRCA2 reverted cells had aberrations and all of them were scorable (Fig. 2c, d). Therefore, BRCA2 is also required for cells to repair DNA damage caused by CNDAC. These cytogenetic studies provide direct evidence for the sensitization of OC cells to CNDAC in the scenario of BRCA1/2 deficiency.
To evaluate the possibility that distinct levels of chromosomal lesion after CNDAC exposure is attributed to differential accumulation of the nucleoside analog in cells, we measured cellular levels of the triphosphate of CNDAC, CNDACTP by anion-exchange HPLC. Both BRCA2-mutant and -revertant cell lines showed a slight dependency of CNDACTP accumulation on drug concentration (1 and 3 μM) and incubation time (2 and 4.5 h). However, there was no significant difference between the mutant and restored cells comparing CNDACTP concentrations detected from whole cell extracts under all conditions (Fig. S1).
Synergistic interaction between CNDAC and PARP inhibitors is selective in BRCA1 and BRCA2 defective ovarian cancer cells
The combination of CNDAC with a PARPi is synergistic in rodent cells with defective homologous recombination (HR) function, such as those lacking XRCC3, RAD51C or RAD51D [30]. We hypothesized that CNDAC potentiates the synthetic lethal condition in HR deficient cells exposed to a PARPi, because CNDAC-induced DNA damage largely depends on the HR pathways for repair [20], whereas PARPi affects both the base excision repair (BER) and HR pathways [31, 32]. To test this combination strategy in human ovarian cancer cells. BRCA1-null OC cells were ~ 80 fold more sensitive to rucaparib (IC50 ~ 13 nM) than were BRCA1 complemented cells (IC50 ~ 1040 nM, Fig. S2A). BRCA2-revertant OC cells were 18-fold more resistant to rucaparib (IC50 ~ 1800 nM) than were BRCA2-mutated cells (IC50 ~ 100 nM, Fig. S2B). These results are consistent with the well-accepted reports of synthetic lethal killing of BRCA-defective cells by PARPis [9, 33].
Combination of rucaparib and CNDAC at a ratio of 10:1 achieved CI < 1 in UWB1.289 cells, indicating synergy in a situation of BRCA1 deficiency (Fig. 3 a). The rucaparib-CNDAC combination in BRCA1-complemented cells (ratio 50:1) also achieved CI < 1 (Fig. 3 b), but in much greater concentration ranges of both agents compared to that used for the deficient cells. When rucaparib and CNDAC were combined at a ratio of 10:1 in BRCA2 deficient PEO1 cells, the synergy was more obvious (CI < 1, Fig. 3 c). Similarly, this combination at a ratio of 50:1 (rucaparib : CNDAC) was synergistic (CI < 1) at greater concentrations of each agent in BRCA2-revertant cells relative to that in the mutated cells (Fig. 3 d). We also tested another potent PARPi, talazoparib in the BRCA2 paired cell lines. Combination of talazoparib with CNDAC (ratio 1:12) resulted in CI < 1 in PEO1 cells (four experiments, same below), showing synergistic interaction between the two agents when lacking BRCA2 (Fig. 3 e). Similar to the rucaparib-CNDAC combination, the talazoparib-CNDAC combination achieved CI < 1 in BRCA2-restored cells at much higher concentrations of both agents than that in the mutated cells (Fig. 3 f). Thus, combinations of CNDAC with rucaparib or talazoparib clearly exerted a synergistic interaction between PARPi and CNDAC. For BRCA1/2-deficient cells, the concentrations of both agents needed were too low to kill the proficient cells, demonstrating selectivity for the BRCA1/2 mutants. However, this selectivity of synergistic cell killing was lost at greater concentrations in BRCA1/2-repleted or -restored cells.
Fig. 3.
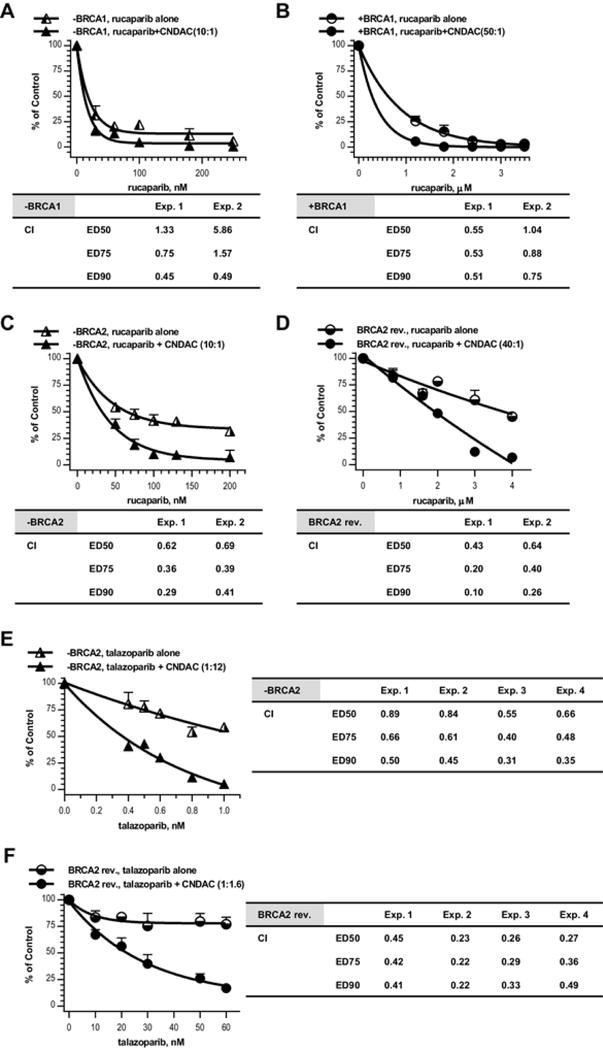
The PARP inhibitors rucaparib and talazoparib synergize with CNDAC in both BRCA1/2 proficient and deficient ovarian cancer cell lines. a BRCA1 deficient (− BRCA1) cells were exposed to rucaparib (30–250 nmol/L) 1 h before addition of CNDAC (3–25 nmol/L). b BRCA1 complemented (+ BRCA1) cells were exposed to rucaparib (1.2–3.5 μmol/L in Exp. 1, or 1– 3 μmol/L in Exp. 2) 1 h before addition of CNDAC (24–70 nmol/L in Exp.1, or 25–75 nmol/L in Exp.2). c BRCA2 deficient (− BRCA2) cells were exposed to rucaparib (5–200 nmol/L) 1 h before addition of CNDAC (5– 20 nmol/L). d BRCA2 revertant (BRCA2 rev.) cells were exposed to rucaparib (0.8–4 μmol/L) 1 h before addition of CNDAC (20–100 nmol/L). a–d 24 h after CNDAC addition, drugs were washed out and cells were incubated in medium with rucaparib at the corresponding concentrations until colonies were fixed. Top, representative of 2 independent experiments, comparison of clonogenicity of − BRCA1 cells after treatment with rucaparib alone versus rucaparib–CNDAC combination at a fixed ratio as indicated. Bottom, CIs from median-effect analysis of the 2 experiments are presented in the accompanying inserts. e BRCA2 deficient (− BRCA2) cells were exposed to talazoparib (0.4–1 nmol/L in Exp. 1 and 2, or 0.4–1.6 nmol/L in Exp. 3 and 4) 1 h before addition of CNDAC (4.8–12 nmol/L in Exp. 1 and 2, or 3.6–14.4 nmol/L in Exp. 3 and 4). f BRCA2 revertant (BRCA2 rev.) cells were exposed to talazoparib (10–60 nmol/L in Exp. 1 and 2, or 15–100 nmol/L in Exp. 3 and 4) 1 h before addition of CNDAC (16–96 nmol/L in Exp. 1 and 2, or 15–100 nmol/L in Exp. 3 and 4). e–f Drugs were washed out 24 h after CNDAC co-incubation, and cells were incubated in medium with talazoparib at the corresponding concentrations until colonies were fixed. Top, representative of four independent experiments, comparison of clonogenicity of BRCA2 deficient cells after treatment with talazoparib alone versus talazoparib–CNDAC combination at a fixed ratio as indicated. Bottom, CIs from median-effect analysis of the four experiments are presented in the accompanying inserts
Combination of CNDAC with platinum compounds
Platinum agents are frequently used in OC chemotherapy regimen. They act by forming intra- and inter-strand crosslinks in DNA. We have reported additive combination effect of cisplatin or oxaliplatin with CNDAC in both HR-proficient and deficient cells [30]. In the context of OC, we sought to investigate the combination effect of CNDAC with cisplatin and oxaliplatin in BRCA1/2 paired OC cell models. In BRCA1-deficient UWB1.289 cells, combination of cisplatin and CNDAC at a ratio of 1.8:1 achieved CI < = 1, while CI for the oxaliplatin–CNDAC combination (6:1) was < 1 (Fig. 4a). Similarly in BRCA1-complemented cells, the cisplatin–CNDAC combination (1.5:1) was additive (CI ~ 1), whereas that of oxaliplatin – CNDAC (5:1) was slightly synergistic (CI < 1, Fig. 4 b). Since the IC50 values of cisplatin and oxaliplatin (single agent) were approximately twice as much in BRCA1-repleted as in BRCA1-defective cells, the combination effect was specific for the defective cells at lower concentrations.
Fig. 4.
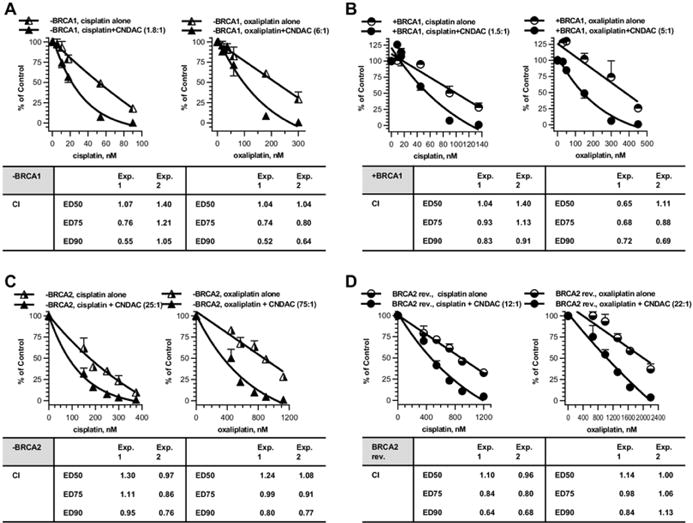
Combinations of platinum compounds cisplatin and oxaliplatin with CNDAC in BRCA1/2 proficient and deficient cells. a BRCA1 deficient OC cells (− BRCA1) were treated concomitantly with cisplatin (5.4– 90 nmol/L) or oxaliplatin (18–300 nmol/L) and then CNDAC (3– 50 nmol/L), and incubated for 24 h before washout. b BRCA1 complemented OC cells (+ BRCA1) were exposed to CNDAC (6– 90 nmol/L) subsequent to cisplatin (9–135 nmol/L) or oxaliplatin (30– 450 nmol/L) for 24 h as in a. c BRCA2 deficient OC cells (− BRCA2) were treated concomitantly with cisplatin (150–375 nmol/L) or oxaliplatin (450–1125 nmol/L) and then CNDAC (6–15 nmol/L), and incubated for 24 h before washout. d BRCA2 revertant OC cells (BRCA2 rev.) were exposed to CNDAC (30–100 nmol/L) subsequent to cisplatin (360– 1200 nmol/L) or oxaliplatin (660–2200 nmol/L) for 24 h as in C. a–d Cells were incubated in drug-free medium until colonies were fixed and quantitated. Top, representative of 2 independent experiments, clonogenicity was compared for treatment with cisplatin alone versus cisplatin–CNDAC combination at a fixed ratio (left graph) or oxaliplatin versus oxaliplatin–CNDAC combination at a fixed ratio (right graph). Bottom, CIs from median-effect analysis of the two experiments are presented in the accompanying inserts
We also compared these combinations in BRCA2 mutated and revertant OC cells. The combination of cisplatin–CNDAC (25:1) in BRCA2 deficient cells was additive (CI < = 1), while that of oxaliplatin–CNDAC (75:1) was synergistic (CI < 1, Fig. 4 c). The trend reversed in BRCA2 restored cells though. The cisplatin–CNDAC combination (12:1) was synergistic (CI < 1), whereas that of oxaliplatin–CNDAC (22:1) was additive (CI < = 1, Fig. 4 d). Since the IC values of cisplatin and oxaliplatin (single agent) were 50 approximately 4–5 fold greater in BRCA2 restored cells than that in BRCA2 defective cells, the combination effect with CNDAC was specific for defective cells at lower concentrations.
Taxane agents and CNDAC have additive effect in cells lacking HR function
Taxanes are another class of agents commonly used in chemotherapy for OC. We have shown additive effect of CNDAC in combination with either of the two well-established tubulin-directed agents, paclitaxel or docetaxel, in HR defective cells and proficient cells as well [30]. Here BRCA1 or BRCA2 deficient OC cells were not sensitized to paclitaxel or docetaxel (Fig. 5 a–d, single agent curves), probably because the taxanes specifically impact on mitotic spindle by suppressing the depolymerization of microtubules. Combination of paclitaxel and CNDAC had additive effect (CI ~ 1) in both BRCA1-null cells (ratio 2:25, Fig. 5 a left panel) and BRCA1-complemented cells (ratio 1:25, Fig. 5 b left panel). Similarly, the docetaxel–CNDAC combination was additive in BRCA1 paired line (ratios 1:40 and 3:250, respectively, Fig. 5 a, b right panels). In the BRCA2 paired cell lines, the combinations of paclitaxel–CNDAC (ratio 3:20 for BRCA2 mutant, 2:75 for BRCA2 revertant) and docetaxel–CNDAC (ratio 3:50 for BRCA2 mutant, 7:500 for BRCA2 revertant) had additive effect with CI ~ 1 (Fig. 5 c, d). This effect is selective in BRCA1/2-deficient cells at lower concentrations of CNDAC that allow survival of proficient cells.
Fig. 5.
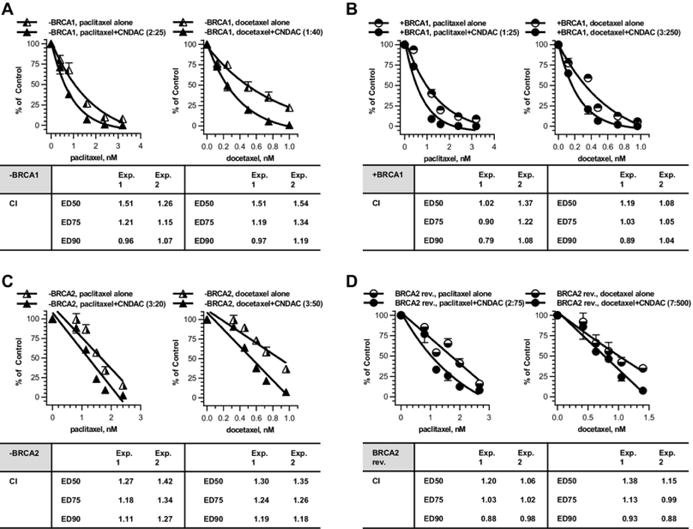
Combinations of taxane compounds paclitaxel and docetaxel with CNDAC in BRCA1/2 proficient and deficient cells. a BRCA1 deficient OC cells (− BRCA1) were treated concomitantly with paclitaxel (0.4–3.2 nmol/L) or docetaxel (0.125–1 nmol/L) and CNDAC (5–40 nmol/L), and incubated for 24 h before washout. b BRCA1 complemented OC cells (+ BRCA1) were exposed concomitantly to paclitaxel (0.4–3.2 nmol/L) or docetaxel (0.12–0.96 nmol/L) and CNDAC (10–80 nmol/L) for 24 h as in a. c BRCA2 deficient OC cells (− BRCA2) were treated concomitantly with paclitaxel (0.81–2.4 nmol/L) or docetaxel (0.324–0.96 nmol/L) and CNDAC (5.4– 16 nmol/L), and incubated for 24 h before washout. d BRCA2 revertant OC cells (BRCA2 rev.) were exposed to CNDAC (30–100 nmol/L) subsequent to paclitaxel (0.8–2.67 nmol/L) or docetaxel (0.42–1.4 nmol/L) for 24 h as in c. a–d Cells were incubated in drug-free medium until colonies were fixed and quantitated. Top, representative of 2 independent experiments, clonogenicity was compared for treatment with paclitaxel alone versus paclitaxel–CNDAC combination at a fixed ratio (left) or docetaxel versus docetaxel–CNDAC combination at a fixed ratio (right). Bottom, CIs from median-effect analysis of the two experiments are presented in the accompanying inserts
Discussion
Clinical trials on CNDAC and sapacitabine have been conducted largely in hematological malignancies. The clinical trials of sapacitabine for the treatment of solid tumors have not passed Phase II yet [34, 35]. Our prior and current studies have indicated activity of CNDAC in solid tumor cell lines. CNDAC has preferential killing effect in OC cells lacking BRCA1 or BRCA2. These deficient cells also manifested more remarkable chromosomal damage before and after exposure to CNDAC, compared with their wild-type counterparts. The BRCA1/2-associated genetic instability is the cause of the sensitization of BRCA1/2 deficient OC cells. Reciprocally, since BRCA1/2 plays a critical role in the repair of CNDAC-induced DNA damage, the absence of BRCA1/2 results in greater levels of chromosomal aberrations in cells treated with CNDAC.
We have demonstrated synergistic interaction between CNDAC and PARPis, as well as additive effects of CNDAC with platinum compounds and taxane agents, in hamster cell model systems [30]. In the current study, we applied these combination strategies in human OC cells and found the effect of combinations largely consistent with that in hamster cells.
Olaparib is the first PARPi which received accelerated approval from FDA in 2014 for ovarian cancer patients with deleterious germline and/or somatic mutations in BRCA1/2 [36]. A priority review has been granted to the new drug application for olaparib as a maintenance therapy in replased patients with platinum-sensitive OC [37]. Another PARPi, rucaparib was approved for OC with deleterious BRCA1/2 mutations in 2016 [38]. A third PARPi, niraparib (MK-4827) was recently approved by FDA for the maintenance treatment of recurrent OC, which does not require testing for BRCA mutation [39]. The synergy between a PARPi and CNDAC in BRCA1/2 deficient cells could be attributed to synthetic lethality in which both the HR and base excision repair (BER) pathway are not functional. PARPi blocks BER by competing the NAD + binding site in PARP, while repair of CNDAC-induced damage requires HR function. We have demonstrated that BER has little influence on cellular response to CNDAC [40]. In other words, inhibition of BER pathway does not enhance the cytotoxicity of CNDAC. Therefore, inhibition of PARP catalytic activity is unlikely the mechanism for synergistic combination of PARPi and CNDAC in BRCA1/2 proficient cells. Talazoparib potentiates the cell-killing effect of CNDAC more efficiently than rucaparib in both HR defective and proficient cells (Fig. 3), Since talazoparib has greater ability in trapping PARP compared with rucaparib and olaparib [41], PARP trapping [42, 43, 44] might outweigh other synergy mechanisms in cells with normal HR function, which is exhausted by significant amount of DNA DSBs after exposure to high concentration of CNDAC (and PARPi as well).
As with other targeted therapies, resistance has arisen in the clinical use of PARP inhibitors. There are multiple mechanisms for the resistance to PARP inhibitors, including restoration of BRCA1/2 function through secondary mutations, upregulation of NHEJ capacity, reduction in PARP1 activity or level, and decreased intracellular concentration of PARPi due to efflux by p-glycoproteins [45, 46]. While complete remission was achieved for years in some high-grade serous OC (HGSOC) patients with somatic biallelic deletion of BRCA1/2 after olaparib therap, resistance also arose in other patients with one copy of intact (WT) BRCA1/2 due to copy number gain and/or upregulation of the remaining functional allele [47]. Therefore, PARPi resistance poses a substantial challenge in OC targeted therapy. We found that at concentrations which allow survival of the majority of wild type cells, the synergistic killing effect of the PARPi-CNDAC combination is selective in BRCA1/2 deficient OC cells. This directs to testing of the combination in animal studies such as using patient-derived xenograft models of OC with BRCA1/2 mutations. In the circumstances without BRCAness, selectivity is lost because higher concentrations are needed for both agents to exert synergy to a similar extent as in the defective cells. However, the concentrations of each agent used in combination are still less compared with those used as a single agent. These findings suggest that combining a PARPi and CNDAC might benefit OC patients broadly, with or without functional BRCA1/2, in overcoming or delaying potential resistance to PARP inhibitors.
Chemotherapy based on platinum and/or taxane compounds plays a crucial role as first-line treatment for OC. Our study showed largely additive effects in the combination of CNDAC with either of the two platinum compounds, cisplatin and oxaliplatin, selective in BRCA1/2 deficient OC cells at relatively lower concentrations. Thus, the cisplatin-CNDAC and oxaliplatin-CNDAC combinations are useful to attenuate the cytotoxicity of platinum. The current study also showed an additive combination effect of CNDAC with paclitaxel and docetaxel used at similar concentrations in BRCA1/2 proficient and deficient OC cells. Such combinations may also be useful for reducing toxicity of the respective taxane agents.
Similar to the case of PARP inhibitors, emergence of resistance to platinum compounds has become a major hurdle in related therapeutic regimens. Progress has been made in nearly 50% of carboplatin-resistant HGSOC by co-therapy with carboplatin and birinapant, a potent cIAP inhibitor, which re-sensitizes cells to carboplatin [48]. As a cisplatin derivative, carboplatin shares a similar mechanism of action. Cisplatin resistant tumors are often not cross-resistant to oxaliplatin, suggesting a distinct action mechanism [49]. Besides cross-resistance among PARP inhibitors, PARPi-resistant cells are resistant to cross-linking agents such as cisplatin and carboplatin, but not to microtubule poisons such as taxanes [50]. Thus, the combination of CNDAC with taxanes could provide a novel treatment modality to deal with the above cross-resistance in OC therapy.
Supplementary Material
Personalised recommendations.
Successful use of Bruton’s kinase inhibitor, ibrutinib, to control paraneoplastic pemphigus in a patient with paraneoplastic autoimmune multiorgan syndrome and Lee, Andrew… Shumack, Stephen Australasian Journal of Dermatology (2017)
Ibrutinib as a Bruton Kinase Inhibitor in the Management of Chronic Lymphocytic Leukemia: A New Agent With Great Promise Foluso, Ogunleye… Jaiyesimi, Ishmael Clinical Lymphoma Myeloma and Leukemia (2015)
Bruton’s tyrosine kinase inhibitors: first and second generation agents for patients with Chronic Lymphocytic Leukemia (CLL)
Acknowledgments
The authors are grateful to Drs. Asha Multani and Jin Ma (T. C. Hsu Molecular Cytogenetics Core at MDACC) for their technical support in cytogenetic studies.
Funding
This work was supported in part by Career Development Award (X. Liu) funded by Ovarian Cancer SPORE of M.D. Anderson Cancer Center (P50 CA83639), Grants CA28596 (W. Plunkett) and Cancer Center Support Grant P30 CA16672 from the National Cancer Institute, Department of Health and Human Services.
Abbreviations
- BER
Base excision repair
- CNDAC
2′-C-cyano-2′-deoxy-1-β- d- arabino-pentofuranosyl-cytosine
- CNDACTP
CNDAC triphosphate
- CNddC
2′-C-cyano-2′,3′-didehydro-2′,3′-dideoxycytidine
- CI
Combination index
- DSB
Double strand break
- FBS
Fetal bovine serum
- HGSOC
High-grade serous ovarian cancer
- HR
Homologous recombination
- OC
Ovarian cancer
- NHEJ
Non-homologous end joining
- PARPi
PARP inhibitor
- PCC
Premature chromosome condensation
- SSB
Single strand break
Footnotes
Electronic supplementary material
The online version of this article (https://doi.org/10.1007/s00280-017-3483-6 (https://doi.org/10.1007/s00280-017-3483-6)) contains supplementary material, which is available to authorized users.
Author contributions
Conception and design: X. Liu, W. Plunkett. Development of methodology: X. Liu, Y. Jiang. Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.): X. Liu, Y. Jiang, B. Nowak, B. Qiang, N. Cheng, Y. Chen. Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): X. Liu, Y. Jiang, W. Plunkett. Writing, review, and/or revision of the manuscript: X. Liu, Y. Jiang, W. Plunkett. Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): X. Liu, Y. Jiang, B. Nowak. Study supervision: W. Plunkett.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Supplementary material
280_2017_3483_MOESM1_ESM.pptx (255 kb)
Supplementary material 1 (PPT 256 KB)
280_2017_3483_MOESM2_ESM.doc (50 kb)
Supplementary material 2 (DOC 50 KB)
References
- 1.Lengyel E. Ovarian cancer development and metastasis. Am J Pathol. 2010;177(3):1053–1064. doi: 10.2353/ajpath.2010.100105. https://doi.org/10.2353/ajpath.2010.100105( https://doi.org/10.2353/ajpath.2010.100105). (pii:S0002-9440(10)60160-5) CrossRef ( https://doi.org/10.2353/ajpath.2010.100105) PubMed ( http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Abstract&list_uids=20651229) PubMedCentral ( http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2928939) Google Scholar ( http://scholar.google.com/scholar_lookup?title=Ovarian%20cancer%20development%20and%20metastasis&author=E.%20_Lengyel&journal=Am%20J%20Pathol&volume=177&issue=3&pages=1053-1064&publication_year=2010&doi=10.2353%2Fajpath.2010.100105) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.CDC. BRCA Gene Mutations. 2015 https://www.cdcgov/cancer/breast/young_women/bringyourbrave/hereditary_breast_cancers_cid=byb_SEM_pd_010&gclid=CI7K6__ApNMCFcO6wAodUr0OGQ( https://www.cdcgov/cancer/breast/young_women/bringyourbrave/hereditary_breast_cancer/brca_gene_mutationshtm?s_cid=byb_SEM_pd_010&gclid=CI7K6__ApNMCFcO6wAodUr0OGQ)
- 3.Berchuck A, Heron KA, Carney ME, Lancaster JM, Fraser EG, Vinson VL, Deffenbaugh AM, Miron A, Marks JR, Futreal PA, Frank TS. Frequency of germline and somatic BRCA1 mutations in ovarian cancer. Clin Cancer Res. 1998;4(10):2433–2437 P. ubMed ( http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Abstract&list_uids=9796975) Google Scholar ( http://scholar.google.com/scholar_lookup?title=Frequency%20of%20germline%20and%20somatic%20BRCA1%20mutations%20in%20ovarian%20cancer&author=A.%20Berchuck&author=KA.%20Heron&author=ME.%20Carney&author=JM.%20Lancaster&author=EG.%20Fraser&author=VL.%20Vinson&author=AM.%20Deffenbaugh&author=A.%20Miron&author=JR.%20Marks&author=PA.%20Futreal&author=TS.%20Frank&journal=Clin%20Cancer%20Res&volume=4&issue=10&pages=2433-2437&publication_year=1998) [PubMed] [Google Scholar]
- 4.Hennessy BT, Timms KM, Carey MS, Gutin A, Meyer LA, Flake DD, 2nd, Abkevich V, Potter J, Pruss D, Glenn P, Li Y, Li J, Gonzalez-Angulo AM, McCune KS, Markman M, Broaddus RR, Lanchbury JS, Lu KH, Mills GB. Somatic mutations in BRCA1 and BRCA2 could expand the number of patients that benefit from poly (ADP ribose) polymerase inhibitors in ovarian cancer. J Clin Oncol. 2010;28(22):3570–3576. doi: 10.1200/JCO.2009.27.2997. https://doi.org/10.1200/JCO.2009.27.2997 ( https://doi.org/10.1200/JCO.2009.27.2997). (pii:JCO.2009.27.2997)CrossRef( https://doi.org/10.1200/JCO.2009.27.2997) PubMed ( http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Abstract&list_uids=20606085) PubMedCentral ( http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2917312) Google Scholar( http://scholar.google.com/scholar_lookup?title=Somatic%20mutations%20in%20BRCA1%20and%20BRCA2%20could%20expand%20the%20number%20of%20patients%20that%20benefit%20from%20poly%20%28ADP%20ribose%29%20polymerase%20inhibitors%20in%20ovarian%20cancer&author=BT.%20Hennessy&author=KM.%20Timms&author=MS.%20Carey&author=A.%20Gutin&author=LA.%20Meyer&author=DD.%20Flake&author=V.%20Abkevich&author=J.%20Potter&author=D.%20Pruss&author=P.%20Glenn&author=Y.%20Li&author=J.%20Li&author=AM.%20Gonzalez-Angulo&author=KS.%20McCune&author=M.%20Markman&author=RR.%20Broaddus&author=JS.%20Lanchbury&author=KH.%20Lu&author=GB.%20Mills&journal=J%20Clin%20Oncol&volume=28&issue=22&pages=3570-3576&publication_year=2010&doi=10.1200%2FJCO.2009.27.2997) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baldwin RL, Nemeth E, Tran H, Shvartsman H, Cass I, Narod S, Karlan BY. BRCA1 promoter region hypermethylation in ovarian carcinoma: a population-based study. Cancer Res. 2000;60(19):5329–5333 P. ubMed ( http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Abstract&list_uids=11034065) Google Scholar ( http://scholar.google.com/scholar_lookup?title=BRCA1%20promoter%20region%20hypermethylation%20in%20ovarian%20carcinoma%3A%20a%20population-based%20study&author=RL.%20Baldwin&author=E.%20Nemeth&author=H.%20Tran&author=H.%20Shvartsman&author=I.%20Cass&author=S.%20Narod&author=BY.%20Karlan&journal=Cancer%20Res&volume=60&issue=19&pages=5329-5333&publication_year=2000) [PubMed] [Google Scholar]
- 6.Dworkin AM, Spearman AD, Tseng SY, Sweet K, Toland AE. Methylation not a frequent “second hit” in tumors with germline BRCA mutations. Fam Cancer. 2009;8(4):339–346. doi: 10.1007/s10689-009-9240-1. https://doi.org/10.1007/s10689-009-9240-1( https://doi.org/10.1007/s10689-009-9240-1) CrossRef ( https://doi.org/10.1007/s10689-009-9240-1) PubMed ( http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Abstract&list_uids=19340607) Google Scholar ( http://scholar.google.com/scholar_lookup?title=Methylation%20not%20a%20frequent%20%E2%80%9Csecond%20hit%E2%80%9D%20in%20tumors%20with%20germline%20BRCA%20mutations&author=AM.%20Dworkin&author=AD.%20Spearman&author=SY.%20Tseng&author=K.%20Sweet&author=AE.%20Toland&journal=Fam%20Cancer&volume=8&issue=4&pages=339346&publication_year=2009&doi=10.1007%2Fs10689-009-9240-1) [DOI] [PubMed] [Google Scholar]
- 7.Bosviel R, Michard E, Lavediaux G, Kwiatkowski F, Bignon YJ, Bernard-Gallon DJ. Peripheral blood DNA methylation detected in the BRCA1 or BRCA2 promoter for sporadic ovarian cancer patients and controls. Clin Chim Acta. 2011;412(15–16):1472–1475. doi: 10.1016/j.cca.2011.04.027. https://doi.org/10.1016/j.cca.2011.04.027 ( https://doi.org/10.1016/j.cca.2011.04.027). (pii:S0009-8981(11)00245-2). CrossRef ( https://doi.org/10.1016/j.cca.2011.04.027) PubMed ( http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Abstract&list_uids=21557934) Google Scholar ( http://scholar.google.com/scholar_lookup?title=Peripheral%20blood%20DNA%20methylation%20detected%20in%20the%20BRCA1%20or%20BRCA2%20promoter%20for%20sporadic%20ovarian%20cancer%20patients%20and%20controls&author=R.%20Bosviel&author=E.%20Michard&author=G.%20Lavediaux&author=F.%20Kwiatkowski&author=YJ.%20Bignon&author=DJ.%20Bernard-Gallon&journal=Clin%20Chim%20Acta&volume=412&issue=15%E2%80%9316&pages=1472-1475&publication_year=2011&doi=10.1016%2Fj.cca.2011.04.027) [DOI] [PubMed] [Google Scholar]
- 8.Hansmann T, Pliushch G, Leubner M, Kroll P, Endt D, Gehrig A, Preisler-Adams S, Wieacker P, Haaf T. Constitutive promoter methylation of BRCA1 and RAD51C in patients with familial ovarian cancer and early-onset sporadic breast cancer. Hum Mol Genet. 2012;21(21):4669–4679. doi: 10.1093/hmg/dds308. https://doi.org/10.1093/hmg/dds308( https://doi.org/10.1093/hmg/dds308). (pii:dds308) CrossRef ( https://doi.org/10.1093/hmg/dds308) PubMed ( http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Abstract&list_uids=22843497) PubMedCentral ( http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3471399) Google Scholar ( http://scholar.google.com/scholar_lookup?title=Constitutive%20promoter%20methylation%20of%20BRCA1%20and%20RAD51C%20in%20patients%20with%20familial%20ovarian%20cancer%20and%20early-onset%20sporadic%20breast%20cancer&author=T.%20Hansmann&author=G.%20Pliushch&author=M.%20Leubner&author=P.%20Kroll&author=D.%20Endt&author=A.%20Gehrig&author=S.%20Preisler-Adams&author=P.%20Wieacker&author=T.%20Haaf&journal=Hum%20Mol%20Genet&volume=21&issue=21&pages=46694679&publication_year=2012&doi=10.1093%2Fhmg%2Fdds308) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, Martin NM, Jackson SP, Smith GC, Ashworth A. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434(7035):917–921. doi: 10.1038/nature03445. https://doi.org/10.1038/nature03445( https://doi.org/10.1038/nature03445). (pii:Nature03445) CrossRef ( https://doi.org/10.1038/nature03445) PubMed ( http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Abstract&list_uids=15829967) Google Scholar ( http://scholar.google.com/scholar_lookup?title=Targeting%20the%20DNA%20repair%20defect%20in%20BRCA%20mutant%20cells%20as%20a%20therapeutic%20strategy&author=H.%20Farmer&author=N.%20McCabe&author=CJ.%20Lord&author=AN.%20Tutt&author=DA.%20Johnson&author=TB.%20Richardson&author=M.%20Santarosa&author=KJ.%20Dillon&author=I.%20Hickson&author=C.%20Knights&author=NM.%20Martin&author=SP.%20Jackson&author=GC.%20Smith&author=A.%20Ashworth&journal=Nature&volume=434&issue=7035&pages=917921&publication_year=2005&doi=10.1038%2Fnature03445) [DOI] [PubMed] [Google Scholar]
- 10.Turner N, Tutt A, Ashworth A. Targeting the DNA repair defect of BRCA tumours. Curr Opin Pharmacol. 2005;5(4):388–393. doi: 10.1016/j.coph.2005.03.006. https://doi.org/10.1016/j.coph.2005.03.006( https://doi.org/10.1016/j.coph.2005.03.006). (pii:S1471-4892(05)00076-7) CrossRef ( https://doi.org/10.1016/j.coph.2005.03.006) PubMed ( http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Abstract&list_uids=15955736) Google Scholar ( http://scholar.google.com/scholar_lookup?title=Targeting%20the%20DNA%20repair%20defect%20of%20BRCA%20tumours&author=N.%20Turner&author=A.%20Tutt&author=A.%20Ashworth&journal=Curr%20Opin%20Pharmacol&volume=5&issue=4&pages=388393&publication_year=2005&doi=10.1016%2Fj.coph.2005.03.006) [DOI] [PubMed] [Google Scholar]
- 11.Lord CJ, Ashworth A. Targeted therapy for cancer using PARP inhibitors. Curr Opin Pharmacol. 2008;8(4):363–369. doi: 10.1016/j.coph.2008.06.016. https://doi.org/10.1016/j.coph.2008.06.016( https://doi.org/10.1016/j.coph.2008.06.016). (pii:S1471-4892(08)00082-9) CrossRef ( https://doi.org/10.1016/j.coph.2008.06.016) PubMed ( http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Abstract&list_uids=18644251) Google Scholar ( http://scholar.google.com/scholar_lookup?title=Targeted%20therapy%20for%20cancer%20using%20PARP%20inhibitors&author=CJ.%20Lord&author=A.%20Ashworth&journal=Curr%20Opin%20Pharmacol&volume=8&issue=4&pages=363369&publication_year=2008&doi=10.1016%2Fj.coph.2008.06.016) [DOI] [PubMed] [Google Scholar]
- 12.Edwards SL, Brough R, Lord CJ, Natrajan R, Vatcheva R, Levine DA, Boyd J, Reis-Filho JS, Ashworth A. Resistance to therapy caused by intragenic deletion in BRCA2. Nature. 2008;451(7182):1111–1115. doi: 10.1038/nature06548. https://doi.org/10.1038/nature06548( https://doi.org/10.1038/nature06548). (pii:nature06548) CrossRef ( https://doi.org/10.1038/nature06548) PubMed ( http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Abstract&list_uids=18264088) Google Scholar ( http://scholar.google.com/scholar_lookup?title=Resistance%20to%20therapy%20caused%20by%20intragenic%20deletion%20in%20BRCA2&author=SL.%20Edwards&author=R.%20Brough&author=CJ.%20Lord&author=R.%20Natrajan&author=R.%20Vatcheva&author=DA.%20Levine&author=J.%20Boyd&author=JS.%20Reis-Filho&author=A.%20Ashworth&journal=Nature&volume=451&issue=7182&pages=1111-1115&publication_year=2008&doi=10.1038%2Fnature06548) [DOI] [PubMed] [Google Scholar]
- 13.Sakai W, Swisher EM, Karlan BY, Agarwal MK, Higgins J, Friedman C, Villegas E, Jacquemont C, Farrugia DJ, Couch FJ, Urban N, Taniguchi T. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature. 2008;451(7182):1116–1120. doi: 10.1038/nature06633. https://doi.org/10.1038/nature06633( https://doi.org/10.1038/nature06633). (pii:nature06633) CrossRef ( https://doi.org/10.1038/nature06633) PubMed ( http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Abstract&list_uids=18264087) PubMedCentral ( http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2577037) Google Scholar ( http://scholar.google.com/scholar_lookup?title=Secondary%20mutations%20as%20a%20mechanism%20of%20cisplatin%20resistance%20in%20BRCA2-mutated%20cancers&author=W.%20Sakai&author=EM.%20Swisher&author=BY.%20Karlan&author=MK.%20Agarwal&author=J.%20Higgins&author=C.%20Friedman&author=E.%20Villegas&author=C.%20Jacquemont&author=DJ.%20Farrugia&author=FJ.%20Couch&author=N.%20Urban&author=T.%20Taniguchi&journal=Nature&volume=451&issue=7182&pages=11161120&publication_year=2008&doi=10.1038%2Fnature06633) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Swisher EM, Sakai W, Karlan BY, Wurz K, Urban N, Taniguchi T. Secondary BRCA1 mutations in BRCA1-mutated ovarian carcinomas with platinum resistance. Cancer Res. 2008;68(8):2581–2586. doi: 10.1158/0008-5472.CAN-08-0088. https://doi.org/10.1158/0008-5472.CAN-08-0088( https://doi.org/10.1158/0008-5472.CAN-08-0088). (pii:68/8/2581) CrossRef ( https://doi.org/10.1158/0008-5472.CAN-08-0088) PubMed ( http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Abstract&list_uids=18413725) PubMedCentral ( http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2674369) Google Scholar ( http://scholar.google.com/scholar_lookup?title=Secondary%20BRCA1%20mutations%20in%20BRCA1-mutated%20ovarian%20carcinomas%20with%20platinum%20resistance&author=EM.%20Swisher&author=W.%20Sakai&author=BY.%20Karlan&author=K.%20Wurz&author=N.%20Urban&author=T.%20Taniguchi&journal=Cancer%20Res&volume=68&issue=8&pages=2581-2586&publication_year=2008&doi=10.1158%2F0008-5472.CAN-08-0088) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang W, Figg WD. Secondary BRCA1 and BRCA2 alterations and acquired chemoresistance. Cancer Biol Ther. 2008;7(7):1004–1005 C. doi: 10.4161/cbt.7.7.6409. rossRef ( https://doi.org/10.4161/cbt.7.7.6409) PubMed ( http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Abstract&list_uids=18720553) PubMedCentral ( http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2731297) Google Scholar ( http://scholar.google.com/scholar_lookup?title=Secondary%20BRCA1%20and%20BRCA2%20alterations%20and%20acquired%20chemoresistance&author=W.%20Wang&author=WD.%20Figg&journal=Cancer%20Biol%20Ther&volume=7&issue=7&pages=10041005&publication_year=2008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sakai W, Swisher EM, Jacquemont C, Chandramohan KV, Couch FJ, Langdon SP, Wurz K, Higgins J, Villegas E, Taniguchi T. Functional restoration of BRCA2 protein by secondary BRCA2 mutations in BRCA2-mutated ovarian carcinoma. Cancer Res. 2009;69(16):6381–6386. doi: 10.1158/0008-5472.CAN-09-1178. https://doi.org/10.1158/0008-5472.CAN-09-1178( https://doi.org/10.1158/0008-5472.CAN-09-1178). (pii:0008-5472.CAN-09-1178) CrossRef ( https://doi.org/10.1158/0008-5472.CAN-09-1178) PubMed ( http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Abstract&list_uids=19654294) PubMedCentral ( http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2754824) Google Scholar ( http://scholar.google.com/scholar_lookup?title=Functional%20restoration%20of%20BRCA2%20protein%20by%20secondary%20BRCA2%20mutations%20in%20BRCA2-mutated%20ovarian%20carcinoma&author=W.%20Sakai&author=EM.%20Swisher&author=C.%20Jacquemont&author=KV.%20Chandramohan&author=FJ.%20Couch&author=SP.%20Langdon&author=K.%20Wurz&author=J.%20Higgins&author=E.%20Villegas&author=T.%20Taniguchi&journal=Cancer%20Res&volume=69&issue=16&pages=6381-6386&publication_year=2009&doi=10.1158%2F0008-5472.CAN-09-1178) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Matsuda A. 2′-C-Cyano-2′-deoxy-1-beta-d-arabinofuranosyl-cytosine(CNDAC): a mechanism-based DNA-strandbreaking antitumor nucleoside. Nucleosides Nucleotides. 1995;14:461–471 C. rossRef ( https://doi.org/10.1080/15257779508012407) Google Scholar ( http://scholar.google.com/scholar_lookup?title=2%E2%80%B2-C-Cyano-2%E2%80%B2-deoxy-1-beta-d-arabinofuranosyl-cytosine%28CNDAC%29%3A%20a%20mechanism-based%20DNA-strandbreaking%20antitumor%20nucleoside&author=A.%20Matsuda&journal=Nucleosides%20Nucleotides&volume=14&pages=461-471&publication_year=1995) [Google Scholar]
- 18.Azuma A, Huang P, Matsuda A, Plunkett W. Cellular pharmacokinetics and pharmacodynamics of the deoxycytidine analog 2′-C-cyano-2′-deoxy-1-beta-d-arabino-pentofuranosylcytosine (CNDAC) Biochem Pharmacol. 2001;61(12):1497– 1507. doi: 10.1016/s0006-2952(01)00617-7. (pii:S0006295201006177) CrossRef ( https://doi.org/10.1016/S0006-2952(01)00617-7) PubMed ( http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Abstract&list_uids=11377379) Google Scholar ( http://scholar.google.com/scholar_lookup?title=Cellular%20pharmacokinetics%20and%20pharmacodynamics%20of%20the%20deoxycytidine%20analog%202%E2%80%B2-C-cyano-2%E2%80%B2-deoxy-1-beta-d-arabino-pentofuranosylcytosine%20%28CNDAC%29&author=A.%20Azuma&author=P.%20Huang&author=A.%20Matsuda&author=W.%20Plunkett&journal=Biochem%20Pharmacol&volume=61&issue=12&pages=14971507&publication_year=2001) [DOI] [PubMed] [Google Scholar]
- 19.Azuma A, Huang P, Matsuda A, Plunkett W. 2′- C-cyano-2′-deoxy-1-beta- d-arabino-pentofuranosylcytosine: a novel anticancer nucleoside analog that causes both DNA strand breaks and G(2) arrest. Mol Pharmacol. 2001;59(4):725–731 P. doi: 10.1124/mol.59.4.725. ubMed ( http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Abstract&list_uids=11259616) Google Scholar ( http://scholar.google.com/scholar_lookup?title=2%E2%80%B2-C-cyano-2%E2%80%B2-deoxy-1-beta-d-arabino-pentofuranosylcytosine%3A%20a%20novel%20anticancer%20nucleoside%20analog%20that%20causes%20both%20DNA%20strand%20breaks%20and%20G%282%29%20arrest&author=A.%20Azuma&author=P.%20Huang&author=A.%20Matsuda&author=W.%20Plunkett&journal=Mol%20Pharmacol&volume=59&issue=4&pages=725731&publication_year=2001) [DOI] [PubMed] [Google Scholar]
- 20.Liu X, Wang Y, Benaissa S, Matsuda A, Kantarjian H, Estrov Z, Plunkett W. Homologous recombination as a resistance mechanism to replication-induced double-strand breaks caused by the antileukemia agent CNDAC. Blood. 2010;116(10):1737–1746. doi: 10.1182/blood-2009-05-220376. https://doi.org/10.1182/blood-2009-05-220376( https://doi.org/10.1182/blood-2009-05-220376). (pii:blood-2009-05-220376) CrossRef ( https://doi.org/10.1182/blood-2009-05-220376) PubMed ( http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Abstract&list_uids=20479284) PubMedCentral ( http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2947394) Google Scholar ( http://scholar.google.com/scholar_lookup?title=Homologous%20recombination%20as%20a%20resistance%20mechanism%20to%20replication-induced%20double-strand%20breaks%20caused%20by%20the%20antileukemia%20agent%20CNDAC&author=X.%20Liu&author=Y.%20Wang&author=S.%20Benaissa&author=A.%20Matsuda&author=H.%20Kantarjian&author=Z.%20Estrov&author=W.%20Plunkett&journal=Blood&volume=116&issue=10&pages=1737-1746&publication_year=2010&doi=10.1182%2Fblood-2009-05-220376) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu XJ, Nowak B, Wang YQ, Plunkett W. Sapacitabine, the prodrug of CNDAC, is a nucleoside analog with a unique action mechanism of inducing DNA strand breaks. Chin J Cancer. 2012;31(8):373–380. doi: 10.5732/cjc.012.10077. https://doi.org/10.5732/cjc.012.10077( https://doi.org/10.5732/cjc.012.10077) CrossRef ( https://doi.org/10.5732/cjc.012.10077) PubMed ( http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Abstract&list_uids=22739266) PubMedCentral ( http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3777512) Google Scholar ( http://scholar.google.com/scholar_lookup?title=Sapacitabine%2C%20the%20prodrug%20of%20CNDAC%2C%20is%20a%20nucleoside%20analog%20with%20a%20unique%20action%20mechanism%20of%20inducing%20DNA%20strand%20breaks&author=XJ.%20Liu&author=B.%20Nowak&author=YQ.%20Wang&author=W.%20Plunkett&journal=Chin%20J%20Cancer&volume=31&issue=8&pages=373380&publication_year=2012&doi=10.5732%2Fcjc.012.10077) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shapiro GI, Cleary JH, Tolaney JM, Ghandi SM, Kwak L, Clark EL, Wolanski JW, Bell A, Schulz T, Frame J, Saladino S, Hogben C, Rodig M, Chiao SJ, Blake JHD. Responses to sequential sapacitabine and seliciclib in patients with brca-deficient solid tumors. Cancer Res. 2013;73(8 Suppl) https://doi.org/10.1158/1538-7445.AM2013-LB-202 ( https://doi.org/10.1158/1538-7445.AM2013-LB-202) [Google Scholar]
- 23.Kantarjian H, Garcia-Manero G, O’Brien S, Faderl S, Ravandi F, Westwood R, Green SR, Chiao JH, Boone PA, Cortes J, Plunkett W. Phase I clinical and pharmacokinetic study of oral sapacitabine in patients with acute leukemia and myelodysplastic syndrome. J Clin Oncol. 2010;28(2):285–291. doi: 10.1200/JCO.2009.25.0209. https://doi.org/10.1200/JCO.2009.25.0209( https://doi.org/10.1200/JCO.2009.25.0209). (pii:JCO.2009.25.0209) CrossRef ( https://doi.org/10.1200/JCO.2009.25.0209) PubMed ( http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Abstract&list_uids=19933907) Google Scholar ( http://scholar.google.com/scholar_lookup?title=Phase%20I%20clinical%20and%20pharmacokinetic%20study%20of%20oral%20sapacitabine%20in%20patients%20with%20acute%20leukemia%20and%20myelodysplastic%20syndrome&author=H.%20Kantarjian&author=G.%20Garcia-Manero&author=S.%20O%E2%80%99Brien&author=S.%20Faderl&author=F.%20Ravandi&author=R.%20Westwood&author=SR.%20Green&author=JH.%20Chiao&author=PA.%20Boone&author=J.%20Cortes&author=W.%20Plunkett&journal=J%20Clin%20Oncol&volume=28&issue=2&pages=285291&publication_year=2010&doi=10.1200%2FJCO.2009.25.0209) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kantarjian H, Faderl S, Garcia-Manero G, Luger S, Venugopal P, Maness L, Wetzler M, Coutre S, Stock W, Claxton D, Goldberg SL, Arellano M, Strickland SA, Seiter K, Schiller G, Jabbour E, Chiao J, Plunkett W. Oral sapacitabine for the treatment of acute myeloid leukaemia in elderly patients: a randomised phase 2 study. Lancet Oncol. 2012;13(11):1096–1104. doi: 10.1016/S1470-2045(12)70436-9. https://doi.org/10.1016/S1470( https://doi.org/10.1016/S1470). (pii:S1470-2045(12)70436-9) CrossRef ( https://doi.org/10.1016/S1470-2045(12)70436-9) PubMed ( http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Abstract&list_uids=23075701) PubMedCentral ( http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4004358) Google Scholar ( http://scholar.google.com/scholar_lookup?title=Oral%20sapacitabine%20for%20the%20treatment%20of%20acute%20myeloid%20leukaemia%20in%20elderly%20patients%3A%20a%20randomised%20phase%202%20study&author=H.%20Kantarjian&author=S.%20Faderl&author=G.%20Garcia-Manero&author=S.%20Luger&author=P.%20Venugopal&author=L.%20Maness&author=M.%20Wetzler&author=S.%20Coutre&author=W.%20Stock&author=D.%20Claxton&author=SL.%20Goldberg&author=M.%20Arellano&author=SA.%20Strickland&author=K.%20Seiter&author=G.%20Schiller&author=E.%20Jabbour&author=J.%20Chiao&author=W.%20Plunkett&journal=Lancet%20Oncol&volume=13&issue=11&pages=1096-1104&publication_year=2012&doi=10.1016%2FS1470) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kantarjian HM, Jabbour EJ, Garcia-Manero G, Kadia TM, DiNardo CD, Daver NG, Borthakur G, Jain N, Waukau J, Kwari M, Anderson BD, Lizuka K, Zhang CJ, Ravandi C, Plunkett FW. Phase I/II study of DFP-10917 in relapsed/refractory AML demonstrates efficacy and safety profile suitable for phase III study. American Society of Hematology 58th Annual Meeting: Abstr# 2822. 2016 Google Scholar ( https://scholar.google.com/scholar?q=Kantarjian%20HM%2C%20Jabbour%20EJ%2C%20Garcia-Manero%20G%2C%20Kadia%20TM%2C%20DiNardo%20CD%2C%20Daver%20NG%2C%20Borthakur%20G%2C%20Jain%20N%2C%20Waukau%20J%2C%20Kwari%20M%2C%20Anderson%20BD%2C%20Lizuka%20K%2C%20Zhang%20CJ%2C%20Ravandi%20C%2C%20Plunkett%20F%20W%20%282016%29%20Phase%20I%2FII%20study%20of%20DFP-10917%20in%20relapsed%2Frefractory%20AML%20demonstrates%20efficacy%20and%20safety%20profile%20suitable%20for%20phase%20III%20study.%20In%3A%20American%20Society%20of%20Hematology%2058th%20Annual%20Meeting%3A%20Abstr%23%202822)
- 26.DelloRusso C, Welcsh PL, Wang W, Garcia RL, King MC, Swisher EM. Functional characterization of a novel BRCA1-null ovarian cancer cell line in response to ionizing radiation. Mol Cancer Res. 2007;5(1):35–45. doi: 10.1158/1541-7786.MCR-06-0234. https://doi.org/10.1158/1541-7786.MCR-06-0234( https://doi.org/10.1158/1541-7786.MCR-06-0234). (pii:5/1/35) CrossRef ( https://doi.org/10.1158/1541-7786.MCR-06-0234) PubMed ( http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Abstract&list_uids=17259345) Google Scholar ( http://scholar.google.com/scholar_lookup?title=Functional%20characterization%20of%20a%20novel%20BRCA1-null%20ovarian%20cancer%20cell%20line%20in%20response%20to%20ionizing%20radiation&author=C.%20DelloRusso&author=PL.%20Welcsh&author=W.%20Wang&author=RL.%20Garcia&author=MC.%20King&author=EM.%20Swisher&journal=Mol%20Cancer%20Res&volume=5&issue=1&pages=35-45&publication_year=2007&doi=10.1158%2F1541-7786.MCR-06-0234) [DOI] [PubMed] [Google Scholar]
- 27.Liu X, Guo Y, Li Y, Jiang Y, Chubb S, Azuma A, Huang P, Matsuda A, Hittelman W, Plunkett W. Molecular basis for G2 arrest induced by 2′-C-cyano-2′-deoxy-1-beta-d-arabino-pentofuranosylcytosine and consequences of checkpoint abrogation. Cancer Res. 2005;65(15):6874–6881. doi: 10.1158/0008-5472.CAN-05-0288. https://doi.org/10.1158/0008-5472.CAN-05-0288( https://doi.org/10.1158/0008-5472.CAN-05-0288). (pii:65/15/6874) CrossRef ( https://doi.org/10.1158/0008-5472.CAN-05-0288) PubMed ( http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Abstract&list_uids=16061671) Google Scholar ( http://scholar.google.com/scholar_lookup?title=Molecular%20basis%20for%20G2%20arrest%20induced%20by%202%E2%80%B2-C-cyano-2%E2%80%B2-deoxy-1-beta-d-arabino-pentofuranosylcytosine%20and%20consequences%20of%20checkpoint%20abrogation&author=X.%20Liu&author=Y.%20Guo&author=Y.%20Li&author=Y.%20Jiang&author=S.%20Chubb&author=A.%20Azuma&author=P.%20Huang&author=A.%20Matsuda&author=W.%20Hittelman&author=W.%20Plunkett&journal=Cancer%20Res&volume=65&issue=15&pages=68746881&publication_year=2005&doi=10.1158%2F0008-5472.CAN-05-0288) [DOI] [PubMed] [Google Scholar]
- 28.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzym Regul. 1984;22:27– 55 C. doi: 10.1016/0065-2571(84)90007-4. rossRef ( https://doi.org/10.1016/0065-2571(84)90007-4) Google Scholar ( http://scholar.google.com/scholar_lookup?title=Quantitative%20analysis%20of%20dose-effect%20relationships%3A%20the%20combined%20effects%20of%20multiple%20drugs%20or%20enzyme%20inhibitors&author=TC.%20Chou&author=P.%20Talalay&journal=Adv%20Enzym%20Regul&volume=22&pages=2755&publication_year=1984) [DOI] [PubMed] [Google Scholar]
- 29.Chou TC. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev. 2006;58(3):621–681. doi: 10.1124/pr.58.3.10. https://doi.org/10.1124/pr.58.3.10( https://doi.org/10.1124/pr.58.3.10). (pii:58/3/621) CrossRef ( https://doi.org/10.1124/pr.58.3.10) PubMed ( http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Abstract&list_uids=16968952) Google Scholar ( http://scholar.google.com/scholar_lookup?title=Theoretical%20basis%2C%20experimental%20design%2C%20and%20computerized%20simulation%20of%20synergism%20and%20antagonism%20in%20drug%20combination%20studies&author=TC.%20Chou&journal=Pharmacol%20Rev&volume=58&issue=3&pages=621681&publication_year=2006&doi=10.1124%2Fpr.58.3.10) [DOI] [PubMed] [Google Scholar]
- 30.Liu X, Jiang Y, Nowak B, Hargis S, Plunkett W. Mechanism-Based Drug Combinations with the DNA Strand-Breaking Nucleoside Analog CNDAC. Mol Cancer Ther. 2016;15(10):2302–2313. doi: 10.1158/1535-7163.MCT-15-0801. https://doi.org/10.1158/1535-7163.MCT-15-0801( https://doi.org/10.1158/1535-7163.MCT-15-0801). (pii:1535-7163.MCT-15-0801) CrossRef ( https://doi.org/10.1158/1535-7163.MCT-15-0801) PubMed ( http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Abstract&list_uids=27474148) PubMedCentral ( http://www.ncbi.nlm.nih.gov/pmc/articles/PMC5050117) Google Scholar ( http://scholar.google.com/scholar_lookup?title=Mechanism-Based%20Drug%20Combinations%20with%20the%20DNA%20Strand-Breaking%20Nucleoside%20Analog%20CNDAC&author=X.%20Liu&author=Y.%20Jiang&author=B.%20Nowak&author=S.%20Hargis&author=W.%20Plunkett&journal=Mol%20Cancer%20Ther&volume=15&issue=10&pages=23022313&publication_year=2016&doi=10.1158%2F1535-7163.MCT-15-0801) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Calabrese CR, Almassy R, Barton S, Batey MA, Calvert AH, Canan-Koch S, Durkacz BW, Hostomsky Z, Kumpf RA, Kyle S, Li J, Maegley K, Newell DR, Notarianni E, Stratford IJ, Skalitzky D, Thomas HD, Wang LZ, Webber SE, Williams KJ, Curtin NJ. Anticancer chemosensitization and radiosensitization by the novel poly(ADP-ribose) polymerase-1 inhibitor AG14361. J Natl Cancer Inst. 2004;96(1):56–67 C. doi: 10.1093/jnci/djh005. rossRef ( https://doi.org/10.1093/jnci/djh005) PubMed ( http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Abstract&list_uids=14709739) Google Scholar ( http://scholar.google.com/scholar_lookup?title=Anticancer%20chemosensitization%20and%20radiosensitization%20by%20the%20novel%20poly%28ADP-ribose%29%20polymerase-1%20inhibitor%20AG14361&author=CR.%20Calabrese&author=R.%20Almassy&author=S.%20Barton&author=MA.%20Batey&author=AH.%20Calvert&author=S.%20Canan-Koch&author=BW.%20Durkacz&author=Z.%20Hostomsky&author=RA.%20Kumpf&author=S.%20Kyle&author=J.%20Li&author=K.%20Maegley&author=DR.%20Newell&author=E.%20Notarianni&author=IJ.%20Stratford&author=D.%20Skalitzky&author=HD.%20Thomas&author=LZ.%20Wang&author=SE.%20Webber&author=KJ.%20Williams&author=NJ.%20Curtin&journal=J%20Natl%20Cancer%20Inst&volume=96&issue=1&pages=5667&publication_year=2004) [DOI] [PubMed] [Google Scholar]
- 32.Guillot C, Favaudon V, Herceg Z, Sagne C, Sauvaigo S, Merle P, Hall J, Chemin I. PARP inhibition and the radiosensitizing effects of the PARP inhibitor ABT-888 in in vitro hepatocellular carcinoma models. BMC Cancer. 2014;14:603. doi: 10.1186/1471-2407-14-603. https://doi.org/10.1186/1471-2407-14-603( https://doi.org/10.1186/1471-2407-14-603). (pii:1471-2407-14-603) CrossRef ( https://doi.org/10.1186/1471-2407-14-603) PubMed ( http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Abstract&list_uids=25139788) PubMedCentral ( http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4153905) Google Scholar ( http://scholar.google.com/scholar_lookup?title=PARP%20inhibition%20and%20the%20radiosensitizing%20effects%20of%20the%20PARP%20inhibitor%20ABT-888%20in%20in%20vitro%20hepatocellular%20carcinoma%20models&author=C.%20Guillot&author=V.%20Favaudon&author=Z.%20Herceg&author=C.%20Sagne&author=S.%20Sauvaigo&author=P.%20Merle&author=J.%20Hall&author=I.%20Chemin&journal=BMC%20Cancer&volume=14&pages=603&publication_year=2014&doi=10.1186%2F1471-2407-14-603) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Helleday T. The underlying mechanism for the PARP and BRCA synthetic lethality: clearing up the misunderstandings. Mol Oncol. 2011;5(4):387–393. doi: 10.1016/j.molonc.2011.07.001. https://doi.org/10.1016/j.molonc.2011.07.001( https://doi.org/10.1016/j.molonc.2011.07.001). (pii:S1574-7891(11)00074-3) CrossRef ( https://doi.org/10.1016/j.molonc.2011.07.001) PubMed ( http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Abstract&list_uids=21821475) PubMedCentral ( http://www.ncbi.nlm.nih.gov/pmc/articles/PMC5528309) Google Scholar ( http://scholar.google.com/scholar_lookup?title=The%20underlying%20mechanism%20for%20the%20PARP%20and%20BRCA%20synthetic%20lethality%3A%20clearing%20up%20the%20misunderstandings&author=T.%20Helleday&journal=Mol%20Oncol&volume=5&issue=4&pages=387-393&publication_year=2011&doi=10.1016%2Fj.molonc.2011.07.001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu X, Kantarjian H, Plunkett W. Sapacitabine for cancer. Expert Opin Investig Drugs. 2012;21(4):541–555. doi: 10.1517/13543784.2012.660249. https://doi.org/10.1517/13543784.2012.660249( https://doi.org/10.1517/13543784.2012.660249) CrossRef ( https://doi.org/10.1517/13543784.2012.660249) PubMed ( http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Abstract&list_uids=22329458) PubMedCentral ( http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3366487) Google Scholar ( http://scholar.google.com/scholar_lookup?title=Sapacitabine%20for%20cancer&author=X.%20Liu&author=H.%20Kantarjian&author=W.%20Plunkett&journal=Expert%20Opin%20Investig%20Drugs&volume=21&issue=4&pages=541-555&publication_year=2012&doi=10.1517%2F13543784.2012.660249) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tolaney SM, Hilton JF, Cleary JM, Gandhi L, Kwak EL, Clark JW. Phase I study of sapacitabine and seliciclib in patients with advanced solid tumors. J Clin Oncol. 2016 https://doi.org/10.1200/JCO.2016.34.15_suppl.2503 ( https://doi.org/10.1200/JCO.2016.34.15_suppl.2503)
- 36.Kim G, Ison G, McKee AE, Zhang H, Tang S, Gwise T, Sridhara R, Lee E, Tzou A, Philip R, Chiu HJ, Ricks TK, Palmby T, Russell AM, Ladouceur G, Pfuma E, Li H, Zhao L, Liu Q, Venugopal R, Ibrahim A, Pazdur R. FDA approval summary: olaparib monotherapy in patients with deleterious germline BRCA-mutated advanced ovarian cancer treated with three or more lines of chemotherapy. Clin Cancer Res. 2015;21(19):4257–4261. doi: 10.1158/1078-0432.CCR-15-0887. https://doi.org/10.1158/1078-0432.CCR-15-0887( https://doi.org/10.1158/1078-0432.CCR-15-0887) CrossRef ( https://doi.org/10.1158/1078-0432.CCR-15-0887) PubMed ( http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Abstract&list_uids=26187614) Google Scholar ( http://scholar.google.com/scholar_lookup?title=FDA%20approval%20summary%3A%20olaparib%20monotherapy%20in%20patients%20with%20deleterious%20germline%20BRCA-mutated%20advanced%20ovarian%20cancer%20treated%20with%20three%20or%20more%20lines%20of%20chemotherapy&author=G.%20Kim&author=G.%20Ison&author=AE.%20McKee&author=H.%20Zhang&author=S.%20Tang&author=T.%20Gwise&author=R.%20Sridhara&author=E.%20Lee&author=A.%20Tzou&author=R.%20Philip&author=HJ.%20Chiu&author=TK.%20Ricks&author=T.%20Palmby&author=AM.%20Russell&author=G.%20Ladouceur&author=E.%20Pfuma&author=H.%20Li&author=L.%20Zhao&author=Q.%20Liu&author=R.%20Venugopal&author=A.%20Ibrahim&author=R.%20Pazdur&journal=Clin%20Cancer%20Res&volume=21&issue=19&pages=42574261&publication_year=2015&doi=10.1158%2F1078-0432.CCR-15-0887) [DOI] [PubMed] [Google Scholar]
- 37.OncLive. FDA grants maintenance olaparib priority review for ovarian cancer. 2017 http://www.onclivecom/web-exclusives/fda-grants-maintenance-olaparib-priority-review-for-ovarian-cancer( http://www.onclivecom/web-exclusives/fda-grants-maintenance-olaparib-priority-review-for-ovarian-cancer)
- 38.FDA. Rucaparib. 2017 https://www.fdagov/drugs/informationondrugs/approveddrugs/ucm533891htm( https://www.fdagov/drugs/informationondrugs/approveddrugs/ucm533891htm)
- 39.OncLive. PARP inhibitor explosion continues in ovarian cancer. 2017 http://www.onclivecom/web-exclusives/parp-inhibitor-explosion-continues-in-ovarian-cancer?p=1( http://www.onclivecom/web-exclusives/parp-inhibitor-explosion-continues-in-ovarian-cancer?p=1)
- 40.Wang Y, Liu X, Matsuda A, Plunkett W. Repair of 2′- C-cyano-2′-deoxy-1-beta-d-arabino-pentofuranosylcytosine-induced DNA single-strand breaks by transcription-coupled nucleotide excision repair. Cancer Res. 2008;68(10):3881–3889. doi: 10.1158/0008-5472.CAN-07-6885. https://doi.org/10.1158/0008-5472.CAN-07-6885( https://doi.org/10.1158/0008-5472.CAN-07-6885). (pii:68/10/3881) CrossRef ( https://doi.org/10.1158/0008-5472.CAN-07-6885) PubMed ( http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Abstract&list_uids=18483273) Google Scholar ( http://scholar.google.com/scholar_lookup?title=Repair%20of%202%E2%80%B2-C-cyano-2%E2%80%B2-deoxy-1-beta-d-arabino-pentofuranosylcytosine-induced%20DNA%20single-strand%20breaks%20by%20transcription-coupled%20nucleotide%20excision%20repair&author=Y.%20Wang&author=X.%20Liu&author=A.%20Matsuda&author=W.%20Plunkett&journal=Cancer%20Res&volume=68&issue=10&pages=38813889&publication_year=2008&doi=10.1158%2F0008-5472.CAN-07-6885) [DOI] [PubMed] [Google Scholar]
- 41.Murai J, Huang SY, Renaud A, Zhang Y, Ji J, Takeda S, Morris J, Teicher B, Doroshow JH, Pommier Y. Stereospecific PARP trapping by BMN 673 and comparison with olaparib and rucaparib. Mol Cancer Ther. 2014;13(2):433–443. doi: 10.1158/1535-7163.MCT-13-0803. https://doi.org/10.1158/1535-7163.MCT-13-0803( https://doi.org/10.1158/1535-7163.MCT-13-0803) CrossRef ( https://doi.org/10.1158/1535-7163.MCT-13-0803) PubMed ( http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Abstract&list_uids=24356813) Google Scholar ( http://scholar.google.com/scholar_lookup?title=Stereospecific%20PARP%20trapping%20by%20BMN%20673%20and%20comparison%20with%20olaparib%20and%20rucaparib&author=J.%20Murai&author=SY.%20Huang&author=A.%20Renaud&author=Y.%20Zhang&author=J.%20Ji&author=S.%20Takeda&author=J.%20Morris&author=B.%20Teicher&author=JH.%20Doroshow&author=Y.%20Pommier&journal=Mol%20Cancer%20Ther&volume=13&issue=2&pages=433-443&publication_year=2014&doi=10.1158%2F1535-7163.MCT-13-0803) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Murai J, Huang SY, Das BB, Renaud A, Zhang Y, Doroshow JH, Ji J, Takeda S, Pommier Y. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res. 2012;72(21):5588–5599. doi: 10.1158/0008-5472.CAN-12-2753. https://doi.org/10.1158/0008-5472.CAN-12-2753( https://doi.org/10.1158/0008-5472.CAN-12-2753) CrossRef ( https://doi.org/10.1158/0008-5472.CAN-12-2753) PubMed ( http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Abstract&list_uids=23118055) PubMedCentral ( http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3528345) Google Scholar ( http://scholar.google.com/scholar_lookup?title=Trapping%20of%20PARP1%20and%20PARP2%20by%20clinical%20PARP%20inhibitors&author=J.%20Murai&author=SY.%20Huang&author=BB.%20Das&author=A.%20Renaud&author=Y.%20Zhang&author=JH.%20Doroshow&author=J.%20Ji&author=S.%20Takeda&author=Y.%20Pommier&journal=Cancer%20Res&volume=72&issue=21&pages=55885599&publication_year=2012&doi=10.1158%2F0008-5472.CAN-12-2753) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pommier Y, O’Connor MJ, de Bono J. Laying a trap to kill cancer cells: PARP inhibitors and their mechanisms of action. Sci Transl Med. 2016;8(362):362p. doi: 10.1126/scitranslmed.aaf9246. s317. https://doi.org/10.1126/scitranslmed.aaf9246( https://doi.org/10.1126/scitranslmed.aaf9246) CrossRef ( https://doi.org/10.1126/scitranslmed.aaf9246) Google Scholar ( http://scholar.google.com/scholar_lookup?title=Laying%20a%20trap%20to%20kill%20cancer%20cells%3A%20PARP%20inhibitors%20and%20their%20mechanisms%20of%20action&author=Y.%20Pommier&author=MJ.%20O%E2%80%99Connor&author=J.%20Bono&journal=Sci%20Transl%20Med&volume=8&issue=362&pages=362ps317&publication_year=2016&doi=10.1126%2Fscitranslmed.aaf9246) [DOI] [PubMed] [Google Scholar]
- 44.Shen Y, Aoyagi-Scharber M, Wang B. Trapping poly(ADP-Ribose) polymerase. J Pharmacol Exp Ther. 2015;353(3):446–457. doi: 10.1124/jpet.114.222448. https://doi.org/10.1124/jpet.114.222448( https://doi.org/10.1124/jpet.114.222448) CrossRef ( https://doi.org/10.1124/jpet.114.222448) PubMed ( http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Abstract&list_uids=25758918) Google Scholar ( http://scholar.google.com/scholar_lookup?title=Trapping%20poly%28ADP-Ribose%29%20polymerase&author=Y.%20Shen&author=M.%20Aoyagi-Scharber&author=B.%20Wang&journal=J%20Pharmacol%20Exp%20Ther&volume=353&issue=3&pages=446-457&publication_year=2015&doi=10.1124%2Fjpet.114.222448) [DOI] [PubMed] [Google Scholar]
- 45.Lord CJ, Ashworth A. Mechanisms of resistance to therapies targeting BRCA-mutant cancers. Nat Med. 2013;19(11):1381–1388. doi: 10.1038/nm.3369. https://doi.org/10.1038/nm.3369( https://doi.org/10.1038/nm.3369) CrossRef ( https://doi.org/10.1038/nm.3369) PubMed ( http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Abstract&list_uids=24202391) Google Scholar ( http://scholar.google.com/scholar_lookup?title=Mechanisms%20of%20resistance%20to%20therapies%20targeting%20BRCA-mutant%20cancers&author=CJ.%20Lord&author=A.%20Ashworth&journal=Nat%20Med&volume=19&issue=11&pages=13811388&publication_year=2013&doi=10.1038%2Fnm.3369) [DOI] [PubMed] [Google Scholar]
- 46.Montoni A, Robu M, Pouliot E, Shah GM. Resistance to PARP-inhibitors in cancer therapy. Front Pharmacol. 2013;4:18. doi: 10.3389/fphar.2013.00018. https://doi.org/10.3389/fphar.2013.00018( https://doi.org/10.3389/fphar.2013.00018) CrossRef ( https://doi.org/10.3389/fphar.2013.00018) PubMed ( http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Abstract&list_uids=23450678) PubMedCentral ( http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3583007) Google Scholar ( http://scholar.google.com/scholar_lookup?title=Resistance%20to%20PARP-inhibitors%20in%20cancer%20therapy&author=A.%20Montoni&author=M.%20Robu&author=E.%20Pouliot&author=GM.%20Shah&journal=Front%20Pharmacol&volume=4&pages=18&publication_year=2013&doi=10.3389%2Ffphar.2013.00018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lheureux S, Bruce JP, Burnier JV, Karakasis K, Shaw PA, Clarke BA, Yang SY, Quevedo R, Li T, Dowar M, Bowering V, Pugh TJ, Oza AM. Somatic BRCA1/2 recovery as a resistance mechanism after exceptional response to poly (ADP-ribose) polymerase inhibition. J Clin Oncol. 2017;35(11):1240–1249. doi: 10.1200/JCO.2016.71.3677. https://doi.org/10.1200/JCO.2016.71.3677( https://doi.org/10.1200/JCO.2016.71.3677) CrossRef ( https://doi.org/10.1200/JCO.2016.71.3677) PubMed ( http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Abstract&list_uids=28221868) Google Scholar ( http://scholar.google.com/scholar_lookup?title=Somatic%20BRCA1%2F2%20recovery%20as%20a%20resistance%20mechanism%20after%20exceptional%20response%20to%20poly%20%28ADP-ribose%29%20polymerase%20inhibition&author=S.%20Lheureux&author=JP.%20Bruce&author=JV.%20Burnier&author=K.%20Karakasis&author=PA.%20Shaw&author=BA.%20Clarke&author=SY.%20Yang&author=R.%20Quevedo&author=T.%20Li&author=M.%20Dowar&author=V.%20Bowering&author=TJ.%20Pugh&author=AM.%20Oza&journal=J%20Clin%20Oncol&volume=35&issue=11&pages=1240-1249&publication_year=2017&doi=10.1200%2FJCO.2016.71.3677) [DOI] [PubMed] [Google Scholar]
- 48.La V, Fujikawa R, Janzen DM, Nunez M, Bainvoll L, Hwang L, Faull K, Lawson G, Memarzadeh S. Birinapant sensitizes platinum-resistant carcinomas with high levels of cIAP to carboplatin therapy. NPJ Precis Oncol. 2017;1 doi: 10.1038/s41698-017-0008-z. https://doi.org/10.1038/s41698-017-0008-z ( https://doi.org/10.1038/s41698-017-0008-z) [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 49.Eckstein N. Platinum resistance in breast and ovarian cancer cell lines. J Exp Clin Cancer Res. 2011;30:91. doi: 10.1186/1756-9966-30-91. https://doi.org/10.1186/1756-9966-30-91( https://doi.org/10.1186/1756-9966-30-91) CrossRef ( https://doi.org/10.1186/1756-9966-30-91) PubMed ( http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Abstract&list_uids=21967738) PubMedCentral ( http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3197542) Google Scholar ( http://scholar.google.com/scholar_lookup?title=Platinum%20resistance%20in%20breast%20and%20ovarian%20cancer%20cell%20lines&author=N.%20Eckstein&journal=J%20Exp%20Clin%20Cancer%20Res&volume=30&pages=91&publication_year=2011&doi=10.1186%2F1756-9966-30-91) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nagasawa S, Sedukhina AS, Nakagawa Y, Maeda I, Kubota M, Ohnuma S, Tsugawa K, Ohta T, Roche-Molina M, Bernal JA, Narvaez AJ, Jeyasekharan AD, Sato K. LSD1 overexpression is associated with poor prognosis in basal-like breast cancer, and sensitivity to PARP inhibition. PLoS One. 2015;10(2):e0118002. doi: 10.1371/journal.pone.0118002. https://doi.org/10.1371/journal.pone.0118002( https://doi.org/10.1371/journal.pone.0118002) CrossRef ( https://doi.org/10.1371/journal.pone.0118002) PubMed ( http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Abstract&list_uids=25679396) PubMedCentral ( http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4332491) Google Scholar ( http://scholar.google.com/scholar_lookup?title=LSD1%20overexpression%20is%20associated%20with%20poor%20prognosis%20in%20basal-like%20breast%20cancer%2C%20and%20sensitivity%20to%20PARP%20inhibition&author=S.%20Nagasawa&author=AS.%20Sedukhina&author=Y.%20Nakagawa&author=I.%20Maeda&author=M.%20Kubota&author=S.%20Ohnuma&author=K.%20Tsugawa&author=T.%20Ohta&author=M.%20Roche-Molina&author=JA.%20Bernal&author=AJ.%20Narvaez&author=AD.%20Jeyasekharan&author=K.%20Sato&journal=PLoS%20One&volume=10&issue=2&pages=e0118002&publication_year=2015&doi=10.1371%2Fjournal.pone.0118002) [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.