

ABSTRACT
Borrelia burgdorferi is a causative agent of Lyme disease, the most common arthropod-borne disease in the United States. B. burgdorferi evades host immune defenses to establish a persistent, disseminated infection. Previous work showed that P66-deficient B. burgdorferi (Δp66) is cleared quickly after inoculation in mice. We demonstrate that the Δp66 strain is rapidly cleared from the skin inoculation site prior to dissemination. The rapid clearance of Δp66 bacteria is not due to inherent defects in multiple properties that might affect infectivity: bacterial outer membrane integrity, motility, chemotactic response, or nutrient acquisition. This led us to the hypothesis that P66 has a role in mouse cathelicidin-related antimicrobial peptide (mCRAMP; a major skin antimicrobial peptide) and/or neutrophil evasion. Neither wild-type (WT) nor Δp66 B. burgdorferi was susceptible to mCRAMP. To examine the role of neutrophil evasion, we administered neutrophil-depleting antibody anti-Ly6G (1A8) to C3H/HeN mice and subsequently monitored the course of B. burgdorferi infection. Δp66 mutants were unable to establish infection in neutrophil-depleted mice, suggesting that the important role of P66 during early infection is through another mechanism. Neutrophil depletion did not affect WT B. burgdorferi bacterial burdens in the skin (inoculation site), ear, heart, or tibiotarsal joint at early time points postinoculation. This was unexpected given that prior in vitro studies demonstrated neutrophils phagocytose and kill B. burgdorferi. These data, together with our previous work, suggest that despite the in vitro ability of host innate defenses to kill B. burgdorferi, individual innate immune mechanisms have limited contributions to controlling early B. burgdorferi infection in the laboratory model used.
KEYWORDS: Borrelia burgdorferi, innate immunity, stress
INTRODUCTION
Lyme disease is the most common arthropod-borne disease in the United States, and it is caused by a group of spirochetes known as Borrelia burgdorferi sensu lato (1). B. burgdorferi is naturally maintained in a vertebrate-arthropod infection cycle and is transmitted to humans through infected ticks of the Ixodes genus (reviewed in references 2 and 3). Lyme disease may cause early localized infection, manifesting as erythema migrans, and/or disseminated infection, manifesting as neuroborreliosis, carditis, or arthritis in the large weight-bearing joints (4–7).
The host's first cellular response to a B. burgdorferi infection occurs in the skin tissue after the tick deposits the bacteria during feeding. In animal models, cellular infiltrates at the site of infection include phagocytic cells such as neutrophils within hours and macrophages within days to weeks of inoculation (8–10). Characterization of the interactions between B. burgdorferi and the cellular infiltrates has been the subject of several previous studies. Macrophages, dendritic cells, and neutrophils have been shown to phagocytose and kill B. burgdorferi in vitro through conventional, coiling, or tube phagocytosis (9, 11–18), leading to the hypothesis that each of these innate immune cells participates in control of early B. burgdorferi infections in vivo. Previous studies have tested this hypothesis by examining the in vivo role of neutrophils in controlling B. burgdorferi infection in mice. Infecting C3H SCID mice with a mutant strain of B. burgdorferi capable of producing and secreting the chemokine KC, a neutrophil chemoattractant, resulted in increased neutrophil recruitment within 6 h to the inoculation site and severely attenuated B. burgdorferi infectivity when assessed 4 weeks postinoculation (19). It has also been shown that BALB/c mice can form neutrophil extracellular traps (NETs) upon tick infestation and that NETs can trap and kill B. burgdorferi in vitro (20). Additionally, treatment of C3H/HeJ mice with neutrophil-depleting anti-RB6-8C5 antibody resulted in increased B. burgdorferi bacterial burden and exacerbated arthritis in the ankles of mice at 7 days after footpad inoculation (21). Although much research has documented how the innate immune system defends against B. burgdorferi infection, the bacteria are still able to overcome these early host responses to establish a persistent, disseminated infection in vivo.
Elucidation of the mechanisms of how B. burgdorferi is able to evade the immune system to establish infection has been a compelling area of investigation for some time. While immune evasion mechanisms of B. burgdorferi are continually being revealed, several mechanisms by which B. burgdorferi can avoid the host immune system have been described. Appropriate regulation of outer membrane protein production is critical for B. burgdorferi to establish infection (reviewed in reference 22). For example, during mammalian infection B. burgdorferi produces outer membrane proteins important for tissue colonization (adhesins) and/or important for innate and adaptive immune system evasion. B. burgdorferi produces CspA, which confers resistance to in vitro serum killing by binding to FH, FHL-1, C7, and C9 (23–26). VlsE is a lipoprotein capable of undergoing antigenic variation as a way to evade the adaptive immune response (27–30). Some outer membrane proteins produced by B. burgdorferi, such as OspC and BBK32, have multiple roles in adhering to host extracellular components and in immune evasion. OspC acts as an adhesin by binding to plasminogen (31) and as an immune evasion factor by facilitating bloodstream survival by competing with binding of complement C2 to C4b and acting as an antiphagocytic factor (32–34). Similarly, BBK32 binds to fibronectin and glycosaminoglycan (35–37) and inhibits the classical complement pathway by blocking C1 activation (38).
Another multifunctional outer membrane protein produced by B. burgdorferi is P66, which forms a pore in the bacterial outer membrane and is a ligand for mammalian integrins (39, 40). P66 is produced during mammalian but not tick infection, suggesting it is a virulence factor specific for mammalian infection (41). This is supported by the infection pattern of B. burgdorferi mutants unable to produce P66 (Δp66): Δp66 mutants are unable to survive in mice longer than 48 h but survive in ticks through the molt (42). The integrin binding function of P66 facilitates both transmigration out of the vasculature and bacterial dissemination but is not critical to establish infection (43, 44). Although the size of the pore has been determined (opening diameter of 1.9 nm with a central constriction of 0.8 nm) (45), the in vivo importance of the pore formed by P66 remains unknown. These data, taken together, suggest that P66 functions to help B. burgdorferi evade the host innate immune system. Previous work investigated the role of P66 in macrophage, dendritic cell, or complement evasion. However, Δp66 mutants are unable to infect TLR2−/−, MyD88−/−, macrophage-depleted, or dendritic cell-depleted mice (42, 43). Additionally, P66 does not have a role in conferring resistance to serum proteins in vitro or in vivo (32, 43). Even though P66 has been well studied, the critical function of P66 during mammalian infection remains to be determined.
In this study, we further investigate the role of P66 during mammalian infection by testing three hypotheses: (i) the loss of P66 may result in B. burgdorferi outer membrane instability, (ii) the pore formed by P66 may be important for acquiring crucial nutrients in the mammalian host, and (iii) P66 may confer resistance to mCRAMP or neutrophils. Major findings from the work presented here are that the loss of P66 does not result in inherent defects in outer membrane stability, motility, chemotactic response, or nutrient acquisition and that Δp66 B. burgdorferi is not susceptible to mCRAMP and does not establish infection in neutrophil-depleted mice. An additional important finding is that the depletion of neutrophils does not affect wild-type B. burgdorferi bacterial burdens in mice. This is similar to previous findings that depletion of macrophages or dendritic cells also do not affect B. burgdorferi burdens in mice (43). Together, these data suggest macrophages, dendritic cells, and neutrophils, individually, have limited contribution to controlling B. burgdorferi infection in mice under the conditions tested despite in vitro experiments documenting phagocytosis and killing of B. burgdorferi by these cell types.
RESULTS
The loss of P66 does not result in B. burgdorferi outer membrane instability.
B. burgdorferi has a relatively fragile outer membrane compared to other Gram-negative bacteria due to a lower density of transmembrane-spanning proteins and lack of lipopolysaccharide (46–49). We hypothesized that the loss of P66 resulted in B. burgdorferi outer membrane instability, which is inconsequential when grown in Barbour-Stoenner-Kelly II (BSKII) but exacerbated and detrimental when the bacteria are moving through tissues in a mammalian host. Detergent susceptibility assays, shear force assays, and osmotic pressure sensitivity assays were conducted to determine if the Δp66 mutants have a more fragile outer membrane than the parental wild-type (WT) strain.
When increasing concentrations of SDS were introduced into BSKII medium, there was no difference in susceptibility between WT and Δp66 bacteria. Both WT and Δp66 bacteria survived in 0.1% SDS, but both were lysed immediately upon introduction to 0.3% SDS (Fig. 1). We also found no difference in susceptibility between WT and Δp66 bacteria to N,N-dimethyldodecylamine N-oxide solution (LDAO) or Triton X-100 (data not shown).
FIG 1.
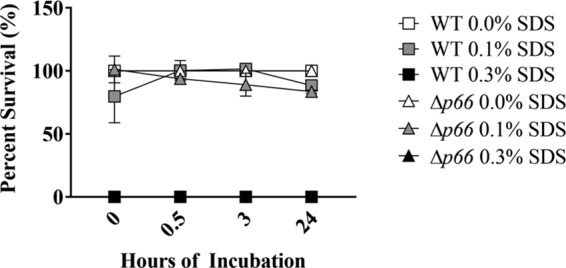
No difference in SDS susceptibility between WT and Δp66 B. burgdorferi. WT or Δp66 cells at 2 × 106 cells ml−1 in 5 ml BSKII supplemented with SDS at final concentrations of 0.0%, 0.1%, and 0.3% were incubated at 33°C up to 24 h. B. burgdorferi survival was quantified through plating for CFU. Percent survival was calculated by comparing each SDS condition to 0.0% SDS at each time point for each B. burgdorferi strain. Data are representative of two independent experiments. Error bars represent SEM. In some cases, the error bar is smaller than the symbols.
B. burgdorferi encounters shear forces during dissemination by moving through the skin and other tissues and by traversing the vasculature. To test if the Δp66 bacteria are less able to endure shear stress than the WT, the bacteria were passed through a 29-gauge needle up to 9 times. Upon repeated needle passages both WT and Δp66 bacteria were killed. Only approximately 50% of cells from each strain survived all 9 passages through the needle (Fig. 2); however, there was no difference in susceptibility to shear forces between WT and Δp66 bacteria following any number of needle passages (Fig. 2).
FIG 2.

No difference in shear force susceptibility between WT and Δp66 B. burgdorferi. WT and Δp66 were suspended in 200 μl of PBS plus 0.2% NMS at a density of 1 × 106 cells ml−1 and passed through a 29-gauge needle up to 9 times. Bacterial survival after each passage was quantified by plating for CFU. Data shown are means from three technical replicates with error bars representing SEM; some error bars are smaller than the symbols. Two-way analysis of variance (ANOVA) with Bonferroni's posttest was used to compare survival of needle passages between B. burgdorferi strains; no significant difference was observed.
Another way to examine the outer membrane stability of bacteria is to introduce osmotic stresses. Since BSKII is made using complex ingredients such as yeastolate, rabbit serum, bovine serum albumin (BSA), and CMRL, it is difficult to make a hypotonic medium suitable for B. burgdorferi growth. Therefore, we elected to grow B. burgdorferi in hypertonic medium by introducing higher concentrations of NaCl to BSKII. Normally, BSKII has approximately 120 mM NaCl. For this experiment, NaCl was added to BSKII for final concentrations of 200 mM, 300 mM, 400 mM, and 500 mM. Both WT and Δp66 bacteria were able to grow in 200 mM NaCl BSKII without inhibition, but they were unable to grow in NaCl concentrations greater than 400 mM (Fig. 3). When grown in 300 mM NaCl BSKII, the Δp66 bacteria were able to grow faster and to higher densities than the WT (Fig. 3). Even though this result is not yet mechanistically understood, the results of this experiment clearly demonstrate that Δp66 bacteria are not more susceptible to osmotic stresses than WT bacteria. Based on the detergent sensitivity assay, the shear force assay, and the osmotic stress assay, it does not appear that the Δp66 bacteria have reduced outer membrane stability compared to that of WT B. burgdorferi.
FIG 3.

B. burgdorferi growth rates in increased salt concentrations. WT and Δp66 cultures were grown to 1 × 107 to 2 × 107 cells ml−1 and then diluted to a starting density of 1 × 104 cells ml−1 in 5 ml of normal BSKII or BSKII supplemented with NaCl to the final concentrations shown. Cultures were incubated at 33°C and counted daily. Data shown are means from three technical replicates with error bars representing SEM; some error bars are smaller than the symbols. Linear regression analysis was used to compare exponential phase of growth curves between B. burgdorferi strains in each NaCl concentration (*, P < 0.05).
P66 mutants are unable to disseminate away from inoculation site.
Δp66 mutants are unable to establish infection in immunocompetent mice, and they are cleared within 48 h (42). In order to gain a better understanding of what is happening to Δp66 B. burgdorferi during this brief period of time in the mice, we conducted a dissemination experiment. Seven- to 9-week-old female C3H/HeN mice were inoculated with 1 × 105 cells of WT, Δp66, or p66cc (p66 restored to the endogenous locus on the chromosome) bacteria (42). After the indicated number of hours or days, mice were euthanized and tissues were collected and placed in culture medium, and cultures were checked for the presence of live B. burgdorferi. WT B. burgdorferi started to disseminate from the site of inoculation by 72 h and became fully disseminated by 14 days postinoculation (Fig. 4). The Δp66 mutants were unable to disseminate from the skin inoculation site prior to being cleared (Fig. 4). The restoration of p66 to the chromosome (p66cc) restored infectivity, albeit with a slight delay in dissemination. However, p66cc was still able to become fully disseminated by 28 days postinoculation (Fig. 4). Since the Δp66 mutant was found only in the skin inoculation site, we focused our efforts on possible interactions between Δp66 bacteria and mouse skin to investigate mechanisms of bacterial clearance.
FIG 4.

Δp66 mutant is unable to disseminate away from the inoculation site prior to being cleared. B. burgdorferi cultures were grown to a density of 5 × 107 to 2 × 108 cells ml−1, washed, and suspended in PBS plus 0.2% NMS. C3H/HeN mice were inoculated subcutaneously with a dose of 1 × 105 organisms. Mice were euthanized, and the indicated tissues were harvested over a time course (3 h to 28 days) postinoculation. The presence of B. burgdorferi in harvested tissues was determined by culturing the tissues in BSKII and incubating at 33°C. Growth of B. burgdorferi in each culture was checked for up to 8 weeks using a dark-field microscope. Tissue coloring: white, uninfected; gray, <50% of mice were infected; black, ≥50% of mice were infected. Data are representative of two independent experiments with 5 mice per group.
Deletion of Δp66 has no impact on motility and chemotaxis.
Recent studies have shown that both motility and chemotaxis are vital for B. burgdorferi to establish a persistent, disseminated infection (50, 51). B. burgdorferi mutants that have a defect in motility or chemotaxis display in vivo phenotypes similar to those of Δp66 bacteria; both are rapidly cleared from the site of inoculation (42, 50, 51). Bacterial motion tracking analysis revealed that the Δp66 strain (15.5 ± 2.3 μm/s; n = 28) and the p66cc complemented strain (12.9 ± 2.9 μm/s; n = 27) did not swim at significantly different speeds. Capillary tube chemotaxis assays were performed to test the hypothesis that the loss of P66 results in an attenuated chemotactic response in B. burgdorferi. A positive chemotactic response was defined as at least twice as many cells entering tubes filled with known attractants (52, 53) compared to the buffer-filled tubes (52, 54). Both WT and Δp66 bacteria had a positive chemotactic response to GlcNAc and rabbit serum (Fig. 5). Based on both experiments, the Δp66 mutants do not display grossly defective motility and chemotaxis under in vitro conditions.
FIG 5.

Loss of P66 does not hinder chemotactic response in B. burgdorferi. Bacterial cells were grown to 5 × 107 to 7 × 107 cells ml−1, harvested, and resuspended in motility buffer. Capillary tubes filled with either the attractant or motility buffer were sealed and inserted into microcentrifuge tubes containing 200 μl of resuspended cells (7 × 108 cells ml−1). After 2 h of incubation at 34°C in a humidified chamber, the solutions were expelled from the capillary tubes and the spirochetes were enumerated. A positive chemotactic response was defined as at least twice as many cells entering the attractant-filled tubes as the buffer-filled tubes. Significance of the fold differences between the two strains was evaluated using an unpaired Student's t test; no significant differences were seen.
Δp66 bacteria are able to grow in mouse skin homogenate ex vivo.
Gram-negative bacteria use porins to bind sugars preferentially to help facilitate diffusion into the cell. For example, ScrY and LamB help facilitate diffusion of sucrose and maltooligosaccharides, respectively, into the cell through binding of the sugars (55–57). Therefore, an additional hypothesis we tested was that the pore formed by P66 is crucial to acquiring critical nutrients from the mouse skin to sustain bacterial growth. This hypothesis was tested through two methods: growing WT and Δp66 strains in glucose-free BSKII medium supplemented with glucose, maltose, lactose, sucrose glycerol, or GlcNAc and by growing Δp66 and WT strains in mouse skin homogenate. There were no differences in growth between WT and Δp66 strains in any of the different sugars tested (see Fig. S1 in the supplemental material).
Preliminary experiments showed that neither WT nor Δp66 bacteria were able to grow in mouse skin homogenate alone. One hypothesis we tested was that the lack of growth in skin homogenate was due to low concentrations of one or more nutrients. Therefore, the homogenate was supplemented with increasing proportions of BSKII. Cultures were grown at 33°C for 10 days, with densities calculated daily using a Petroff-Hausser counting chamber under dark-field microscopy. Both WT and Δp66 strains were able to grow in media containing up to 40% skin homogenate at the same rate and to the same densities as those grown in 100% BSKII (Fig. 6). Both strains reached stationary phase earlier in higher concentrations of skin homogenate, but overall there was no difference in growth between WT and Δp66 strains at any concentration of skin homogenate tested.
FIG 6.

Loss of P66 does not result in impaired growth in mouse skin homogenate. WT and Δp66 strains were grown to mid-log phase and then diluted to a starting density of 1 × 105 cells ml−1 at the indicated ratios of BSKII to mouse skin homogenate (top) or BSKII to homogenate buffer (bottom). The cultures were incubated at 33°C and counted daily. Data shown are means from three technical replicates with error bars representing SEM. Some error bars are smaller than the symbols. Linear regression analysis was used to compare the exponential phase of growth curves between B. burgdorferi strains grown in each skin homogenate concentration; no significant differences were seen.
Interestingly, growth was inhibited for both B. burgdorferi strains in medium containing over 40% mouse skin homogenate, but both strains were able to grow well in medium containing over 40% of the homogenate buffer (Fig. 6). This indicated that the early growth arrest is not due to lack of nutrients but due to some component of the mouse skin having detrimental effects on B. burgdorferi growth.
We therefore tested a second hypothesis arising from the lack of growth in skin homogenate. One component of mouse skin we examined was the mouse cathelicidin-related antimicrobial peptide (mCRAMP). To test the hypothesis that mCRAMP is detrimental to either the WT or Δp66 strain, both strains were incubated with increasing concentrations of the peptide. mCRAMP was unable to kill either B. burgdorferi strain even at a concentration of 200 μg ml−1 (51.5 μM) (Fig. 7), which is about 5 times the concentration of mCRAMP found in the skin (3 to 10 μM) following injury (58). A laboratory Escherichia coli strain was used to demonstrate that the mCRAMP peptide was active, and it was killed at mCRAMP concentrations as low as 6.25 μg ml−1 (1.6 μM) (Fig. 7). This result indicated that mCRAMP is not responsible for killing the WT or Δp66 strain. Therefore, we next investigated the role of a critical innate immune cell type at the site of inoculation that might be responsible for clearing the Δp66 cells early after inoculation.
FIG 7.

P66 does not confer cathelicidin resistance. B. burgdorferi cultures were grown to a density between 5 × 107 and 3 × 108 cells ml−1 and then incubated with increasing concentrations of mCRAMP at 33°C for 3 h. Viable bacteria were enumerated by plating. Vehicle control replicated the ratio of 0.01% acetic acid plus 0.2% BSA to MHB, as found under the 200-μg ml−1 mCRAMP condition. Dose-dependent killing of E. coli was determined in parallel. Percent survival was calculated by comparing each mCRAMP concentration to the 0-μg ml−1 mCRAMP condition for each bacterial strain. Two-way ANOVA with Bonferroni's posttest was used to compare survival of B. burgdorferi strains incubated at different mCRAMP concentrations (*, P < 0.05).
Neutrophils have limited contribution to controlling early B. burgdorferi infection.
The Δp66 strain is unable to establish infection in TLR2−/−, MyD88−/−, macrophage-depleted, or dendritic cell-depleted mice (42, 43), but the role of neutrophils in clearing the mutant bacteria has not been examined in vivo. To test the hypothesis that P66 confers resistance to neutrophils, we depleted neutrophils in C3H/HeN mice using an anti-Ly6G (clone 1A8) antibody (59). Following depletion, mice were inoculated subcutaneously with the WT or Δp66 strain at a dose of 1 × 105 cells. Following mouse euthanasia at 3 h, 3 days, and 7 days postinoculation, blood samples were collected to determine the extent of neutrophil depletion in each mouse. At both 3 h and 3 days after B. burgdorferi inoculation, all mice treated with anti-Ly6G antibody had significantly reduced circulating neutrophils compared to the mice treated with an isotype control antibody (P < 0.05) (Fig. 8A). At 7 days postinoculation, Δp66- and sham-inoculated mice treated with anti-Ly6G antibody had significantly reduced circulating neutrophils as well. While there was a reduction of circulating neutrophils in the anti-Ly6G-treated mice infected with the WT compared to isotype control-treated mice 7 days postinoculation, the difference was not statistically significant (P = 0.0898) (Fig. 8A).
FIG 8.

Δp66 mutant is unable to establish infection in neutrophil-depleted C3H/HeN mice. (A) Mice were injected intraperitoneally (i.p.) with anti-Ly6G (1A8) or isotype control antibodies (500 μg/mouse) every 48 h, starting at 24 h prior to B. burgdorferi inoculation. Mice were euthanized at the indicated time points postinoculation. Blood was collected in the presence of an anticoagulant solution. Red blood cells were lysed, and white blood cells were stained with anti-GR-1 and anti-F4/80 antibody conjugates. Flow cytometry was performed using the Guava EasyCyte Mini, and data were analyzed using FlowJo (version 9.7.6). Data are shown as the absolute number of side-scatter (SSC)-high, GR-1-positive, and F4/80-negative cells per microliter of blood. Data were analyzed using unpaired Student's t test (*, P < 0.05). (B) WT and Δp66 strains were inoculated subcutaneously into neutrophil-depleted mice at a dose of 1 × 105 cells. Mice were euthanized and the indicated tissues were collected. The presence of live B. burgdorferi in each tissue was determined by culturing the tissue in BSKII. Tissue coloring: white, uninfected; gray, <50% of mice were infected; black, ≥50% of mice were infected.
To determine if the depletion of neutrophils allowed the Δp66 strain to establish infection, tissues (inoculation site, ear, heart, bladder, and tibiotarsal joint) were collected at the time of euthanasia for both culturing and quantitative PCR (qPCR). The presence of live B. burgdorferi was determined by culturing each tissue in BSKII. Cultures were checked for the presence of B. burgdorferi by direct observation under dark-field microscopy for up to 8 weeks postharvest. WT cells fully disseminated by 7 days in both anti-Ly6G- and isotype control-treated mice (Fig. 8B). It is interesting that WT B. burgdorferi had a different pattern of dissemination in anti-Ly6G treated mice than the isotype control-treated mice. At 3 days postinoculation, WT cells were found at the inoculation site and in the heart in the isotype control mice, while in the anti-Ly6G-treated mice, WT bacteria were found at the inoculation site and the ear. The depletion of neutrophils did not rescue the infectivity phenotype of the Δp66 strain, which was cleared within 3 days from both anti-Ly6G-treated mice and isotype control-treated mice (Fig. 8B). Interestingly, at 3 h postinoculation the Δp66 mutant was found in the ear of one of the anti-Ly6G-treated mice but not in any of the isotype control-treated mice (Fig. 8B).
To determine whether neutrophils play a role in controlling B. burgdorferi infection in vivo, the bacterial burdens in each tissue were determined using qPCR. The loss of neutrophils did not affect the bacterial loads of WT bacteria in the tissues examined (Fig. 9). This is consistent with previous results that showed that depletion of macrophages or dendritic cells did not have an effect on B. burgdorferi bacterial loads (43).
FIG 9.

Neutrophil depletion did not impact the bacterial burden in mice infected with WT or Δp66 B. burgdorferi. Mice were injected i.p. with anti-Ly6G (1A8) or isotype control antibodies (500 μg/mouse) every 48 h starting 24 h prior to B. burgdorferi inoculations. Mice were inoculated with 1 × 105 cells of WT or Δp66 bacteria. Sham-inoculated (PBS plus 0.2% NMS) mice were used as controls. Mice were euthanized at the indicated time points postinoculation, and the indicated tissues were collected for qPCR. Results from qPCR are represented as B. burgdorferi genomes per 104 mouse genomes. The limits of detection were six B. burgdorferi genomes and 17 mouse genomes per qPCR. Data are presented as mean bacterial load with error bars representing standard deviations. Data were analyzed using unpaired Student's t test to compare bacterial burdens between anti-Ly6G and isotype control treated mice within each infection group (*, P < 0.05).
DISCUSSION
Establishment of a persistent, disseminated infection is critical for B. burgdorferi to maintain its enzootic infection cycle. P66 is necessary for B. burgdorferi to establish infection in mice (42, 43). P66 has two functions identified in vitro: integrin binding and pore formation in the bacterial outer membrane (39, 40). The integrin binding function of P66 facilitates dissemination and transmigration out of the vasculature in vivo but is not necessary for establishment of infection (43, 44). This suggests that either the pore-forming function or an unknown role of P66 is critical for mammalian infection. Here, we tested several hypotheses that investigated the possible nature of this function.
First, we tested the hypothesis that the Δp66 strain has some sort of physical defect compared to the WT. Since B. burgdorferi has a fragile outer membrane, we hypothesized that the deletion of P66, an integral outer membrane protein, results in an outer membrane that is incapable of withstanding the stresses present in the mammalian environment. We tested this hypothesis through detergent sensitivity, shear force, and osmolarity assays. Our results indicated that the loss of P66 does not result in bacteria that are hypersensitive to detergent, shear force, or osmolarity pressures compared to the WT. From this set of experiments, we have found no evidence suggesting that the reason the Δp66 strain is unable to establish mammalian infection is due to an outer membrane integrity defect. Regarding shear force in particular, this is consistent with the equivalent motility of WT and Δp66 bacterial cells in mouse skin shortly after inoculation (43).
One known in vitro function of P66 is pore formation in the bacterial outer membrane (39). While the diameter of the pore has been estimated experimentally, 1.9 nm with a central constriction of 0.8 nm, it is still unclear what physiologically relevant substrates pass through the pore (45). Gram-negative bacteria use porins to allow diffusion of nutrients across the outer membrane. Some porins bind sugars preferentially to help facilitate diffusion into the cell. For example, ScrY and LamB help facilitate diffusion of sucrose and maltooligosaccharides, respectively, into the cell through binding of the sugars (55–57). In this study, we tested the hypothesis that the pore formed by P66 is critical for diffusion of sugars or other undefined nutrients in the skin homogenate across the bacterial outer membrane and into the cell. While it is known that the Δp66 strain survives and replicates as well as the WT inside a dialysis chamber inserted into the rat peritoneum (42), there are two caveats to this experiment. First, the rat peritoneum is not the same environment that B. burgdorferi encounters early during natural infection in the skin. Second, it is possible that the dialysis chamber (8,000-molecular-weight cutoff) retained components of BSKII medium, providing a more nutrient-rich environment compared to natural infection. Therefore, we investigated a potential role for P66 in the experimentally tractable question of sugar transport.
Here, we examined the ability of Δp66 bacteria to grow in glucose-free BSKII supplemented with different sugars to determine if P66 is necessary for diffusion of specific sugars into the cell. The Δp66 mutants are capable of growing as well as the WT in glucose-free BSKII supplemented with maltose and GlcNAc, two carbon sources known to sustain B. burgdorferi growth in vitro (60). In our study, neither WT nor Δp66 B. burgdorferi was capable of growing above glucose-free BSKII levels in BSKII supplemented with glycerol (see Fig. S1 in the supplemental material). This is different from what von Lackum and Stevenson (60) observed. One possible explanation for this difference is that we supplemented BSKII with 0.25% glycerol, while von Lackum and Stevenson supplemented with 0.4% (60). Furthermore, neither WT nor Δp66 bacteria were able to grow in lactose or sucrose, two sugars not tested by von Lackum and Stevenson, above the levels observed in glucose-free BSKII medium, demonstrating that B. burgdorferi is unable to use either of these sugars as carbon sources (60). However, there is no difference between WT and Δp66 bacterial growth in diluted BSKII (Fig. 6), suggesting that one or more host factors not present in the medium serve as the critical discriminator between WT and Δp66 bacteria. These data demonstrate the pore formed by P66 is not necessary for the acquisition of any of these carbon sources tested at the concentrations examined. It is possible that a difference in growth rate between WT and Δp66 bacteria would be observed with different concentrations of these carbon sources, but all those tested here were used at the concentration of glucose in conventional BSK II medium.
Since BSKII does not precisely recapitulate the nutritional environment to which B. burgdorferi is exposed in mammalian skin, we investigated whether the loss of P66 resulted in decreased growth in mouse skin homogenate. In preliminary experiments, neither WT nor Δp66 bacteria grew in mouse skin homogenate alone. Therefore, skin homogenate was supplemented with various levels of BSKII. There was no significant difference in growth between Δp66 and WT bacteria at any of the concentrations of mouse skin homogenate in the BSKII mixture. These experiments, along with the dialysis chamber growth experiment (42), demonstrate that Δp66 bacteria do not have an inherent defect in acquiring or metabolizing any of the tested nutrients. However, in addition to previously mentioned caveats for the dialysis membrane chamber experiment, another caveat is that the base medium for B. burgdorferi culture, CMRL, is not available devoid of amino acids or other nutrient classes. It remains possible that P66 is critical for the diffusion of untested nutrients across the bacterial outer membrane.
Lyme disease is systemic, with the causative agent, B. burgdorferi, infecting a variety of tissues, including the skin, heart, brain, and joints. B. burgdorferi encounters different host defense mechanisms in different tissues. For example, invariant natural killer T cells (iNKT) isolated from mouse joint, but not the liver or spleen, can lethally attack B. burgdorferi through a granzyme B-dependent pathway (61). In order to determine if Δp66 cells are being cleared by a tissue-specific host defense mechanism, we tracked the dissemination of the mutant up to 28 days postinoculation. In our experiment, the WT disseminated into the blood by day three and was fully disseminated by 14 days postinoculation, which is similar to previously reported results (62). The restoration of p66 to the chromosome restored infectivity, albeit with a slight delay in dissemination. However, the Δp66 strain did not disseminate to other tissues prior to being cleared.
Motility and chemotaxis are both critical for efficient B. burgdorferi infection of mice and dissemination (50, 51, 63). Even though the Δp66 strain is able to swim in vitro at speeds similar to those of the p66cc strain and in the skin inoculation site at speeds similar to those of the WT (43), it remains to be determined whether these movements are coordinated or random. We hypothesized Δp66 bacteria are unable to disseminate due to a defect in chemotaxis, i.e., the pore formed by P66 may be acting as a portal for chemoattractant or chemorepellent substrates to enter the bacteria. We found that the Δp66 mutant is chemotactic toward both rabbit serum and GlcNAc. While P66 may be necessary for unknown chemotactic substrates to enter the periplasm in vivo, our data demonstrate that the loss of P66 does not result in a grossly defective chemotactic response.
B. burgdorferi is clearly very adept at evading the host immune system. Dissemination away from the inoculation site to distal tissues and development of a persistent infection in immunocompetent hosts are evidence of the bacterium's ability to evade host defenses. Currently, the mechanisms of immune evasion are not fully understood. Since Δp66 bacteria are unable to disseminate away from the skin inoculation site, we investigated the role of P66 in evasion of host defenses found in the skin.
Previous work showed that macrophages, dendritic cells, and neutrophils can phagocytose and kill B. burgdorferi in vitro using conventional, coiling, or tube phagocytosis (9, 11–18). Additional studies examined the in vivo role of neutrophils in controlling B. burgdorferi infection at the inoculation site and showed that increased neutrophil recruitment to the site of inoculation of engineered B. burgdorferi severely attenuates B. burgdorferi infectivity (19). It has also been shown that BALB/c mice can form neutrophil extracellular traps (NETs) upon tick infestation and that NETs can trap and kill B. burgdorferi in vitro (20). One study previously investigated the effect of depleting neutrophils in mice. Neutrophil depletion in C3H/HeJ mice using an anti-RB6-8C5 antibody resulted in increased bacterial burden in the ankle 7 days after footpad inoculation, and it exacerbated arthritis in the ankle of these mice (21). Therefore, the prevailing thought was that early infection control was dependent upon phagocytosis and killing by neutrophils, macrophages, and dendritic cells.
However, our work, in a model that more closely resembles natural infection, provides evidence that each cell type, individually, makes a limited contribution to controlling B. burgdorferi infection in mice under conditions examined. Our previous studies investigated the role of P66 in macrophage or dendritic cell evasion; however, depletion of either cell type did not restore Δp66 infectivity in mice (in the C57BL/6 background), and it did not have an effect on WT bacterial burdens (43). Since the Δp66 mutant is cleared within 48 h postinoculation, we hypothesized that P66 is responsible for neutrophil evasion in vivo. To test this, we depleted neutrophils in mice using an anti-Ly6G (1A8) antibody (59). While depletion experiments are unable to completely rid the mice of a particular cell type, we observed significantly fewer circulating neutrophils in all mice treated with anti-Ly6G antibody compared to isotype control mice at 3 h and 3 days after B. burgdorferi inoculation (P < 0.05) (Fig. 8A). At 7 days postinoculation, Δp66- and sham-inoculated mice treated with anti-Ly6G antibody had significantly reduced circulating neutrophils as well. There was not a statistically significant difference of neutrophils between anti-Ly6G- and isotype control-treated mice infected with the WT (P = 0.0898). However, based on the definition of severe neutropenia in human (normal neutrophil range, 1,800 to 7,700 cells/μl blood; severe neutropenia, less than 500 cells/μl blood [64, 65]; correlating to a 72% to 93% reduction of normal neutrophil levels), the anti-Ly6G-treated mice were neutropenic compared to isotype control-treated mice. There was a 97% reduction in mean circulating neutrophil following treatment with anti-Ly6G antibody in WT-inoculated mice at 7 days postinoculation (Table S1). Additionally, if the human definition of neutropenia is applied to mice, all anti-Ly6G-treated mice would be considered neutropenic. On average, female C3H/HeJ mice have 470 to 850 neutrophils/μl blood (http://phenome.jax.org/projects/Jaxpheno4). Using the percentage of neutrophil reduction in human neutropenia (72% to 93%), a neutropenic mouse would have under 132 neutrophils per microliter of blood. All anti-Ly6G-treated mice inoculated with WT or Δp66 B. burgdorferi have neutrophil levels of <132/μl of blood, with the majority having <50 neutrophils/μl blood (Fig. 8).
Culture and qPCR results from the neutrophil depletion experiment showed that Δp66 bacteria were unable to establish infection in neutropenic mice. Additionally, the qPCR results showed that the loss of neutrophils did not influence WT bacterial burdens in tissues, even though there was a significant decrease of circulating neutrophils in anti-Ly6G antibody-treated mice compared to isotype control antibody-treated mice. These results suggest that neutrophils have a limited role in controlling B. burgdorferi infection in mice in the conventional laboratory model system tested. Therefore, while macrophages, dendritic cells, and neutrophils have been shown to phagocytose B. burgdorferi in vitro using conventional, coiling, or tube phagocytosis (9, 11–18), none of these innate immune effector cell types individually appears to have a major role in controlling B. burgdorferi infection in mice under conditions tested here or in our previous work (43). One caveat is that these cells types may have redundant phagocytic functions, so it is possible that one cell type can compensate for depletion of another in these experiments. However, all of the anti-Ly6G-treated mice had similar or lower mean white blood cell (WBC) levels than isotype control-treated mice, indicating that these mice did not compensate for the loss of neutrophils by overproducing more WBCs (Fig. S2). Also, there was not an observed increased number of other host defense cells at the site of inoculation in mice depleted of macrophages or dendritic cells (43). Additionally, we only used one dosage of B. burgdorferi in this experiment, so it is possible that using a lower dosage of B. burgdorferi could reveal effects of neutrophil depletion not observed here.
Our data may appear to contradict a recent report that shows that hyperglycemic mice have higher B. burgdorferi burdens in tissues than normal mice 4 weeks postinoculation (66). The authors of that study showed that the higher bacterial burdens were associated with impaired neutrophil function in the hyperglycemic mice by demonstrating the neutrophils isolated from hyperglycemic mice were impaired in phagocytosis and killing of B. burgdorferi ex vivo (66). This study differed from our study in that they quantified bacterial burdens at 4 weeks postinoculation, while we investigated the role of neutrophils at early time points. Additionally, it has been documented that chronically hyperglycemic rodents have impairments of multiple components of the immune system. Bone marrow-derived dendritic cells isolated from chronically hyperglycemic mice have reduced phagocytic ability toward Burkholderia pseudomallei (67), a bacterial infection to which it is well known that human diabetics are more susceptible (68, 69). In the same study, both isolated bone marrow-derived dendritic cells and peritoneally elicited macrophages from chronically hyperglycemic mice had a reduced ability to kill internalized B. pseudomallei (67). Peritoneal macrophages isolated from hyperglycemic rats had impaired catabolism of immune complexes compared to those of peritoneal macrophages isolated from control animals (70). Additionally, hyperglycemic mice had a delay in the adaptive immune response compared to that of control mice when infected with Mycobacterium tuberculosis (71). This suggests that the hyperglycemic mice may have multiple impairments in the immune system that could be contributing to the increases in bacterial burdens.
The data presented here show that, individually, neutrophils have limited contribution to controlling B. burgdorferi infection in C3H/HeN mice after subcutaneous inoculation of known numbers of B. burgdorferi. Previous work similarly showed that macrophages and dendritic cells have limited contribution to controlling B. burgdorferi infection in mice (43). Overall, we conclude that there is either redundancy between macrophages, dendritic cells, and neutrophils in their role in controlling B. burgdorferi infection or that B. burgdorferi is adept at evading all of these cells types in vivo. Supporting our conclusion, B. burgdorferi-elicited interleukin-10 (IL-10) suppresses production of proinflammatory mediators and phagocyte-associated events in isolated macrophages and dendritic cells from C57BL/6 mice in vitro, suggesting that suppression of some innate responses is one of many mechanisms B. burgdorferi employs to establish infection (72). Ixodes tick saliva also has an immunosuppressive effect on the host that could play a role in helping B. burgdorferi establish a persistent, disseminated infection within the host under the natural course of infection (73–78). Therefore, it is possible that removing macrophages, dendritic cells, or neutrophils does not affect bacterial burdens because WT B. burgdorferi employs multiple mechanisms to evade host defenses. Further investigation into how B. burgdorferi is able to interact with innate host defenses to establish a persistent, disseminated infection in living animals will likely reveal additional interesting and novel mechanisms of pathogenesis of Lyme disease spirochetes.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
Borrelia burgdorferi B31 A3 (79) and derivative strains (42) were grown in Barbour-Stoenner-Kelly II (BSKII) (80) medium at 33°C, unless otherwise noted. For B. burgdorferi growth on plates, BSKII medium was supplemented with 6.8 g liter−1 agarose (81) and incubated at 33°C with 2% CO2 for 2 weeks. Prior to mouse infections, B. burgdorferi cultures were subjected to multiplex PCR to ensure retention of all genomic plasmids found in the parental strain (79, 82). BSKII medium was supplemented with rifampin to 50 μg ml−1, amphotericin B to 2.5 μg ml−1, and phosphomycin to 20 μg ml−1 to culture B. burgdorferi from mouse tissues as described previously (42).
Mouse strains and housing conditions.
C3H/HeN mice were purchased from Charles River Laboratories (Wilmington, MA). All mice were fed and watered ad libitum throughout the experiments. All experiments involving animals were approved by the IACUC at the Medical College of Wisconsin.
Mouse infections.
B. burgdorferi strains were grown to mid- or late-log phase as indicated in the figure legends. Prior to inoculation, bacteria were washed and concentrated in phosphate-buffered saline (PBS; 150 mM NaCl, 17 mM K2HPO4, 5 mM KH2PO4, pH 7.4) plus 0.2% heat-inactivated normal mouse serum (NMS) as described previously (42). After washing, the bacteria were diluted to a density of 1 × 106 cells ml−1 in PBS plus 0.2% NMS. Mice were syringe inoculated subcutaneously (100 μl) between the scapulae with a dose of 1 × 105 cells. Tissues were collected for culturing and qPCR at the time points postinoculation indicated in the figure legends. To determine the presence of live B. burgdorferi, tissues were placed individually in BSKII medium and incubated up to 8 weeks at 33°C. Each culture was checked for B. burgdorferi growth by direct observation under dark-field microscopy. If, after 8 weeks, the culture did not show signs of B. burgdorferi growth, the tissue was deemed uninfected. The bacterial burdens in each tissue were determined by qPCR as described previously (42). Briefly, DNA was extracted from collected mouse tissues, and samples were stored at −80°C until the day of analysis. All DNA extractions for qPCR analysis were performed using the Qiagen QIAmp minikit. B31 A3 was grown in BSKII medium to mid-log phase, and DNA was extracted for the B. burgdorferi standard curve. Mouse DNA was extracted from uninfected mouse livers for the mouse standard curve. Tenfold dilutions of each standard were made in elution buffer the day of analysis. One hundred nanograms of mouse DNA was added to each well of the B. burgdorferi standard curve to mimic the conditions of the experimental samples. All standards and samples were run in triplicate. A standard curve was generated using Bio-Rad CFX Manager software, and the number of genomes per sample was calculated from the threshold cycle (CT) values. Data are presented as number of B. burgdorferi genomes per 10,000 mouse genomes. A value of 1 was added to all normalized values to allow representation of zero values on log-scale graphs.
Neutrophil depletion.
To deplete mice of neutrophils, 8-week-old, female C3H/HeN mice were injected intraperitoneally with a dose of 500 μg of anti-Ly6G clone 1A8 (BE0075-1; BioXcell) or isotype control antibody (BE0089; BioXcell) 24 h prior to bacterial inoculation and every 48 h after the first dose. To determine the extent of neutrophil depletion in each mouse, blood was collected in anticoagulant (60 mM sodium citrate and 40 mM citric acid)-treated syringes and placed in microcentrifuge tubes containing 50 μl anticoagulant. To lyse red blood cells, 500 μl of blood was incubated with 6 ml red blood cell lysis buffer (155 mM ammonium chloride and 10 mM potassium bicarbonate) for 3 min at room temperature. Cells were pelleted at 500 × g for 5 min. The supernatant was discarded, and repeated incubations and centrifugations were performed until the pellet was white (white blood cells). The cells were resuspended in fluorescence-activated cell sorting (FACS)/flow buffer (PBS, 25 mM HEPES, 5% fetal bovine serum [FBS]; pH 8.5) and incubated at room temperature for 1 h to block. The number of WBCs per microliter of blood was determined by using a hemocytometer. The cells were pelleted and resuspended in FACS/flow buffer at a density of 1 × 106 cells ml−1. Cells (1 × 105) were incubated with GR-1-phycoerythrin-conjugated antibody (108407; BioLegend) and F4/80-fluorescein isothiocyanate-conjugated antibody (11-4801-82; eBioscience) (both at 1:50 dilutions) for 1 h at 4°C in the dark. The cells were washed in FACS/flow buffer and fixed in 2% paraformaldehyde for 10 min. Cells were pelleted and resuspended in 200 μl FACS/flow buffer. Flow cytometry was performed using Guava EasyCyte Mini, and data were analyzed using FlowJo (version 9.7.6). Absolute numbers of high-side-scatter (SSCHi), GR-1+, F4/80− cells per microliter of blood were calculated by multiplying the percentage of SSCHi, GR-1+, F4/80− cells, determined by flow cytometry, by the concentration of WBCs.
B. burgdorferi growth curves.
For all B. burgdorferi growth curves, cultures were initially grown to densities between 1 × 107 and 5 × 107 cells ml−1 and then diluted in media tailored to test the effects of NaCl, various sugars, and mouse skin homogenate on B. burgdorferi survival and growth.
NaCl.
Cultures were diluted to 1 × 104 cells ml−1 in 5 ml of normal BSKII or BSKII supplemented with NaCl to final concentrations of 200 mM, 300 mM, 400 mM, and 500 mM. Cultures were incubated at 33°C and counted daily using a Petroff-Hausser counting chamber under dark-field microscopy. If an individual culture had a bacterial density below the limit of detection at a specific time point, that sample was omitted from analysis. There were three technical replicates for each B. burgdorferi strain under each condition.
Mouse skin homogenate.
Uninfected adult female C3H/HeN mice were euthanized and shaved along the dorsal side to remove all hair. The skin was cleaned using betadine followed by 70% ethanol. The dorsal skin was collected and stored on ice. The skin homogenate was made in homogenate buffer (100 mM HEPES, 150 mM NaCl, pH 7.6) at a ratio of 0.053 g ml−1 using a Tissue Tearor homogenizer. Homogenate was spun at 500 × g for 10 min to remove remaining hair and debris. The supernatant was sterilized through a 0.22-μm filter and combined with BSKII in the following BSKII percentages: 100%, 80%, 60%, 40%, and 20%. BSKII was also combined with homogenate buffer at identical percentages. Cultures were diluted to a starting density of 1 × 105 cells ml−1 in 5-ml cultures. The cultures were incubated at 33°C and counted daily using a Petroff-Hausser counting chamber under dark-field microscopy.
Membrane perturbation susceptibility assays. (i) SDS.
Cultures of WT or Δp66 B. burgdorferi were grown to densities between 5 × 107 and 2 × 108 cells ml−1 and then diluted to 2 × 106 cells ml−1 in BSKII supplemented with increasing concentrations of sodium dodecyl sulfate (SDS) to final concentrations of 0%, 0.1%, and 0.3%. Cultures were incubated at 33°C for up to 24 h, and bacterial survival was determined through plating of CFU in BSKII plating medium.
(ii) Shear force.
WT and Δp66 B. burgdorferi cultures were grown to 1 × 108 to 2 × 108 cells ml−1. Cells were harvested by centrifugation at 1,500 × g for 30 min, washed in PBS plus 0.2% NMS, spun at 5,900 × g for 8 min, resuspended, and diluted to 1 × 106 cells ml−1 in PBS plus 0.2% NMS. Cells were passed through a 29-gauge needle up to 9 times. A participant blind to experimental conditions performed the needle passages, and B. burgdorferi survival was determined through plating for CFU in BSKII plating medium after 0, 1, 3, 5, 7, and 9 passages through the needle.
mCRAMP susceptibility.
B. burgdorferi susceptibility to mCRAMP was determined essentially as described before (83). Briefly, WT or Δp66 B. burgdorferi cells (1 × 105) were incubated with increasing concentrations of mCRAMP (61305; AnaSpec, reconstituted in 1 mg ml−1 in 0.01% acetic acid plus 0.2% BSA) at 33°C for 3 h. Mueller-Hinton broth (MHB) was used as the assay medium with a total volume of 100 μl. Viable bacteria were enumerated by plating in BSKII plating medium. Vehicle control replicated the ratio of 0.01% acetic acid plus 0.2% BSA in MHB as found under the 200-μg ml−1 mCRAMP condition. Dose-dependent killing of E. coli was determined in a similar fashion at 37°C, and surviving bacteria were quantified as CFU on LB agar plates. E. coli plates were incubated overnight at 37°C.
Bacterial motion tracking and chemotaxis assays.
A previously described computer-based bacterial motion tracking system was used to measure B. burgdorferi cell velocity in 1% methylcellulose (52, 84). For each strain, at least 10 bacterial cells were tracked for at least 30 s, and average cell velocities and standard errors of the means (SEM) were calculated. Capillary assays were carried out as previously described, with modifications (52, 54, 85). Briefly, B. burgdorferi was grown to a density of 5 × 107 to 7 × 107 cells ml−1. Cells were concentrated by centrifugation (1,800 × g) at room temperature and resuspended in motility buffer (52). Capillary tubes (80 μl) filled with either the attractant (0.1 M GlcNAc or 1% rabbit serum dissolved in motility buffer) or motility buffer alone (negative control) were sealed and inserted into microcentrifuge tubes containing 200 μl of resuspended cells (7 × 108 cells ml−1). After 2 h of incubation at 34°C in a humidified chamber, the solutions were expelled from the capillary tubes and the spirochete cells were enumerated using Petroff-Hausser counting chambers under dark-field microscopy. A positive chemotactic response was defined as at least twice as many cells entering tubes filled with known attractants as the buffer-filled tubes, as previously described (52, 54).
Statistical tests.
The unpaired Student's t test was used to compare differences in bacterial burdens and neutrophil levels between mice treated with anti-Ly6G antibody and isotype control antibody within each infection group (WT, Δp66, or sham). Linear regression analysis was used to compare differences between the exponential phase of the growth curves of the different B. burgdorferi strains. For chemotaxis assays, the unpaired Student's t test was used to compare differences between strains.
Supplementary Material
ACKNOWLEDGMENTS
We thank Allison Reeme for training on Guava EasyCyte Mini and FlowJo (version 9.7.6) and Hiromi Sato for advice and assistance with preparation of figures.
This work was supported by NIH grants R01-AI084873 and R01-AI078958.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/IAI.00186-17.
REFERENCES
- 1.Burgdorfer W, Barbour AG, Hayes SF, Benach JL, Grunwaldt E, Davis JP. 1982. Lyme disease-a tick-borne spirochetosis? Science 216:1317–1319. doi: 10.1126/science.7043737. [DOI] [PubMed] [Google Scholar]
- 2.Radolf JD, Caimano MJ, Stevenson B, Hu LT. 2012. Of ticks, mice and men: understanding the dual-host lifestyle of Lyme disease spirochaetes. Nat Rev Microbiol 10:87–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Piesman J, Schwan TG. 2010. Ecology of Borreliae and their arthropod vectors, p 251–278. In Samuels DS, Radolf JD (ed), Borrelia: molecular biology, host interaction and pathogenesis. Caister Academic Press, Norfolk, United Kingdom. [Google Scholar]
- 4.Steere AC, Batsford WP, Weinberg M, Alexander J, Berger HJ, Wolfson S, Malawista SE. 1980. Lyme carditis: cardiac abnormalities of Lyme disease. Ann Intern Med 93:8–16. doi: 10.7326/0003-4819-93-1-8. [DOI] [PubMed] [Google Scholar]
- 5.Steere AC, Bartenhagen NH, Craft JE, Hutchinson GJ, Newman JH, Rahn DW, Sigal LH, Spieler PN, Stenn KS, Malawista SE. 1983. The early clinical manifestations of Lyme disease. Ann Intern Med 99:76–82. doi: 10.7326/0003-4819-99-1-76. [DOI] [PubMed] [Google Scholar]
- 6.Steere AC, Schoen RT, Taylor E. 1987. The clinical evolution of Lyme arthritis. Ann Intern Med 107:725–731. doi: 10.7326/0003-4819-107-5-725. [DOI] [PubMed] [Google Scholar]
- 7.Nadelman RB, Nowakowski J, Forseter G, Goldberg NS, Bittker S, Cooper D, Aguero-Rosenfeld M, Wormser GP. 1996. The clinical spectrum of early Lyme borreliosis in patients with culture-confirmed erythema migrans. Am J Med 100:502–508. doi: 10.1016/S0002-9343(95)99915-9. [DOI] [PubMed] [Google Scholar]
- 8.Antonara S, Ristow L, McCarthy J, Coburn J. 2010. Effect of Borrelia burgdorferi OspC at the site of inoculation in mouse skin. Infect Immun 78:4723–4733. doi: 10.1128/IAI.00464-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Benach JL, Habicht GS, Gocinski BL, Coleman JL. 1984. Phagocytic cell responses to in vivo and in vitro exposure to the Lyme disease spirochete. Yale J Biol Med 57:599–605. [PMC free article] [PubMed] [Google Scholar]
- 10.Hovius JWR, Bijlsma MF, van der Windt GJW, Wiersinga WJ, Boukens BJD, Coumou J, Oei A, de Beer R, de Vos AF, Veer Cvt, van Dam AP, Wang P, Fikrig E, Levi MM, Roelofs JJTH, van der Poll T. 2009. The urokinase receptor (uPAR) facilitates clearance of Borrelia burgdorferi. PLoS Pathog 5:e1000447. doi: 10.1371/journal.ppat.1000447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Peterson PK, Clawson CC, Lee DA, Garlich DJ, Quie PG, Johnson RC. 1984. Human phagocyte interactions with the Lyme disease spirochete. Infect Immun 46:608–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rittig MG, Krause A, Häupl T, Schaible UE, Modolell M, Kramer MD, Lütjen-Drecoll E, Simon MM, Burmester GR. 1992. Coiling phagocytosis is the preferential phagocytic mechanism for Borrelia burgdorferi. Infect Immun 60:4205–4212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Montgomery RR, Nathanson MH, Malawista SE. 1993. The fate of Borrelia burgdorferi, the agent for Lyme disease, in mouse macrophages. Destruction, survival, recovery. J Immunol 150:909–915. [PubMed] [Google Scholar]
- 14.Linder S, Heimerl C, Fingerle V, Aepfelbacher M, Wilske B. 2001. Coiling phagocytosis of Borrelia burgdorferi by primary human macrophages is controlled by CDC42Hs and Rac1 and involves recruitment of Wiskott-Aldrich Syndrome Protein and Arp2/3 complex. Infect Immun 69:1739–1746. doi: 10.1128/IAI.69.3.1739-1746.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Montgomery RR, Lusitani D, de Boisfleury Chevance A, Malawista SE. 2002. Human phagocytic cells in the early innate immune response to Borrelia burgdorferi. J Infect Dis 185:1773–1779. doi: 10.1086/340826. [DOI] [PubMed] [Google Scholar]
- 16.Filgueira L, Nestlé FO, Rittig M, Joller HI, Groscurth P. 1996. Human dendritic cells phagocytose and process Borrelia burgdorferi. J Immunol 157:2998–3005. [PubMed] [Google Scholar]
- 17.Suhonen J, Komi J, Soukka J, Lassila O, Viljanen MK. 2003. Interaction between Borrelia burgdorferi and immature human dendritic cells. Scand J Immunol 58:67–75. doi: 10.1046/j.1365-3083.2003.01284.x. [DOI] [PubMed] [Google Scholar]
- 18.Tuominen-Gustafsson H, Penttinen M, Hytönen J, Viljanen M. 2006. Use of CFSE staining of borreliae in studies on the interaction between borreliae and human neutrophils. BMC Microbiol 6:92. doi: 10.1186/1471-2180-6-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xu Q, Seemanapalli SV, Reif KE, Brown CR, Liang FT. 2007. Increasing the recruitment of neutrophils to the site of infection dramatically attenuates Borrelia burgdorferi infectivity. J Immunol 178:5109–5115. doi: 10.4049/jimmunol.178.8.5109. [DOI] [PubMed] [Google Scholar]
- 20.Menten-Dedoyart C, Faccinetto C, Golovchenko M, Dupiereux I, Van Lerberghe P-B, Dubois S, Desmet C, Elmoualij B, Baron F, Rudenko N, Oury C, Heinen E, Couvreur B. 2012. Neutrophil extracellular traps entrap and kill Borrelia burgdorferi sensu stricto spirochetes and are not affected by Ixodes ricinus tick saliva. J Immunol 189:5393–5401. doi: 10.4049/jimmunol.1103771. [DOI] [PubMed] [Google Scholar]
- 21.Brown CR, Blaho VA, Loiacono CM. 2004. Treatment of mice with the neutrophil-depleting antibody RB6-8C5 results in early development of experimental Lyme arthritis via the recruitment of Gr-1-polymorphonuclear leukocyte-like cells. Infect Immun 72:4956–4965. doi: 10.1128/IAI.72.9.4956-4965.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Samuels DS. 2011. Gene regulation in Borrelia burgdorferi. Annu Rev Microbiol 65:479–499. doi: 10.1146/annurev.micro.112408.134040. [DOI] [PubMed] [Google Scholar]
- 23.Brooks CS, Vuppala SR, Jett AM, Alitalo A, Meri S, Akins DR. 2005. Complement regulator-acquiring surface protein 1 imparts resistance to human serum in Borrelia burgdorferi. J Immunol 175:3299–3308. doi: 10.4049/jimmunol.175.5.3299. [DOI] [PubMed] [Google Scholar]
- 24.Kenedy MR, Vuppala SR, Siegel C, Kraiczy P, Akins DR. 2009. CspA-mediated binding of human factor H inhibits complement deposition and confers serum resistance in Borrelia burgdorferi. Infect Immun 77:2773–2782. doi: 10.1128/IAI.00318-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hallström T, Siegel C, Mörgelin M, Kraiczy P, Skerka C, Zipfel PF. 2013. CspA from Borrelia burgdorferi inhibits the terminal complement pathway. mBio 4:e00481-. doi: 10.1128/mBio.00481-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kraiczy P, Hellwage J, Skerka C, Becker H, Kirschfink M, Simon MM, Brade V, Zipfel PF, Wallich R. 2004. Complement resistance of Borrelia burgdorferi correlates with the expression of BbCRASP-1, a novel linear plasmid-encoded surface protein that interacts with human factor H and FHL-1 and is unrelated to Erp proteins. J Biol Chem 279:2421–2429. doi: 10.1074/jbc.M308343200. [DOI] [PubMed] [Google Scholar]
- 27.Zhang J-R, Hardham JM, Barbour AG, Norris SJ. 1997. Antigenic variation in Lyme disease Borreliae by promiscuous recombination of VMP-like sequence cassettes. Cell 89:275–285. doi: 10.1016/S0092-8674(00)80206-8. [DOI] [PubMed] [Google Scholar]
- 28.Bankhead T, Chaconas G. 2007. The role of VlsE antigenic variation in the Lyme disease spirochete: persistence through a mechanism that differs from other pathogens. Mol Microbiol 65:1547–1558. doi: 10.1111/j.1365-2958.2007.05895.x. [DOI] [PubMed] [Google Scholar]
- 29.Rogovskyy AS, Casselli T, Tourand Y, Jones CR, Owen JP, Mason KL, Scoles GA, Bankhead T. 2015. Evaluation of the importance of VlsE antigenic variation for the enzootic cycle of Borrelia burgdorferi. PLoS One 10:e0124268. doi: 10.1371/journal.pone.0124268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang J-R, Norris SJ. 1998. Kinetics and in vivo induction of genetic variation of vlsE in Borrelia burgdorferi. Infect Immun 66:3689–3697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Önder Ö Humphrey PT, McOmber B, Korobova F, Francella N, Greenbaum DC, Brisson D. 2012. OspC is potent plasminogen receptor on surface of Borrelia burgdorferi. J Biol Chem 287:16860–16868. doi: 10.1074/jbc.M111.290775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Caine JA, Coburn J. 2015. A short-term Borrelia burgdorferi infection model identifies tissue tropisms and bloodstream survival conferred by adhesion proteins. Infect Immun 83:3184–3194. doi: 10.1128/IAI.00349-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Caine JA, Lin Y-P, Kessler JR, Sato H, Leong JM, Coburn J. 26 September 2017. Borrelia burgdorferi outer surface protein C (OspC) binds complement component C4b and confers bloodstream survival. Cell Microbiol doi: 10.1111/cmi.12786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Carrasco SE, Troxell B, Yang Y, Brandt SL, Li H, Sandusky GE, Condon KW, Serezani CH, Yang XF. 2015. Outer surface protein OspC is an antiphagocytic factor that protects Borrelia burgdorferi from phagocytosis by macrophages. Infect Immun 83:4848–4860. doi: 10.1128/IAI.01215-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Probert WS, Johnson BJB. 1998. Identification of a 47 kDa fibronectin-binding protein expressed by Borrelia burgdorferi isolate B31. Mol Microbiol 30:1003–1015. doi: 10.1046/j.1365-2958.1998.01127.x. [DOI] [PubMed] [Google Scholar]
- 36.Fischer JR, LeBlanc KT, Leong JM. 2006. Fibronectin binding protein BBK32 of the Lyme disease spirochete promotes bacterial attachment to glycosaminoglycans. Infect Immun 74:435–441. doi: 10.1128/IAI.74.1.435-441.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lin Y-P, Chen Q, Ritchie JA, Dufour NP, Fischer JR, Coburn J, Leong JM. 2015. Glycosaminoglycan binding by Borrelia burgdorferi adhesin BBK32 specifically and uniquely promotes joint colonization. Cell Microbiol 17:860–875. doi: 10.1111/cmi.12407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Garcia BL, Zhi H, Wager B, Höök M, Skare JT. 2016. Borrelia burgdorferi BBK32 inhibits the classical pathway by blocking activation of the C1 complement complex. PLoS Pathog 12:e1005404. doi: 10.1371/journal.ppat.1005404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Skare JT, Mirzabekov TA, Shang ES, Blanco DR, Erdjument-Bromage H, Bunikis J, Berström S, Tempst P, Kagan BL, Miller JN, Lovett MA. 1997. The Oms66 (p66) is a Borrelia burgdorferi porin. Infect Immun 65:3654–3661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Coburn J, Chege W, Magoun L, Bodary SC, Leong JM. 1999. Characterization of a candidate Borrelia burgdorferi β3-chain integrin ligand identified using a phage display library. Mol Microbiol 34:926–940. doi: 10.1046/j.1365-2958.1999.01654.x. [DOI] [PubMed] [Google Scholar]
- 41.Cugini C, Medrano M, Schwan TG, Coburn J. 2003. Regulation of expression of the Borrelia burgdorferi β3-chain integrin ligand, P66, in ticks and in culture. Infect Immun 71:1001–1007. doi: 10.1128/IAI.71.2.1001-1007.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ristow LC, Miller HE, Padmore LJ, Chettri R, Salzman N, Caimano MJ, Rosa PA, Coburn J. 2012. The β3-integrin ligand of Borrelia burgdorferi is critical for infection of mice but not ticks. Mol Microbiol 85:1105–1118. doi: 10.1111/j.1365-2958.2012.08160.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ristow LC, Bonde M, Lin Y-P, Sato H, Curtis M, Wesley E, Hahn BL, Fang J, Wilcox DA, Leong JM, Bergström S, Coburn J. 2015. Integrin binding by Borrelia burgdorferi P66 facilitates dissemination but is not required for infectivity. Cell Microbiol 17:1021–1036. doi: 10.1111/cmi.12418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kumar D, Ristow LC, Shi M, Mukherjee P, Caine JA, Lee W-Y, Kubes P, Coburn J, Chaconas G. 2015. Intravital imaging of vascular transmigration by the Lyme spirochete: requirement for the integrin binding residues of the B. burgdorferi P66 protein. PLoS Pathog 11:e1005333. doi: 10.1371/journal.ppat.1005333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bárcena-Uribarri I, Thein M, Maier E, Bonde M, Bergström S, Benz R. 2013. Use of nonelectrolytes reveals the channel size and oligomeric constitution of the Borrelia burgdorferi P66 porin. PLoS One 8:e78272. doi: 10.1371/journal.pone.0078272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Radolf JD, Bourell KW, Akins DR, Brusca JS, Norgard MV. 1994. Analysis of Borrelia burgdorferi membrane architecture by freeze-fracture electron microscopy. J Bacteriol 176:21–31. doi: 10.1128/jb.176.1.21-31.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bledsoe HA, Carroll JA, Whelchel TR, Farmer MA, Dorward DW, Gherardini FC. 1994. Isolation and partial characterization of Borrelia burgdorferi inner and outer membranes by using isopycnic centrifugation. J Bacteriol 176:7447–7455. doi: 10.1128/jb.176.24.7447-7455.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lugtenberg B, Van Alphen L. 1983. Molecular architecture and functioning of the outer membrane of Escherichia coli and other gram-negative bacteria. Biochim Biophys Acta 737:51–115. [DOI] [PubMed] [Google Scholar]
- 49.Fraser CM, Casjens S, Huang WM, Sutton GG, Clayton R, Lathigra R, White O, Ketchum KA, Dodson R, Hickey EK, Gwinn M, Dougherty B, Tomb J-F, Fleischmann RD, Richardson D, Peterson J, Kerlavage AR, Quackenbush J, Salzberg S, Hanson M, van Vugt R, Palmer N, Adams MD, Gocayne J, Weidman J, Utterback T, Watthey L, McDonald L, Artiach P, Bowman C, Garland S, Fujii C, Cotton MD, Horst K, Roberts K, Hatch B, Smith HO, Venter JC. 1997. Genomic sequence of a Lyme disease spirochaete, Borrelia burgdorferi. Nature 390:580–586. doi: 10.1038/37551. [DOI] [PubMed] [Google Scholar]
- 50.Sze CW, Zhang K, Kariu T, Pal U, Li C. 2012. Borrelia burgdorferi needs chemotaxis to establish infection in mammals and to accomplish its enzootic cycle. Infect Immun 80:2485–2492. doi: 10.1128/IAI.00145-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Novak EA, Sekar P, Xu H, Moon KH, Manne A, Wooten RM, Motaleb MA. 2016. The Borrelia burgdorferi CheY3 response regulator is essential for chemotaxis and completion of its natural infection cycle. Cell Microbiol 18:1782–1799. doi: 10.1111/cmi.12617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bakker RG, Li C, Miller MR, Cunningham C, Charon NW. 2007. Identification of specific chemoattractants and genetic complementation of a Borrelia burgdorferi chemotaxis mutant: flow cytometry-based capillary tube chemotaxis assay. Appl Environ Microbiol 73:1180–1188. doi: 10.1128/AEM.01913-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shi W, Yang Z, Geng Y, Wolinsky LE, Lovett MA. 1998. Chemotaxis in Borrelia burgdorferi. J Bacteriol 180:231–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang K, Liu J, Tu Y, Xu H, Charon NW, Li C. 2012. Two CheW coupling proteins are essential in a chemosensory pathway of Borrelia burgdorferi. Mol Microbiol 85:782–794. doi: 10.1111/j.1365-2958.2012.08139.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schmid K, Ebner R, Jahreis K, Lengeler JW, Titgemeyer F. 1991. A sugar-specific porin, ScrY, is involved in sucrose uptake in enteric bacteria. Mol Microbiol 5:941–950. doi: 10.1111/j.1365-2958.1991.tb00769.x. [DOI] [PubMed] [Google Scholar]
- 56.Schülein K, Schmid K, Benzl R. 1991. The sugar-specific outer membrane channel ScrY contains functional characteristics of general diffusion pores and substrate-specific porins. Mol Microbiol 5:2233–2241. doi: 10.1111/j.1365-2958.1991.tb02153.x. [DOI] [PubMed] [Google Scholar]
- 57.Ishii JN, Okajima Y, Nakae T. 1981. Characterization of lamB protein from the outer membrane of Escherichia coli that forms diffusion pores selective for maltose-maltodextrins. FEBS Lett 134:217–220. doi: 10.1016/0014-5793(81)80605-9. [DOI] [PubMed] [Google Scholar]
- 58.Dorschner RA, Pestonjamasp VK, Tamakuwala S, Ohtake T, Rudisill J, Nizet V, Agerberth B, Gudmundsson GH, Gallo RL. 2001. Cutaneous injury induces the release of cathelicidin anti-microbial peptides active against group A Streptococcus. J Investig Dermatol 117:91–97. doi: 10.1046/j.1523-1747.2001.01340.x. [DOI] [PubMed] [Google Scholar]
- 59.Daley JM, Thomay AA, Connolly MD, Reichner JS, Albina JE. 2008. Use of Ly6G-specific monoclonal antibody to deplete neutrophils in mice. J Leukoc Biol 83:64–70. doi: 10.1189/jlb.0407247. [DOI] [PubMed] [Google Scholar]
- 60.von Lackum K, Stevenson B. 2005. Carbohydrate utilization by the Lyme borreliosis spirochete, Borrelia burgdorferi. FEMS Microbiol Lett 243:173–179. doi: 10.1016/j.femsle.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 61.Lee W-Y, Sanz M-J, Wong CHY, Hardy P-O, Salman-Dilgimen A, Moriarty TJ, Chaconas G, Marques A, Krawetz R, Mody CH, Kubes P. 2014. Invariant natural killer T cells act as an extravascular cytotoxic barrier for joint-invading Lyme Borrelia. Proc Natl Acad Sci U S A 111:13936–13941. doi: 10.1073/pnas.1404769111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Barthold SW, Persing DH, Armstrong AL, Peeples RA. 1991. Kinetics of Borrelia burgdorferi dissemination and evolution of disease after intradermal inoculation of mice. Am J Pathol 139:263–273. [PMC free article] [PubMed] [Google Scholar]
- 63.Sultan SZ, Manne A, Stewart PE, Bestor A, Rosa PA, Charon NW, Motaleb MA. 2013. Motility is crucial for the infectious life cycle of Borrelia burgdorferi. Infect Immun 81:2012–2021. doi: 10.1128/IAI.01228-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dale DC. 2016. How I diagnose and treat neutropenia. Curr Opin Hematol 23:1–4. doi: 10.1097/MOH.0000000000000208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rolston KVI. 2004. The infectious disease society of America 2002 guidelines for the use of antimicrobial agents in patients with cancer and neutropenia: salient features and comments. Clin Infect Dis 39:S44–S48. doi: 10.1086/383053. [DOI] [PubMed] [Google Scholar]
- 66.Javid A, Zlotnikov N, Pětrošová H, Tang TT, Zhang Y, Bansal AK, Ebady R, Parikh M, Ahmed M, Sun C, Newbigging S, Kim YR, Santana Sosa M, Glogauer M, Moriarty TJ. 2016. Hyperglycemia impairs neutrophil-mediated bacterial clearance in mice infected with the Lyme disease pathogen. PLoS One 11:e0158019. doi: 10.1371/journal.pone.0158019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Williams NL, Morris JL, Rush C, Govan BL, Ketheesan N. 2011. Impact of streptozotocin-induced diabetes on functional responses of dendritic cells and macrophages towards Burkholderia pseudomallei. FEMS Immunol Med Microbiol 61:218–227. doi: 10.1111/j.1574-695X.2010.00767.x. [DOI] [PubMed] [Google Scholar]
- 68.Suputtamongkol Y, Chaowagul W, Chetchotisakd P, Lertpatanasuwun N, Intaranongpai S, Ruchutrakool T, Budhsarawong D, Mootsikapun P, Wuthiekanun V, Teerawatasook N, Lulitanond A. 1999. Risk factors for melioidosis and bacteremic melioidosis. Clin Infect Dis 29:408–413. doi: 10.1086/520223. [DOI] [PubMed] [Google Scholar]
- 69.Kingsley PV, Leader M, Nagodawithana NS, Tipre M, Sathiakumar N. 2016. Melioidosis in Malaysia: a review of case reports. PLoS Negl Trop Dis 10:e0005182. doi: 10.1371/journal.pntd.0005182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Abrass CK, Hori M. 1984. Alterations in Fc receptor function of macrophages from streptozotocin-induced diabetic rats. J Immunol 133:1307–1312. [PubMed] [Google Scholar]
- 71.Vallerskog T, Martens GW, Kornfeld H. 2010. Diabetic mice display a delayed adaptive immune response to Mycobacterium tuberculosis. J Immunol 184:6275–6282. doi: 10.4049/jimmunol.1000304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chung Y, Zhang N, Wooten RM. 2013. Borrelia burgdorferi elicited-IL-10 suppresses the production of inflammatory mediators, phagocytosis, and expression of co-stimulatory receptors by murine macrophages and/or dendritic cells. PLoS One 8:e84980. doi: 10.1371/journal.pone.0084980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kern A, Collin E, Barthel C, Michel C, Jaulhac B, Boulanger N. 2011. Tick saliva represses innate immunity and cutaneous inflammation in a murine model of Lyme disease. Vector Borne Zoonotic Dis 11:1343–1350. doi: 10.1089/vbz.2010.0197. [DOI] [PubMed] [Google Scholar]
- 74.Horká H, Černá-Kýčková K, Skallová A, Kopecký J. 2009. Tick saliva affects both proliferation and distribution of Borrelia burgdorferi spirochetes in mouse organs and increases transmission of spirochetes to ticks. Int J Med Microbiol 299:373–380. doi: 10.1016/j.ijmm.2008.10.009. [DOI] [PubMed] [Google Scholar]
- 75.Montgomery RR, Lusitani D, de Boisfleury Chevance A, Malawista SE. 2004. Tick saliva reduces adherence and area of human neutrophils. Infect Immun 72:2989–2994. doi: 10.1128/IAI.72.5.2989-2994.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zeidner NS, Schneider BS, Nuncio MS, Gern L, Piesman J. 2002. Coinoculation of Borrelia spp. with tick salivary gland lysate enhances spirochete load in mice and is tick species-specific. J Parasitol 88:1276–1278. doi: 10.1645/0022-3395(2002)088[1276:COBSWT]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 77.Ramamoorthi N, Narasimhan S, Pal U, Bao F, Yang XF, Fish D, Anguita J, Norgard MV, Kantor FS, Anderson JF, Koski RA, Fikrig E. 2005. The Lyme disease agent exploits a tick protein to infect the mammalian host. Nature 436:573–577. doi: 10.1038/nature03812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hovius JW, Schuijt TJ, de Groot KA, Roelofs JJTH, Oei GA, Marquart JA, de Beer R, van't Veer C, van der Poll T, Ramamoorthi N, Fikrig E, van Dam AP. 2008. Preferential protection of Borrelia burgdorferi sensu stricto by a Salp15 homologue in Ixodes ricinus saliva. J Infect Dis 198:1189–1197. doi: 10.1086/591917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Elias AF, Stewart PE, Grimm D, Caimano MJ, Eggers CH, Tilly K, Bono JL, Akins DR, Radolf JD, Schwan TG, Rosa P. 2002. Clonal polymorphism of Borrelia burgdorferi strain B31 MI: implications for mutagenesis in an infectious strain background. Infect Immun 70:2139–2150. doi: 10.1128/IAI.70.4.2139-2150.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Barbour AG. 1984. Isolation and cultivation of Lyme disease spirochetes. Yale J Biol Med 57:521–525. [PMC free article] [PubMed] [Google Scholar]
- 81.Samuels DS. 1995. Electrotransformation of the spirochete Borrelia burgdorferi, p 253–259. In Nickoloff JA. (ed), Electroporation protocols for microorganisms. Humana Press, Totowa, NJ. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bunikis I, Kutschan-Bunikis S, Bonde M, Bergström S. 2011. Multiplex PCR as a tool for validating plasmid content of Borrelia burgdorferi. J Microbiol Methods 86:243–247. doi: 10.1016/j.mimet.2011.05.004. [DOI] [PubMed] [Google Scholar]
- 83.Sarkar A, Tilly K, Stewart P, Bestor A, Battisti JM, Rosa PA. 2009. Borrelia burgdorferi resistance to a major skin antimicrobial peptide is independent of outer surface lipoprotein content. Antimicrob Agents Chemother 53:4490–4494. doi: 10.1128/AAC.00558-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Li C, Xu H, Zhang K, Liang FT. 2010. Inactivation of a putative flagellar motor switch protein FliG1 prevents Borrelia burgdorferi from swimming in highly viscous media and blocks its infectivity. Mol Microbiol 75:1563–1576. doi: 10.1111/j.1365-2958.2010.07078.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Li C, Bakker RG, Motaleb MA, Sartakova ML, Cabello FC, Charon NW. 2002. Asymmetrical flagellar rotation in Borrelia burgdorferi nonchemotactic mutants. Proc Natl Acad Sci U S A 99:6169–6174. doi: 10.1073/pnas.092010499. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.