

Abstract
Pan-Assay Interference Compounds (PAINS) are very familiar to medicinal chemists who have spent time fruitlessly trying to optimize these nonprogressible compounds. Electronic filters formulated to recognize PAINS can process hundreds and thousands of compounds in seconds and are in widespread current use to identify PAINS in order to exclude them from further analysis. However, this practice is fraught with danger because such black box treatment is simplistic. Here, we outline for the first time all necessary considerations for the appropriate use of PAINS filters.
In 2003, one of us (J.B.) established a general purpose high throughput screening (HTS) library, numbering some 100 000 compounds selected from around four different and well-known vendors. Our guiding philosophy was to include reasonably lead-like and optimizable compounds (MW 150–400; rings 1–4; HBA < 8 and HBD < 5). We also excluded very large numbers of compounds that were deemed to be too similar (>85%) to others already selected and undertook extensive curation to exclude unwanted functional groups prior to purchase. The point to be made here is that the compounds in our library would have been similar to those in other academic screening collections.
Random inspection of several hundred structures corresponding to compounds selected for purchase raised relatively few alarms and the HTS library was duly established. We ran assays in the presence of detergent (0.01–0.05% Tween-20) to avoid aggregate interference, and so excitement was considerable when we started identifying numerous potent and selective hits for our various targets. However, much time in the ensuing first few years was wasted on hits that turned out to be translational cul-de-sacs. In the most straightforward cases, biological activity was not reproduced in resynthesized or repurified samples. In other cases, early and promising structure–activity relationships (SARs) dissipated, ending in flat or otherwise uninterpretable SARs. We observed that similar looking but not necessarily the same compounds kept appearing in different screens, and so we developed the notion of classes of intrinsically promiscuous compounds sharing common substructural motifs. Identification of the offending substructures was not straightforward, but we eventually settled on definitions with which we were satisfied.
On searching the literature, we were struck by the observation that many of these problematic chemotypes were known to exhibit varying forms of chemical reactivity and apparent biological activity. We surmised that such behavior could result in positive readouts in biochemical assays via a variety of mechanisms but that such readouts were not associated with compounds that could be regarded as optimizable and progressible. It was for this reason that we coined the term Pan-Assay Interference Compounds but it is important to realize that this nomenclature is class-based and individual compounds recognized by a PAINS substructure do not necessarily exhibit broad spectrum interference.
In 2010, our investigation was published in the Journal of Medicinal Chemistry.1 Some 7 years later, with 1035 citations (Google Scholar, October 18, 2017), the response to this publication has been larger than we had anticipated. It is the recognition of these nuisance compound classes rather than just specific compounds by medicinal chemists worldwide that has in our view largely driven the attention that the PAINS issue has garnered.
The implementation of PAINS substructures as electronic filters allows for near immediate identification of PAINS even given the input of many thousands of compound structures.2,3 However, with such ease of use comes the danger that the appropriate degree of intellectual rigor and scrutiny of the screening context is not applied to this important process of compound triage. In recent years, we have discussed many aspects of PAINS characteristics that should be brought into consideration during any triage process,1,4−15 but it has become increasingly clear that overzealous or simplistic use of these filters may inappropriately exclude a useful compound from consideration and inappropriately tag a useless compound as worthy of development.
Since in no single place have these important considerations related to PAINS assessment been summarized, we feel it is timely to do so, and that is the purpose of this Review.
It is useful to start with a few observations around the evolution of the PAINS concept over the past seven years, and to separate this from discussion around the identification of PAINS specifically by the electronic filters as originally published.
PAINS comprise classes of compounds defined by a common substructural motif that encodes for an increased chance of any member registering as a hit in any given assay, that may be independent of platform technology. Such compounds are concomitantly less likely to be optimizable toward a useful compound.
A common reason why PAINS register as hits in assays is that the substructure can confer an ability to interfere in biochemical assays. Mechanisms are various and include, for example, reactivity with biological and bioassay nucleophiles such as thiols and amines; photoreactivity with any protein functionality; metal chelation that can interfere with proteins, assay reagents, or through bringing in heavy metal contaminants; redox cycling and redox activity; physicochemical interference such as micelle formation, or; having photochromic properties that might interfere with typically used assay signaling such as absorption and fluorescence.
A small proportion (ca. 5%) of FDA-approved drugs contain PAINS-recognized substructures, these comprising both natural products and synthetic drugs. Regardless, this observation has no relevance to low micromolar screening hits even if they belong to the same PAINS class as that of an FDA-approved drug, and cannot be used to suggest that such a screening hit is necessarily progressible. This is because the relatively few known PAINS-containing drugs were discovered in a traditional manner after observation of potent downstream efficacy, not through target-based screening. We have discussed this in detail elsewhere.6,10,15
The derivation of the electronic PAINS filters has been purely observational, and they have in no way been derived actively from known toxicophores or groups conveying unattractive physicochemical or pharmacokinetic properties. Although these phenomena do appear to be quite commonly associated,1,10 it would be completely wrong to assume for example that a PAIN must have poor pharmacokinetic properties or that a toxicophore must also be a PAIN.
Nor are these filters comprehensive—a compound not recognized as a PAIN may still be a PAIN in its behavior. The reason for this limitation is that PAINS were defined based on a curated screening library. Though it reasonably represents other screening libraries of vendor-derived compounds, it was subjected to prefiltering to exclude a large number of functional groups defined as problematic screening hits in several publications emanating from the pharmaceutical industry at the time. An electron-deficient and reactive epoxide (1), aziridine (2), or nitroalkene (3), for example, will not be recognized by the electronic filters because all epoxides, aziridines, and nitro groups were excluded from our general purpose screening library from which PAINS chemotypes were derived (Figure 1).
Figure 1.

Epoxide 1, aziridine 2, and nitroalkene 3 unrecognized by PAINS electronic filters because such compounds were not included in the inaugural WEHI HTS library. The dicyanoalkene 4, however, is a recognized PAINS chemotype, but the corresponding electronic filter ene_cyano_A would not recognize consequently plausible PAIN 5 simply because 5 was a substructure not represented by any compound in the initial HTS library from which PAINS were defined. Other reactive compounds such as β-aminoketone 6, isothiazolones 7, and toxoflavins like 8 are not recognized by PAINS filters because their PAINS behavior was only subsequently identified after filter definition.
A compound with a PAINS chemotype recognized by the electronic filters and that exhibits PAINS behavior may be partnered with an analogue that is not recognized by the filters if that analogue represents a variant that, however slight or peripheral, was absent in any analogue present in the original general purpose 100 000 HTS library, which is effectively a “training set.” This is exemplified by 4 and 5 in Figure 1.
Apart from the structural bias in the underlying data set, we must further consider the relatively small and specific set of data on which PAINS were defined. This comprised approximately 100 000 compounds and six HTS campaigns against protein–protein interactions using a single assay technology (AlphaScreen). We parenthetically comment that we have found AlphaScreen to be a highly robust yet sensitive assay technology returning hit rates as well as any other technology16 if properly optimized. Wider analysis of additional data sets has since led to recognition of newer classes of protein-reactive PAINS, such as β-aminoketones 6, isothiazolones 7, and toxoflavins 8 in Figure 1, which had not been so prominent in the earlier screens, and consequently these do not feature in the original set of filters.6,15
A change of technology platform or even changes within the same technology platform can introduce numerous ways of interference in signaling that may not have been apparent in the original AlphaScreen data set. An example is the specific and unexpected interference of salicylates in FRET technology17 or acetamides in assays using what turned out to be insufficiently selective antiacetyllysine antibodies, for example.13 Such compounds will not of course be recognized by the electronic PAINS filters.
Assay conditions will play a role as well. The assays used to define PAINS were run under detergent-containing conditions designed to minimize interference by aggregates, a phenomenon identified by Shoichet and coworkers.18 So although aggregation could be regarded as PAINS-type behavior, the PAINS electronic filters will not necessarily recognize aggregate-forming compounds. Additionally, the test concentration employed in the early screens from which PAINS were derived was relatively high and generally 50 μM. This will likely have emphasized bad behavior that is unlikely to translate proportionally to settings with a significantly lower test concentration. This would also obviously require redefinition of what constitutes a frequent hitting class in any PAINS class performance analyses of HTS data derived at lower test concentrations. Unfortunately, such fundamental principles are sometimes not taken into account in studies of PAINS filter performance.21
There does not seem to be an industry-accepted nomenclature or ontology of anomalous binding behavior. Offending compounds have been termed frequent hitters and have been variously subdivided into false positives and promiscuous binders,19 or specific and nonspecific groupings.16 In Figure 2, we suggest pictorially how the terms most commonly associated with such discussions, whether these be micelle formers, false positives, false hits, bad actors, frequent hitters, true positives, or true hits, can be appropriately applied at different stages of hit triage, and we have annotated the image with approaches commonly used to characterize such hits.
Figure 2.

Simplified ontology of hits and false-positives. Red boxes indicate potential approaches to identify different types of hits in a cascade of assays. The type and order of assays used in the cascade needs to be considered based on the expected hit rates and achievable throughputs of the relevant assays. The term “frequent hitter” in a sense lies outside this system as it presumes a body of associated historical screening data.
In brief, we suggest it is useful to first segregate actives arising from a screen into target modulators and readout modulators, and that the term “true positives” be assigned to the former and “false positives” to the latter. For a variety of subversive reasons, target modulators may not always be progressible, and so we suggest the term “false hit” is usefully applied to such target modulators, to differentiate these from the remainder, these therefore being termed “true hits.” The term “bad actor” would fit nicely into the category of “false hit” in this regime. On the other hand, a “frequent hitter” can simply be any individual compound or chemotype associated with a body of screening data that suggests higher than expected active readout—in other words, the blue box in Figure 2—in a statistically significant manner. A “frequent hitter” can be a true hit, false hit, or false positive depending on the context of the specific assay in which it is tested.
With an initial intention for maximal capture of subversive compounds, we did not run assay technology-specific counterscreens to exclude compounds from analysis. For all these reasons, a limitation of the electronic filters is that some aspects may be specific toward the conditions outlined above. Most obviously, some compounds will simply interfere in AlphaScreen signaling such as dialkylaniline 9 in Figure 3 that probably quenches singlet oxygen. Others, such as sulforhodamine, trypan blue, malachite green, and Chicago Skye Blue, absorb light at wavelengths in the 576–618 nm range that we have shown to represent signaling interference. However, while many PAINS classes contain some member compounds that registered as hits in all the assays analyzed and that therefore could be AlphaScreen-specific signal interference compounds, most compounds in such classes signal in only a portion of assays. For these, chemical reactivity that is only induced in some assays is a plausible mechanism for platform-independent assay interference.1,20 Hence in the language used around Figure 2, PAINS can be considered to represent frequent hitter chemotypes derived from a particular data set but with broader relevance to the screening community, and they can be true or false hits, or false positives depending on the context of the assay in which they are tested.
Figure 3.
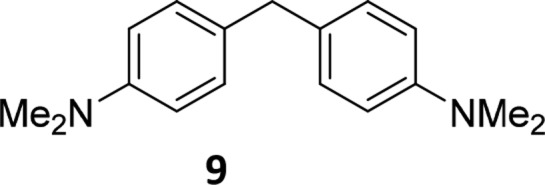
Interference by alkylanilines such as 9 with AlphaScreen signaling, probably through reaction with singlet oxygen, routinely returning an apparent IC50 value of around 3 μM.
Just as PAINS classes may contain compounds that registered as hits in all assays studied, and compounds that registered as hits in a portion of assays studied, so do they contain large numbers of compounds that were entirely clean. That is, a distribution of activity is observed for every class of PAIN. Independent analysis by one of the authors (J.W.M.N) has confirmed that incidence of PAIN behavior in classes of compounds sharing a substructural motif is generally elevated, but not observed throughout.16 It should therefore be emphasized that the presence of a PAINS substructure does not convey PAINS behavior in all instances, but merely reflects a risk of PAINS behavior, the risk increasing as that class is shown to be relatively more promiscuous. Apparently clean members of PAINS chemotypes may simply have needed more assays to reveal their problematic behavior, or conversely they might be genuinely benign. In light of the reflections above, the observation21,22 that more extensively studied compounds may be consistently inactive even with a PAINS-recognized core is not surprising. In this context, recently identified alterations that confer benign behavior to an otherwise promiscuous PAINS core are of particular interest.22 Physicochemical properties such as hydrophilicity are not captured by simple substructures yet may contribute to amelioration of assay interference.16 Further work along these lines is to be encouraged because until we understand these rules, elaboration of a PAIN that may be a clean compound or one that is thought to simply represent signal interference is a risky venture if it cannot be predicted when PAINS behavior will arise for any given analogue.
An aspect of PAINS that is much harder to predict is the influence of commonly associated contaminants or decomposition products. In such cases, activity of a hit may vanish on purification. Purity checks as well as orthogonal resynthesis of hits should therefore be considered as part of a hit validation cascade. We have little understanding of the effect of such contaminants on the derivation of the original set of electronic filters, but it may have led to highlighting of classes for reasons that could be considered spurious for properly treated samples where this is not a problem. Contamination by heavy metals, in particular, is a significant issue in this context.23
The set of data originally used has been relatively small and has limited statistical significance of the results, and this was recognized by classifying the filters into three families. While PAINS substructures represented in the filters number some 480, a large number (58%) of compounds are encoded by just 16 substructures that represent highly exemplified chemistry (“Family_Filter_A”, each substructure with 150 or more analogues), and the significant remainder (27%) in just another 55 substructures (“Family_Filter_B”, each of the 55 substructures encoding for 15–149 analogues). The vast majority of filters (409, or 85%) is contained in Family_Filter_C, each with 1–14 analogues, only representing 15% of PAINS-recognized compounds and being very poorly populated. Therefore, discussion and analyses of the veracity of PAINS filters and the concept should focus on Family Filter A, to a varying extent Family Filter B, but arguably not Family Filter C, until such a time as the latter family of substructures is better understood. It cannot be said that a compound recognized by Family Filter C is necessarily a PAIN until more is understood about this set of filters.
Technology has thrown a spanner in the works as well when it comes to PAINS recognition. Users of PAINS filters translated from the original language (SLN) into more common languages such as SMARTS are cautioned that the two sets of filters (SMARTS compared with SLN) might return different results. This is exemplified in 10 and 11 in Figure 4, inappropriately identified as belonging to hzone_phenol_B and dyes5a PAINS classes, respectively (Duncan Beniston, ChemBridge Corporation, personal communication). We would recommend use of the FAF-Drugs3 implementation, but discrepancies may still arise.2
Figure 4.

SMARTS implementations of the original SLN PAINS filter may inappropriately identify non-PAINS, shown here for 10 and 11 inappropriately identified as belonging to hzone_phenol_B and dyes5a PAINS classes, respectively.
Finally, the distinction needs to be made between a PAINS chemotype and a privileged scaffold, as both may present data suggestive of promiscuity. For example, the 2-aminopyrimidine-based scaffold is a well-known kinase inhibition motif, and its screening data may reflect a bias toward being screened in kinase assays, for example, as a result of subset screening. The data can therefore be suggestive of promiscuous and nonprogressible behavior solely due to the higher incidence of activity from such targeted campaigns.16 Comparison across different target classes and different assay technologies would distinguish between the two cases. Other scaffolds are more generally privileged across different target classes.19 For these cases, analogue-specific target selectivity and potency would be a distinguishing feature from PAINS behavior, which is core-driven and generally most prevalent at micromolar concentrations.
PAINS Are Recognized by Other, Independently Developed Promiscuity Filters
PAINS that are recognized as interference compounds by other methods have been commented on extensively.1 Examples of alternative means of detection and annotation comprise the ALARM NMR assay, reported in 2005 by Abbott Laboratories,24 as well as promiscuity filters published by Bristol-Myer Squibb25 and Hofmann La-Roche.26 We felt this was an important observation because PAINS were defined by us based on a combination of medicinal chemistry recognition, first-hand experience, primary HTS data, and recognition of poorly performing compound classes in the literature.
The corroboration of PAINS classes by such independent efforts provides strong support for the structural filters and subsequent recognition and awareness of poorly performing compound classes in the literature. It is instructive therefore to introduce two more recent and fully statistically validated frequent-hitter analytical methods that are assay platform-independent. The first was reported in 2014 by AstraZeneca16 and the second in 2016 by academic researchers and called Badapple.27
A compound can be defined as a frequent hitter agnostic of substructure and setting if its activity is significantly higher than expected, and this is the approach that AstraZeneca takes in interrogation of their own extensive corporate database.16 It is possible therefore to take a PAINS core and undertake a substructure search and observe the cumulative promiscuity behavior of all compounds containing that substructure, which can be reported as an “incidence.” A random set of compounds from the collection has an expected incidence of 6.5% of apparently promiscuous compounds, and the more a substructure class deviates upward relative to this value, the more promiscuous it can be said to be. On the other hand, Badapple requires input of a compound of interest and then undertakes a hierarchical scaffold analysis and reports on the promiscuity score (pScore) of the different predefined scaffolds recognized in the compound of interest, where a pScore of 0–100 suggests no indication, 100–300 is a moderate score suggesting weak indication of promiscuity, and >300 is a high pScore with a strong suggestion of promiscuity.
In Tables 1 and 2, we have taken the 16 most highly populated PAINS substructures—those that represent at least 150 analogues in the original HTS library—as reported in the original publication that comprise the family A set of filters and analyzed them using the AstraZeneca database as recently described16 but updated to take into account minor improvements in accuracy for SLN to SMARTS conversion. Where a relevant scaffold is also encoded for in Badapple, we have included a pScore as well. In Table 1 are listed the 13 PAINS substructures that are convincingly problematic when assessed by these other, independent approaches, while in Table 2 are listed the three PAINS substructures where this is not the case.
Table 1. Some of the Most Common PAINS Generally Recognized by Other Measures of Promiscuitya.
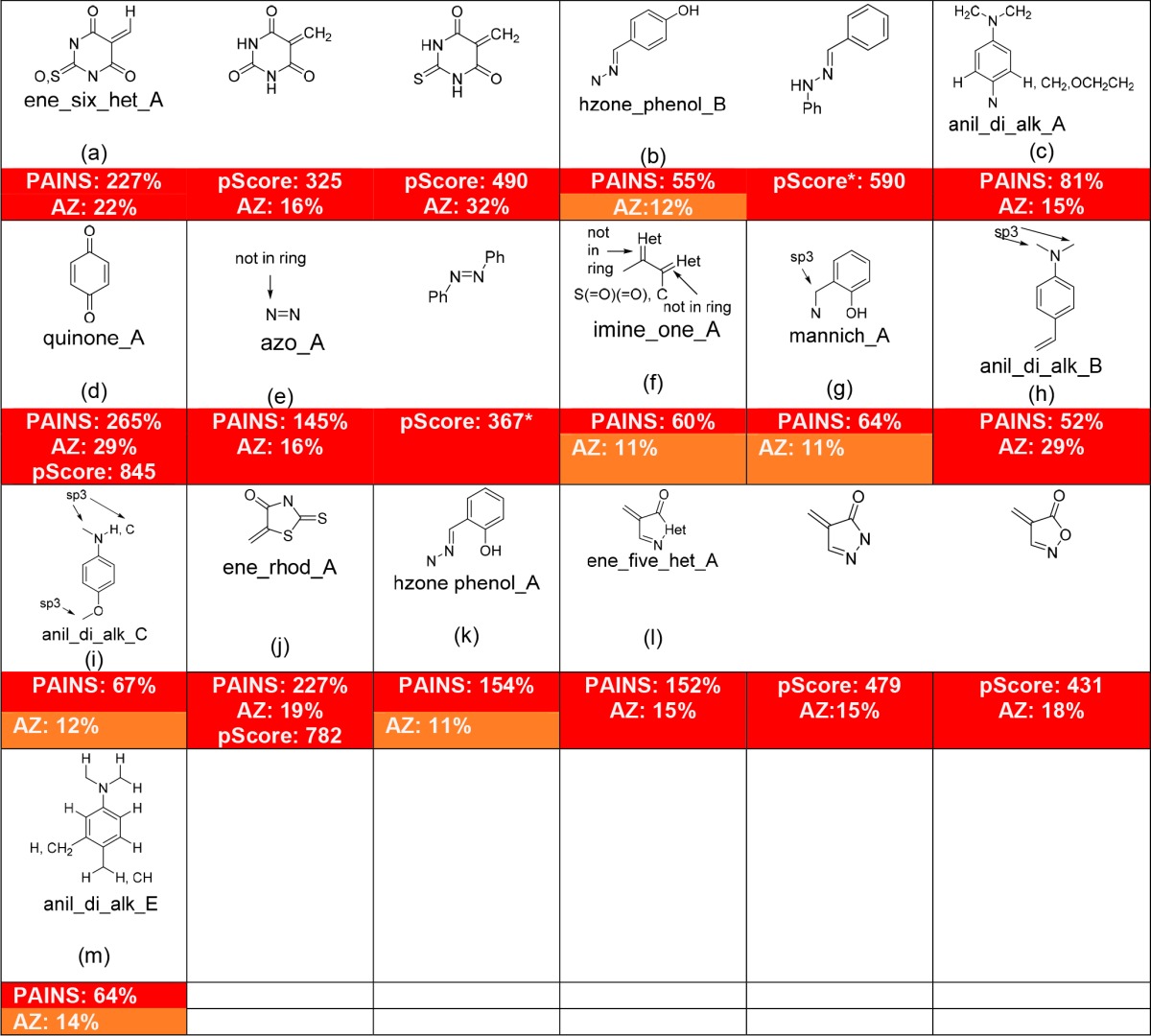
These are in order according to the original Family_Filter_A. PAINS are characterized by an enrichment factor, defined as the number of analogues of a given class that registered as active in between 2 and 6 of the 6 HTS campaigns analyzed, expressed as a percentage of the number of analogues of that class that did not register as active in any of the 6 HTS campaigns. The AstraZeneca approach (AZ incidence) interrogates the AZ corporate database and reports the incidence of bioactivity of any compound relative to that expected from a random selection (6.5%). We have arbitrarily selected <10%, <15%, and ≥15% as the criteria for color coding green (benign), orange (raised), red (promiscuous). Badapple requires input of a compound of interest and then undertakes a hierarchical scaffold analysis and reports on the promiscuity score (pScore) of the different scaffolds that make up the compound of interest, where a pScore of 0–100 suggests no indication (green), 100–300 is a moderate score suggesting weak indication of promiscuity (orange), and >300 is a high pScore with a strong suggestion of promiscuity (red). Because Badapple is scaffold-centric, some substructures where substituents are part of the definition cannot be sensibly analyzed (no pScore shown) or are incompletely analyzed (pScore*).
Table 2. Substructures Defined in PAINS Filters Not Generally Recognized As Promiscuous by Other Measuresa.

Immediately obvious are the most problematic and readily identified PAINS such as alkylidene barbiturates (a), rhodanines (j), and related heterocycles (l), as well as quinones (d). A previous discussion explains1 the reasons why quinones and alkylidene Michael acceptors are PAINS. Some other less discussed classes merit some further comment based on the results shown in these tables.
Hydroxyphenylhydrazones (b and k) also exhibit PAINS-like behavior1 but are observed by AstraZeneca to be moderately promiscuous, as are phenolic Mannich bases (g). This may be a reflection of structural bias toward “better” compounds in the collection. Badapple recognizes the broader N′-phenyl–phenylhydrazone scaffold as strongly promiscuous, with a pScore of 590, but cannot provide a finer detailed breakdown for more accurate comparison because it only identifies scaffolds and does not consider substituent effects. This is a limitation in this method.
We have found azo containing compounds (e) to be frequent hitters and have wondered whether singlet oxygen conferred an AlphaScreen-specific interference component.1 However, AstraZeneca analysis also concurs that the azo group is promiscuous, with an incidence of 16%. Badapple requires elaboration to an azobenzene scaffold before recognition is possible but does identify this moiety as highly promiscuous with a pScore of 367. Azo groups are commonly colored and their photoreactivity exploited in their use as photochemical switches, but these attributes do not necessarily wholly account for this apparent promiscuity. We have previously discussed the drug Eltrombopag’s origins as an azo-containing screening hit,8 and clarification of the nature of azo group PAINS mechanism(s) would be a useful future endeavor.
It is observed that an aniline-based PAINS substructure like b in Table 2 is assessed by AstraZeneca to be entirely benign. This observation is in keeping with our previous discussion1 that such compounds could selectively interfere with AlphaScreen signaling through reaction with singlet oxygen. It is therefore particularly interesting that the aniline-based PAINS substructure shown as c and h and to a lesser extent i and m in Table 1 are determined by AstraZeneca analysis to be frequent hitters. In their excellent analysis of Eli Lilly HTS data that discussed issues ranging from contaminant activity to apparent promiscuity in privileged scaffolds, Bruns and Watson also confirmed that aniline-based structure h was highly enriched in sets of screening hits.28 This suggests that among the aniline-based PAINS, while some may be AlphaScreen interference compounds, others are more generally promiscuous, the nature of which remains to be determined. Badapple cannot be used for analysis of any of the aniline-based queries because of the substituent-inclusive nature of the substructure definitions. A key observation remains that even in sets of very promiscuous substructures, significant numbers of specific compounds are contained therein that do not themselves give rise to an anomalous hit rate in HTS screens.
It is for all the above reasons that in our Table of Contents graphic, we have deliberately selected structures to most strongly exemplify our message. However, it is important to note that this is not a quantitative representation of the performance of PAINS filters, the veracity of which, at least for the major FAMILY_FILTER_A, is underscored by the data presented in Table 1.
Finally, for the reasons discussed earlier, a word of caution needs to be reiterated when comparing the results of SMARTS promiscuity filters applied to screening data derived from a compound library such as AstraZeneca’s that may differ in unknown ways to the compound library from which those promiscuity filters originated, such as the WEHI library and associated screening data that gave rise to the SLN PAINS filters.
Future Directions
Experienced medicinal chemists will generally agree that a screening hit should not be assumed to be useful unless supported by strong structure–activity relationships accompanied by hit-to-lead optimization. Too many high profile publications continue to focus on excessive downstream interrogation of unoptimized hits unsupported by SAR. Experienced medicinal chemists recognize that screening hits readily give readouts in biochemical and cell-based assays and that this does not mean such compounds are progressible or that cell-based activity can be assumed to be linked to the biochemical activity. A compound that belongs to a class with a history of promiscuity enabled via various mechanisms is a high risk option to select for optimization.
It is perhaps because this issue is so readily understood by medicinal chemists that approximately 2 years ago the Journal of Medicinal Chemistry implemented requirements of authors to provide more than the usual amount of data for manuscripts containing compounds recognized by SMARTS-based PAINS filters. The filters are advantageous for researchers less experienced in medicinal chemistry as it diminishes the need for structure recognition experience. Earlier this year, we witnessed a significant and welcomed expansion in ACS editorial policy, published simultaneously in nine ACS journals, calling on researchers to be more aware of the problem of PAINS as well as aggregators.29
In this Review, we emphasize the point that PAINS filters can be used to identify compounds that have a higher likelihood than others of being anomalous screening actives, but that the extent to which one views a compound as nonprogressible is very much dependent on the PAINS substructure it belongs to, and an understanding of how that chemotype may be promiscuous, if indeed there is any understanding at play. It is clear that some PAINS chemotypes are far more promiscuous–and hence suspect—than others.
If accompanied by appropriate evidence and wording, publication of PAINS may be appropriate and summary rejection of such efforts entirely inappropriate. A recent and extreme example of how even a quinone-based screening hit, which may be regarded as among the most troublesome of all PAINS chemotypes, can be a viable starting point for optimization, is a case in point. Here, researchers took a quinoid HTS hit (12) against poly(ADP-ribose) glycohydrolase and transformed it into a potent, selective, nonquinoid, advanced compound (13, Figure 5).30 Circumstances were unusual: it was the only hit from screening 1 400 000 compounds and therefore allowed for more focused attention, which revealed plausible on-target cell-based activity. Whether screening data suggested it was a benign subclass of this type of PAIN was not reported, but it is noteworthy that 86 out of 370 quinones in our own original study were clean. SPR and structural biology demonstrated credible and stoichiometric binding, leading to in silico scaffold hopping and clear early SAR. Above all, there was acute awareness that this core was a PAIN, and this was factored in for all early data interpretation, leading to clarity in compound progression criteria (A. Jordan, University of Manchester, personal communication). Publication of the early work, if based just around the quinoid-type screening hit, would not have been acceptable given the troublesome history of this compound class, but becomes so after such demonstration of SAR and optimization with successful scaffold hopping. It remains to be determined whether this team has inadvertently discovered a quinoid subtype with specific structural features that render it inert. The key is to remain evidence-based, and in our experience some medicinal chemists are inappropriately regarding non-PAINS as PAINS on the basis of molecular similarity alone.
Figure 5.

An example of successful scaffold hop during optimization of a PAIN to a non-PAIN chemotype.
Another comment we frequently encounter and very relevant to this journal is that PAINS may not be appropriate for drug development but may still comprise useful tool compounds. This is not so, as tool compounds need to be much more pharmacologically precise in order that the biological responses they invoke can be unambiguously interpreted.4
Given our discussion documented herein, the development of an optimized set of PAINS filters in the universally used SMARTS format and derived from a universally accessible set of data, such as PubChem,31 and with input from corporate sources, is clearly an attractive proposition, and we will report on our efforts here in due course. Further, the moderated ACS editorial stance most recently adopted for PAINS and related compounds would seem to be entirely appropriate.29 Indeed, one might suggest that journal judgment be best focused on the more universally recognized PAINS cores than simply all those encoded for by the electronic filters.
In summary, we have previously discussed a variety of issues key to interpretation of PAINS filter outputs, ranging from HTS library design and screening concentration, relevance of PAINS-bearing FDA-approved drugs, issues in SMARTS to SLN conversion, the reality of nonfrequent hitter PAINS, as well as PAINS and non-PAINS that are respectively not recognized or recognized in the PAINS filters as originally published. However, nowhere has a discussion around these key principles been summarized in one article, and that is the point of the current article. Had this been the case, we believe some recent contributions to the literature would have been more thoughtfully directed.21,32
Affirmation of PAINS promiscuity using independent methods developed by AstraZeneca or as scripted within BadApple is compelling verification of PAINS filter utility, but the structure-based positive identification of a PAIN can never be regarded as a black-and-white issue. Authors whose chemotypes are questioned by colleagues, reviewers, or editors should be conversant with the nuances discussed in this article. This knowledge will allow for formulation of appropriate and convincing counterarguments in support of their work, or prevent investigators being led astray.
Acknowledgments
The National Health and Medical Research Council of Australia (NHMRC) is thanked for Fellowship support for J.B. (2012–2016 Senior Research Fellowship #1020411, 2017–Principal Research Fellowship #1117602). Acknowledged is Australian Federal Government Education Investment Fund Super Science Initiative and the Victorian State Government, Victoria Science Agenda Investment Fund for infrastructure support and the facilities and the scientific and technical assistance of the Australian Translational Medicinal Chemistry Facility (ATMCF), Monash Institute of Pharmaceutical Sciences (MIPS). ATMCF is supported by Therapeutic Innovation Australia (TIA). TIA is supported by the Australian Government through the National Collaborative Research Infrastructure Strategy (NCRIS) program. We also thank J. Dahlin for useful input to Figure 2.
The authors declare no competing financial interest.
References
- Baell J. B.; Holloway G. A. (2010) New Substructure Filters for Removal of Pan Assay Interference Compounds (PAINS) from Screening Libraries and for Their Exclusion in Bioassays. J. Med. Chem. 53, 2719–2740. 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- Lagorce D.; Sperandio O.; Baell J. B.; Miteva M. A.; Villoutreix B. O. (2015) FAF-Drugs3: a web server for compound property calculation and chemical library design. Nucleic Acids Res. 43, W200–207. 10.1093/nar/gkv353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saubern S.; Guha R.; Baell J. B. (2011) KNIME Workflow to Assess PAINS Filters in SMARTS Format. Comparison of RDKit and Indigo Cheminformatics Libraries. Mol. Inf. 30, 847–850. 10.1002/minf.201100076. [DOI] [PubMed] [Google Scholar]
- Arrowsmith C. H.; Audia J. E.; Austin C.; Baell J.; Bennett J.; Blagg J.; Bountra C.; Brennan P. E.; Brown P. J.; Bunnage M. E.; Buser-Doepner C.; Campbell R. M.; Carter A. J.; Cohen P.; Copeland R. A.; Cravatt B.; Dahlin J. L.; Dhanak D.; Edwards A. M.; Frederiksen M.; Frye S. V.; Gray N.; Grimshaw C. E.; Hepworth D.; Howe T.; Huber K. V.; Jin J.; Knapp S.; Kotz J. D.; Kruger R. G.; Lowe D.; Mader M. M.; Marsden B.; Mueller-Fahrnow A.; Muller S.; O’Hagan R. C.; Overington J. P.; Owen D. R.; Rosenberg S. H.; Roth B.; Ross R.; Schapira M.; Schreiber S. L.; Shoichet B.; Sundstrom M.; Superti-Furga G.; Taunton J.; Toledo-Sherman L.; Walpole C.; Walters M. A.; Willson T. M.; Workman P.; Young R. N.; Zuercher W. J. (2015) The promise and peril of chemical probes. Nat. Chem. Biol. 11, 536–541. 10.1038/nchembio.1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baell J.; Walters M. A. (2014) Chemistry: Chemical con artists foil drug discovery. Nature 513, 481–483. 10.1038/513481a. [DOI] [PubMed] [Google Scholar]
- Baell J. B. (2010) Observations on screening-based research and some concerning trends in the literature. Future Med. Chem. 2, 1529–1546. 10.4155/fmc.10.237. [DOI] [PubMed] [Google Scholar]
- Baell J. B. (2011) Redox-active nuisance screening compounds and their classification. Drug Discovery Today 16, 840–841. 10.1016/j.drudis.2011.06.011. [DOI] [Google Scholar]
- Baell J. B. (2015) Screening-based translation of public research encounters painful problems. ACS Med. Chem. Lett. 6, 229–234. 10.1021/acsmedchemlett.5b00032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baell J. B. (2013) Broad coverage of commercially available lead-like screening space with fewer than 350,000 compounds. J. Chem. Inf. Model. 53, 39–55. 10.1021/ci300461a. [DOI] [PubMed] [Google Scholar]
- Baell J. B. (2016) Feeling Nature’s PAINS: Natural Products, Natural Product Drugs, and Pan Assay Interference Compounds (PAINS). J. Nat. Prod. 79, 616–628. 10.1021/acs.jnatprod.5b00947. [DOI] [PubMed] [Google Scholar]
- Dahlin J. L., Baell J., and Walters M. A. (2004) Assay Interference by Chemical Reactivity, in Assay Guidance Manual (Sittampalam G. S., Coussens N. P., Nelson H., Arkin M., Auld D., Austin C., Bejcek B., Glicksman M., Inglese J., Iversen P. W., Li Z., McGee J., McManus O., Minor L., Napper A., Peltier J. M., Riss T., Trask O. J. Jr., and Weidner J., Eds.), Eli Lilly & Company and the National Center for Advancing Translational Sciences, Bethesda, MD. [PubMed] [Google Scholar]
- Devine S. M.; Mulcair M. D.; Debono C. O.; Leung E. W.; Nissink J. W.; Lim S. S.; Chandrashekaran I. R.; Vazirani M.; Mohanty B.; Simpson J. S.; Baell J. B.; Scammells P. J.; Norton R. S.; Scanlon M. J. (2015) Promiscuous 2-aminothiazoles (PrATs): a frequent hitting scaffold. J. Med. Chem. 58, 1205–1214. 10.1021/jm501402x. [DOI] [PubMed] [Google Scholar]
- Falk H.; Connor T.; Yang H.; Loft K. J.; Alcindor J. L.; Nikolakopoulos G.; Surjadi R. N.; Bentley J. D.; Hattarki M. K.; Dolezal O.; Murphy J. M.; Monahan B. J.; Peat T. S.; Thomas T.; Baell J. B.; Parisot J. P.; Street I. P. (2011) An Efficient High-Throughput Screening Method for MYST Family Acetyltransferases, a New Class of Epigenetic Drug Targets. J. Biomol. Screening 16, 1196–1205. 10.1177/1087057111421631. [DOI] [PubMed] [Google Scholar]
- Lackovic K.; Lessene G.; Falk H.; Leuchowius K. J.; Baell J.; Street I. (2014) A perspective on 10-years HTS experience at the Walter and Eliza Hall Institute of Medical Research - eighteen million assays and counting. Comb. Chem. High Throughput Screening 17, 241–252. 10.2174/1386207317666140109122450. [DOI] [PubMed] [Google Scholar]
- Baell J. B.; Ferrins L.; Falk H.; Nikolakopoulos G. (2013) PAINS: Relevance to Tool Compound Discovery and Fragment-Based Screening. Aust. J. Chem. 66, 1483–1494. 10.1071/CH13551. [DOI] [Google Scholar]
- Nissink J. W. M.; Blackburn S. (2014) Quantification of frequent-hitter behavior based on historical high-throughput screening data. Future Med. Chem. 6, 1113–1126. 10.4155/fmc.14.72. [DOI] [PubMed] [Google Scholar]
- Hanley R. P.; Horvath S.; An J.; Hof F.; Wulff J. E. (2016) Salicylates are interference compounds in TR-FRET assays. Bioorg. Med. Chem. Lett. 26, 973–977. 10.1016/j.bmcl.2015.12.050. [DOI] [PubMed] [Google Scholar]
- Coan K. E.; Shoichet B. K. (2008) Stoichiometry and physical chemistry of promiscuous aggregate-based inhibitors. J. Am. Chem. Soc. 130, 9606–9612. 10.1021/ja802977h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider P.; Schneider G. (2017) Privileged Structures Revisited. Angew. Chem., Int. Ed. 56, 7971–7974. 10.1002/anie.201702816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlin J. L.; Nissink J. W. M.; Strasser J. M.; Francis S.; Higgins L.; Zhou H.; Zhang Z. G.; Walters M. A. (2015) PAINS in the Assay: Chemical Mechanisms of Assay Interference and Promiscuous Enzymatic Inhibition Observed during a Sulfhydryl-Scavenging HTS. J. Med. Chem. 58, 2091–2113. 10.1021/jm5019093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capuzzi S. J.; Muratov E. N.; Tropsha A. (2017) Phantom PAINS: Problems with the Utility of Alerts for Pan-Assay INterference CompoundS. J. Chem. Inf. Model. 57, 417–427. 10.1021/acs.jcim.6b00465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jasial S.; Hu Y.; Bajorath J. (2017) How Frequently Are Pan-Assay Interference Compounds Active? Large-Scale Analysis of Screening Data Reveals Diverse Activity Profiles, Low Global Hit Frequency, and Many Consistently Inactive Compounds. J. Med. Chem. 60, 3879–3886. 10.1021/acs.jmedchem.7b00154. [DOI] [PubMed] [Google Scholar]
- Hermann J. C.; Chen Y.; Wartchow C.; Menke J.; Gao L.; Gleason S. K.; Haynes N. E.; Scott N.; Petersen A.; Gabriel S.; Vu B.; George K. M.; Narayanan A.; Li S. H.; Qian H.; Beatini N.; Niu L.; Gan Q. F. (2013) Metal impurities cause false positives in high-throughput screening campaigns. ACS Med. Chem. Lett. 4, 197–200. 10.1021/ml3003296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huth J. R.; Mendoza R.; Olejniczak E. T.; Johnson R. W.; Cothron D. A.; Liu Y. Y.; Lerner C. G.; Chen J.; Hajduk P. J. (2005) ALARM NMR: A rapid and robust experimental method to detect reactive false positives in biochemical screens. J. Am. Chem. Soc. 127, 217–224. 10.1021/ja0455547. [DOI] [PubMed] [Google Scholar]
- Pearce B. C.; Sofia M. J.; Good A. C.; Drexler D. M.; Stock D. A. (2006) An empirical process for the design of high-throughput screening deck filters. J. Chem. Inf. Model. 46, 1060–1068. 10.1021/ci050504m. [DOI] [PubMed] [Google Scholar]
- Roche O.; Schneider P.; Zuegge J.; Guba W.; Kansy M.; Alanine A.; Bleicher K.; Danel F.; Gutknecht E. M.; Rogers-Evans M.; Neidhart W.; Stalder H.; Dillon M.; Sjogren E.; Fotouhi N.; Gillespie P.; Goodnow R.; Harris W.; Jones P.; Taniguchi M.; Tsujii S.; von der Saal W.; Zimmermann G.; Schneider G. (2002) Development of a virtual screening method for identification of ″frequent hitters″ in compound libraries. J. Med. Chem. 45, 137–142. 10.1021/jm010934d. [DOI] [PubMed] [Google Scholar]
- Yang J. J.; Ursu O.; Lipinski C. A.; Sklar L. A.; Oprea T. I.; Bologa C. G. (2016) Badapple: promiscuity patterns from noisy evidence. J. Cheminf. 8, 29. 10.1186/s13321-016-0137-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruns R. F.; Watson R. A. (2012) Rules for identifying potentially reactive or promiscuous Compounds. J. Med. Chem. 55 (2012), 9763–9772. 10.1021/jm301008n. [DOI] [PubMed] [Google Scholar]
- Aldrich C.; Bertozzi C.; Georg G. I.; Kiessling L.; Lindsley C.; Liotta D.; Merz K. M. Jr.; Schepartz A.; Wang S. (2017) The Ecstasy and Agony of Assay Interference Compounds. ACS Med. Chem. Lett. 8, 379–382. 10.1021/acsmedchemlett.7b00056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James D. I.; Smith K. M.; Jordan A. M.; Fairweather E. E.; Griffiths L. A.; Hamilton N. S.; Hitchin J. R.; Hutton C. P.; Jones S.; Kelly P.; McGonagle A. E.; Small H.; Stowell A. I.; Tucker J.; Waddell I. D.; Waszkowycz B.; Ogilvie D. J. (2016) First-in-Class Chemical Probes against Poly(ADP-ribose) Glycohydrolase (PARG) Inhibit DNA Repair with Differential Pharmacology to Olaparib. ACS Chem. Biol. 11, 3179–3190. 10.1021/acschembio.6b00609. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Bryant S. H.; Cheng T.; Wang J.; Gindulyte A.; Shoemaker B. A.; Thiessen P. A.; He S.; Zhang J. (2017) PubChem BioAssay: 2017 update. Nucleic Acids Res. 45, D955–D963. 10.1093/nar/gkw1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenny P. W. (2017) Comment on ‘The Ecstasy and Agony of Assay Interference Compounds’. J. Chem. Inf. Model. 57, 2640. 10.1021/acs.jcim.7b00313. [DOI] [PubMed] [Google Scholar]