

Abstract
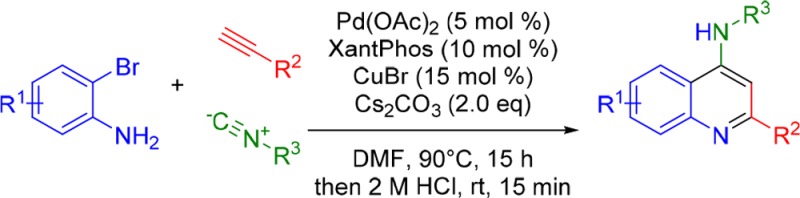
We developed a one-pot, two-stage synthetic route to substituted 4-aminoquinolines involving an imidoylative Sonogashira coupling followed by acid-mediated cyclization. This three-component reaction affords pharmaceutically valuable 4-aminoquinolines in a one-pot procedure from readily available starting materials. The reaction tolerates various substituents on the arene as well as the use of secondary and even primary isocyanides. Additionally, the wide tolerance for functionalized isocyanides allows for the one-pot synthesis of various substituted chloroquine analogues as well as other medicinally relevant products.
Introduction
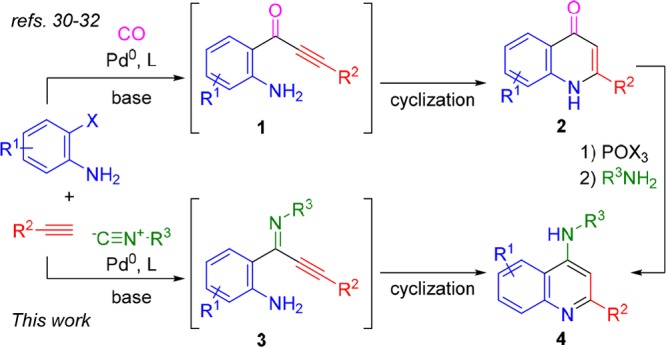
4-Aminoquinolines (4-AQs) have been widely investigated for their pharmaceutical potential, in particular, their antimalarial properties.1−5 Recently, B-ring-substituted (in particular, 7-halo) 4-AQs were shown to display enhanced activities against highly resistant strains of Plasmodium falciparum.6 Consequently, this scaffold is still of high interest for the development of novel antimalarials.7 Additionally, chloroquine and amodiaquine (Figure 1) have recently been flagged as leads against Ebola and Marburg viruses.8,9 These known antimalarials also inhibit endosomal Toll-like receptors (TLRs).10 Recently, other applications of 4-AQs have also been reported, as diversely substituted 4-AQs have been identified as promising antifilarials,11 translocator protein biomarker ligands,12 tuberculosis ATP synthase inhibitors,13 and as nociceptin receptor antagonists.14,15 Additionally, 4-AQs bearing 2-(hetero)aryl functionalities also show promise as non-nucleoside HIV-1 inhibitors.16 Not surprisingly, there is great interest to efficiently access the 4-AQ core, and over the past decades, several cascade reactions toward these medicinally valuable scaffolds have been developed (Scheme 1).17−22 For example, 4-AQs can be obtained from the reaction of ynamides and nitrilium species, resulting from either arylation of nitriles17 or dehydration of anilides.18 The Cu(I)-catalyzed cyclization of imidoylacetylenes and sulfonyl azides affords 4-AQs, as well.19 A carbonylative Sonogashira reaction of 2-ethynylanilines with in situ amination at C4 also provides access to 4-AQs.21 Finally, we recently showed that palladium-catalyzed oxidative isocyanide insertion of N-aryl imines by double C–H activation also affords 4-AQs.22 However, these methods rely on rather specialized starting materials17−20 and/or afford the desired 4-AQs in disappointing yields.18,22 Most diversity-oriented routes toward 4-AQs rely on the amination of 4- haloquinolines,23,24 in turn derived from the corresponding 4-quinolones. Such multisubstituted quinolones (2) are generally difficult to synthesize by traditional condensation methods.25−29 However, quinolones 2 are accessible via a carbonylative Sonogashira coupling (Scheme 2), using organic base to mediate the postcoupling cyclization (Scheme 2).30−32 To the best of our knowledge, only one example of isocyanide insertion in a Sonogashira coupling has been reported, where the product ynimines are hydrolyzed in situ to the corresponding ynones.33,34 Based on this, we envisioned a direct, one-pot synthesis of 4-AQs 4 via an imidoylative Sonogashira coupling and subsequent cycloaromatization (Scheme 2). Such an approach prevents the need for toxic CO gas and avoids the two-step derivatization of quinolones 2.
Figure 1.
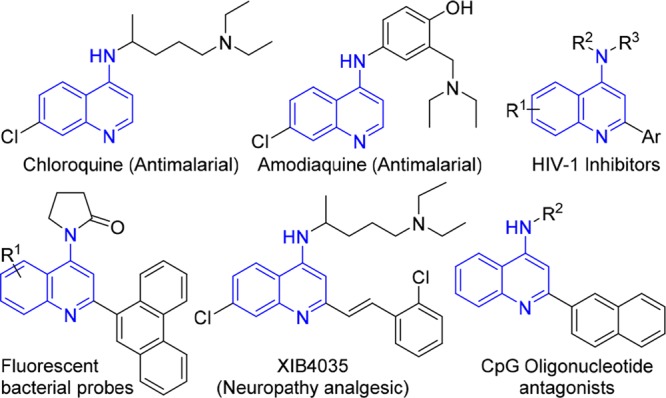
Examples of medicinally relevant 4-aminoquinolines.
Scheme 1. Previous Routes to 4-Aminoquinolines.

Scheme 2. Carbonylative and Imidoylative Sonogashira Coupling to 4-Aminoquinolines.
Results and Discussion
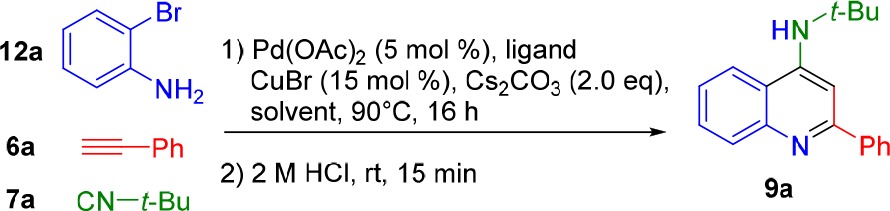
Our initial studies showed that such an imidoylative Sonogashira approach using 5, 6a, and 7a afforded a range of products as a difficult to analyze mixture under typical conditions (Scheme 3). Most notably, with temperatures under 50 °C or CuBr loadings approaching stoichiometric amounts, only the alkyne homocoupling product 10 was isolated. Additionally, SFC-MS analysis indicated that the use of o-iodoaniline promotes intramolecular Buchwald–Hartwig-type reactivity with double isocyanide insertion, resulting in the formation of isatin diimine 11 (Scheme 3). If o-bromoaniline is employed, formation of 11 is suppressed to trace amounts, which is known for the synthesis of these isatinimines.35 Surprisingly, these initial experiments revealed that the main product in all cases was the intermediate imidoylative Sonogashira product 8, with only trace amounts of the desired cyclization product 9. Direct formation of 4-AQs 9 from these previously underexplored ynimines 8 appears to be surprisingly difficult. Consequently, we focused our attention on the optimization of this cycloaromatization. Multiple cyclization conditions were investigated for the formation of aminoquinoline 9 from the proposed intermediate ynimine 8. Neither strong nor weak bases proved effective in facilitating this cyclization (Table 1, entries 1 and 2). Weak acids (SiO2, AcOH) were applied to activate the imine toward cyclization but only afforded cyclization product 9a in trace quantities (entries 3 and 4). However, stronger acids like TFA (pKa = 0.23), phenylphosphinic acid (pKa = 1.85), and methanesulfonic acid (pKa = −1.9) rapidly afforded 4-aminoquinoline 9a, even after short reaction times at ambient temperature (Table 1, entries 8–10). Interestingly, HCl also facilitated the cycloaromatization, even when employed as an aqueous solution. Curiously, acids with non-nucleophilic conjugate bases also afford aminoquinoline 9a in high to quantitative yield (Table 1, entries 8 and 10), implying either an intermolecular Michael/Michael/retro-Michael mechanism30−32 or a direct intramolecular Michael reaction. Regardless, postcoupling addition of aqueous hydrochloric acid was selected as the optimal cyclization condition. While the addition of 1 M aqueous HCl readily facilitated cycloaromatization of 8 to 9a, we opted for slightly harsher conditions (2 M aqueous HCl (4 mL), 15 min, rt) for the in situ cyclization following the imidoylative Sonogashira reaction to account for excess Cs2CO3 as well as any residual starting materials. With the optimized cyclization protocol in hand, we turned our attention to the optimization of the imidoylative Sonogashira coupling. Using standard conditions (5 mol % of Pd(PPh3)4, 15 mol % of CuBr, 2.0 equiv of Cs2CO3, DMSO, 90 °C, 16 h), we obtained the desired 4-AQ 9a in 61% yield (Table 1, entry 2). When the Cu(I)Br cocatalyst was omitted, only isatin diimine 11 was obtained (Table 2, entry 1). Subsequent solvent screening indicated that the transformation proceeds most efficiently in polar aprotic solvents, with DMF giving superior results (entries 2–5). As expected, a heterogeneous palladium catalyst (Pd/C) did not catalyze the reaction (entry 6). Switching from Pd(PPh3)4 to an in situ formed Pd0 complex allowed for a ligand screening (entries 7–10). Interestingly, in addition to leading to the highest 4-AQ yield, the use of Xantphos as a ligand completely inhibited the formation of isatin diimine 11 (Table 1, entry 10). Changing the base from Cs2CO3 to other inorganic or organic bases (KOtBu, K2CO3, Et3N, DBU) did not lead to an increase in yield (entries 11–14). Finally, as anticipated, 9a was not formed when either the palladium source or the ligand was omitted (entries 15 and 16).
Scheme 3. Initial Imidoylative Sonogashira Studies toward 4-AQs and Side Products.
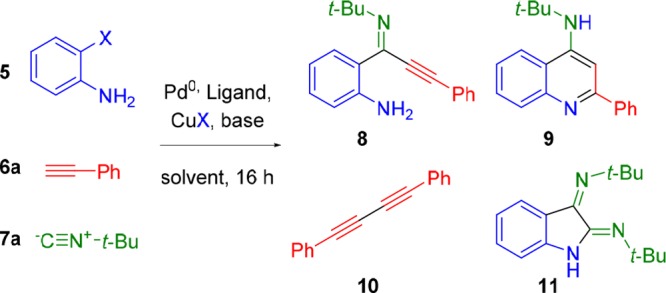
Table 1. Ynimine Cyclization Optimization.

| entrya | additive (equiv) | yield of 9 (%)b |
|---|---|---|
| 1 | NaOEt (2.0) | nr |
| 2 | HNEt3 (2.0) | nr |
| 3c | SiO2 | trace |
| 4 | AcOH (4.0) | trace |
| 5 | HCl in H2O (4.0) | 100 |
| 6 | HCl in MeOH (4.0) | 96 |
| 7 | HCl in dioxane (4.0) | 99 |
| 8 | TFA (4.0) | 80 |
| 9 | PhP(OH)2 (4.0) | 85 |
| 10 | MeSO3H (4.0) | 100 |
Conditions: ynimine 8 (0.55 mmol) in DMF (1 mL), additive, rt, 1 h.
Yield determined by NMR using an internal standard (1,3,5-trimethoxybenzene).
Using 0.150 g of SiO2; nr = no reaction, TFA = trifluoroacetic acid.
Table 2. Optimization of the Imidoylative Sonogashira/Cyclization Conditionsa.
| entry | Pd source | ligand | solvent | 9a (%)b |
|---|---|---|---|---|
| 1c | Pd(PPh3)4 | none | DMSO | nr |
| 2 | Pd(PPh3)4 | none | DMSO | 61 |
| 3 | Pd(PPh3)4 | none | toluene | 27 |
| 4 | Pd(PPh3)4 | none | dioxane | 13 |
| 5 | Pd(PPh3)4 | none | DMF | 70 |
| 6 | Pd/C | none | DMF | nr |
| 7 | Pd(OAc)2 | PPh3 | DMF | 82 |
| 8 | Pd(OAc)2 | Bu3P | DMF | nr |
| 9 | Pd(OAc)2 | DPEPhos | DMF | 82 |
| 10 | Pd(OAc)2 | XantPhos | DMF | 91 |
| 11d | Pd(OAc)2 | XantPhos | DMF | nr |
| 12e | Pd(OAc)2 | XantPhos | DMF | 56 |
| 13f | Pd(OAc)2 | XantPhos | DMF | nr |
| 14g | Pd(OAc)2 | XantPhos | DMF | 31 |
| 15 | Pd(OAc)2 | none | DMF | nr |
| 16 | none | XantPhos | DMF | nr |
Reaction conditions: 2-bromoaniline (12a, 0.5 mmol, 1 equiv), phenylacetylene (6a, 1 mmol, 2 equiv), tert-butyl isocyanide (7a, 0.625 mmol, 1.25 equiv), catalyst (5 mol %), ligand (monodentate: 15 mol %, bidentate: 10 mol %), CuBr (15 mol %), and Cs2CO3 (1 mmol, 2 equiv) in solvent (3.0 mL) were stirred at 100 °C for 16–20 h under N2 atmosphere.
Yields determined by 1H NMR analysis using 2,5-dimethylfuran as internal standard.
Reaction performed in the absence of CuBr.
KOtBu (2.0 equiv) employed as base.
K2CO3 (2.0 equiv) employed as base.
Et3N (2.0 equiv) employed as base.
DBU (2.0 equiv) employed as base.
With the optimized reaction conditions in hand, we set out to explore the compatibility of diversely substituted o-bromoanilines and terminal alkynes with our three-component reaction (Scheme 4). The reaction proved compatible with neutral and moderately electron-donating substituents (9a,b). The use of halo-substituted o-bromoanilines did not lead to lower yields (9c–g). Decoration of the o-bromoaniline or the arylalkyne with electron-donating functionalities afforded the substituted 4-AQs in high yields (9h–k). However, o-bromoanilines bearing electron-withdrawing substituents displayed lower compatibility with the three-component cascade protocol. When 3-bromo-4-aminobenzoate was used, the corresponding aminoquinoline 9l was isolated in lower yield. Similar results were observed for 9m, where the introduction of an additional distal electron acceptor on the opposite side of the ynimine renders the alkyne susceptible to 5-exo-dig cyclization. This cyclization to an aza-aurone side product most likely accounts for the diminished yields, as it does not require external acid activation and can therefore take place during the reaction. Additionally, it is possible that withdrawing electron density through substituent effects renders ynimines 8 more susceptible to hydrolysis during the cyclization stage. Gratifyingly, the developed conditions proved to tolerate aliphatic alkynes, as well, as demonstrated by the isolation of 9n and 9o, with somewhat lower yield for 9n as a result of the volatility of 1-hexyne.
Scheme 4. Alkyne and o-Bromoaniline Variation.
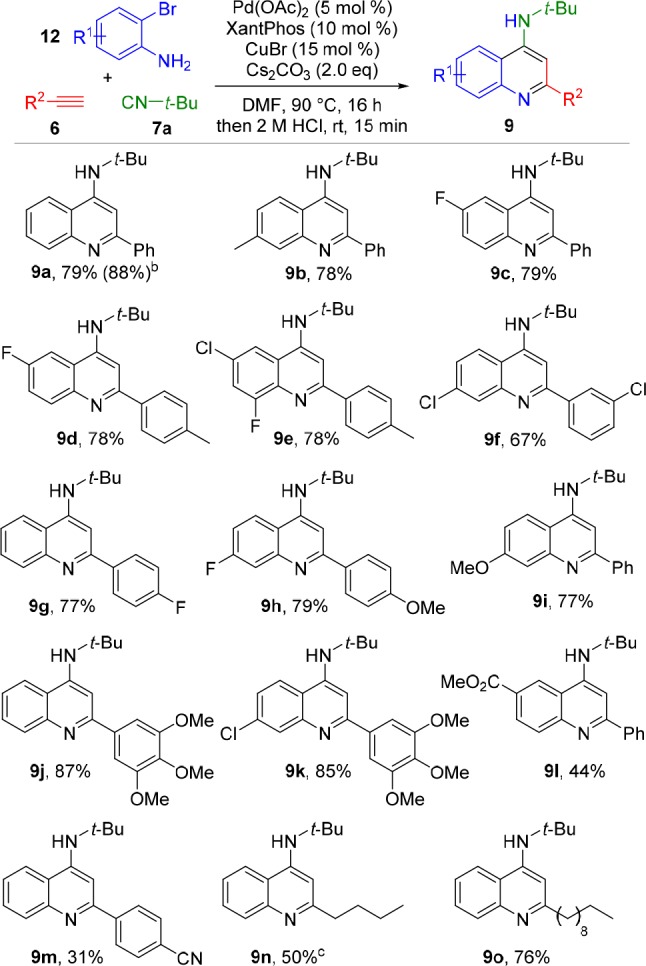
Conditions: o-bromoaniline (12, 1.0 mmol), tert-butyl isocyanide (7a, 1.25 mmol), alkyne (6, 2.0 mmol), Pd(OAc)2 (0.05 mmol), Xantphos (0.1 mmol), CuBr (0.15 mmol), Cs2CO3 (2.0 mmol) in DMF (8.0 mL).
Reaction performed on 10.0 mmol scale.
Closed vial, 5.0 mmol of 1-hexyne.
Interestingly, this palladium-catalyzed imidoylative Sonogashira coupling displayed high tolerance with regard to the isocyanide input (Scheme 5). Secondary isocyanides, which are known to undergo double insertion more readily under palladium catalysis,36,37 proved fully compatible with this protocol (9p–r). Satisfyingly, even primary isocyanide insertion afforded aminoquinoline 9s in acceptable yield. The use of bromoanilines and arylalkynes bearing electron-withdrawing substituents afforded the corresponding products in lower yields, in accordance with earlier results (9t,u). Unfortunately, the use of 4-methoxyphenyl isocyanide did not afford isolable quantities of the corresponding 4-arylaminoquinoline 9v. We then investigated the utility of our method in the synthesis of pharmaceutically relevant 4-AQs. The use of readily accessible N,N-diethyl-4-isocyanopentan-1-amine provided access to a variety of chloroquine analogues (9w–9y) in high yields. Similarly, the use of the isocyanide derived from the known vasoconstrictor octodrine38 afforded 9aa in good yield. Analogues of this highly fluorescent 2-naphthyl-4-AQ have been identified as potent immunostimulatory CpG-oligonucleotide antagonists.39,40 Furthermore, the use of 2-bromo-5-fluoroaniline, combined with N,N-diethyl-4-isocyanopentan-1-amine and cyclohexylacetylene, furnishes 4-amino-2-cyclohexyl-7-fluoroquinoline (9z). Structures of this type have been described as promising antileishmanials.41 Finally, ethyl 4-isocyanopiperidine-1-carboxylate can be used to construct 9ab in 68% yield, providing a handle for further functionalization of these medicinally valuable scaffolds.
Scheme 5. Isocyanide Variation.
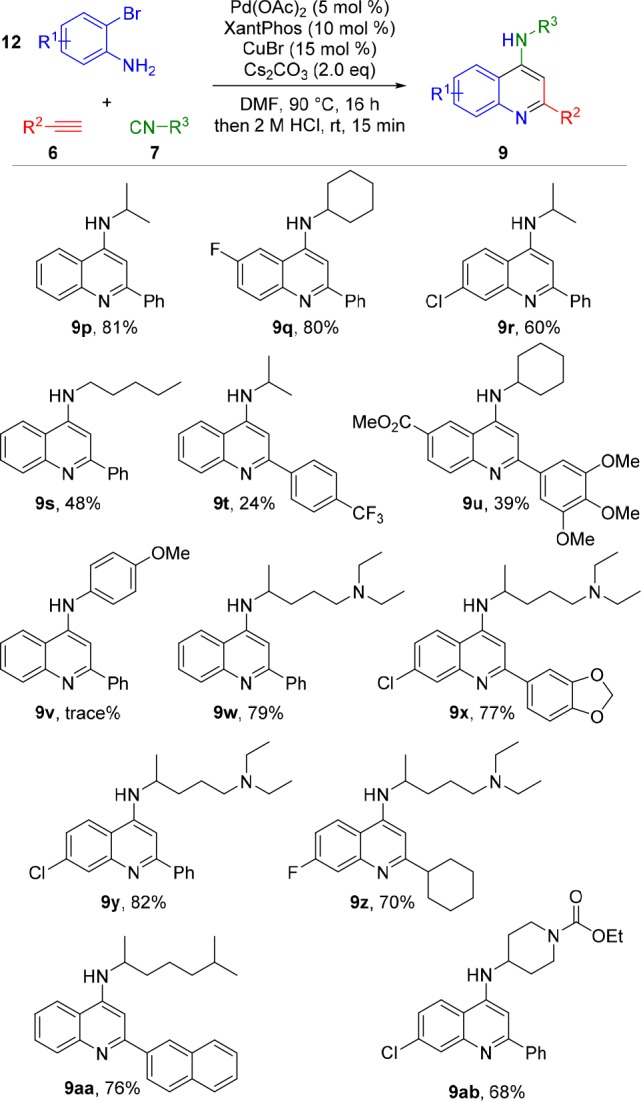
Conditions: o-bromoaniline (1.0 mmol), isocyanide (1.75 mmol), alkyne (2.0 mmol), Pd(OAc)2 (0.05 mmol), Xantphos (0.1 mmol), CuBr (0.15 mmol), Cs2CO3 (2.0 mmol) in DMF (8.0 mL).
In conclusion, we have developed a novel one-pot imidoylative Sonogashira cross-coupling/acid-mediated cyclization toward 4-aminoquinolines. The devised methodology is highly compatible with electronically diverse o-bromoanilines, arylalkynes, and aliphatic alkynes, although electron-withdrawing substituents on either arene lead to lower yields. The transformation is compatible with tertiary, secondary, and even primary aliphatic isocyanides. Additionally, using functionalized alkyl isocyanides, this method can be used to directly synthesize 2-substituted 4-aminoquinolines of high medicinal relevance.
Experimental Section
General Information
Chemicals were purchased from Sigma-Aldrich or Fluorochem and were used without purification. Solvents were purchased from VWR Chemicals (CH2Cl2) or Sigma-Aldrich (toluene, dioxane, DMSO) and used without purification, unless stated otherwise. Dry solvents were dried over an inert PS-MD-5 solvent purification system, equipped with an activated alumina/copper wire column. 1H NMR measurements were acquired on a Bruker Avance 300 (300.13 MHz) or Bruker Avance 500 (500.23 MHz) spectrometer. 13C NMR measurements were acquired on a Bruker Avance 500 (125.78 MHz) spectrometer. Chemical shifts are reported in parts per million downfield of tetramethylsilane and are corrected according to solvent. Mass analysis was performed using a Bruker MicrOTOF-Q instrument on a positive ion polarity mode for ESI (electrospray ionization). Capillary charge: 4000 V. Melting points were measured using a Büchi M-565 melting point apparatus. SiO2 column chromatography was performed using Merck silica gel C60 (particle size 40–60 μm). TLC chromatography was performed on Merck silica gel C60 F254 plates (silica coat on aluminum support). All isolated yields are corrected for present impurities (if present).
General Procedure 1 for the Synthesis of 4-tert-Butylaminoquinolines from Bromoanilines, Alkynes, and tert-Butyl Isocyanide
A solution of Pd(OAc)2 (0.011 g, 0.05 mmol) and Xantphos (0.058 g, 0.10 mmol) in dry DMF (8 mL) was stirred for 20 min at rt under N2 atmosphere, forming a yellow suspension. To this mixture were added consecutively Cs2CO3 (0.651 g, 2.0 mmol), o-bromoaniline (1.0 mmol), tert-butyl isocyanide (0.141 mL, 1.25 mmol), CuBr (0.021 g, 0.15 mmol), and alkyne (2.0 mmol). The resulting mixture was stirred at 90 °C for 16 h. Hereafter, the reaction was allowed to cool to rt, after which 4 mL of 2 M HCl was added and stirred for 15 min. The mixture was diluted with CH2Cl2, filtered over a pad of diatomaceous earth, and washed with 1 M HCl and saturated NaHCO3, before being dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. Purification was performed by column chromatography (gradient, cHex/EtOAc/Et3N 19:1:0.01–1:1:0.01, unless stated otherwise) to afford the pure 4-aminoquinoline derivative.
General Procedure 2 for the Synthesis of 4-Aminoquinolines from Bromoanilines, Alkynes, and Isocyanides
A solution of Pd(OAc)2 (0.011 g, 0.05 mmol) and Xantphos (0.058 g, 0.10 mmol) in dry DMF (8 mL) was stirred for 20 min at rt under N2 atmosphere, forming a yellow suspension. To this mixture were added consecutively Cs2CO3 (0.651 g, 2.0 mmol), o-bromoaniline (1.0 mmol), isocyanide (1.75 mmol), CuBr (0.021 g, 0.15 mmol), and alkyne (2.0 mmol). The resulting mixture was stirred at 90 °C for 16 h. Hereafter, the reaction was allowed to cool to rt, after which 4 mL of 2 M HCl was added and stirred for 15 min. The mixture was diluted with CH2Cl2, filtered over a pad of diatomaceous earth, and washed with 1 M HCl and saturated NaHCO3, before being dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. Purification was performed by column chromatography (gradient, cHex/EtOAc/Et3N 19:1:0.01–1:1:0.01, unless stated otherwise) to afford the pure 4-aminoquinoline derivative.
N-(tert-Butyl)-2-phenylquinolin-4-amine (9a)
Synthesized in accordance with general procedure 1. Isolated as a yellow solid (0.234 g, 79%): Rf = 0.19 (cHex/EtOAc/Et3N 4:1:0.05); mp 139 °C; 1H NMR (500.23 MHz, CDCl3) δ 8.10–8.03 (m, 3H), 7.68 (dd, J = 1.5, 8.5 Hz, 1H), 7.63 (ddd, J = 1.5, 7.0, 8.5 Hz, 1H), 7.51 (t, J = 7.5 Hz, 2H), 7.47–7.38 (m, 2H), 7.12 (s, 1H), 5.03 (bs, 1H), 1.60 (s, 9H); 13C NMR (125.97 MHz, CDCl3) δ 157.9, 148.8, 148.3, 141.2, 130.5, 129.0, 128.8, 128.6, 127.5, 124.3, 118.9, 118.5, 99.1, 51.5, 29.4; HRMS (ESI) m/z calcd for C19H21N2+ [M + H]+ 277.1695, found 277.1707.
N-(tert-Butyl)-7-methyl-2-phenylquinolin-4-amine (9b)
Synthesized in accordance with general procedure 1. Isolated as a yellow solid (0.237 g, 78%): Rf = 0.33 (cHex/EtOAc/Et3N 4:1:0.05); mp 134 °C; 1H NMR (500.23 MHz, CDCl3) δ 8.05 (dd, J = 1.5, 7.5 Hz, 2H), 7.85 (s, 1H), 7.57 (d, J = 8.5 Hz, 1H), 7.50 (t, J = 7.5 Hz, 2H), 7.47–7.40 (m, 1H), 7.23 (dd, J = 7.5, 8.5 Hz, 1H), 7.07 (d, J = 1.5 Hz, 1H), 4.99 (bs, 1H), 2.52 (s, 3H), 1.59 (s, 9H); 13C NMR (125.97 MHz, CDCl3) δ 157.9, 149.0, 148.3, 141.3, 139.0, 129.7, 128.7, 128.6, 127.5, 126.4, 118.6, 116.4, 98.7, 51.4, 29.4, 21.5; HRMS (ESI) m/z calcd for C20H23N2+ [M + H]+ 291.1851, found 291.1854.
N-(tert-Butyl)-6-fluoro-2-phenylquinolin-4-amine (9c)
Synthesized in accordance with general procedure 1. Isolated as a light brown solid (0.232 g, 79%): Rf = 0.44 (cHex/EtOAc/Et3N 4:1:0.05); mp 163 °C; 1H NMR (500.23 MHz, CDCl3) δ 8.14–8.06 (m, 3H), 7.54 (t, J = 7.5 Hz, 2H), 7.50–7.44 (m, 1H), 7.48–7.37 (m, 1H), 7.36 (dd, J = 2.5, 10.0 Hz, 1H), 7.17 (s, 1H), 4.79 (bs, 1H), 1.57 (s, 9H); 13C NMR (125.97 MHz, CDCl3) δ 159.5 (d, J = 245.0 Hz), 157.3 (d, J = 2.5 Hz), 147.9 (d, J = 4.5 Hz), 145.8, 140.9, 132.8 (d, J = 8.5 Hz), 128.8, 128.6, 127.4, 118.9 (d, J = 8.0 Hz), 118.6 (d, J = 24.5 Hz), 103.3 (d, J = 23.0 Hz), 99.4, 51.6, 29.3; HRMS (ESI) m/z calcd for C19H20N2F+ [M + H]+ 295.1601, found 295.1604.
N-(tert-Butyl)-6-fluoro-2-(p-tolyl)quinolin-4-amine (9d)
Synthesized in accordance with general procedure 1. Isolated as a light brown solid (0.241 g, 78%): Rf = 0.43 (cHex/EtOAc/Et3N 4:1:0.05); mp 155 °C; 1H NMR (500.23 MHz, CDCl3) δ 8.04 (dd, J = 5.5, 9.0 Hz, 1H), 7.95 (d, J = 8.0 Hz, 2H), 7.42–7.36 (m, 1H), 7.34–7.27 (m, 3H), 7.12 (s, 1H), 4.74 (bs, 1H), 2.43 (s, 3H), 1.59 (s, 9H); 13C NMR (125.97 MHz, CDCl3) δ 159.5 (d, J = 245.0 Hz), 157.3 (d, J = 2.5 Hz), 147.8 (d, J = 5.0 Hz), 145.8, 138.8, 138.0, 132.7 (d, J = 8.5 Hz), 129.4, 127.3, 118.9 (d, J = 8.0 Hz), 118.5 (d, J = 25.0 Hz), 103.3 (d, J = 23.0 Hz), 99.3, 51.5, 29.3, 21.3; HRMS (ESI) m/z calcd for C20H22N2F+ [M + H]+ 309.1757, found 309.1769.
N-(tert-Butyl)-6-chloro-8-fluoro-2-(p-tolyl)quinolin-4-amine (9e)
Synthesized in accordance with general procedure 1. Isolated as a yellow solid (0.253 g, 78%): Rf = 0.69 (cHex/EtOAc/Et3N 4:1:0.01); mp 166–167 °C; 1H NMR (500.23 MHz, CDCl3) δ 7.98 (dd, J = 8.0, 2.0 Hz, 1H), 7.42 (s, 1H), 7.34–7.28 (m, 3H), 7.17 (s, 1H), 4.86 (s, 0H), 2.42 (s, 3H), 1.59 (s, 9H); 13C NMR (125.97 MHz, CDCl3) δ 159.8, 157.8 (d, J = 18.5 Hz), 147.3 (d, J = 3.5 Hz), 138.4 (d, J = 235.5 Hz), 138.2 (d, J = 11.0 Hz), 129.4, 128.5 (d, J = 11.0 Hz), 127.37, 120.6 (d, J = 3.5 Hz), 114.7 (d, J = 23.0 Hz), 114.4 (d, J = 4.5 Hz), 100.1, 51.8, 29.3, 21.4; HRMS (ESI) m/z calcd for C20H21N2ClF+ [M + H]+ 343.1368, found 343.1376.
N-(tert-Butyl)-7-chloro-2-(3-chlorophenyl)quinolin-4-amine (9f)
Synthesized in accordance with general procedure 1. Isolated as a yellow solid (0.231 g, 67%): Rf = 0.58 (cHex/EtOAc/Et3N 4:1:0.01); mp 51–54 °C; 1H NMR (500.23 MHz, CDCl3) δ 8.08–7.98 (m, 2H), 7.89 (d, J = 6.5 Hz, 1H), 7.59 (d, J = 9.0 Hz, 1H), 7.47–7.39 (m, 2H), 7.34 (d, J = 9.0 Hz, 1H), 7.04 (s, 1H), 5.03 (bs, 1H), 1.59 (s, 9H); 13C NMR (125.97 MHz, CDCl3) δ 157.3, 149.5, 148.6, 142.5, 135.0, 134.7, 129.9, 129.3, 129.0, 127.6, 125.5, 125.2, 120.5, 117.0, 98.9, 51.7, 29.3; HRMS (ESI) m/z calcd for C19H19N2Cl2+ [M + H]+ 345.0917, found 345.0925.
N-(tert-Butyl)-2-(4-fluorophenyl)quinolin-4-amine (9g)
Synthesized in accordance with general procedure 1. Isolated as a yellow solid (0.252 g, 77%): Rf = 0.33 (cHex/EtOAc/Et3N 4:1:0.05); mp 138 °C; 1H NMR (500.23 MHz, CDCl3) δ 8.09–8.01 (m, 3H), 7.68 (d, J = 8.5 Hz, 1H), 7.67–7.60 (m, 1H), 7.44–7.37 (m, 1H), 7.19 (t, J = 8.5 Hz, 2H), 7.06 (s, 1H), 5.05 (bs, 1H), 1.60 (s, 9H); 13C NMR (125.97 MHz, CDCl3) δ 163.4 (d, J = 248.0 Hz), 156.8, 148.8, 148.4, 137.3 (d, J = 3.0 Hz), 130.4, 129.2 (d, J = 8.5 Hz), 129.1, 124.4, 118.9, 118.4, 115.5 (d, J = 21.5 Hz), 98.7; HRMS (ESI) m/z calcd for C19H20N2F+ [M + H]+ 295.1601, found 295.1601.
N-(tert-Butyl)-7-fluoro-2-(4-methoxyphenyl)quinolin-4-amine (9h)
Synthesized in accordance with general procedure 1. Isolated as a yellow solid (0.257 g, 79%): Rf = 0.38 (cHex/EtOAc/Et3N 4:1:0.01); mp 47–49 °C; 1H NMR (500.23 MHz, CDCl3) δ 8.11–7.99 (m, 2H), 7.78–7.57 (m, 2H), 7.13 (t, J = 8.5 Hz, 1H), 7.08–6.93 (m, 3H), 4.97 (bs, 1H), 3.88 (s, 3H), 1.59 (s, 9H); 13C NMR (125.97 MHz, CDCl3) δ 163.0 (d, J = 248.0 Hz), 160.5, 158.5, 150.3 (d, J = 12 Hz), 148.4, 133.3, 128.8, 121.1 (d, J = 10.0 Hz), 114.0, 113.7 (d, J = 19.5 Hz), 113.6 (d, J = 25.0 Hz), 98.3, 55.4, 51.6, 29.4); HRMS (ESI) m/z calcd for C20H22N2FO+ [M + H]+ 325.1706, found 325.1723.
N-(tert-Butyl)-7-methoxy-2-phenylquinolin-4-amine (9i)
Synthesized in accordance with general procedure 1. Isolated as a bright yellow solid (0.236 g, 77%): Rf = 0.29 (cHex/EtOAc/Et3N 4:1:0.01); mp 56–61 °C; 1H NMR (500.23 MHz, CDCl3) δ 8.06 (m, 2H), 7.57 (d, J = 9.0 Hz, 1H), 7.54–7.47 (m, 2H), 7.46–7.41 (m, 2H), 7.04 (dd, J = 2.5, 9.0 Hz, 1H), 7.02 s, 1H), 4.96 (bs, 1H), 3.94 (s, 3H), 1.58 (s, 9H); 13C NMR (125.97 MHz, CDCl3) δ 160.2, 158.4, 150.7, 148.5, 141.3, 128.7, 128.6, 127.4, 120.2, 116.7, 112.9, 108.6, 98.2, 55.4, 51.5, 29.4; HRMS (ESI) m/z calcd for C20H23N2O+ [M + H]+ 307.1800, found 307.1788.
N-(tert-Butyl)-2-(3,4,5-trimethoxyphenyl)quinolin-4-amine (9j)
Synthesized in accordance with general procedure 1. Isolated as a yellow solid (0.319 g, 87%): Rf = 0.10 (cHex/EtOAc/Et3N 4:1:0.01); mp 67–69 °C; 1H NMR (500.23 MHz, CDCl3) δ 8.06 (d, J = 8.5 Hz, 1H), 7.68 (d, J = 8.5 Hz, 1H), 7.63 (t, J = 7.5 Hz, 1H), 7.40 (t, J = 7.5 Hz, 1H), 7.26 (s, 2H), 7.03 (s, 1H), 5.06 (bs, 1H), 3.99 (s, 6H), 3.90 (s, 3H), 1.59 (s, 9H); 13C NMR (125.97 MHz, CDCl3) δ 157.8, 153.3, 184.6, 148.6, 148.4, 138.9, 130.3, 129.1, 124.4, 118.9, 118.4, 104.9, 99.1, 60.3, 56.2, 51.5, 29.4; HRMS (ESI) m/z calcd for C22H27N2O3+ [M + H]+ 367.2010, found 367.2026.
N-(tert-Butyl)-7-chloro-2-(3,4,5-trimethoxyphenyl)quinolin-4-amine (9k)
Synthesized in accordance with general procedure 1. Isolated as a yellow solid (0.328 g, 82%): Rf = 0.28 (cHex/EtOAc/Et3N 4:1:0.01); mp 68–70 °C; 1H NMR (500.23 MHz, CDCl3) δ 8.03 (s, 1H), 7.60 (dd, J = 2.0, 9.0 Hz, 1H), 7.33 (dt, J = 2.0, 9.0 Hz, 1H), 7.25 (d, J = 1.5 Hz, 2H), 7.02 (d, J = 1.5 Hz, 1H), 5.00 (bs, 1H), 3.98 (s, 6H), 3.91 (s, 3H), 1.59 (s, 9H); 13C NMR (125.97 MHz, CDCl3) δ 158.7, 153.3, 149.5, 148.4, 139.1, 136.6, 134.9, 129.2, 124.9, 120.5, 116.9, 104.8, 99.2, 60.9, 56.3, 51.7, 29.3; HRMS (ESI) m/z calcd for C22H26N2O3Cl+ [M + H]+ 401.1621, found 401.1634.
Methyl 4-(tert-Butylamino)-2-phenylquinoline-6-carboxylate (9l)
Synthesized in accordance with general procedure 1. Isolated as a yellow solid (0.147 g, 44%): Rf = 0.38 (cHex/EtOAc/Et3N 4:1:0.01); mp 131–134 °C; 1H NMR (500.23 MHz, CDCl3) δ 8.49 (s, 1H), 8.19 (dd, J = 2.0, 8.5 Hz, 1H), 8.11–8.02 (m, 3H), 7.56–7.49 (m, 2H), 7.49–7.45 (m, 1H), 7.15 (s, 1H), 5.27 (bs, 1H), 3.99 (s, 3H), 1.62 (s, 9H); 13C NMR (125.97 MHz, CDCl3) δ 167.0, 159.8, 151.3, 149.4, 140.7, 130.5, 129.3, 128.7, 128.6, 127.6, 125.2, 122.5, 117.7, 99.5, 52.3, 51.9, 29.3; HRMS (ESI) m/z calcd for C21H23N2O2+ [M + H]+ 335.1749, found 335.1759.
4-(4-(tert-Butylamino)quinolin-2-yl)benzonitrile (9m)
Synthesized in accordance with general procedure 1. Isolated as a yellow solid (0.090 g, 31%): Rf = 0.33 (cHex/EtOAc/Et3N 4:1:0.01); mp 79–81 °C; 1H NMR (500.23 MHz, CDCl3) δ 8.18 (d, J = 8.5 Hz, 2H), 8.04 (dd, J = 1.5, 8.5 Hz, 1H), 7.79 (d, J = 8.5 Hz, 2H), 7.70 (dd, J = 1.5, 8.5 Hz, 1H), 7.66 (ddd, J = 1.5, 7.0, 8.5 Hz, 1H), 7.45 (ddd, J = 1.5, 7.0, 8.5 Hz, 1H), 7.09 (s, 1H), 5.13 (bs, 1H), 1.61 (s, 9H); 13C NMR (125.97 MHz, CDCl3) δ 155.6, 148.7, 145.4, 132.5, 130.6, 129.4, 128.1, 125.1, 119.0, 118.9, 118.6, 112.2, 98.7, 51.7, 29.3; HRMS (ESI) m/z calcd for C20H20N3+ [M + H]+ 302.1648, found 302.1656.
N-(tert-Butyl)-2-butylquinolin-4-amine (9n)
Synthesized in accordance with general procedure 1. Isolated as a brown solid (0.129 g, 50%): Rf = 0.53 (EtOAc/Et3N 100:1); mp 68–70 °C; 1H NMR (500.23 MHz, CDCl3) δ 7.91 (d, J = 8.5 Hz, 1H), 7.62 (d, J = 8.5 Hz, 1H), 7.59–7.53 (m, 1H), 7.35 (ddd, J = 1.0, 7.0, 8.5 Hz, 1H), 6.57 (s, 1H), 4.91 (bs, 1H), 2.87–2.79 (m, 2H), 1.81–1.72 (m, 2H), 1.54 (s, 9H), 1.51–1.40 (m, 2H), 0.96 (t, J = 7.5 Hz, 3H); 13C NMR (125.97 MHz, CDCl3) δ 163.0, 148.4, 147.8, 129.5, 128.7, 123.7, 118.8, 118.1, 100.8, 51.4, 39.6, 32.4, 29.3, 22.7, 14.1; HRMS (ESI) m/z calcd for C17H25N2+ [M + H]+ 257.2007, found 257.2023.
N-(tert-Butyl)-2-decylquinolin-4-amine (9o)
Synthesized in accordance with general procedure 1. Isolated as a red-brown wax (0.255 g, 75%): Rf = 0.62 (EtOAc:Et3N 100:1); 1H NMR (500.23 MHz, CDCl3) δ 7.92 (d, J = 8.5 Hz, 1H), 7.62 (d, J = 8.5 Hz, 1H), 7.56 (ddd, J = 1.5, 7.0, 8.5 Hz, 1H), 7.34 (t, J = 1.5, 7.5, 7.0 Hz, 1H), 6.57 (s, 1H), 4.91 (bs, 1H), 2.82 (t, J = 8.0 Hz, 2H), 1.78 (p, J = 8.0 Hz, 2H), 1.53 (s, 9H), 1.46–1.38 (m, 2H), 1.38–1.32 (m, 2H), 1.31–1.21 (m, 10H), 0.87 (t, J = 7.0 Hz, 3H); 13C NMR (125.97 MHz, CDCl3) δ 163.0, 148.4, 147.8, 129.5, 128.6, 123.7, 118.8, 118.1, 100.8, 51.3, 39.9, 31.9, 30.2, 29.62, 29.58, 29.56, 29.3, 29.2, 22.6, 14.1; HRMS (ESI) m/z calcd for C23H37N2+ [M + H]+ 341.2943, found 341.2965.
N-Isopropyl-2-phenylquinolin-4-amine (9p)
Synthesized in accordance with general procedure 2. Isolated as a red-brown solid (0.216 g, 81%): Rf = 0.16 (cHex/EtOAc/Et3N 4:1:0.05); mp 176 °C; 1H NMR (500.23 MHz, CDCl3) δ 8.12 (m, 3H), 7.72 (d, J = 8.5 Hz, 1H), 7.68–7.61 (m, 1H), 7.51 (t, J = 7.5 Hz, 2H), 7.48–7.38 (m, 2H), 6.89 (s, 1H), 4.87 (bd, J = 7.0 Hz, 1H), 4.04–3.92 (m, 1H), 1.40 (dd, J = 1.5, 6.5 Hz, 6H); 13C NMR (125.97 MHz, CDCl3) δ 158.4, 149.0, 148.7, 141.1, 130.3, 129.1, 128.7, 128.5, 127.5, 124.2, 119.0, 117.8, 97.0, 43.9, 22.5; HRMS (ESI) m/z calcd for C18H19N2+ [M + H]+ 263.1539, found 263.1556.
N-Cyclohexyl-6-fluoro-2-phenylquinolin-4-amine (9q)
Synthesized in accordance with general procedure 2. Isolated as a yellow solid (0.229 g, 80%): Rf = 0.50 (cHex/EtOAc/Et3N 4:1:0.01); mp 146–149 °C; 1H NMR (500.23 MHz, CDCl3) δ 8.09–8.06 (m, 1H), 8.04 (d, J = 8.0 Hz, 2H), 7.51 (d, J = 7.0 Hz, 2H), 7.47–7.37 (m, 2H), 7.34 (dd, J = 2.0, 10.0 Hz, 1H), 6.86 (s, 1H), 4.71 (bd, J = 7.5 Hz, 1H), 3.66–3.56 (m, 1H), 2.20 (dd, J = 4.0, 13.0 Hz, 2H), 1.89–1.81 (m, 2H), 1.76–1.70 (m, 1H), 1.53–1.44 (m, 2H), 1.42–1.30 (m, 3H); 13C NMR (125.97 MHz, CDCl3) δ 159.5 (d, J = 245.0), 157.9 (d, J = 2.5 Hz), 148.6 (d, J = 5.0 Hz), 145.7, 140.8, 132.5 (d, J = 8.5 Hz), 128.9, 128.6, 127.5, 118.8 (d, J = 25.0 Hz), 118.2 (d, J = 8.0 Hz), 103.4 (d, J = 23.0 Hz), 97.3, 51.3, 32.8, 25.7, 24.8; HRMS (ESI) m/z calcd for C21H22N2F+ [M + H]+ 321.1757, found 321.1776.
7-Chloro-N-isopropyl-2-phenylquinolin-4-amine (9r)
Synthesized in accordance with general procedure 2. Isolated as a yellow solid (0.175 g, 60%): Rf = 0.47 (cHex/EtOAc/Et3N 4:1:0.01); mp 147–148 °C; 1H NMR (500.23 MHz, CDCl3) δ 8.11–8.00 (m, 3H), 7.65 (d, J = 9.0 Hz, 1H), 7.53–7.48 (m, 2H), 7.47–7.43 (m, 1H), 7.34 (dd, J = 2.0, 9.0 Hz, 1H), 6.85 (s, 1H), 4.88 (bs, 1H), 4.04–3.91 (m, 1H), 1.40 (d, J = 6.5 Hz, 6H); 13C NMR (125.97 MHz, CDCl3) δ 159.3, 149.4, 149.2, 140.5, 135.0, 129.1, 129.0, 128.6, 127.5, 124.9, 120.6, 116.2, 97.2, 44.1, 22.5; HRMS (ESI) m/z calcd for C18H18N2Cl+ [M + H]+ 297.1150, found 297.1163.
N-Pentyl-2-phenylquinolin-4-amine (9s)
Synthesized in accordance with general procedure 2. Isolated as a red-brown wax (0.131 g, 48%): Rf = 0.35 (cHex/EtOAC/Et3N 4:1:0.01); 1H NMR (500.23 MHz, CDCl3) δ 8.14–8.06 (m, 3H), 7.72 (d, J = 8.5 Hz, 1H), 7.65 (t, J = 7.5 Hz, 1H), 7.51 (t, J = 7.5 Hz, 2H), 7.47–7.38 (m, 2H), 6.86 (s, 1H), 5.00 (bt, J = 5.0 Hz, 1H), 3.36 (q, J = 6.5 Hz, 2H), 1.79 (p, J = 7.5 Hz, 2H), 1.53–1.37 (m, 4H), 0.96 (t, J = 7.0 Hz, 3H); 13C NMR (125.97 MHz, CDCl3) δ 158.4, 150.1, 148.6, 141.0, 130.2, 129.1, 128.8, 128.5, 127.5, 124.3, 119.0, 117.8, 96.6, 43.2, 29.3 28.6, 22.4, 14.0; HRMS (ESI) m/z calcd for C20H23N2+ [M + H]+ 291.1851, found 291.1846.
N-Isopropyl-2-(4-(trifluoromethyl)phenyl)quinolin-4-amine (9t)
Synthesized in accordance with general procedure 2. Isolated as a yellow solid (0.074 g, 24%): Rf = 0.39 (cHex/EtOAc/Et3N 4:1:0.01); mp 60–63 °C; 1H NMR (500.23 MHz, CDCl3) δ 8.18 (d, J = 8.0 Hz, 2H), 8.07 (d, J = 8.5 Hz, 1H), 7.79–7.70 (m, 3H), 7.67 (t, J = 7.5 Hz, 1H), 7.44 (t, J = 7.5 Hz, 1H), 6.86 (s, 1H), 4.93 (bd, J = 7.0 Hz, 1H), 4.05–3.96 (m, 1H), 1.41 (d, J = 6.5 Hz, 6H); 13C NMR (125.97 MHz, CDCl3) δ 156.8, 149.3, 148.7, 144.4, 130.6 (q, J = 32.5 Hz), 130.4, 129.4, 127.8, 125.5 (q, J = 4.0 Hz), 124.8, 124.3 (q, J = 271.5 Hz), 119.0, 117.9, 96.8, 40.0, 22.5; HRMS (ESI) m/z calcd for C19H18N2F3+ [M + H]+ 331.1413, found 331.1429.
Methyl 4-(Cyclohexylamino)-2-(3,4,5-trimethoxyphenyl)quinoline-6-carboxylate (9u)
Synthesized in accordance with general procedure 2. Isolated as a dark yellow solid (0.167 g, 39%): Rf = 0.13 (cHex/EtOAc/Et3N 4:1:0.01); mp 141–143 °C; 1H NMR (500.23 MHz, CDCl3) δ 8.52 (d, J = 2.0 Hz, 1H), 8.20 (dd, J = 2.0, 9.0 Hz, 1H), 8.03 (d, J = 9.0 Hz, 1H), 7.27 (s, 2H), 6.80 (s, 1H), 5.19 (bd, J = 7.0 Hz), 4.11–3.95 (m, 9H), 3.91 (s, 3H), 3.63 (dtt, J = 3.5, 7.0, 10.0 Hz, 1H), 2.24–2.17 (m, 2H), 1.97–1.82 (m, 2H), 1.81–1.68 (m, 2H), 1.52–1.37 (m, 4H); 13C NMR (125.97 MHz, CDCl3) δ 167.0, 160.3, 153.3, 151.1, 149.9, 139.3, 136.5, 130.2, 128.8, 125.1, 122.7, 117.0, 105.1, 97.1, 60.9, 56.3, 52.3, 51.5, 32.7, 25.6, 24.9; HRMS (ESI) m/z calcd for C26H31N2O5+ [M + H]+ 451.2220, found 451.2236.
N1,N1-Diethyl-N4-(2-phenylquinolin-4-yl)pentane-1,4-diamine (9w)
Synthesized in accordance with general procedure 2. Isolated as a red-brown wax (0.287 g, 79%): Rf = 0.09 (EtOAc/Et3N 100:1); 1H NMR (500.23 MHz, CDCl3) δ 8.09–8.02 (m, 3H), 7.76 (d, J = 8.0 Hz, 1H), 7.67–7.60 (m, 1H), 7.50 (t, J = 7.5 Hz, 2H), 7.47–7.37 (m, 2H), 6.86 (s, 1H), 5.18 (bd, J = 7.5 Hz, 1H), 3.90–3.80 (m, 1H), 2.57 (q, J = 7.0 Hz, 4H), 2.50 (t, J = 7.0 Hz, 2H), 1.85–1.75 (m, 1H), 1.73–1.63 (m, 2H), 1.37 (d, J = 6.5 H, 3H), 1.03 (t, J = 7.0 Hz, 6H); 13C NMR (125.97 MHz, CDCl3) δ 158.5, 149.3, 148.8, 141.2, 130.3, 129.1, 128.7, 128.6, 127.5, 124.2, 119.2, 117.9, 96.9, 52.6, 48.2, 46.8, 34.7, 23.8, 20.4, 11.4; HRMS (ESI) m/z calcd for C24H32N3+ [M + H]+ 362.2584, found 362.2595.
N4-(3-(Benzo[d][1,3]dioxol-5-yl)-6-chloronaphthalen-1-yl)-N1,N1-diethylpentane-1,4-diamine (9x)
Synthesized in accordance with general procedure 2. Isolated as a yellow-brown solid (0.327 g, 78%): Rf = 0.15 (EtOAc/Et3N 99:1); mp 48–50 °C; 1H NMR (500.23 MHz, CDCl3) δ 7.98 (d, J = 2.0 Hz, 1H), 7.63 (d, J = 9.0 Hz, 1H), 7.60 (d, J = 2.0 Hz, 1H), 7.56 (d, J = 8.0 Hz, 1H), 7.29 (d, J = 9.0 Hz, 1H), 6.91 (d, J = 8.0 Hz, 1H), 6.76 (s, 1H), 6.01 (s, 2H), 5.25 (bd, J = 7.0 Hz, 1H), 3.79 (hept, J = 6.5 Hz, 1H), 2.53 (q, J = 7.0 Hz, 4H), 2.46 (t, J = 7.0 Hz, 2H), 1.81–1.65 (m, 2H), 1.65–1.59 (m, 2H), 1.34 (d, J = 6.5 Hz, 3H), 1.00 (t, J = 7.0 Hz, 6H); 13C NMR (125.97 MHz, CDCl3) δ 158.6, 149.5, 149.3, 148.5, 148.0, 135.1, 134.8, 128.9, 124.4, 121.5, 120.9, 116.3, 108.2, 107.9, 101.2, 96.5, 52.4, 48.2, 46.7, 34.5, 23.8, 20.2, 11.3; HRMS (ESI) m/z calcd for C25H31N3O2Cl+ [M + H]+ 440.2093, found 440.2092.
N4-(7-Chloro-2-phenylquinolin-4-yl)-N1,N1-diethylpentane-1,4-diamine (9y)
Synthesized in accordance with general procedure 2. Isolated as a red-brown wax (0.326 g, 82%): Rf = 0.14 (EtOAc/Et3N 99:1); 1H NMR (500.23 MHz, CDCl3) δ 8.08–8.02 (m, 3H), 7.67 (d, J = 9.0 Hz, 1H), 7.50 (t, J = 7.0 Hz, 2H), 7.48–7.40 (m, 1H), 7.33 (dd, J = 2.0, 9.0 Hz, 1H), 6.85 (s, 1H), 5.29 (bd, J = 7.0 Hz, 1H), 3.82 (hept, J = 6.5, 1H), 2.54 (q, J = 7.0 Hz, 4H), 2.47 (t, J = 7.0 Hz, 2H), 1.83–1.74 (m, 1H), 1.74–1.60 (m, 3H), 1.36 (d, J = 6.5 Hz, 3H), 1.01 (t, J = 7.0 Hz, 6H); 13C NMR (125.97 MHz, CDCl3) δ 159.4, 149.6, 149.5, 140.7, 134.9, 129.1, 129.0, 128.6, 127.5, 124.7, 120.9, 116.4, 97.1, 52.5, 48.3, 46.8, 34.6, 23.8, 20.3, 11.3; HRMS (ESI) m/z calcd for C24H31N3Cl+ [M + H]+ 396.2195, found 396.2213.
N4-(2-Cyclohexyl-7-fluoroquinolin-4-yl)-N1,N1-diethylpentane-1,4-diamine (9z)
Synthesized in accordance with general procedure 2. Isolated as a red-brown wax (0.268 g, 70%): Rf = 0.14 (EtOAc/Et3N 99:1); 1H NMR (500.23 MHz, CDCl3) δ 7.71–7.64 (m, 1H), 7.53 (dt, J = 2.0, 10.5 Hz, 1H), 7.06 (t, J = 8.5 Hz, 1H), 6.28 (d, J = 2.0 Hz, 1H), 5.16 (bd, J = 7.0 Hz, 1H), 3.72 (p, J = 6.5 Hz, 1H), 2.72 (td, J = 3.0, 12.0 Hz, 1H), 2.52 (q, J = 7.0 Hz, 4H), 2.45 (t, J = 7.0 Hz, 2H), 2.01–1.94 (m, 2H), 1.87–1.81 (m, 2H), 1.77–1.69 (m, 2H), 1.65–1.50 (m, 5H), 1.42 (q, J = 11.5 Hz, 2H), 1.34–1.24 (m, 1H), 1.30 (d, J = 6.5 Hz, 3H), 1.00 (td, J = 2.0, 7.0, 6H); 13C NMR (125.97 MHz, CDCl3) δ 168.7, 162.9 (d, J = 247.5 Hz), 149.6 (d, J = 12.5 Hz), 149.3, 121.5 (d, J = 10.5 Hz), 114.8, 113.1 (d, J = 25.0 Hz), 112.8 (d, J = 20.0 Hz), 96.3, 52.5, 48.0, 47.9, 46.7, 34.5, 32.89, 32.86, 26.5, 26.0, 23.7, 20.2, 11.2; HRMS (ESI) m/z calcd for C24H37N3F+ [M + H]+ 386.2958, found 386.2970.
N-(6-Methylheptan-2-yl)-2-(naphthalen-2-yl)quinolin-4-amine (9aa)
Synthesized in accordance with general procedure 2. Isolated as a red-brown solid (0.294 g, 77%): Rf = 0.26 (cHex/EtOAc/Et3N 4:1:0.01); mp 128–129 °C; 1H NMR (500.23 MHz, CDCl3) δ 8.57–8.53 (m, 1H), 8.26 (dd, J = 2.8, 8.5 Hz, 1H), 8.13 (d, J = 8.5 Hz, 1H), 8.01–7.98 m, 1H), 7.98 (d, J = 9.0 Hz, 1H), 7.92–7.87 (m, 1H), 7.76–7.72 (m, 1H), 7.67 (ddd, J = 1.5, 7.0, 8.5 Hz, 1H), 7.54–7.49 (m, 2H), 7.44 (ddd, J = 1.5, 7.0, 8.5 Hz, 1M), 7.01 (s, 1H), 4.90 (bd, J = 7.5 Hz, 1H), 3.89 (hept, J = 6.5 Hz, 1H), 1.81–1.73 (m, 1H), 1.70–1.53 (m, 3H), 1.53–1.44 (m, 2H), 1.39 (d, J = 6.5 Hz, 3H), 1.30–1.24 (m, 2H), 0.90 (d, J = 1.5 Hz, 3H), 0.89 (d, J = 1.5 Hz, 3H); 13C NMR (125.97 MHz, CDCl3) δ 158.2, 149.4, 148.8, 138.3, 133.7, 133.4, 130.3, 129.3, 128.7, 128.2, 127.7, 126.9, 126.4, 126.1, 125.4, 124.4, 119.0, 117.9, 97.0, 48.3, 38.9, 37.1, 27.9, 24.0, 22.60, 22.58, 20.4; HRMS (ESI) m/z calcd for C27H31N2+ [M + H]+ 383.2475, found 383.2490.
Ethyl 4-((7-Chloro-2-phenylquinolin-4-yl)amino)piperi-dine-1-carboxylate (9ab)
Synthesized in accordance with general procedure 2. Isolated as a yellow-orange solid (0.277 g, 68%): Rf = 0.11 (cHex/EtOAc/Et3N 4:1:0.01); mp 186–187 °C; 1H NMR (500.23 MHz, CDCl3) δ 8.07 (d, J = 2.0 Hz, 1H), 8.05–8.03 (m, 1H), 8.03–8.01 (m, 1H), 7.65 d, J = 9.0 Hz, 1H), 7.68–7.62 (m, 2H), 7.50–7.42 (m, 1H), 7.35 (dd, J = 2.0, 9.0 Hz, 1H), 6.85 (s, 1H), 4.95 (bs, 1H), 4.32–4.25 m, 2H), 4.17 (q, J = 7.0 Hz, 2H), 3.80 (tdt, J = 4.0, 7.5, 10.5 Hz, 1H), 3.08 (t, J = 12.5 Hz, 2H), 2.28–2.14 (m, 2H), 1.64–1.48 (m, 2H), 1.29 (t, J = 7.0 Hz, 3H); 13C NMR (125.97 MHz, CDCl3): 159.3, 155.4, 148.7, 140.3, 135.3, 129.3, 129.1,128.7, 127.6, 125.2, 120.6, 116.2, 97.2, 61.6, 49.7, 42.6, 31.7, 14.7; HRMS (ESI) m/z calcd for C23H25N3O2Cl+ [M + H]+ 410.1625, found 410.1630.
N,N-Diethyl-4-isocyanopentan-1-amine (7w)
A solution of N1,N1-diethylpentane-1,4-diamine (3.87 mL, 20 mmol) in ethyl formate (4.82 mL, 60 mmol) was refluxed for 16 h, after which the solvent was removed in vacuo. To a solution of the crude product (3.00 g, 16.1 mmol) in dry CH2Cl2 (80 mL) was added DIPEA (7.01 mL, 40.25 mmol) and cooled to −40 °C. Triphosgene (1.67 g, 5.64 mmol) was added portionwise, after which the mixture was allowed to warm to 0 °C and stirred for 2 h. The crude product was poured in water (80 mL), and the organic layer was dried over Na2SO4, filtered, and concentrated in vacuo to afford the title compound as a pale yellow liquid (2.370 g, 88%): Rf = 0.11 (EtOAc/MeOH/Et3N 9:1:0.09); 1H NMR (500.23 MHz, CDCl3) δ 3.71–3.59 (m, 1H), 2.52 (q, J = 7.0 Hz, 4H), 2.44 (t, J = 7.0 Hz, 2H), 1.70–1.52 (m, 4H), 1.37 (dt, JHN = 2.0 Hz, JHH = 7.0 Hz, 3H), 1.02 (t, J = 7.0 Hz, 3H); 13C (125.97 MHz, CDCl3) δ 154.2 (t, JC(sp)N = 5.0 Hz), 52.0, 50.3 (t, JC(sp3)N = 5.5 Hz), 46.8, 34.8, 23.3, 21.8, 11.6; HRMS (ESI) m/z calcd for C10H21N2+ [M + H]+ 169.1695, found 169.1705.
Ethyl 4-Isocyanopiperidine-1-carboxylate (7ab)
Ethyl 4-aminopiperidine-1-carboxylate (4.03 mL, 29.0 mmol) was refluxed in ethyl formate (7.01 mL, 87.1 mmol) for 16 h, after which the solvent was removed in vacuo. To a solution of the crude formamide (3.10 g, 15.5 mmol) in dry CH2Cl2 (45 mL) was added Et3N (4.32 mL, 31 mmol). The solution was cooled to −40 °C, after which triphosgene (1.61 g, 5.52 mmol) was added portionwise. The resulting mixture was slowly warmed to room temperature and stirred for 3 h. Hereafter, the reaction was quenched with ice–water (50 mL), and the resulting biphasic system was separated. The organic layer was washed with 1 M HCl, saturated NaHCO3, and water, dried over Na2SO4, filtered, and concentrated in vacuo, affording the target isocyanide as a pale yellow oil (2.471 g, 88%): Rf = 0.16 (cHex/EtOAc/Et3N 4:1:0.01); 1H NMR (500.23 MHz, CDCl3) δ 4.12 (q, J = 7.0 Hz, 2H), 3.89–3.81 (m, 1H), 3.60 (ddd, J = 3.5, 8.0, 14.0 Hz, 2H), 3.54–3.44 (m, 1H), 1.93–1.83 (m, 2H), 1.83–1.75 (m, 2H), 1.25 (t, J = 7.0 Hz, 3H); 13C (125.97 MHz, CDCl3) δ 156.2 (t, J = 5.0 Hz), 155.2, 61.6, 49.3 (t, J = 6.0 Hz), 40.0, 31.2, 14.6; HRMS (ESI) m/z calcd for C9H14N2O2Na+ [M + Na]+ 205.0944, found 205.0955.
Acknowledgments
We thank Elwin Janssen for NMR support and maintenance. The research leading to these findings has received financial support from The Netherlands Organization for Scientific Research (NWO).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.joc.7b02844.
1H and 13C NMR spectra for all new compounds (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Fidock D. A. Nature 2010, 465, 297. 10.1038/465297a. [DOI] [PubMed] [Google Scholar]
- Ridley R. G. Nature 2002, 415, 686. 10.1038/415686a. [DOI] [PubMed] [Google Scholar]
- Wellems T. E. Nature 1992, 355, 108. 10.1038/355108a0. [DOI] [PubMed] [Google Scholar]
- Gregson A.; Plowe C. V. Pharmacol. Rev. 2005, 57, 117. 10.1124/pr.57.1.4. [DOI] [PubMed] [Google Scholar]
- Goulopoulou S.; McCarthy C. G.; Webb R. C. Pharmacol. Rev. 2016, 68, 142. 10.1124/pr.114.010090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madrid P. B.; Sherrill J.; Liou A. P.; Weisman J. L.; DeRisi J. L.; Guy R. K. Bioorg. Med. Chem. Lett. 2005, 15, 1015. 10.1016/j.bmcl.2004.12.037. [DOI] [PubMed] [Google Scholar]
- Miller L. H.; Ackerman H. C.; Su X.-z.; Wellems T. E. Nat. Med. 2013, 19, 156. 10.1038/nm.3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gignoux E.; Azman A. S.; de Smet M.; Azuma P.; Massaquoi M.; Job D.; Tiffany A.; Petrucci R.; Sterk E.; Potet J.; Suzuki M.; Kurth A.; Cannas A.; Bocquin A.; Strecker T.; Logue C.; Pottage T.; Yue C.; Cabrol J.-C.; Serafini M.; Ciglenecki I. N. Engl. J. Med. 2016, 374, 23. 10.1056/NEJMoa1504605. [DOI] [PubMed] [Google Scholar]
- Madrid P. B.; Chopra S.; Manger I. D.; Gilfillan L.; Keepers T. R.; Shurtleff A. C.; Green C. E.; Iyer L. V.; Dilks H. H.; Davey R. A.; Kolokoltsov A. A.; Carrion R. Jr.; Patterson J. L.; Bavari S.; Panchal R. G.; Warren T. K.; Wells J. B.; Moos W. H.; Burke R. L.; Tanga M. J. PLoS One 2013, 8, e60579. 10.1371/journal.pone.0060579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kužnik A.; Benčina M.; Švajger U.; Jeras M.; Rozman B.; Jerala R. J. Immunol. 2011, 186, 4794. 10.4049/jimmunol.1000702. [DOI] [PubMed] [Google Scholar]
- Tewari S.; Chauhan P. M. S.; Bhaduri A. P.; Fatima N.; Chatterjee R. K. Bioorg. Med. Chem. Lett. 2000, 10, 1409. 10.1016/S0960-894X(00)00255-9. [DOI] [PubMed] [Google Scholar]
- Brouwer C.; Jenko K.; Zoghbi S. S.; Innis R. B.; Pike V. W. J. Med. Chem. 2014, 57, 6240. 10.1021/jm5007947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh S.; Roy K. K.; Khan S. R.; Kashyap V. K.; Sharma A.; Jaiswal S.; Sharma S. K.; Krishnan M. Y.; Chaturvedi V.; Lal J.; Sinha S.; Dasgupta A.; Srivastava R.; Saxena A. K. Bioorg. Med. Chem. 2015, 23, 742. 10.1016/j.bmc.2014.12.060. [DOI] [PubMed] [Google Scholar]
- Shinkai H.; Ito T.; Iida T.; Kitao Y.; Yamada H.; Uchida I. J. Med. Chem. 2000, 43, 4667. 10.1021/jm0002073. [DOI] [PubMed] [Google Scholar]
- Sestili I.; Borioni A.; Mustazza C.; Rodomonte A.; Turchetto L.; Sbraccia M.; Riitano D.; Del Giudice M. R. Eur. J. Med. Chem. 2004, 39, 1047. 10.1016/j.ejmech.2004.09.009. [DOI] [PubMed] [Google Scholar]
- Kireev D. B.; Chrétien J. R.; Raevsky O. A. Eur. J. Med. Chem. 1995, 30, 395. 10.1016/0223-5234(96)88249-3. [DOI] [Google Scholar]
- Oh K. H.; Kim J. G.; Park J. K. Org. Lett. 2017, 19, 3994. 10.1021/acs.orglett.7b01701. [DOI] [PubMed] [Google Scholar]
- Wezeman T.; Zhong S.; Nieger M.; Bräse S. Angew. Chem., Int. Ed. 2016, 55, 3823. 10.1002/anie.201511385. [DOI] [PubMed] [Google Scholar]
- Li Y.; Zhang L.; Zhang L.; Wu Y.; Gong Y. Org. Biomol. Chem. 2013, 11, 7267. 10.1039/c3ob41658e. [DOI] [PubMed] [Google Scholar]
- Dhanapal R.; Perumal P. T.; Damodiran M.; Ramprasath C.; Mathivanan N. Bioorg. Med. Chem. Lett. 2012, 22, 6494. 10.1016/j.bmcl.2012.08.039. [DOI] [PubMed] [Google Scholar]
- Abbiati G.; Arcadi A.; Canevari V.; Capezzuto L.; Rossi E. J. Org. Chem. 2005, 70, 6454. 10.1021/jo050882q. [DOI] [PubMed] [Google Scholar]
- Vlaar T.; Maes B. U. W.; Ruijter E.; Orru R. V. A. Chem. Heterocycl. Compd. 2013, 49, 902. 10.1007/s10593-013-1324-z. [DOI] [Google Scholar]
- Tsai J.-Y.; Chang C.-S.; Huang Y.-F.; Chen H.-S.; Lin S.-K.; Wong F. F.; Huang L.-J.; Kuo S.-C. Tetrahedron 2008, 64, 11751. 10.1016/j.tet.2008.09.100. [DOI] [Google Scholar]
- Ren J.; Zhao J.; Zhou Y.-S.; Liu X.-H.; Chen X.; Hu K. Med. Chem. Res. 2013, 22, 2855. 10.1007/s00044-012-0283-8. [DOI] [Google Scholar]
- Tollari S.; Cenini S.; Ragaini F.; Cassar L. J. Chem. Soc., Chem. Commun. 1994, 15, 1741. 10.1039/c39940001741. [DOI] [Google Scholar]
- Li L.; Wang H.-K.; Kuo S.-C.; Wu T.-S.; Lednicer D.; Lin C. M.; Hamel E.; Lee K.-H. J. Med. Chem. 1994, 37, 1126. 10.1021/jm00034a010. [DOI] [PubMed] [Google Scholar]
- Sun F.; Zhao X.; Shi D. Tetrahedron Lett. 2011, 52, 5633. 10.1016/j.tetlet.2011.08.089. [DOI] [Google Scholar]
- Carta D.; Bortolozzi R.; Hamel E.; Basso G.; Moro S.; Viola G.; Ferlin M. G. J. Med. Chem. 2015, 58, 7991. 10.1021/acs.jmedchem.5b00805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strekowski L.; Say M.; Henary M.; Ruiz P.; Manzel L.; Macfarlane D. E.; Bojarski A. J. J. Med. Chem. 2003, 46, 1242. 10.1021/jm020374y. [DOI] [PubMed] [Google Scholar]
- Torii S.; Okumoto H.; Xu L. H. Tetrahedron Lett. 1991, 32, 237. 10.1016/0040-4039(91)80864-3. [DOI] [Google Scholar]
- Torii S.; Okumoto H.; Xu L. H.; Sadakane M.; Shostakovsky M. V.; Ponomaryov A. B.; Kalinin V. N. Tetrahedron 1993, 49, 6773. 10.1016/S0040-4020(01)80421-X. [DOI] [Google Scholar]
- Åkerbladh L.; Nordeman P.; Wejdemar M.; Odell L. R.; Larhed M. J. Org. Chem. 2015, 80, 1464. 10.1021/jo502400h. [DOI] [PubMed] [Google Scholar]
- Tang T.; Fei X.-D.; Ge Z.-Y.; Chen Z.; Zhu Y.-M.; Ji S.-J. J. Org. Chem. 2013, 78, 3170. 10.1021/jo4001096. [DOI] [PubMed] [Google Scholar]
- Kosugi M.; Ogata T.; Tamura H.; Sano H.; Migita T. Chem. Lett. 1986, 15, 1197. 10.1246/cl.1986.1197. [DOI] [Google Scholar]
- Pan Y.-Y.; Wu Y.-N.; Chen Z.-Z.; Hao W.-J.; Li G.; Tu S.-J.; Jiang B. J. Org. Chem. 2015, 80, 5764. 10.1021/acs.joc.5b00727. [DOI] [PubMed] [Google Scholar]
- Whitby R. J.; Saluste C. G.; Furber M. Org. Biomol. Chem. 2004, 2, 1974. 10.1039/B408673M. [DOI] [PubMed] [Google Scholar]
- Vlaar T.; Cioc R. C.; Mampuys P.; Maes B. U. W.; Orru R. V. A.; Ruijter E. Angew. Chem., Int. Ed. 2012, 51, 13058. 10.1002/anie.201207410. [DOI] [PubMed] [Google Scholar]
- Craciunescu D. G.; Doadrio A.; Furlani A.; Scarcia V. Chem.-Biol. Interact. 1982, 42, 153. 10.1016/0009-2797(82)90129-6. [DOI] [PubMed] [Google Scholar]
- Manzel L.; Strekowski L.; Ismail F. M. D.; Smith J. C.; Macfarlane D. E. J. Pharmacol. Exp. Ther. 1999, 291, 1337. [PubMed] [Google Scholar]
- Strekowski L.; Zegrocka O.; Henary M.; Say M.; Mokrosz M. J.; Kotecka B. M.; Manzel L.; Macfarlane D. E. Bioorg. Med. Chem. Lett. 1999, 9, 1819. 10.1016/S0960-894X(99)00291-7. [DOI] [PubMed] [Google Scholar]
- Herrera L.; Stephens D. E.; D’Avila A.; George K. G.; Arman H.; Zhang Y.; Perry G.; Lleonart R.; Larionov O. V.; Fernandez P. L. Org. Biomol. Chem. 2016, 14, 7053. 10.1039/C6OB01149G. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.