

ABSTRACT
In every epidemic some individuals become sick and some may die, whereas others recover from illness and still others show no signs or symptoms of disease. These differences highlight a fundamental question of microbial pathogenesis: why are some individuals susceptible to infectious diseases while others who acquire the same microbe remain well? For most of human history, the answer assumed the hand of providence. With the advent of the germ theory of disease, the focus on disease causality became the microbe, but this did not explain how there can be different outcomes of infection in different individuals with the same microbe. Here we examine the attributes of susceptibility in the context of the “damage-response framework” of microbial pathogenesis. We identify 11 attributes that, although not independent, are sufficiently distinct to be considered separately: microbiome, inoculum, sex, temperature, environment, age, chance, history, immunity, nutrition, and genetics. We use the first letter of each to create the mnemonic MISTEACHING, underscoring the need for caution in accepting dogma and attributing disease causality to any single factor. For both populations and individuals, variations in the attributes that assemble into MISTEACHING can create an enormity of combinations that can in turn translate into different outcomes of host-microbe encounters. Combinatorial diversity among the 11 attributes makes identifying “signatures” of susceptibility possible. However, with their inevitable uncertainties and propensity to change, there may still be a low likelihood for prediction with regard to individual host-microbe interactions, although probabilistic prediction may be possible.
KEYWORDS: host resistance, pathogenesis, virulence
INTRODUCTION
In a series of essays spanning almost 2 decades, we have written about microbial virulence and pathogenicity in the context of an organizing concept that we call the damage-response framework (DRF). The first article was published in Infection and Immunity in 1999 (1), and the concept was further developed in subsequent essays (2–5). The DRF is based on three assumptions that are self-evident and incontrovertible: (i) there are two entities, a host and a microbe, and they interact to create the microbial states of commensalism, colonization, and disease; (ii) the states differ in the degree of damage that the host incurs, and this can range from benefit (no damage) in commensalism (e.g., due to presence of normal microbiota) to that which compromises homeostasis and results in disease; and (iii) the damage can come from the host response, the microbe, or both (5). Although the DRF emerged from class notes in a microbial pathogenesis course at the Albert Einstein College of Medicine (1) and was developed originally to explain the great diversity of infectious diseases, it has proven to be highly flexible and able to account for all known states that stem from host-microbe interaction, commensalism, colonization, latency, and disease. The DRF has also provided a new lens to consider such questions as the nature of pathogens and infectiveness (6, 7).
Recently we updated the DRF to incorporate the microbiome and provided a definition for “host” (8). During the review process, one referee suggested expanding the essay to include host differences with regard to individual susceptibility. Given the importance and complexity of this question, we opted to consider this issue in a separate essay focused on the attributes of disease susceptibility that bear on the question of why it is that in any host-microbe encounter, some individuals get sick while others remain asymptomatic. In this essay, we consider the factors governing individual susceptibility and identify 11 attributes that affect the outcome of host-microbe interaction: microbiome, inoculum, sex, temperature, environment, age, chance, history, immunity, nutrition, and genetics. The first letters of these attributes can be assembled into the acronym MISTEACHING, which conveys the additional meaning that teaching microbial pathogenesis is difficult as it requires adjustments and change when new information that challenges previous thought and dogma emerges. In addition, it can be fraught with potential foibles, such as confusion associated with its lexicon, such as the words infection and pathogen (9). Also, MISTEACHING was the only single word we could form with the 11 letters. Given rapidly accumulating knowledge in the field of microbial pathogenesis, it is likely that more attributes will be identified. Hence, while the mnemonic may not stand the test of time, at this moment it underscores how the concept that microbial pathogenesis is driven solely by microbial or host factors, or even both, does not incorporate the complex, often unpredictable, variables that affect the susceptibility of individuals or populations.
SUSCEPTIBILITY ATTRIBUTES
The 11 attributes that are discussed below, although separable, are not independent. Age, history, and immune function are necessarily interrelated. For others, recent findings highlight new connections. For example, in amphibians, disruption of the microbiome in early life results in susceptibility to parasites later in life (6), illustrating connections between age, microbiome, time, and susceptibility. For humans, there is increasing evidence that antibiotics in early life may predispose children to obesity (10, 11). Despite being interrelated, the 11 attributes are sufficiently distinct to be considered separable factors affecting susceptibility. We caution that these are enormous areas, which we are condensing to mere paragraphs. Some readers may prefer to consider the variables that constitute each attribute in the context of different attributes, and we encourage such experimentation. Our goal was not to be exhaustive but rather to introduce the breadth of factors that may influence susceptibility and resistance to infectious diseases in the context of the DRF. The attributes we have identified, whether inherent or external to the host, interact in complex ways to generate a susceptibility composite for each host that is almost certainly unique and specific to that host. In this regard, the composite of factors for a given individual constitutes a “signature” that may or may not also apply to populations exhibiting a similar composition of factors.
Microbiome.
The use of antimicrobial drugs is known to be associated with the development of certain infectious diseases, such as colitis and candidiasis, through disruption of the host-associated microbiome. In fact, another term starting with “m,” metabolome, has emerged as an important signature of infectious diseases (12). Indeed, the metabolome and microbiome are linked, with the latter providing the fuel for generation of important biomolecules (13). In recent years, numerous studies have unequivocally established that resistance and susceptibility to microbial diseases can be determined by the composition of the resident microbiome. The ability of the microbiome to determine host susceptibility to disease was established in insects by showing that the ability of Bacillus thuringiensis to kill gypsy moth larvae was dependent on the presence of certain gut bacteria (14). There are now numerous examples of this phenomenon, including a report that human susceptibility to Campylobacter is associated with certain microbiome compositions (15) and another providing a mechanistic explanation for how certain colonic species maintain a state of anaerobiosis that inhibits facultative anaerobes and aerobes via the colonic epithelium (16). Microbiome influence on host susceptibility is not limited to bacterium-bacterium interaction, as exemplified by interference of bacteria with Plasmodium infection in mosquitoes (17) and the finding that mouse susceptibility to an enteric virus was dependent on the presence of certain bacteria in the gut (18). In other invertebrate systems, microbiome bacteria produce antifungal compounds that reduce susceptibility to fungal pathogens (19). Emerging evidence has also identified a role for fungal (20) and viral (21) microbiomes in health and disease. Each individual host is likely to have a different microbiome, since the microbiota species composition and diversity can be affected by and change with time, diet, health, and disease.
Immunity.
A properly functioning immune system is essential for reducing susceptibility to many infectious diseases. This was first formally recognized with the emergence of the so-called “opportunistic diseases” in patients with defective immunity due to immunosuppressive therapies (22). Then, HIV infection and AIDS highlighted how an acquired immune deficiency could render the host vulnerable to many infectious diseases that rarely affected hosts with intact immunity. Recognizing that the “reach” of immunity includes intact mucosal barriers, in addition to cellular and humoral components, is important for understanding and assessing risks for catheter-related and skin and soft tissue infections. Today, the literature is full of papers that link use of immune-suppressing agents, from chemotherapy to cytokine-blocking monoclonal antibodies, to risk for infectious diseases. Similarly, recognizing the impact of disease states, such as diabetes and malnutrition, and perturbations of the microbiome on immune function is critical for assessing the contribution of immune impairment to risk for infectious diseases. Although an absolute measure of what constitutes a healthy immune system has not been defined and the immune system features many redundancies and complexities, impaired immune function, primary or acquired, is unequivocally associated with susceptibility to infection and disease. Furthermore, host immunity can differ from individual to individual by the history of an individual's previous interactions with microbes (see “History” below) as well as the fact that immune responses of different individuals may vary in intensity and with time.
Sex.
The sex of a host can be a major determinant of susceptibility to infectious diseases. Numerous infectious diseases exhibit marked differences in the frequency at which they affect the sexes (23). The mechanisms responsible for sex-related differences in susceptibility include anatomical, hormonal, and immune differences (23, 24). Another way in which sex contributes to susceptibility to infectious disease is via sexual transmission of certain microbes. While sexual transmission of certain agents, such as Neisseria gonorrhea, Treponema pallidum (causing syphilis), herpesviruses, and agents causing other so-called sexually transmittable diseases, has been long recognized, it is now apparent that some arboviruses, including chikungunya and Zika viruses (25, 26), in addition to Ebola virus, a filovirus, can be sexually transmitted and remain in seminal fluid for prolonged periods of time (27).
Temperature.
Host temperature can have a major effect on host susceptibility since it can create a thermal exclusionary zone for the growth of certain microbes (28). For example, many bat species in the North American continent become susceptible to the fungus Pseudogymnoascus destructans during the torpor of winter hibernation, when their core temperatures drop to 10 to 12°C (29). Bats can be cured of the disease by waking and feeding them, which increases temperature and controls the fungal disease (30). In this regard, endogenous and induced fevers serve to increase metabolism and, for some microbes, create a thermal exclusionary zone (31). For ectotherms, numerous studies have established that the ability to induce fever correlates with survival (32). However, it is noteworthy that endotherms and homeotherms, such as mammals, differ in temperature depending on the body part. The skin and mucosal surfaces are generally cooler than core regions. Consequently, some microorganisms that lack high thermal tolerances, such as dermatophytes, can cause skin disease but do not cause systemic disease.
Environment.
The attribute of the environment includes all conditions that may influence host-microbe interaction, including vitally important entities such as nutrition, climate, crowding, hygiene, and the presence of environmental toxins. This is a far-reaching topic, and we can provide only a few examples to illustrate environmental effects on susceptibility to infection: frog eggs have increased susceptibility to fungal infection after exposure to deicing salts (33), fish exposed to the herbicide glyphosate are more susceptible to bacterial infection (34), and daphnia exposed to pesticides become rapidly resistant to the chemical but have increased susceptibility to parasites (35). Human examples include the distribution of vector-borne diseases, which parallels the distribution of the vector, which in turn reflects the availability of environmental conditions that enable the vector to survive and reproduce. Climate change may have effects on the host, microbe, and vectors that transmit microbes that may in turn affect susceptibility to infectious diseases (36–38). The attribute of environment also includes social, political, and psychological factors that influence the conditions under which individuals and populations live, which in turn influence microbial exposure.
Age.
Many infectious diseases are associated with high mortality at the extremes of age, while others appear to occur most commonly in young, healthy persons. In humans, invasive Haemophilus influenzae type B infection was a common and deadly early childhood disease before the advent of the conjugate vaccine, yet resistance to disease increased rapidly with age (39). Similarly, diarrheal diseases are major killers of young children. In contrast, the probability of reactivation of latent herpes zoster virus increases with age. At younger ages increased susceptibility can reflect immaturity of the immune system, while at older ages increased susceptibility can reflect immune senescence. Hence, the attribute of age incorporates the element of time, as the immune response changes over time from ontogeny to puberty to adulthood to senescence. In some instances, susceptibility reflects exuberant inflammatory responses, which in turn may be age related. For example, the 1918 and 2009 influenza pandemics, the 2003 severe acute respiratory syndrome (SARS) outbreak, and adenovirus pneumonia in military recruits were often more severe in younger people, who can mount robust inflammatory responses. Age is also an important variable in affecting risk though the environment. For example, crowding and close quarters, such as in schools, camps, and barracks, in which younger people often find themselves, can increase the spread of transmissible microbes. Similarly, older individuals in nursing homes find themselves in conditions propitious for communicable diseases. Notably, the attribute of age (and aging) incorporates social, cultural, political, and psychological factors, which may be important determinants of microbial exposure.
Chance.
At a fundamental level, many natural process are stochastic, and thus the element of chance plays a role in who lives and dies. Chance can affect susceptibility in its own right as well as by affecting other variables. An unexpected encounter with a rabid animal reflects the element of chance, and the encounter can be a nonevent in the life of the host or can be followed by a rapidly progressive infection, depending on the extent of injuries incurred. Risk can emerge as an unintended consequence of construction and curiosity. For example, outbreaks of histoplasmosis have followed tree cutting (40), exploration of caves (41), and home renovations (42). On the other hand, the history of a host, the size of an infective inoculum, and the host genetics are usually the summary of chance events, each of which can have a major effect on host susceptibility. The finding that the outcome of host-microbe interactions is influenced by circadian rhythms (43, 44) adds the dimension of timing to the element of change.
History.
Two individuals can reach the same age with very different timelines, or histories. It is well known that a prior immune response to a microbe can affect host susceptibility and resistance to the same microbe as well as other microbes. This observation dates to the late 18th century, when Jenner observed that milkmaids exposed to cowpox were less likely to develop smallpox, presumably because infection with cowpox virus reduced susceptibility to disease with variola virus. This observation became the basis of smallpox vaccination, with the word vaccine deriving from the Latin word for cow, vacca. On the other hand, infection with one serotype of dengue virus predisposes to hemorrhagic disease upon infection with a different serotype, with the severe outcome reflecting a form of antibody-mediated enhancement of infection. Similarly, recent epidemics have demonstrated that infection with one arbovirus can influence the immune response to another (45). This phenomenon is a variation on the concept of “original antigenic sin,” whereby an individual mounts an immune response to a new microbe, albeit with related antigenic determinants, as if it were to the original microbe (46). New tools for dissecting the antibody response have made it possible to identify defined antibodies that bind an originally encountered (priming) microbe and have beneficial functional activity against the newly acquired microbe (47). On the other hand, it is also known that infection with some microbes, such as measles virus, can directly impair host immunity such that the individual is more susceptible to subsequent infections with unrelated organisms. However, what is not clearly appreciated is that infection with one microbe can affect the outcome of infection with an unrelated microbe by changing the immunological state of the host. This phenomenon is known as “heterologous immunity” and has been dissected by studying in animal models the effects of certain viruses on subsequent infection with different viruses (for a review, see reference 48). In other words, having an infectious disease can change the host such that its susceptibility to a new infection with an antigenically unrelated host can depend on its immunological history. Having an infectious disease can also increase long-term mortality. For example, patients who recover from pneumonia have increased long-term mortality (49). The attribute of history also includes prior vaccinations and resolved infections, with the latter providing protection to subsequent infection or, as described above, sometimes enhancing susceptibility to severe disease.
Inoculum.
A major factor influencing susceptibility is the microbial inoculum, which has been known to be a determinant of the outcome of infection since the earliest days of the germ theory. In fact, differences in the pathogenicity of microbes are often expressed as the lethal dose that kills 50% of the infected hosts (LD50). In general, microbes with virulence factors that allow them to undermine host physiology have the capacity to cause disease in hosts in fewer numbers than microbes lacking such factors. For example, a toxigenic encapsulated bacterium such as Bacillus anthracis is able to cause kill mice at much lower doses than other bacteria lacking those characteristics (50). On the other hand, large inocula of certain fungi, e.g., Histoplasma capsulatum and Coccidioides spp., cause disease in apparently normal people who lack immunological risk factors present in those who develop reactivation disease after a period of latency. Hence, for any given host-microbe encounter, the infecting inoculum is likely to make a decisive contribution to the outcome of the interaction. Furthermore, the inoculum that leads to disease will be microbe and host dependent, being affected by the identity of the microbe and such host characteristics as genetics, immunological function, and nutritional status. In this regard, vaccines have been proposed to work by neutralizing the infecting inoculum such that the immune response of the host is able to reduce the effectiveness of the inoculum to the point that no disease develops (51). This viewpoint is important, because it posits that vaccines mediate protection not by preventing infection but by inducing an immune response that renders the inoculum insufficient to result in disease once infection occurs (51). Finally, we note that additional microbial variables are also related to the inoculum and different environments within the host, including the fitness of the microbe in the host, the portal of entry into the host, and tissue tropism.
Nutrition.
Proper nutrition is critical for homeostasis, and nutritional deficits are likely to have protean effects on physiology, including immune function. For example, Pneumocystis pneumonia, a disease that is often associated with AIDS and impaired immunity, was first described in association with malnourished infants, although some have postulated other causes (52, 53). Malnourished infants are also more susceptible to life-threatening diarrheal diseases, and diarrheal diseases can promote malnutrition (54, 55). Although nutrition is generally considered to be an important variable for infectious diseases, it is important to acknowledge that the relationship between nutritional status and many infectious disease has not been carefully studied. Iron overload is known to predispose to Yersinia enterocolitica (56). Less commonly known is that improved nourishment may exacerbate the course of certain infectious diseases, a fact noted before modern medicine, when it was observed that smallpox had greater mortality among the wealthy (57). The association between obesity and certain infectious diseases has been known for some time, and there are indications that the relationship is reciprocal, such that obesity predisposes to disease while certain infections may contribute to obesity (58).
Genetics.
The genetic makeup of the host is known to be an important determinant of host susceptibility for decades (59). Susceptibility and resistance to disease may have strong genetic components. In mice, major histocompatibility loci have been associated with susceptibility to Theiler's virus (60), Mycobacterium tuberculosis (61), and Rickettsia conorii (62), to name a few examples. In humans, the death of native peoples following the Columbian exchange from putative measles and smallpox may have been the result of the fact that these populations had no experience with those viruses, and thus susceptibility was much greater than in Eurasian populations, which were under virus selection for millennia. Some human genetic diseases, such as sickle cell disease and thalassemia, have been associated with resistance to certain microbes, such as malaria parasites. Furthermore, some HLA haplotypes have been associated with susceptibility to certain postinfectious diseases, such as reactive arthritis (Reiter's syndrome) (63). For some viruses, such as HIV, the dissection of their infective cycles at the molecular levels has shown that coreceptor molecules can determine susceptibility, since CCR5-deficient individuals are not susceptible to productive infection with HIV (64). Today, there are multiple infectious diseases for which genes that enhance susceptibility have been identified, which contribute to individual variability (65). However, while some diseases might exhibit monogenetic inheritance, more recently genetic mutations have been identified as cofactors or additional variables contributing to disease pathogenesis as part of a “multihit” theory, as put forth for cryptococcosis in apparently normal persons (66).
INDIVIDUAL SUSCEPTIBILITY AND THE DAMAGE-RESPONSE FRAMEWORK
The basic relationship of the DRF is depicted by a parabola with damage on the y axis and host response on the x axis (Fig. 1). The central concept in the DRF is that all host-microbe interactions have the potential to result in a host response. One item that was not clarified in our prior essays was whether these curves applied to humans or populations. Here we illustrate how the basic parabolic relationship applies to individuals and, by the aggregate of individual relationships, to populations. To illustrate these relationships, below we work through several examples of microbe-host interactions. We consider human populations in the context of interactions with three microbes of various pathogenic potential, i.e., Saccharomyces cerevisiae, M. tuberculosis, and hepatitis C virus (HCV), to illustrate the applicability of these concepts to very different types of host-microbe interactions.
FIG 1.

DRF parabolic curves representative of populations. We consider three host-microbe interactions involving humans and propose that in each case the curve emerges from the points contributed by individuals manifesting certain damage from their responses. In this idealized formulation, each point on the curve represents one individual. The position of the apex of the parabola differs in that the asymptomatic condition is accompanied by different degrees of host damage in each interaction, ranging from none to minimal with S. cerevisiae colonization to local granuloma damage in latent tuberculosis to chronic hepatitis in asymptomatic HCV infection. The line for disease threshold (DT) denotes a point at which host damage is sufficient to affect homeostasis, with those above and below the line colored red and blue, respectively, to denote those with and without clinical symptoms.
S. cerevisiae-human interactions (Fig. 1).
Saccharomyces cerevisiae, or brewer's yeast, is not considered a microbe with the ability to cause disease. Nonetheless, there are numerous reports of in the literature of its association with disease. Clinical and nonclinical isolates of S. cerevisiae manifest phenotypic and genetic traits that are associated with virulence in the former group, such as the ability to grow at mammalian temperatures (67, 68). S. cerevisiae is a relatively common member of the human microbiome, especially in individuals with impaired immunity (69). Association between humans and S. cerevisiae is not surprising given its use in brewing and baking. However, less appreciated is the fact that this organism can cause disease in certain hosts. For example, early in the HIV epidemic, cases of invasive disease were reported in patients with the AIDS (70, 71). S. cerevisiae is also a cause of fungal vaginitis in certain women and can trigger severe inflammation after inhalation (72). An example of the latter is a baker who required a lung resection for granulomas associated with yeast cells (73). Considering these interactions in the context of the DRF allows us to see them as continuous (Fig. 1). Although the majority of individuals who acquire S. cerevisiae suffer no clinical symptoms, disease can occur at the extremes of the immune response. In patients with AIDS, weak responses allow invasion, whereas strong responses produce tissue damage from allergy and severe inflammatory responses. Whether vaginitis is a result of an overexuberant response to fungal antigens or reflects some undetected immune abnormality is not currently known. Given that the pathogenesis of Candida albicans vaginitis is currently thought to be the result of a strong immune response to fungal antigens (74), it is possible that S. cerevisiae vaginitis has the same pathogenesis. However, we note that a strong response to fungal antigens may stem from a dysregulated immune response and thus reflect an abnormal immune response.
M. tuberculosis-human interactions (Fig. 1).
Human infection with Mycobacterium tuberculosis can lead to several outcomes, including eradication, latency, or disease, and manifestations of disease can differ from focal pneumonia to dissemination, including meningitis. Most human infections are asymptomatic, and in many individuals infection is followed by the establishment of a latent state that is marked by delayed hypersensitivity. Latent disease can reactivate, and reactivation may follow changes in the immune status of an individual, such as those due to HIV infection or the administration of immunosuppressive drugs such as corticosteroids. Tuberculosis has protean disease manifestations which include caseous necrosis in the lung following a strong inflammatory response and disseminated disease which can present as miliary tuberculosis, with the name coming from the radiologic appearance of small granulomas in the lung. Given that each individual will have a different MISTEACHING composite, the interaction of each individual with M. tuberculosis when the attributes are combined can be plotted on the DRF parabola. For example, we consider three individual hosts who manifest different aspects of M. tuberculosis disease (Fig. 2). Host 1 is an individual with asymptomatic latent M. tuberculosis infection who becomes immunosuppressed as a consequence of HIV infection, leading to reactivation of mycobacterial infection resulting in disseminated disease. Host 2 is an individual with disseminated tuberculosis who responds to antimicrobial therapy combined with gamma interferon. In contrast, Host 3 represents a case of tuberculous meningitis, a disease where inflammation contributes to damage and therapy includes the use of immunosuppressive agents in the form of corticosteroids. We note that the profound differences in these states in regard to immunity and inoculum pose a significant challenge to vaccine development, as a vaccine that stimulates a strong immune response may prevent infection in a naive person but may induce a detrimental inflammatory response in a person with latent tuberculosis.
FIG 2.

DRF parabolic curves representative of individuals. We consider three hosts infected with M. tuberculosis to illustrate how events and medical interventions can move the position of an individual along the parabola. Host 1 has latent infection characterized by a positive tuberculin test and is asymptomatic. Infection with HIV results in a damaged immune system that cannot contain the latent infection, which disseminates. Treatment with antiretroviral therapy restores immunity, which then attacks host tissues where residual mycobacterial antigens are found to cause the immune reconstitution syndrome. Host 2 has miliary tuberculosis but is treated with antimicrobial agents, which reduce burden, and adjunctive gamma interferon, which stimulates cellular immunity. The treatment enhances the host response and moves the individual to the right, resulting in cure. Host 3 has tuberculous meningitis, a condition where the inflammatory response is a major contributor to brain damage. This host is treated with a combination of antimicrobial agents that lower the bacterial burden and corticosteroids to reduce inflammation. The treatment reduces the host response and moves the individual to the left, resulting in cure. DT, disease threshold.
HCV-human interactions (Fig. 1).
Hepatitis C virus (HCV) infection in humans has two major outcomes: eradiation in a minority of individuals or chronic infection causing hepatitis that can progress to cirrhosis and hepatocellular carcinoma. Some individuals are able to forestall clinical disease for decades with mild hepatitis while remaining infected and viremic with HCV, which presumably reflects an ability of the host response to prevent liver failure (75). There is considerable evidence that liver damage leading to cirrhosis is a consequence of chronic inflammation and that progression to end-stage liver disease is associated with strong inflammatory responses to persistent hepatocyte infection. On the other hand, coinfection with HIV, which produces immunosuppression, is also associated with rapid liver disease (76–78). Although the mechanism by which HIV accelerates HCV-mediated liver disease is unknown and currently an area of intense study, it is reasonable to assume that the immunosuppression that follows HIV infection is an important variable in this phenomenon.
When the DRF was first introduced in 1999, we classified pathogenic microbes into six groups based on the most common type of clinical presentation, each with its own damage-response curve, and used the parabolic shape for what we then called a type 3 pathogen (1). Over the years our thinking has evolved such that we now regard the parabola as the central damage-response relationship that underpins all host-microbe interactions. In support of this concept, the original six curves (1), which were derived by tipping the parabola toward the left or right, produce a parabola when combined. Thus, we now use an “idealized” parabola to depict host-microbe interaction, with the knowledge that the attributes which constitute MISTEACHING introduce significant deviations from the idealized curve. In other words, each host-microbe interaction produces its own distinct parabolic curve, which is a derivative of the basic parabola. We feel that the use of one basic curve enhances the flexibility of the DRF such that new information or ways in which host-microbe interaction translate into host damage can easily be depicted. Finally, the use of one basic curve also makes it possible to separate the DRF from the outcome of infection with specific microbes (which were depicted by the six curves). Nonetheless, the original curves (1) remain useful for understanding host-microbe relationships for specific microbes and disease syndromes, as exemplified by a recent analysis that interpreted the different forms of candidiasis as different curves (79). We posit that each host-microbe interaction produces a different version of the damage-response parabola, tipped to the right or left or adjusted up or down, as a function of the attributes of MISTEACHING. These curves are almost certainly unique, given the multitude of possible combinations. Notably, a single host could have different damage-response relationships with different microbes and vice versa, depending on which MISTEACHING attributes are driving the relevant host-microbe interaction (Fig. 3). This also explains how one microbe can exist in a symbiotic, commensal, or pathogenic relationship in one host depending on the MISTEACHING factor combination (Fig. 3).
FIG 3.
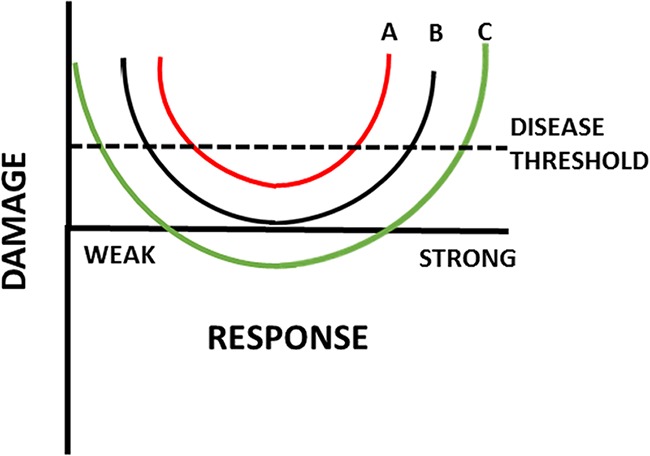
Different host-microbe interactions result in different damage-response relationships, idealized as parabolic curves. The three curves (A to C) can denote the damage-response relationships of a single microbe with three hosts or those of a single host with three different microbes. During intermediate responses, none of these interactions is sufficient to cause diseases, since the amount of damage incurred by the host is insufficient to affect homeostasis. However, in the setting of weak or strong responses, each of these host-microbe interactions has the potential to cause sufficient damage to cross the disease threshold. The position of the parabola vertex is determined by the particular combination of MISTEACHING factors that applies for the host-microbe interaction and a particular time. For curve A, the interaction results in some host damage at all response intensities, but in intermediate responses the vertex lies below the disease threshold. For curve B, the vertex lies on the axis during intermediate responses, resulting in an interaction where no damage occurs in the state of commensalism. For curve C, the vertex lies below the axis, representing an interaction that is potentially symbiotic in some MISTEACHING combinations with the potential for host virulence when the combination results in weak or strong responses.
ON PREDICTABILITY
Can we predict susceptibility and resistance to disease? Given the complexity of the 11 attributes put forth above, it is possible many would conclude that prediction is not possible. However, to make this claim would ignore ample evidence that the risk for certain diseases is fairly predictable. For example, a person without immunity to vaccine-preventable diseases such as varicella, measles, mumps, and rubella has a high likelihood of acquiring these viruses and exhibiting disease after an exposure. This is why herd protection is so important to maintain protection against vaccine-preventable diseases that are further prevented by herd immunity. Similarly, it would ignore evidence that infection with HIV can be prevented by behavioral factors, such as use of condoms and not sharing (or using) needles for injection of drugs. HIV transmission via blood transfusion was eliminated by screening the blood supply. At the genetic level, susceptibility to HIV infection is related to the ability of the virus to replicate in tissue, which requires cell entry via the CCR5 coreceptor. Humans lacking this protein are resistant to infection. Notably, these examples concern infectious diseases for which acquisition of the microbe is highly likely to lead to disease. Less difficult to predict with current knowledge is the outcome of infection for microbes that can result in no disease or variable outcomes, such as M. tuberculosis, and microbes that affect large populations at once, where diversity among the attributes of MISTEACHING often precludes assessments of individual risk.
There are several levels to prediction. One is whether the microbe will be encountered at all. This is something that cannot always be predicted. For example, transmission of Ebola virus and the chikungunya and Zika viruses exploded in West Africa and the Americas, respectively, in the early 2010s for reasons that were not sufficiently appreciated at the time. Now that these are better understood, public health measures and education aimed at reducing risk of exposure are recognized as critical components of disease prevention. In addition, these epidemics revealed previously unrecognized outcomes of infection, e.g., the ability of Zika virus and Ebola virus to be transmitted sexually and the ability of Zika virus to infect fetal neural tissue. High temperatures in mammals, which prevent the growth of most fungal species, eliminating their ability to serve as hosts, make it possible to predict that acquisition of such species is not likely to result in disease. However, for many host-microbe interactions, no dominant variable can reliably predict host susceptibility and resistance. For example, even patients with AIDS and profound CD4 T cell depletion do not manifest all the infectious diseases that can complicate profound immunodeficiency. Therefore, we believe that assessing risk for disease as a function of the attributes of MISTEACHING provides a roadmap for a rational assessment of susceptibility in individuals and populations. Nevertheless, MISTEACHING includes attributes that are interrelated, such as genetics, age, nutrition, microbiome, and immunity, as well as attributes that are independent of the host, such as inoculum, temperature, and some components of environment, further underscoring that the MISTEACHING signature will differ for each host-microbe encounter within and between individuals and populations. Hence, the MISTEACHING attributes will make it possible to predict the outcome of some host-microbe interactions but not others, although assessment of the attributes that are known may allow probabilistic predictions for both individuals and populations.
ACKNOWLEDGMENTS
A.C. is supported by NIH grants AI033142, AI052733, and HL059842. L.P. is supported by NIH grants AG045044, AI097096, and AI123654.
REFERENCES
- 1.Casadevall A, Pirofski L. 1999. Host-pathogen interactions: redefining the basic concepts of virulence and pathogenicity. Infect Immun 67:3703–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Casadevall A, Pirofski L. 2000. Host-pathogen interactions: the basic concepts of microbial commensalism, colonization, infection, and disease. Infect Immun 68:6511–6518. doi: 10.1128/IAI.68.12.6511-6518.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pirofski L, Casadevall A. 2002. The meaning of microbial exposure, infection, colonisation, and disease in clinical practice. Lancet Infect Dis 2:628. doi: 10.1016/S1473-3099(02)00398-5. [DOI] [PubMed] [Google Scholar]
- 4.Casadevall A, Pirofski L. 2001. Host-pathogen interactions: the attributes of virulence. J Infect Dis 184:337–344. doi: 10.1086/322044. [DOI] [PubMed] [Google Scholar]
- 5.Casadevall A, Pirofski L. 2003. The damage-response framework of microbial pathogenesis. Nat Microbiol Rev 1:17–24. doi: 10.1038/nrmicro732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pirofski LA, Casadevall A. 2015. What is infectiveness and how is it involved in infection and immunity? BMC Immunol 16:13. doi: 10.1186/s12865-015-0076-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pirofski LA, Casadevall A. 2012. Q and A: what is a pathogen? A question that begs the point. BMC Biol 10:6. doi: 10.1186/1741-7007-10-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Casadevall A, Pirofski LA. 2015. What is a host? Incorporating the microbiota into the damage-response framework. Infect Immun 83:2–7. doi: 10.1128/IAI.02627-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Casadevall A, Pirofski LA. 2014. Microbiology: ditch the term pathogen. Nature 516:165–166. doi: 10.1038/516165a. [DOI] [PubMed] [Google Scholar]
- 10.Blaser MJ. 2014. The microbiome revolution. J Clin Invest 124:4162–4165. doi: 10.1172/JCI78366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cho I, Yamanishi S, Cox L, Methe BA, Zavadil J, Li K, Gao Z, Mahana D, Raju K, Teitler I, Li H, Alekseyenko AV, Blaser MJ. 2012. Antibiotics in early life alter the murine colonic microbiome and adiposity. Nature 488:621–626. doi: 10.1038/nature11400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shrinet J, Shastri JS, Gaind R, Bhavesh NS, Sunil S. 2016. Serum metabolomics analysis of patients with chikungunya and dengue mono/co-infections reveals distinct metabolite signatures in the three disease conditions. Sci Rep 6:36833. doi: 10.1038/srep36833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Buffie CG, Bucci V, Stein RR, McKenney PT, Ling L, Gobourne A, No D, Liu H, Kinnebrew M, Viale A, Littmann E, van den Brink MR, Jenq RR, Taur Y, Sander C, Cross JR, Toussaint NC, Xavier JB, Pamer EG. 2015. Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile. Nature 517:205–208. doi: 10.1038/nature13828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Broderick NA, Raffa KF, Handelsman J. 2006. Midgut bacteria required for Bacillus thuringiensis insecticidal activity. Proc Natl Acad Sci U S A 103:15196–15199. doi: 10.1073/pnas.0604865103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dicksved J, Ellstrom P, Engstrand L, Rautelin H. 2014. Susceptibility to Campylobacter infection is associated with the species composition of the human fecal microbiota. mBio 5:e01212-14. doi: 10.1128/mBio.01212-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Byndloss MX, Olsan EE, Rivera-Chavez F, Tiffany CR, Cevallos SA, Lokken KL, Torres TP, Byndloss AJ, Faber F, Gao Y, Litvak Y, Lopez CA, Xu G, Napoli E, Giulivi C, Tsolis RM, Revzin A, Lebrilla CB, Baumler AJ. 2017. Microbiota-activated PPAR-gamma signaling inhibits dysbiotic Enterobacteriaceae expansion. Science 357:570–575. doi: 10.1126/science.aam9949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cirimotich CM, Dong Y, Clayton AM, Sandiford SL, Souza-Neto JA, Mulenga M, Dimopoulos G. 2011. Natural microbe-mediated refractoriness to Plasmodium infection in Anopheles gambiae. Science 332:855–858. doi: 10.1126/science.1201618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kuss SK, Best GT, Etheredge CA, Pruijssers AJ, Frierson JM, Hooper LV, Dermody TS, Pfeiffer JK. 2011. Intestinal microbiota promote enteric virus replication and systemic pathogenesis. Science 334:249–252. doi: 10.1126/science.1211057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Florez LV, Biedermann PH, Engl Kaltenpoth TM. 2015. Defensive symbioses of animals with prokaryotic and eukaryotic microorganisms. Nat Prod Rep 32:904–936. doi: 10.1039/C5NP00010F. [DOI] [PubMed] [Google Scholar]
- 20.Huffnagle GB, Noverr MC. 2013. The emerging world of the fungal microbiome. Trends Microbiol 21:334–341. doi: 10.1016/j.tim.2013.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Robinson CM, Pfeiffer JK. 2014. Viruses and the microbiota. Annu Rev Virol 1:55–69. doi: 10.1146/annurev-virology-031413-085550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Armstrong D. 1993. History of opportunistic infection in the immunocompromised host. Clin Infect Dis 17(Suppl 2):S318–S321. doi: 10.1093/clinids/17.Supplement_2.S318. [DOI] [PubMed] [Google Scholar]
- 23.vom Steeg LG, Klein SL. 2016. SeXX matters in infectious disease pathogenesis. PLoS Pathog 12:e1005374. doi: 10.1371/journal.ppat.1005374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Klein SL, Flanagan KL. 2016. Sex differences in immune responses. Nat Rev Immunol 16:626–638. doi: 10.1038/nri.2016.90. [DOI] [PubMed] [Google Scholar]
- 25.Bandeira AC, Campos GS, Rocha VF, Souza BS, Soares MB, Oliveira AA, Abreu YC, Menezes GS, Sardi SI. 2016. Prolonged shedding of Chikungunya virus in semen and urine: a new perspective for diagnosis and implications for transmission. IDCases 6:100–103. doi: 10.1016/j.idcr.2016.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Althaus CL, Low N. 2016. How relevant is sexual transmission of Zika virus? PLoS Med 13:e1002157. doi: 10.1371/journal.pmed.1002157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Abbate JL, Murall CL, Richner H, Althaus CL. 2016. Potential impact of sexual transmission on Ebola virus epidemiology: Sierra Leone as a case study. PLoS Negl Trop Dis 10:e0004676. doi: 10.1371/journal.pntd.0004676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Robert VA, Casadevall A. 2009. Vertebrate endothermy restricts most fungi as potential pathogens. J Infect Dis 200:1623–1626. doi: 10.1086/644642. [DOI] [PubMed] [Google Scholar]
- 29.Blehert DS, Hicks AC, Behr M, Meteyer CU, Berlowski-Zier BM, Buckles EL, Coleman JT, Darling SR, Gargas A, Niver R, Okoniewski JC, Rudd RJ, Stone WB. 2009. Bat white-nose syndrome: an emerging fungal pathogen? Science 323:227. doi: 10.1126/science.1163874. [DOI] [PubMed] [Google Scholar]
- 30.Meteyer CU, Valent M, Kashmer J, Buckles EL, Lorch JM, Blehert DS, Lollar A, Berndt D, Wheeler E, White CL, Ballmann AE. 2011. Recovery of little brown bats (Myotis lucifugus) from natural infection with Geomyces destructans, white-nose syndrome. J Wildl Dis 47:618–626. doi: 10.7589/0090-3558-47.3.618. [DOI] [PubMed] [Google Scholar]
- 31.Casadevall A. 2016. Thermal restriction as an antimicrobial function of fever. PLoS Pathog 12:e1005577. doi: 10.1371/journal.ppat.1005577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kluger MJ, Kozak W, Conn CA, Leon LR, Soszynski D. 1998. Role of fever in disease. Ann N Y Acad Sci 856:224–233. doi: 10.1111/j.1749-6632.1998.tb08329.x. [DOI] [PubMed] [Google Scholar]
- 33.Karraker NE, Ruthig GR. 2009. Effect of road deicing salt on the susceptibility of amphibian embryos to infection by water molds. Environ Res 109:40–45. doi: 10.1016/j.envres.2008.09.001. [DOI] [PubMed] [Google Scholar]
- 34.Kreutz LC, Barcellos LJ, Marteninghe A, Dos Santos ED, Zanatta R. 2010. Exposure to sublethal concentration of glyphosate or atrazine-based herbicides alters the phagocytic function and increases the susceptibility of silver catfish fingerlings (Rhamdia quelen) to Aeromonas hydrophila challenge. Fish Shellfish Immunol 29:694–697. doi: 10.1016/j.fsi.2010.06.003. [DOI] [PubMed] [Google Scholar]
- 35.Jansen M, Stoks R, Coors A, van Doorslaer W, de Meester L. 2011. Collateral damage: rapid exposure-induced evolution of pesticide resistance leads to increased susceptibility to parasites. Evolution 65:2681–2691. doi: 10.1111/j.1558-5646.2011.01331.x. [DOI] [PubMed] [Google Scholar]
- 36.Wu X, Lu Y, Zhou S, Chen L, Xu B. 2016. Impact of climate change on human infectious diseases: Empirical evidence and human adaptation. Environ Int 86:14–23. doi: 10.1016/j.envint.2015.09.007. [DOI] [PubMed] [Google Scholar]
- 37.Lafferty KD, Mordecai EA. 2016. The rise and fall of infectious disease in a warmer world. 5:F1000. doi: 10.12688/f1000research.8766.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hjelle B, Glass GE. 2000. Outbreak of hantavirus infection in the Four Corners region of the United States in the wake of the 1997-1998 El Nino-southern oscillation. J Infect Dis 181:1569–1573. doi: 10.1086/315467. [DOI] [PubMed] [Google Scholar]
- 39.Fothergill LD, Wright J. 1933. Influenzal meningitis: the relation of age incidence to the bactericidal power of blood against the causal organism. J Immunol 24:273–284. [Google Scholar]
- 40.Davies SF, Colbert RL. 1990. Concurrent human and canine histoplasmosis from cutting decayed wood. Ann Intern Med 113:252–253. doi: 10.7326/0003-4819-113-3-252. [DOI] [PubMed] [Google Scholar]
- 41.Lyon GM, Bravo AV, Espino A, Lindsley MD, Gutierrez RE, Rodriguez I, Corella A, Carrillo F, McNeil MM, Warnock DW, Hajjeh RA. 2004. Histoplasmosis associated with exploring a bat-inhabited cave in Costa Rica, 1998-1999. Am J Trop Med Hyg 70:438–442. [PubMed] [Google Scholar]
- 42.Anonymous. 2014. Histoplasmosis outbreak associated with the renovation of an old house—Quebec, Canada, 2013. MMWR Morb Mortal Wkly Rep 62:1041–1044. [PMC free article] [PubMed] [Google Scholar]
- 43.Bhardwaj V, Meier S, Petersen LN, Ingle RA, Roden LC. 2011. Defence responses of Arabidopsis thaliana to infection by Pseudomonas syringae are regulated by the circadian clock. PLoS One 6:e26968. doi: 10.1371/journal.pone.0026968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kiessling S, Dubeau-Laramee G, Ohm H, Labrecque N, Olivier M, Cermakian N. 2017. The circadian clock in immune cells controls the magnitude of Leishmania parasite infection. Sci Rep 7:10892. doi: 10.1038/s41598-017-11297-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dejnirattisai W, Supasa P, Wongwiwat W, Rouvinski A, Barba-Spaeth G, Duangchinda T, Sakuntabhai A, Cao-Lormeau VM, Malasit P, Rey FA, Mongkolsapaya J, Screaton GR. 2016. Dengue virus sero-cross-reactivity drives antibody-dependent enhancement of infection with zika virus. Nat Immunol 17:1102–1108. doi: 10.1038/ni.3515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Henry C, Palm AE, Krammer F, Wilson PC. 2017. From original antigenic sin to the universal influenza virus vaccine. Trends Immunol doi: 10.1016/j.it.2017.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Robbiani DF, Bozzacco L, Keeffe JR, Khouri R, Olsen PC, Gazumyan A, Schaefer-Babajew D, Avila-Rios S, Nogueira L, Patel R, Azzopardi SA, Uhl LFK, Saeed M, Sevilla-Reyes EE, Agudelo M, Yao KH, Golijanin J, Gristick HB, Lee YE, Hurley A, Caskey M, Pai J, Oliveira T, Wunder EA Jr, Sacramento G, Nery N Jr, Orge C, Costa F, Reis MG, Thomas NM, Eisenreich T, Weinberger DM, de Almeida ARP, West AP Jr, Rice CM, Bjorkman PJ, Reyes-Teran G, Ko AI, MacDonald MR, Nussenzweig MC. 2017. Recurrent potent human neutralizing antibodies to Zika virus in Brazil and Mexico. Cell 169:597–609.e11. doi: 10.1016/j.cell.2017.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Welsh RM, Che JW, Brehm MA, Selin LK. 2010. Heterologous immunity between viruses. Immunol Rev 235:244–266. doi: 10.1111/j.0105-2896.2010.00897.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Marrie TJ, Michelakis E. 2003. Increased long-term mortality after an episode of community-acquired pneumonia—time to move beyond descriptive studies. Clin Infect Dis 37:1625–1628. doi: 10.1086/379723. [DOI] [PubMed] [Google Scholar]
- 50.Casadevall A. 2017. The pathogenic potential of a microbe. mSphere 2:e00015-17. doi: 10.1128/mSphere.00015-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Robbins JB, Schneerson R, Szu SC. 1995. Perspective: hypothesis: serum IgG antibody is sufficient to confer protection against infectious diseases by inactivating the inoculum. J Infect Dis 171:1387–1398. doi: 10.1093/infdis/171.6.1387. [DOI] [PubMed] [Google Scholar]
- 52.Goldman AS, Goldman LR, Goldman DA. 2005. What caused the epidemic of Pneumocystis pneumonia in European premature infants in the mid-20th century? Pediatrics 115:e725–e736. doi: 10.1542/peds.2004-2157. [DOI] [PubMed] [Google Scholar]
- 53.Hughes WT, Price RA, Sisko F, Havron WS, Kafatos AG, Schonland M, Smythe PM. 1974. Protein-calorie malnutrition. A host determinant for Pneumocystis carinii infection. Am J Dis Child 128:44–52. [DOI] [PubMed] [Google Scholar]
- 54.Platts-Mills JA, Taniuchi M, Uddin MJ, Sobuz SU, Mahfuz M, Gaffar SA, Mondal D, Hossain MI, Islam MM, Ahmed AS, Petri WA, Haque R, Houpt ER, Ahmed T. 2017. Association between enteropathogens and malnutrition in children aged 6-23 mo in Bangladesh: a case-control study. Am J Clin Nutr 105:1132–1138. doi: 10.3945/ajcn.116.138800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chhagan MK, Kauchali S. 2006. Comorbidities and mortality among children hospitalized with diarrheal disease in an area of high prevalence of human immunodeficiency virus infection. Pediatr Infect Dis J 25:333–338. doi: 10.1097/01.inf.0000207400.93627.4c. [DOI] [PubMed] [Google Scholar]
- 56.Bottone EJ. 1997. Yersinia enterocolitica: the charisma continues. Clin Microbiol Rev 10:257–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Murray J, Murray A. 1977. Suppression of infection by famine and its activation by refeeding—a paradox? Perspect Biol Med 20:471–483. doi: 10.1353/pbm.1977.0037. [DOI] [PubMed] [Google Scholar]
- 58.Hainer V, Zamrazilova H, Kunesova M, Bendlova B, Aldhoon-Hainerova I. 2015. Obesity and infection: reciprocal causality. Physiol Res 64(Suppl 2):S105–S119. [DOI] [PubMed] [Google Scholar]
- 59.Hill AV. 1996. Genetic susceptibility to malaria and other infectious diseases: from the MHC to the whole genome. Parasitology 112(Suppl):S75–S84. doi: 10.1017/S003118200007668X. [DOI] [PubMed] [Google Scholar]
- 60.Patick AK, Pease LR, David CS, Rodriguez M. 1990. Major histocompatibility complex-conferred resistance to Theiler's virus-induced demyelinating disease is inherited as a dominant trait in B10 congenic mice. J Virol 64:5570–5576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bian Y, Shang S, Siddiqui S, Zhao J, Joosten SA, Ottenhoff THM, Cantor H, Wang CR. 2017. MHC Ib molecule Qa-1 presents Mycobacterium tuberculosis peptide antigens to CD8+ T cells and contributes to protection against infection. PLoS Pathog 13:e1006384. doi: 10.1371/journal.ppat.1006384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fang R, Ismail N, Soong L, Popov VL, Whitworth T, Bouyer DH, Walker DH. 2007. Differential interaction of dendritic cells with Rickettsia conorii: impact on host susceptibility to murine spotted fever rickettsiosis. Infect Immun 75:3112–3123. doi: 10.1128/IAI.00007-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Selmi C, Gershwin ME. 2014. Diagnosis and classification of reactive arthritis. Autoimmun Rev 13:546–549. doi: 10.1016/j.autrev.2014.01.005. [DOI] [PubMed] [Google Scholar]
- 64.Zimmerman PA, Buckler-White A, Alkhatib G, Spalding T, Kubofcik J, Combadiere C, Weissman D, Cohen O, Rubbert A, Lam G, Vaccarezza M, Kennedy PE, Kumaraswami V, Giorgi JV, Detels R, Hunter J, Chopek M, Berger EA, Fauci AS, Nutman TB, Murphy PM. 1997. Inherited resistance to HIV-1 conferred by an inactivating mutation in CC chemokine receptor 5: studies in populations with contrasting clinical phenotypes, defined racial background, and quantified risk. Mol Med 3:23–36. [PMC free article] [PubMed] [Google Scholar]
- 65.Casanova JL. 2015. Human genetic basis of interindividual variability in the course of infection. Proc Natl Acad Sci U S A 112:E7118–E7127. doi: 10.1073/pnas.1521644112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Panackal AA, Rosen LB, Uzel G, Davis MJ, Hu G, Adeyemo A, Tekola-Ayele F, Lisco A, Diachok C, Kim JD, Shaw D, Sereti I, Stoddard J, Niemela J, Rosenzweig SD, Bennett JE, Williamson PR. 2017. Susceptibility to cryptococcal meningoencephalitis associated with idiopathic CD4+ lymphopenia and secondary germline or acquired defects. Open Forum Infect Dis 4:ofx082. doi: 10.1093/ofid/ofx082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Clemons KV, McCusker JH, Davis RW, Stevens DA. 1994. Comparative pathogenesis of clinical and nonclinical isolates of Saccharomyces cerevisiae. J Infect Dis 169:859–867. doi: 10.1093/infdis/169.4.859. [DOI] [PubMed] [Google Scholar]
- 68.McCusker JH, Clemons KV, Stevens DA, Davis RW. 1994. Genetic characterization of pathogenic Saccharomyces cerevisiae isolates. Genetics 136:1261–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Clemons KV, Salonen JH, Issakainen J, Nikoskelainen J, McCullough MJ, Jorge JJ, Stevens DA. 2010. Molecular epidemiology of Saccharomyces cerevisiae in an immunocompromised host unit. Diagn Microbiol Infect Dis 68:220–227. doi: 10.1016/j.diagmicrobio.2010.06.010. [DOI] [PubMed] [Google Scholar]
- 70.Doyle MG, Pickering LK, O'Brien N, Hoots K, Benson JE. 1990. Saccharomyces cerevisiae infection in a patient with acquired immunodeficiency syndrome. Pediatr Infect Dis J 9:850–851. doi: 10.1097/00006454-199011000-00015. [DOI] [PubMed] [Google Scholar]
- 71.Tawfik OW, Papasian CJ, Dixon AY, Potter LM. 1989. Saccharomyces cerevisiae pneumonia in a patient with acquired immune deficiency syndrome. J Clin Microbiol 27:1689–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nyirjesy P, Vazquez JA, Ufberg DD, Sobel JD, Boikov DA, Buckley HR. 1995. Saccharomyces cerevisiae vaginitis: transmission from yeast used in baking. Obstet Gynecol 86:326–329. doi: 10.1016/0029-7844(95)00174-P. [DOI] [PubMed] [Google Scholar]
- 73.Ren P, Sridhar S, Chaturvedi V. 2004. Use of paraffin-embedded tissue for identification of Saccharomyces cerevisiae in a baker's lung nodule by fungal PCR and nucleotide sequencing. J Clin Microbiol 42:2840–2842. doi: 10.1128/JCM.42.6.2840-2842.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Fidel PL., Jr 2007. History and update on host defense against vaginal candidiasis. Am J Reprod Immunol 57:2–12. doi: 10.1111/j.1600-0897.2006.00450.x. [DOI] [PubMed] [Google Scholar]
- 75.Murphy N, O'Mahony B, Flanagan P, Noone D, White B, Bergin C, Norris S, Thornton L. 2017. Progression of hepatitis C in the haemophiliac population in Ireland, after 30 years of infection in the pre-DAA treatment era. Haemophilia doi: 10.1111/hae.13244. [DOI] [PubMed] [Google Scholar]
- 76.Shmagel KV, Saidakova EV, Shmagel NG, Korolevskaya LB, Chereshnev VA, Robinson J, Grivel JC, Douek DC, Margolis L, Anthony DD, Lederman MM. 2016. Systemic inflammation and liver damage in HIV/hepatitis C virus coinfection. HIV Med 17:581–589. doi: 10.1111/hiv.12357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Garcia-Samaniego J, Soriano V, Castilla J, Bravo R, Moreno A, Carbo J, Iniguez A, Gonzalez J, Munoz F. 1997. Influence of hepatitis C virus genotypes and HIV infection on histological severity of chronic hepatitis C. The Hepatitis/HIV Spanish Study Group. Am J Gastroenterol 92:1130–1134. [PubMed] [Google Scholar]
- 78.Kirk GD, Mehta SH, Astemborski J, Galai N, Washington J, Higgins Y, Balagopal A, Thomas DL. 2013. HIV, age, and the severity of hepatitis C virus-related liver disease: a cohort study. Ann Intern Med 158:658–666. doi: 10.7326/0003-4819-158-9-201305070-00604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Jabra-Rizk MA, Kong EF, Tsui C, Nguyen MH, Clancy CJ, Fidel PL Jr, Noverr M. 2016. Candida albicans pathogenesis: fitting within the host-microbe damage response framework. Infect Immun 84:2724–2739. doi: 10.1128/IAI.00469-16. [DOI] [PMC free article] [PubMed] [Google Scholar]