

An Inside Job
Enantioselective phthalide synthesis is achieved through internal redox allylation of o-phthalaldehydes. Oxidative esterification is balanced by reductive carbonyl addition to achieve an overall redox neutral process. Using this method, formal syntheses of ent-spirolaxine methyl ether and CJ-12,954 are achieved.

Keywords: Enantioselective, C-C Bond Formation, Iridium, Hydrogen Transfer, Homogenous Catalysis
Isobenzofuranones or “phthalides” occur frequently in nature and, due to their wide ranging biological activities, are common architectures in medicinal chemistry.[1] As illustrated in the Hauser-Kraus annulation, phthalides are also important building blocks in chemical synthesis.[2] Despite considerable progress, there are relatively few catalytic asymmetric methods for phthalide synthesis that are atom-efficient and do not employ stoichiometric metals.[3–8] These include ruthenium catalyzed transfer hydrogenation of ortho-keto-benzoates,[3] biocatalytic reduction of ortho-keto-benzonitriles,[4] intramolecular ketone hydroacylation,[5] [2+2+2] cycloadditions,[6] as well as organocatalyzed aldol additions[7] and arylboronic acid additions[8] to ortho-formyl-benzoates. Our interest in the development of alcohol-mediated C-C coupling[9] led us to consider a catalytic enantioselective phthalide synthesis based on the redox neutral allylation of aromatic ortho-dialdehydes or “phthalaldehydes” (Figure 1).[10] Specifically, a hydrogen auto-transfer process was envisioned in which asymmetric aldehyde allylation would trigger formation of an iridium lactolate, which upon β-hydride elimination would form the phthalide and an iridium hydride.[11] β-Hydride elimination from the homoallylic iridium alkoxide (not shown) is suppressed by coordination of the adjacent alkene. Deprotonation of the iridium hydride followed by ionization of allyl acetate would regenerate the π-allyliridium nucleophile to close the catalytic cycle. Beyond the mechanistic novelty of this process, we were motivated by the fact that despite enormous advances made in the field of enantioselective carbonyl allylation,[9g,12] application of asymmetric allylation to phthalide formation is limited to a small number of reports involving additions to ortho-formyl-benzoates for which relatively modest levels of enantiomeric enrichment were observed.[13,14]
Figure 1.

Redox neutral allylation of phthalaldehydes.
In an initial series of experiments, the parent phthalaldehyde 1a (100 mol%) was exposed to the chromatographically isolated π-allyliridium C,O-benzoate complexes (S)-Ir-I to (S)-Ir-VI in the presence of allyl acetate 2a (200 mol%) and Cs2CO3 (100 mol%) in THF (0.1 M) at 100 °C (Table 1, entries 1–6). This initial screen revealed that the proposed phthalide formation was indeed feasible and that the iridium catalyst (S)-Ir-VI modified by the “Roche ligand”[15] enforced the highest levels of enantioselectivity, providing phthalide 3a in 85% yield and 84% enantiomeric excess (Table 1, entry 6). As illustrated in the reactions catalyzed by (S)-Ir-II, (S)-Ir-V and (S)-Ir-VI, the addition of water (100 mol%) significantly enhances enantiomeric excess (Table 1, entries 7–9). Water might help solubilize the base, Cs2CO3, which maintains the structural integrity of the C,O-benzoate by promoting cyclometallation. The C,O-benzoate moiety is not kinetically inert under the reaction conditions and the “non-cyclometalated” catalyst, although less efficient, promotes formation of the opposite enantiomer.10b As illustrated in reactions catalyzed by (S)-Ir-VI, the use of other bases (K3PO4 and K2CO3) did not diminish yield or enantioselectivity (Table 1, entries 10, 11). Finally, if the reaction is run at higher concentration (0.2 M), the loading of catalyst (S)-Ir-VI could be decreased (3 mol%) without any erosion in performance (Table 1, entry 12).
Table 1.
Selected optimization experiments in the enantioselective iridium catalyzed internal redox allylation of phthalaldehyde 1a to form phthalide 3a.a

| ||||||
|---|---|---|---|---|---|---|
| Entry | [Ir] | Base | [THF] | H2O | Yield% 3a | ee% |
| 1 | (S)-Ir-I | Cs2CO3 | 0.1 M | --- | 93 | 65 |
| 2 | (S)-Ir-II | Cs2CO3 | 0.1 M | --- | 92 | 80 |
| 3 | (S)-Ir-III | Cs2CO3 | 0.1 M | --- | 68 | 62 |
| 4 | (S)-Ir-IV | Cs2CO3 | 0.1 M | --- | 73 | 60 |
| 5 | (S)-Ir-V | Cs2CO3 | 0.1 M | --- | 80 | 74 |
| 6 | (S)-Ir-VI | Cs2CO3 | 0.1 M | --- | 85 | 84 |
| 7 | (S)-Ir-II | Cs2CO3 | 0.1 M | 100 mol% | 88 | 96 |
| 8 | (S)-Ir-V | Cs2CO3 | 0.1 M | 100 mol% | 82 | 93 |
| 9 | (S)-Ir-VI | Cs2CO3 | 0.1 M | 100 mol% | 95 | 96 |
| 10 | (S)-Ir-VI | K3PO4 | 0.1 M | 100 mol% | 93 | 97 |
| 11 | (S)-Ir-VI | K2CO3 | 0.1 M | 100 mol% | 96 | 96 |
| ⇨12 | (S)-Ir-VIb | K2CO3 | 0.2 M | 100 mol% | 98 | 96 |
|
| ||||||
|
| ||||||
Yields are of material isolated by silica gel chromatography. Enantioselectivities were determined by chiral stationary phase HPLC analysis.
(S)-Ir-VI (3 mol%). See Supporting Information for further experimental details.
To assess the scope of this process, optimal conditions identified for the formation of phthalide 3a were applied to symmetric phthalaldehydes 1a–1f (Table 2). The desired phthalides 3a–3f were formed in good yield with generally high levels of enantioselectivity. These results inspired us to explore the use of α-substituted allyl donors (Table 3). Toward this end, o-phthalaldehyde 1a was exposed to allyl pronucleophiles 2b–2j in the presence of chiral catalyst (S)-Ir-VI. Here, lower temperatures are required to preserve kinetic selectivity in the formation of the (E)-σ-allyliridium haptomer[10c] and to enhance enantioselectivity. Thus, notwithstanding minor changes in reaction temperature, the standard conditions enabled access to products of crotylation 3g,[10c,i] cyclopropylallylation 3h,[10o] trimethylsilylallylation 3i,[10g] as well as adducts incorporating aliphatic (3j), aromatic (3k–3n) and heteroaromatic (3o) side chains. In each case, complete levels of branched regioselectivity were accompanied by good to complete levels of anti-diastereoselectivity and uniformly high levels of enantioselectivity.
Table 2.
Enantioselective iridium catalyzed internal redox allylation of symmetric dialdehydes 1a–1f to form phthalides 3a–3f.a

|
Yields are of material isolated by silica gel chromatography. Enantioselectivities were determined by chiral stationary phase HPLC analysis. See Supporting Information for further experimental details.
Table 3.
Enantioselective iridium catalyzed internal redox allylation of phthalaldehyde 1a using substituted allylic acetates 2b–2j to form phthalides 3g–3o.a

|
Yields are of material isolated by silica gel chromatography. Enantioselectivities were determined by chiral stationary phase HPLC analysis. See Supporting Information for further experimental details.
2d (300 mol%).
THF (1 M).
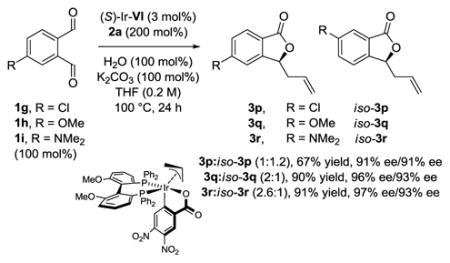
Site-selective internal redox allylation of non-symmetric o-phthalaldehydes requires discrimination between aldehyde moieties in the C-C bond forming event. To assess the influence of remote electronic effects on the chemoselectivity of such processes, the 4-substituted phthalaldehydes 1g–1i were subjected to standard conditions for internal redox allylation (eq. 1). For 1g–1i, the mesomeric effect of the remote 4-substituent increases and, in the case of 1h and 1i, was anticipated to deactivate the p-substituted aldehyde moiety to favor formation of 3q and 3r over the constitutionally isomeric products iso-3q and iso-3r, respectively. However, the observed levels of regioselectivity were quite modest. In contrast, the ortho-methoxy substituted phthalaldehydes 1j and 1kcould be engaged with significantly higher levels of selectivity (eq. 2). By exploiting synergistic steric and electronic effects, the 3,5-dimethoxy-substituted phthalaldehyde 1k was converted to 3t with good levels of site-selectivity. In the internal redox allylation of the ortho-methoxy substituted phthalaldehydes 1j and 1k, the π-allyliridium C,O-benzoate complex derived from 3,4-dinitrobenzoic acid and the “Roche ligand,”[15] (S)-Ir-VII, displayed superior performance compared to the catalysts (S)-Ir-I through (S)-Ir-VI. The assignment of absolute stereochemistry for adducts 3a–3t was made in analogy to phthalide 3l, which was determined by single crystal X-ray diffraction, and phthalide 3t, which was evaluated by comparison of its optical rotation to that of an authentic sample.[16] Underscoring the utility of this method in target oriented synthesis, the preparation of 3t constitutes a formal synthesis of two different phthalide natural products: ent-spirolaxine methyl ether[17] and CJ-12,954 (Figure 2).[18] Both compounds have been shown to exhibit antibiotic activity against the Gram-negative bacteria Helicobacter pylori.[19]
Figure 2.

Formal syntheses of ent-spirolaxine methyl ether and CJ-12,954.
 |
(eq 1) |
 |
(eq 2) |
In summary, we report an enantioselective hydrogen auto-transfer[20] reaction that converts o-phthalaldehydes to phthalides. In this processes, asymmetric aldehyde allylation trigger formation of an iridium lactolate, which upon β-hydride elimination delivers the phthalide. Thus, reductive carbonyl addition is balanced by oxidative esterification to achieve an overall redox neutral process. Beyond the parent enantioselective allylation, diverse substituted allyl donors were found to participate in this process. Additionally, as illustrated by the formation of phthalide 3t, the synergistic steric and electronic influence of substituents of the o-phthalaldehydes enable site-selective internal redox allylation. Finally, the formation of phthalide 3t represents formal asymmetric syntheses of two related natural products, ent-spirolaxine methyl ether and CJ-12,954, highlighting the synthetic utility of this process.
Supplementary Material
Footnotes
The Welch Foundation (F-0038), the NIH (RO1-GM069445) and the UT Austin Center for Green Chemistry and Catalysis are acknowledged for partial support of this research. The Deutsche Forschungsgemeinschaft (DFG) is acknowledged for postdoctoral fellowship support (JT).
References
- 1.For selected reviews on natural occurrence and synthesis of phthalides, see: Lin G, Chan SS-K, Chung H-S, Li S-L. In: Studies in Natural Products Chemistry: Bioactive Natural Products. Part L. Atta-ur-Rahman, editor. Vol. 32. Elsevier; Amsterdam: 2005. pp. 611–669.Karmakar R, Pahari P, Mal D. Chem Rev. 2014;114:6213. doi: 10.1021/cr400524q.
- 2.For selected reviews on the Hauser-Kraus annulation, see: Mitchell AS, Russell RA. Tetrahedron. 1995;51:5207.Rathwell K, Brimble MA. Synthesis. 2007:643.Mal D, Pahari P. Chem Rev. 2007;107:1892. doi: 10.1021/cr068398q.
- 3.a) Kitamura M, Ohkuma T, Inoue S, Sayo N, Kumobayashi H, Akutagawa S, Ohta T, Takaya H, Noyori R. J Am Chem Soc. 1988;110:629. [Google Scholar]; b) Ohkuma T, Kitamura M, Noyori R. Tetrahedron Lett. 1990;31:5509. [Google Scholar]; c) Everaere K, Scheffler JL, Mortreuz A, Carpentier JF. Tetrahedron Lett. 2001;42:1899. [Google Scholar]; d) Zhang B, Xu MH, Lin GQ. Org Lett. 2009;11:4712. doi: 10.1021/ol901674k. [DOI] [PubMed] [Google Scholar]
- 4.Mangas-Sánchez J, Busto E, Gotor-Fernández V, Gotor V. Org Lett. 2012;14:1444. doi: 10.1021/ol300191s. [DOI] [PubMed] [Google Scholar]
- 5.a) Phan DHT, Kim B, Dong VM. J Am Chem Soc. 2009;131:15608. doi: 10.1021/ja907711a. [DOI] [PubMed] [Google Scholar]; b) Yang J, Yoshikai N. J Am Chem Soc. 2014;136:16748. doi: 10.1021/ja509919x. [DOI] [PubMed] [Google Scholar]
- 6.Tanaka K, Osaka T, Noguchi K, Hirano M. Org Lett. 2007;9:1307. doi: 10.1021/ol070179j. [DOI] [PubMed] [Google Scholar]
- 7.Zhang H, Zhang S, Liu L, Luo G, Duan W, Wang W. J Org Chem. 2010;75:368. doi: 10.1021/jo902118x. [DOI] [PubMed] [Google Scholar]
- 8.Yohda M, Yamamoto Y. Org Biomol Chem. 2015;13:10874. doi: 10.1039/c5ob01661d. [DOI] [PubMed] [Google Scholar]
- 9.For recent reviews on alcohol-mediated carbonyl addition, see: Ketcham JM, Shin I, Montgomery TP, Krische MJ. Angew Chem Int Ed. 2014;53:9142. doi: 10.1002/anie.201403873.Angew Chem. 2014;126:9294.Dechert-Schmitt AMR, Schmitt DC, Gao X, Itoh T, Krische MJ. Nat Prod Rep. 2014;31:504. doi: 10.1039/c3np70076c.Feng J, Kasun ZA, Krische MJ. J Am Chem Soc. 2016;138:5467. doi: 10.1021/jacs.6b02019.Perez F, Oda S, Geary LM, Krische MJ. Top Curr Chem. 2016;374:365. doi: 10.1007/s41061-016-0028-0.Shin I, Krische MJ. Top Curr Chem. 2016;372:85. doi: 10.1007/128_2015_651.Nguyen KD, Park BY, Luong T, Sato H, Garza VJ, Krische MJ. Science. 2016;354:aah5133. doi: 10.1126/science.aah5133.Kim SW, Zhang W, Krische MJ. Acc Chem Res. 2017;50:2371. doi: 10.1021/acs.accounts.7b00308.
- 10.For selected alcohol-mediated enantioselective carbonyl allylations catalyzed by iridium, see: Kim IS, Ngai MY, Krische MJ. J Am Chem Soc. 2008;130:6340. doi: 10.1021/ja802001b.Kim IS, Ngai MY, Krische MJ. J Am Chem Soc. 2008;130:14891. doi: 10.1021/ja805722e.Kim IS, Han SB, Krische MJ. J Am Chem Soc. 2009;131:2514. doi: 10.1021/ja808857w.Han SB, Kim IS, Han H, Krische MJ. J Am Chem Soc. 2009;131:6916. doi: 10.1021/ja902437k.Han SB, Han H, Krische MJ. J Am Chem Soc. 2010;132:1760. doi: 10.1021/ja9097675.Zhang YJ, Yang JH, Kim SH, Krische MJ. J Am Chem Soc. 2010;132:4562. doi: 10.1021/ja100949e.Han SB, Gao X, Krische MJ. J Am Chem Soc. 2010;132:9153. doi: 10.1021/ja103299f.Hassan A, Zbieg JR, Krische MJ. Angew Chem Int Ed. 2011;50:3493. doi: 10.1002/anie.201100646.Angew Chem. 2011;123:3555.Gao X, Townsend IA, Krische MJ. J Org Chem. 2011;76:2350. doi: 10.1021/jo200068q.Gao X, Zhang YJ, Krische MJ. Angew Chem Int Ed. 2011;50:4173. doi: 10.1002/anie.201008296.Angew Chem. 2011;123:4259.Hassan A, Townsend IA, Krische MJ. Chem Comm. 2011:10028. doi: 10.1039/c1cc14392a.Moran J, Smith AG, Carris RM, Johnson JS, Krische MJ. J Am Chem Soc. 2011;133:18618. doi: 10.1021/ja2090993.Feng J, Garza VJ, Krische MJ. J Am Chem Soc. 2014;136:8911. doi: 10.1021/ja504625m.Wang G, Franke J, Ngo CQ, Krische MJ. J Am Chem Soc. 2015;137:7915. doi: 10.1021/jacs.5b04404.Tsutsumi R, Hong S, Krische MJ. Chem Eur J. 2015;21:12903. doi: 10.1002/chem.201502499.
- 11.For an authoritative review on iridium catalyzed alcohol oxidation, see: Suzuki T. Chem Rev. 2011;111:1825. doi: 10.1021/cr100378r.
- 12.For selected reviews on enantioselective carbonyl allylation, see: Ramachandran PV. Aldrichim Acta. 2002;35:23.Denmark SE, Fu J. Chem Rev. 2003;103:2763. doi: 10.1021/cr020050h.Yu CM, Youn J, Jung HK. Bull Korean Chem Soc. 2006;27:463.Marek I, Sklute G. Chem Commun. 2007:1683. doi: 10.1039/b615042j.Hall DG. Synlett. 2007:1644.Lachance H, Hall DG. Org React. 2008;73:1.Yus M, Gonzalez-Gomez JC, Foubelo F. Chem Rev. 2011;111:7774. doi: 10.1021/cr1004474.
- 13.a) Mirabdolbaghi R, Dudding T. Org Lett. 2012;14:3748. doi: 10.1021/ol301566f. [DOI] [PubMed] [Google Scholar]; b) Mirabdolbaghi R, Dudding T. Tetrahedron. 2013;69:3287. [Google Scholar]
- 14.For an isolated example of asymmetric NHK allylation of ortho-formyl-benzoates, see: Huang X, Pan X, Lee G, Chen C. Adv Synth Catal. 2011;353:1949.
- 15.a) Schmid R, Cereghetti M, Heiser B, Schönholzer P, Hansen HJ. Helv Chim Acta. 1988;71:897. [Google Scholar]; b) Schmid R, Foricher J, Cereghetti M, Schönholzer P. Helv Chim Acta. 1991;74:370. [Google Scholar]
- 16.Keaton KA, Phillips AJ. Org Lett. 2007;9:2717. doi: 10.1021/ol0710111. [DOI] [PubMed] [Google Scholar]
- 17.a) Robinson JE, Brimble MA. Chem Commun. 2005:1560. doi: 10.1039/b418106a. [DOI] [PubMed] [Google Scholar]; b) Keaton KA, Phillips AJ. Org Lett. 2007;9:2717. doi: 10.1021/ol0710111. [DOI] [PubMed] [Google Scholar]; c) Gadakh SK, Sudalai A. Tetrahedron Lett. 2016;57:25. [Google Scholar]
- 18.Brimble MA, Bryant CJ. Chem Commun. 2006:4506. doi: 10.1039/b612757f. [DOI] [PubMed] [Google Scholar]
- 19.a) Dekker KA, Inagaki T, Gootz TD, Kaneda K, Nomura E, Sakakibara T, Sakemi S, Sugi Y, Yamauchi Y, Yoshikawa N, Kojima N. J Antibiotics. 1997;50:833. doi: 10.7164/antibiotics.50.833. [DOI] [PubMed] [Google Scholar]; b) Radcliff FJ, Fraser JD, Wilson ZE, Heapy AM, Robinson JE, Bryant CJ, Flowers CL, Brimble MA. Bioorg Med Chem. 2008;16:6179. doi: 10.1016/j.bmc.2008.04.037. [DOI] [PubMed] [Google Scholar]
- 20.For selected reviews on related hydrogen auto-transfer or so-called “borrowing hydrogen” reactions, see: Guillena G, Ramón DJ, Yus M. Angew Chem Int Ed. 2007;46:2358. doi: 10.1002/anie.200603794.Angew Chem. 2007;119:2410.Hamid MHSA, Slatford PA, Williams JMJ. Adv Synth Catal. 2007;349:1555.Nixon TD, Whittlesey MK, Williams JMJ. Dalton Trans. 2009:753. doi: 10.1039/b813383b.Dobereiner GE, Crabtree RH. Chem Rev. 2010;110:681. doi: 10.1021/cr900202j.Guillena G, Ramón DJ, Yus M. Chem Rev. 2010;110:1611. doi: 10.1021/cr9002159.Yang Q, Wang Q, Yu Z. Chem Soc Rev. 2015;44:2305. doi: 10.1039/c4cs00496e.Nandakumar A, Midya SP, Landge VG, Balaraman E. Angew Chem Int Ed. 2015;54:11022. doi: 10.1002/anie.201503247.Angew Chem. 2015;127:11174.Huang F, Liu Z, Yu Z. Angew Chem Int Ed. 2016;55:862. doi: 10.1002/anie.201507521.Angew Chem. 2016;128:872.Quintard A, Rodriguez J. Chem Comm. 2016;52:10456. doi: 10.1039/c6cc03486a.Quintard A, Rodriguez J. ChemSusChem. 2016;9:28. doi: 10.1002/cssc.201501460.Chelucci G. Coord Chem Rev. 2017;331:37.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.